
The Role of Astrocytes in the Molecular Pathophysiology of Schizophrenia: Between Neurodevelopment and Neurodegeneration

 , , ,
, , ,  and
and 
Abstract
1. Introduction
- 1.
- Could astrocytes represent a point of convergence between neurodevelopment and neurodegeneration in schizophrenia due to aberrant programming during early development?
- 2.
- What are the molecular mechanisms at the tripartite synapse that may influence the schizophrenia pathophysiology?
- 3.
- Could astrocytes serve as a potential therapeutic target for schizophrenia?
2. Search Strategy for the Comprehensive Review
3. Astrocyte–Neuron Crosstalk in Neurodevelopmental and Neurodegenerative Processes: Inference for Schizophrenia
3.1. The Role of Astrocytes in Schizophrenia: Molecular Basis and Clinical Evidence

{kind=link}
{kind=link}
| Genetic or Pharmacological Animal Model | Species | Main Molecular or Behavioral Findings | Implication for Schizophrenia | References |
|---|---|---|---|---|
| DISC1 KD | Mice | Reduced astrocyte density and altered levels of GLAST that result in glutamate/GABA imbalance | Excitatory/inhibitory imbalance | [58] |
| GLT-1 KO | Mice | Increased excitotoxicity and schizophrenia-like behavior | Glutamate dysregulation | [63] |
| MIA model | Rat | Astrocyte activation with increased IL-6 and IL-1β; long-term changes in synaptic pruning | Neuroinflammation relevant for schizophrenia vulnerability | [64] |
| EAAT2 blockade, via dihydrokainic acid | Rat | Anhedonia and impaired spatial memory | Dysfunctions that model schizophrenia’s negative and cognitive symptoms | [65] |
| Inducible expression of mutant DISC1 (ΔC-hDISC1) in astrocytes | Mice | Reduction in D-serine levels in the mice brains | Defects in astrocyte maturation related to NMDAR hypofunction during neurodevelopment | [53] |
| FABP7 KO | Mice | Reduction in number of synapses in mPFC; reduction in both amplitude and frequency of mEPSCs | Impaired astrocyte lipid signaling affects postnatal cognition; hyperactivity and anxiety-related phenotypes | [66,67] |
3.2. Astrocytes and Neurodevelopment: Focus on Molecular Mechanism in Schizophrenia
3.2.1. Astrocytes as Regulators of Synaptic Formation and Neurotransmission: Relevance for Schizophrenia Pathophysiology
| Domain | Astrocyte Role | Molecular Mechanisms | Functional Effect | References |
|---|---|---|---|---|
| Neurodevelopment | Regulation of synaptogenesis and dendritic development | BDNF/TrkB—TGF-β/TBR1/2 | Synaptogenesis | [66,90,101,102] |
| NLG | Synaptic pruning | |||
| FABP7 | Dendritic maturation | |||
| Control of BBB development and integrity | AQP4 | BBB development | [103,104,105] | |
| Claudin-5 and ICAM-1 via Shh pathway | BBB integrity | |||
| Neurotransmission | Glutamate | EAAT1/2 | Extracellular glutamate level regulation and prevention of excitotoxicity | [94,95,96] |
| NMDAR | ||||
| Glutamine synthetase | ||||
| GABA | A2aR activation through the gephyrin pathway | Improvement of GABAergic stability | [106] | |
| Dopamine | Ca2⁺ signaling | Dopamine vesicle transport and reuptake in glial cells | [107,108] | |
| VMAT2 and OCT3 | ||||
| Neurodegeneration | Metabolic and ionic homeostasis | Kir1.4 and Na⁺/K⁺ ATPase | Stabilization in ionic gradients | [109,110] |
| Oxidative stress and neuroinflammation | IL-1β, TNF-α, NF-κB, NLRP3, COX-2 | Driving inflammation | [111,112,113] | |
| ROS and STAT3 | Stress response regulation |
3.2.2. Disruptions in Astrocytic Function and Implications for Schizophrenia Pathophysiology
3.2.3. Astrocyte–Neuron Interactions: The Tripartite Synapse
3.2.4. Environmental and Genetic Factors Influencing Astrocyte Function
3.3. The Role of Astrocytes in Neurodegeneration: The Focus on Molecular Mechanisms in Schizophrenia
Astrocyte–Microglia Crosstalk in Schizophrenia: Relevance for Neuroinflammation in Neurodegeneration Processes
3.4. From the Tripartite to Tetrapartite Synapse Conceptualization: Insights into the Neurobiology of Schizophrenia
3.4.1. Postsynaptic Density Structure and Schizophrenia
3.4.2. Regulation of Synaptic Plasticity: Pre- and Postsynaptic Terminals, Glial Cells, and Extracellular Matrix
3.4.3. Astrocytes’ Role in Dopamine–Glutamate Interplay: Relevance for Schizophrenia
3.5. Aberrant Astrocyte Programming in Neurodevelopment: Implications for Schizophrenia
3.6. Astrocyte-Related Intracellular Mechanisms as Putative Pharmacological Targets
| Drug | Doses | Duration of Treatment | Preclinical Setting | Astrocytes’ Effects | Molecular Effect | References |
|---|---|---|---|---|---|---|
| Clozapine | 30 μM | 48 h | Primary astrocyte cultures from the cerebral cortex of 1-day-old Sprague Dawley rats | Astrocyte activation and glutamate modulation | Reduction in GLT-1 levels of about 50%; no changes in GLAST levels | [317,318] |
| unknown | unknown | hiPSC-derived astrocytes from schizophrenia patients | Reduction in L-serine levels | [307] | ||
| 3 μM | 7 days | Primary astrocyte cultures | Increased Cx43 expression | [306] | ||
| Brexpiprazole | 0.3 μM | Astrocytic glutamate modulation | ||||
| Quetiapine | 1 μM | Astrocytic glutamate release | ||||
| Risperidone | 0.1, 1, and 10 μM | acute | C6 glial cell line | Glutamate uptake and antioxidant defense | Increased GSH levels | [305] |
| Haloperidol | 10 μM | Increased ROS levels | ||||
| 20–40 μM | acute | Gibco® human astrocytes | Astrocyte toxicity and glutamate transport | Increased Ca2+ levels | [311] | |
| Lumateperone | 3 mg/kg | acute | LPS-injected C57BL/6 mice | BBB integrity | Modulation of claudin-5 and intercellular adhesion molecule 1 | [315] |
| Xanomeline | 300 nM | 10 min | Prefrontal brain slices of mice | Neuroprotection | Reduction in ROS production and inhibition of astrocyte-mediated apoptosis | [316] |
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Howes, O.D.; McCutcheon, R.; Agid, O.; de Bartolomeis, A.; van Beveren, N.J.; Birnbaum, M.L.; Bloomfield, M.A.; Bressan, R.A.; Buchanan, R.W.; Carpenter, W.T.; et al. Treatment-Resistant Schizophrenia: Treatment Response and Resistance in Psychosis (TRRIP) Working Group Consensus Guidelines on Diagnosis and Terminology. Am. J. Psychiatry 2017, 174, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Remington, G.; Addington, D.; Honer, W.; Ismail, Z.; Raedler, T.; Teehan, M. Guidelines for the Pharmacotherapy of Schizophrenia in Adults. Can. J. Psychiatry 2017, 62, 604–616. [Google Scholar] [CrossRef]
- Weinberger, D.R. On the plausibility of “the neurodevelopmental hypothesis” of schizophrenia. Neuropsychopharmacology 1996, 14 (Suppl. 1), 1s–11s. [Google Scholar] [CrossRef] [PubMed]
- Bayer, T.A.; Falkai, P.; Maier, W. Genetic and non-genetic vulnerability factors in schizophrenia: The basis of the “two hit hypothesis”. J. Psychiatr. Res. 1999, 33, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Eyre, H.; Jacka, F.N.; Dodd, S.; Dean, O.; McEwen, S.; Debnath, M.; McGrath, J.; Maes, M.; Amminger, P.; et al. A review of vulnerability and risks for schizophrenia: Beyond the two hit hypothesis. Neurosci. Biobehav. Rev. 2016, 65, 185–194. [Google Scholar] [CrossRef]
- Keshavan, M.S. Development, disease and degeneration in schizophrenia: A unitary pathophysiological model. J. Psychiatr. Res. 1999, 33, 513–521. [Google Scholar] [CrossRef]
- Keshavan, M.S.; Hogarty, G.E. Brain maturational processes and delayed onset in schizophrenia. Dev. Psychopathol. 1999, 11, 525–543. [Google Scholar] [CrossRef]
- Maynard, T.M.; Sikich, L.; Lieberman, J.A.; LaMantia, A.S. Neural development, cell-cell signaling, and the “two-hit” hypothesis of schizophrenia. Schizophr. Bull. 2001, 27, 457–476. [Google Scholar] [CrossRef]
- Bouet, V.; Percelay, S.; Leroux, E.; Diarra, B.; Léger, M.; Delcroix, N.; Andrieux, A.; Dollfus, S.; Freret, T.; Boulouard, M. A new 3-hit mouse model of schizophrenia built on genetic, early and late factors. Schizophr. Res. 2021, 228, 519–528. [Google Scholar] [CrossRef]
- Jarskog, L.F.; Gilmore, J.H.; Lieberman, J.A. Neurodegenerative models of schizophrenia. In Neurodevelopment and Schizophrenia; Keshavan, M.S., Kennedy, J.L., Murray, R.M., Eds.; Cambridge University Press: Cambridge, UK, 2004; pp. 373–389. [Google Scholar] [CrossRef]
- Stone, W.S.; Phillips, M.R.; Yang, L.H.; Kegeles, L.S.; Susser, E.S.; Lieberman, J.A. Neurodegenerative model of schizophrenia: Growing evidence to support a revisit. Schizophr. Res. 2022, 243, 154–162. [Google Scholar] [CrossRef]
- McDonald, J.W.; Silverstein, F.S.; Johnston, M.V. Neurotoxicity of N-methyl-D-aspartate is markedly enhanced in developing rat central nervous system. Brain Res. 1988, 459, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W.; Farber, N.B. Glutamate receptor dysfunction and schizophrenia. Arch. Gen. Psychiatry 1995, 52, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidou, C.; Bosch, F.; Miksa, M.; Bittigau, P.; Vöckler, J.; Dikranian, K.; Tenkova, T.I.; Stefovska, V.; Turski, L.; Olney, J.W. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 1999, 283, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, S.R.; Das, I.; Garey, L.J.; de Belleroche, J. A pivotal role for glutamate in the pathogenesis of schizophrenia, and its cognitive dysfunction. Pharmacol. Biochem. Behav. 1997, 56, 797–802. [Google Scholar] [CrossRef]
- Grace, A.A. Cortical regulation of subcortical dopamine systems and its possible relevance to schizophrenia. J. Neural Transm. Gen. Sect. 1993, 91, 111–134. [Google Scholar] [CrossRef]
- Li, Z.; Chen, J.; Yu, H.; He, L.; Xu, Y.; Zhang, D.; Yi, Q.; Li, C.; Li, X.; Shen, J.; et al. Genome-wide association analysis identifies 30 new susceptibility loci for schizophrenia. Nat. Genet. 2017, 49, 1576–1583. [Google Scholar] [CrossRef]
- Periyasamy, S.; John, S.; Padmavati, R.; Rajendren, P.; Thirunavukkarasu, P.; Gratten, J.; Vinkhuyzen, A.; McRae, A.; Holliday, E.G.; Nyholt, D.R.; et al. Association of Schizophrenia Risk With Disordered Niacin Metabolism in an Indian Genome-wide Association Study. JAMA Psychiatry 2019, 76, 1026–1034. [Google Scholar] [CrossRef]
- Ayalew, M.; Le-Niculescu, H.; Levey, D.F.; Jain, N.; Changala, B.; Patel, S.D.; Winiger, E.; Breier, A.; Shekhar, A.; Amdur, R.; et al. Convergent functional genomics of schizophrenia: From comprehensive understanding to genetic risk prediction. Mol. Psychiatry 2012, 17, 887–905. [Google Scholar] [CrossRef]
- Jaaro-Peled, H.; Ayhan, Y.; Pletnikov, M.V.; Sawa, A. Review of Pathological Hallmarks of Schizophrenia: Comparison of Genetic Models With Patients and Nongenetic Models. Schizophr. Bull. 2009, 36, 301–313. [Google Scholar] [CrossRef]
- De la Torre, R.; De Sola, S.; Pons, M.; Duchon, A.; de Lagran, M.M.; Farré, M.; Fitó, M.; Benejam, B.; Langohr, K.; Rodriguez, J.; et al. Epigallocatechin-3-gallate, a DYRK1A inhibitor, rescues cognitive deficits in Down syndrome mouse models and in humans. Mol. Nutr. Food Res. 2014, 58, 278–288. [Google Scholar] [CrossRef]
- Vasile, F.; Dossi, E.; Rouach, N. Human astrocytes: Structure and functions in the healthy brain. Brain Struct. Funct. 2017, 222, 2017–2029. [Google Scholar] [CrossRef] [PubMed]
- Nichols, N.R.; Day, J.R.; Laping, N.J.; Johnson, S.A.; Finch, C.E. GFAP mRNA increases with age in rat and human brain. Neurobiol. Aging 1993, 14, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Oberheim, N.A.; Takano, T.; Han, X.; He, W.; Lin, J.H.; Wang, F.; Xu, Q.; Wyatt, J.D.; Pilcher, W.; Ojemann, J.G.; et al. Uniquely hominid features of adult human astrocytes. J. Neurosci. 2009, 29, 3276–3287. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, A.; Olszewski, J. Glia/nerve cell index for cortex of the whale. Science 1957, 126, 76–77. [Google Scholar] [CrossRef]
- Pelvig, D.P.; Pakkenberg, H.; Stark, A.K.; Pakkenberg, B. Neocortical glial cell numbers in human brains. Neurobiol. Aging 2008, 29, 1754–1762. [Google Scholar] [CrossRef]
- Mederos, S.; González-Arias, C.; Perea, G. Astrocyte-Neuron Networks: A Multilane Highway of Signaling for Homeostatic Brain Function. Front. Synaptic Neurosci. 2018, 10, 45. [Google Scholar] [CrossRef]
- Adermark, L.; Lagström, O.; Loftén, A.; Licheri, V.; Havenäng, A.; Loi, E.A.; Stomberg, R.; Söderpalm, B.; Domi, A.; Ericson, M. Astrocytes modulate extracellular neurotransmitter levels and excitatory neurotransmission in dorsolateral striatum via dopamine D2 receptor signaling. Neuropsychopharmacology 2022, 47, 1493–1502. [Google Scholar] [CrossRef]
- Requie, L.M.; Gómez-Gonzalo, M.; Speggiorin, M.; Managò, F.; Melone, M.; Congiu, M.; Chiavegato, A.; Lia, A.; Zonta, M.; Losi, G.; et al. Astrocytes mediate long-lasting synaptic regulation of ventral tegmental area dopamine neurons. Nat. Neurosci. 2022, 25, 1639–1650. [Google Scholar] [CrossRef]
- Sidoryk-Wegrzynowicz, M.; Wegrzynowicz, M.; Lee, E.; Bowman, A.B.; Aschner, M. Role of astrocytes in brain function and disease. Toxicol. Pathol. 2011, 39, 115–123. [Google Scholar] [CrossRef]
- Kim, Y.; Park, J.; Choi, Y.K. The Role of Astrocytes in the Central Nervous System Focused on BK Channel and Heme Oxygenase Metabolites: A Review. Antioxidants 2019, 8, 121. [Google Scholar] [CrossRef]
- Niess, F.; Strasser, B.; Hingerl, L.; Bader, V.; Frese, S.; Clarke, W.T.; Duguid, A.; Niess, E.; Motyka, S.; Krššák, M.; et al. Whole-brain deuterium metabolic imaging via concentric ring trajectory readout enables assessment of regional variations in neuronal glucose metabolism. Hum. Brain Mapp. 2024, 45, e26686. [Google Scholar] [CrossRef] [PubMed]
- Townsend, L.; Pillinger, T.; Selvaggi, P.; Veronese, M.; Turkheimer, F.; Howes, O. Brain glucose metabolism in schizophrenia: A systematic review and meta-analysis of (18)FDG-PET studies in schizophrenia. Psychol. Med. 2023, 53, 4880–4897. [Google Scholar] [CrossRef] [PubMed]
- Iasevoli, F.; D’Ambrosio, L.; Ciccarelli, M.; Barone, A.; Gaudieri, V.; Cocozza, S.; Pontillo, G.; Brunetti, A.; Cuocolo, A.; de Bartolomeis, A.; et al. Altered Patterns of Brain Glucose Metabolism Involve More Extensive and Discrete Cortical Areas in Treatment-resistant Schizophrenia Patients Compared to Responder Patients and Controls: Results From a Head-to-Head 2-[18F]-FDG-PET Study. Schizophr. Bull. 2023, 49, 474–485. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; Iasevoli, F.; Barone, A.; Gaudieri, V.; Cuocolo, A.; Ciccarelli, M.; Pappatà, S.; de Bartolomeis, A. Addressing brain metabolic connectivity in treatment-resistant schizophrenia: A novel graph theory-driven application of (18)F-FDG-PET with antipsychotic dose correction. Schizophrenia 2024, 10, 116. [Google Scholar] [CrossRef]
- Knight, S.R.; Abbasova, L.; Zeighami, Y.; Hansen, J.Y.; Martins, D.; Zelaya, F.; Dipasquale, O.; Liu, T.; Shin, D.; Bossong, M.; et al. Transcriptional and neurochemical signatures of cerebral blood flow alterations in schizophrenia and individuals at clinical high-risk for psychosis. Biol. Psychiatry 2025, in press. [CrossRef]
- Verkhratsky, A.; Bush, N.A.O.; Nedergaard, M.; Butt, A. The Special Case of Human Astrocytes. Neuroglia 2018, 1, 21–29. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Volterra, A.; Meldolesi, J. Astrocytes, from brain glue to communication elements: The revolution continues. Nat. Rev. Neurosci. 2005, 6, 626–640. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Katayama, T.; Prat, A. Glial influence on the blood brain barrier. Glia 2013, 61, 1939–1958. [Google Scholar] [CrossRef]
- Kolomeets, N.S. [Astroglia of the hippocampus in schizophrenia]. Zh. Nevrol. Psikhiatr. Im. S S Korsakova 2008, 108, 70–76. [Google Scholar]
- Tarasov, V.V.; Svistunov, A.A.; Chubarev, V.N.; Sologova, S.S.; Mukhortova, P.; Levushkin, D.; Somasundaram, S.G.; Kirkland, C.E.; Bachurin, S.O.; Aliev, G. Alterations of Astrocytes in the Context of Schizophrenic Dementia. Front. Pharmacol. 2019, 10, 1612. [Google Scholar] [CrossRef] [PubMed]
- Benes, F.M.; Davidson, J.; Bird, E.D. Quantitative cytoarchitectural studies of the cerebral cortex of schizophrenics. Arch. Gen. Psychiatry 1986, 43, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Cotter, D.R.; Pariante, C.M.; Everall, I.P. Glial cell abnormalities in major psychiatric disorders: The evidence and implications. Brain Res. Bull. 2001, 55, 585–595. [Google Scholar] [CrossRef]
- Shan, D.; Lucas, E.K.; Drummond, J.B.; Haroutunian, V.; Meador-Woodruff, J.H.; McCullumsmith, R.E. Abnormal expression of glutamate transporters in temporal lobe areas in elderly patients with schizophrenia. Schizophr. Res. 2013, 144, 1–8. [Google Scholar] [CrossRef]
- Feresten, A.H.; Barakauskas, V.; Ypsilanti, A.; Barr, A.M.; Beasley, C.L. Increased expression of glial fibrillary acidic protein in prefrontal cortex in psychotic illness. Schizophr. Res. 2013, 150, 252–257. [Google Scholar] [CrossRef]
- Trépanier, M.O.; Hopperton, K.E.; Mizrahi, R.; Mechawar, N.; Bazinet, R.P. Postmortem evidence of cerebral inflammation in schizophrenia: A systematic review. Mol. Psychiatry 2016, 21, 1009–1026. [Google Scholar] [CrossRef]
- Zhang, L.; Verwer, R.W.H.; Lucassen, P.J.; Huitinga, I.; Swaab, D.F. Prefrontal cortex alterations in glia gene expression in schizophrenia with and without suicide. J. Psychiatr. Res. 2020, 121, 31–38. [Google Scholar] [CrossRef]
- Katsel, P.; Byne, W.; Roussos, P.; Tan, W.; Siever, L.; Haroutunian, V. Astrocyte and glutamate markers in the superficial, deep, and white matter layers of the anterior cingulate gyrus in schizophrenia. Neuropsychopharmacology 2011, 36, 1171–1177. [Google Scholar] [CrossRef]
- Toker, L.; Mancarci, B.O.; Tripathy, S.; Pavlidis, P. Transcriptomic Evidence for Alterations in Astrocytes and Parvalbumin Interneurons in Subjects With Bipolar Disorder and Schizophrenia. Biol. Psychiatry 2018, 84, 787–796. [Google Scholar] [CrossRef]
- Ramaker, R.C.; Bowling, K.M.; Lasseigne, B.N.; Hagenauer, M.H.; Hardigan, A.A.; Davis, N.S.; Gertz, J.; Cartagena, P.M.; Walsh, D.M.; Vawter, M.P.; et al. Post-mortem molecular profiling of three psychiatric disorders. Genome Med. 2017, 9, 72. [Google Scholar] [CrossRef]
- González-Peñas, J.; Costas, J.; Villamayor, M.J.G.; Xu, B. Enrichment of rare genetic variants in astrocyte gene enriched co-expression modules altered in postmortem brain samples of schizophrenia. Neurobiol. Dis. 2019, 121, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.M.; Abazyan, S.; Abazyan, B.; Nomura, J.; Yang, C.; Seshadri, S.; Sawa, A.; Snyder, S.H.; Pletnikov, M.V. Pathogenic disruption of DISC1-serine racemase binding elicits schizophrenia-like behavior via D-serine depletion. Mol. Psychiatry 2013, 18, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Terrillion, C.E.; Abazyan, B.; Yang, Z.; Crawford, J.; Shevelkin, A.V.; Jouroukhin, Y.; Yoo, K.H.; Cho, C.H.; Roychaudhuri, R.; Snyder, S.H.; et al. DISC1 in Astrocytes Influences Adult Neurogenesis and Hippocampus-Dependent Behaviors in Mice. Neuropsychopharmacology 2017, 42, 2242–2251. [Google Scholar] [CrossRef]
- Facal, F.; Costas, J. Evidence of association of the DISC1 interactome gene set with schizophrenia from GWAS. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 95, 109729. [Google Scholar] [CrossRef]
- Niwa, M.; Cash-Padgett, T.; Kubo, K.I.; Saito, A.; Ishii, K.; Sumitomo, A.; Taniguchi, Y.; Ishizuka, K.; Jaaro-Peled, H.; Tomoda, T.; et al. DISC1 a key molecular lead in psychiatry and neurodevelopment: No-More Disrupted-in-Schizophrenia 1. Mol. Psychiatry 2016, 21, 1488–1489. [Google Scholar] [CrossRef]
- Ryan, N.M.; Lihm, J.; Kramer, M.; McCarthy, S.; Morris, S.W.; Arnau-Soler, A.; Davies, G.; Duff, B.; Ghiban, E.; Hayward, C.; et al. DNA sequence-level analyses reveal potential phenotypic modifiers in a large family with psychiatric disorders. Mol. Psychiatry 2018, 23, 2254–2265. [Google Scholar] [CrossRef]
- Shevelkin, A.V.; Terrillion, C.E.; Hasegawa, Y.; Mychko, O.A.; Jouroukhin, Y.; Sawa, A.; Kamiya, A.; Pletnikov, M.V. Astrocyte DISC1 contributes to cognitive function in a brain region-dependent manner. Hum. Mol. Genet. 2020, 29, 2936–2950. [Google Scholar] [CrossRef]
- Karlsson, R.M.; Tanaka, K.; Heilig, M.; Holmes, A. Loss of glial glutamate and aspartate transporter (excitatory amino acid transporter 1) causes locomotor hyperactivity and exaggerated responses to psychotomimetics: Rescue by haloperidol and metabotropic glutamate 2/3 agonist. Biol. Psychiatry 2008, 64, 810–814. [Google Scholar] [CrossRef]
- Karlsson, R.M.; Tanaka, K.; Saksida, L.M.; Bussey, T.J.; Heilig, M.; Holmes, A. Assessment of glutamate transporter GLAST (EAAT1)-deficient mice for phenotypes relevant to the negative and executive/cognitive symptoms of schizophrenia. Neuropsychopharmacology 2009, 34, 1578–1589. [Google Scholar] [CrossRef]
- Matsugami, T.R.; Tanemura, K.; Mieda, M.; Nakatomi, R.; Yamada, K.; Kondo, T.; Ogawa, M.; Obata, K.; Watanabe, M.; Hashikawa, T.; et al. From the Cover: Indispensability of the glutamate transporters GLAST and GLT1 to brain development. Proc. Natl. Acad. Sci. USA 2006, 103, 12161–12166. [Google Scholar] [CrossRef]
- Windrem, M.S.; Osipovitch, M.; Liu, Z.; Bates, J.; Chandler-Militello, D.; Zou, L.; Munir, J.; Schanz, S.; McCoy, K.; Miller, R.H.; et al. Human iPSC Glial Mouse Chimeras Reveal Glial Contributions to Schizophrenia. Cell Stem Cell 2017, 21, 195–208.e6. [Google Scholar] [CrossRef] [PubMed]
- Rimmele, T.S.; Rosenberg, P.A. GLT-1: The elusive presynaptic glutamate transporter. Neurochem. Int. 2016, 98, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Al-Shami, A.S.; Abd Elkader, H.A.E.; Moussa, N.; Essawy, A.E.; Haroun, M. Early-life bisphenol A exposure causes neuronal pyroptosis in juvenile and adult male rats through the NF-κB/IL-1β/NLRP3/caspase-1 signaling pathway: Exploration of age and dose as effective covariates using an in vivo and in silico modeling approach. Mol. Cell Biochem. 2024, 480, 2301–2330. [Google Scholar] [CrossRef] [PubMed]
- Bechtholt-Gompf, A.J.; Walther, H.V.; Adams, M.A.; Carlezon, W.A., Jr.; Ongür, D.; Cohen, B.M. Blockade of astrocytic glutamate uptake in rats induces signs of anhedonia and impaired spatial memory. Neuropsychopharmacology 2010, 35, 2049–2059. [Google Scholar] [CrossRef]
- Ebrahimi, M.; Yamamoto, Y.; Sharifi, K.; Kida, H.; Kagawa, Y.; Yasumoto, Y.; Islam, A.; Miyazaki, H.; Shimamoto, C.; Maekawa, M.; et al. Astrocyte-expressed FABP7 regulates dendritic morphology and excitatory synaptic function of cortical neurons. Glia 2016, 64, 48–62. [Google Scholar] [CrossRef]
- Shimamoto, C.; Ohnishi, T.; Maekawa, M.; Watanabe, A.; Ohba, H.; Arai, R.; Iwayama, Y.; Hisano, Y.; Toyota, T.; Toyoshima, M.; et al. Functional characterization of FABP3, 5 and 7 gene variants identified in schizophrenia and autism spectrum disorder and mouse behavioral studies. Hum. Mol. Genet. 2014, 23, 6495–6511. [Google Scholar] [CrossRef]
- Jonas, K.G.; Lencz, T.; Li, K.; Malhotra, A.K.; Perlman, G.; Fochtmann, L.J.; Bromet, E.J.; Kotov, R. Schizophrenia polygenic risk score and 20-year course of illness in psychotic disorders. Transl. Psychiatry 2019, 9, 300. [Google Scholar] [CrossRef]
- Elgueta, D.; Murgas, P.; Riquelme, E.; Yang, G.; Cancino, G.I. Consequences of Viral Infection and Cytokine Production During Pregnancy on Brain Development in Offspring. Front. Immunol. 2022, 13, 816619. [Google Scholar] [CrossRef]
- Ding, S.; Hu, Y.; Luo, B.; Cai, Y.; Hao, K.; Yang, Y.; Zhang, Y.; Wang, X.; Ding, M.; Zhang, H.; et al. Age-related changes in neuroinflammation and prepulse inhibition in offspring of rats treated with Poly I:C in early gestation. Behav. Brain Funct. 2019, 15, 3. [Google Scholar] [CrossRef]
- Waddington, J.L.; Brown, A.S.; Lane, A.; Schaefer, C.A.; Goetz, R.R.; Bresnahan, M.; Susser, E.S. Congenital anomalies and early functional impairments in a prospective birth cohort: Risk of schizophrenia-spectrum disorder in adulthood. Br. J. Psychiatry 2008, 192, 264–267. [Google Scholar] [CrossRef]
- Weinstock, M. The long-term behavioural consequences of prenatal stress. Neurosci. Biobehav. Rev. 2008, 32, 1073–1086. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.; Towers-Evans, H.; MacCabe, J. The aetiology of schizophrenia: What have the Swedish Medical Registers taught us? Soc. Psychiatry Psychiatr. Epidemiol. 2015, 50, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.; Bray, N.J. Schizophrenia Genomics: Convergence on Synaptic Development, Adult Synaptic Plasticity, or Both? Biol. Psychiatry 2022, 91, 709–717. [Google Scholar] [CrossRef]
- Tromp, A.; Mowry, B.; Giacomotto, J. Neurexins in autism and schizophrenia-a review of patient mutations, mouse models and potential future directions. Mol. Psychiatry 2021, 26, 747–760. [Google Scholar] [CrossRef]
- Singh, T.; Poterba, T.; Curtis, D.; Akil, H.; Al Eissa, M.; Barchas, J.D.; Bass, N.; Bigdeli, T.B.; Breen, G.; Bromet, E.J.; et al. Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature 2022, 604, 509–516. [Google Scholar] [CrossRef]
- Trubetskoy, V.; Pardiñas, A.F.; Qi, T.; Panagiotaropoulou, G.; Awasthi, S.; Bigdeli, T.B.; Bryois, J.; Chen, C.Y.; Dennison, C.A.; Hall, L.S.; et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022, 604, 502–508. [Google Scholar] [CrossRef]
- Schmitt, A.; Malchow, B.; Hasan, A.; Falkai, P. The impact of environmental factors in severe psychiatric disorders. Front. Neurosci. 2014, 8, 19. [Google Scholar] [CrossRef]
- Weinberger, D.R. The pathogenesis of schizophrenia: A neurodevelopmental theory. In Neurology of Schizophrenia; Elsevier: Amsterdam, The Netherlands, 1986; pp. 387–405. [Google Scholar]
- Hof, P.R.; Haroutunian, V.; Friedrich, V.L., Jr.; Byne, W.; Buitron, C.; Perl, D.P.; Davis, K.L. Loss and altered spatial distribution of oligodendrocytes in the superior frontal gyrus in schizophrenia. Biol. Psychiatry 2003, 53, 1075–1085. [Google Scholar] [CrossRef]
- Schmitt, A.; Steyskal, C.; Bernstein, H.G.; Schneider-Axmann, T.; Parlapani, E.; Schaeffer, E.L.; Gattaz, W.F.; Bogerts, B.; Schmitz, C.; Falkai, P. Stereologic investigation of the posterior part of the hippocampus in schizophrenia. Acta Neuropathol. 2009, 117, 395–407. [Google Scholar] [CrossRef]
- Falkai, P.; Malchow, B.; Wetzestein, K.; Nowastowski, V.; Bernstein, H.G.; Steiner, J.; Schneider-Axmann, T.; Kraus, T.; Hasan, A.; Bogerts, B.; et al. Decreased Oligodendrocyte and Neuron Number in Anterior Hippocampal Areas and the Entire Hippocampus in Schizophrenia: A Stereological Postmortem Study. Schizophr. Bull. 2016, 42 (Suppl. 1), S4–S12. [Google Scholar] [CrossRef]
- Falkai, P.; Raabe, F.; Bogerts, B.; Schneider-Axmann, T.; Malchow, B.; Tatsch, L.; Huber, V.; Slapakova, L.; Dobrowolny, H.; Schmitz, C.; et al. Association between altered hippocampal oligodendrocyte number and neuronal circuit structures in schizophrenia: A postmortem analysis. Eur. Arch. Psychiatry Clin. Neurosci. 2020, 270, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Constantine-Paton, M. Effects of NMDA receptor antagonists on the developing brain. Psychopharmacol. Bull. 1994, 30, 561–565. [Google Scholar] [PubMed]
- Zecevic, N.; Bourgeois, J.P.; Rakic, P. Changes in synaptic density in motor cortex of rhesus monkey during fetal and postnatal life. Brain Res. Dev. Brain Res. 1989, 50, 11–32. [Google Scholar] [CrossRef]
- Johnston, M.V. Neurotransmitters and vulnerability of the developing brain. Brain Dev. 1995, 17, 301–306. [Google Scholar] [CrossRef]
- Coyle, J.T. The glutamatergic dysfunction hypothesis for schizophrenia. Harv. Rev. Psychiatry 1996, 3, 241–253. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Sloan, S.A.; Barres, B.A. Mechanisms of astrocyte development and their contributions to neurodevelopmental disorders. Curr. Opin. Neurobiol. 2014, 27, 75–81. [Google Scholar] [CrossRef]
- Chiareli, R.A.; Carvalho, G.A.; Marques, B.L.; Mota, L.S.; Oliveira-Lima, O.C.; Gomes, R.M.; Birbrair, A.; Gomez, R.S.; Simão, F.; Klempin, F.; et al. The Role of Astrocytes in the Neurorepair Process. Front. Cell Dev. Biol. 2021, 9, 665795. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Li, Y.; Shen, M.; Stockton, M.E.; Zhao, X. Hippocampal deficits in neurodevelopmental disorders. Neurobiol. Learn. Mem. 2019, 165, 106945. [Google Scholar] [CrossRef]
- Sonnewald, U.; Schousboe, A. Introduction to the Glutamate-Glutamine Cycle. Adv. Neurobiol. 2016, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lalo, U.; Koh, W.; Lee, C.J.; Pankratov, Y. The tripartite glutamatergic synapse. Neuropharmacology 2021, 199, 108758. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef] [PubMed]
- Sattler, R.; Rothstein, J.D. Regulation and dysregulation of glutamate transporters. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2006; pp. 277–303. [Google Scholar] [CrossRef]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The glutamate/GABA-glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef]
- Spangaro, M.; Bosia, M.; Zanoletti, A.; Bechi, M.; Mariachiara, B.; Pirovano, A.; Lorenzi, C.; Bramanti, P.; Smeraldi, E.; Cavallaro, R. Exploring effects of EAAT polymorphisms on cognitive functions in schizophrenia. Pharmacogenomics 2014, 15, 925–932. [Google Scholar] [CrossRef]
- Ramsey, A.J. NR1 knockdown mice as a representative model of the glutamate hypothesis of schizophrenia. Prog. Brain Res. 2009, 179, 51–58. [Google Scholar] [CrossRef]
- Takahashi, K.; Kong, Q.; Lin, Y.; Stouffer, N.; Schulte, D.A.; Lai, L.; Liu, Q.; Chang, L.C.; Dominguez, S.; Xing, X.; et al. Restored glial glutamate transporter EAAT2 function as a potential therapeutic approach for Alzheimer’s disease. J. Exp. Med. 2015, 212, 319–332. [Google Scholar] [CrossRef]
- Tan, C.X.; Eroglu, C. Cell adhesion molecules regulating astrocyte-neuron interactions. Curr. Opin. Neurobiol. 2021, 69, 170–177. [Google Scholar] [CrossRef]
- Dang, R.; Liu, A.; Zhou, Y.; Li, X.; Wu, M.; Cao, K.; Meng, Y.; Zhang, H.; Gan, G.; Xie, W.; et al. Astrocytic neuroligin 3 regulates social memory and synaptic plasticity through adenosine signaling in male mice. Nat. Commun. 2024, 15, 8639. [Google Scholar] [CrossRef]
- Hanske, S.; Dyrna, F.; Bechmann, I.; Krueger, M. Different segments of the cerebral vasculature reveal specific endothelial specifications, while tight junction proteins appear equally distributed. Brain Struct. Funct. 2017, 222, 1179–1192. [Google Scholar] [CrossRef]
- Ikeshima-Kataoka, H. Neuroimmunological Implications of AQP4 in Astrocytes. Int. J. Mol. Sci. 2016, 17, 1306. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonnière, L.; Bernard, M.; et al. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Castro, F.; Zappettini, S.; Pressey, J.C.; Silva, C.G.; Russeau, M.; Gervasi, N.; Figueiredo, M.; Montmasson, C.; Renner, M.; Canas, P.M.; et al. Convergence of adenosine and GABA signaling for synapse stabilization during development. Science 2021, 374, eabk2055. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Dallérac, G.; Pucci, L.; Calì, C.; Zehnder, T.; Sultan, S.; Lecca, S.; Chicca, A.; Ivanov, A.; Asensio, C.S.; et al. Dysfunction of homeostatic control of dopamine by astrocytes in the developing prefrontal cortex leads to cognitive impairments. Mol. Psychiatry 2020, 25, 732–749. [Google Scholar] [CrossRef]
- Vaarmann, A.; Gandhi, S.; Abramov, A.Y. Dopamine induces Ca2+ signaling in astrocytes through reactive oxygen species generated by monoamine oxidase. J. Biol. Chem. 2010, 285, 25018–25023. [Google Scholar] [CrossRef]
- Kucheryavykh, Y.V.; Kucheryavykh, L.Y.; Nichols, C.G.; Maldonado, H.M.; Baksi, K.; Reichenbach, A.; Skatchkov, S.N.; Eaton, M.J. Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia 2007, 55, 274–281. [Google Scholar] [CrossRef]
- Nanitsos, E.K.; Nguyen, K.T.; St’astný, F.; Balcar, V.J. Glutamatergic hypothesis of schizophrenia: Involvement of Na+/K+-dependent glutamate transport. J. Biomed. Sci. 2005, 12, 975–984. [Google Scholar] [CrossRef]
- Almeida, P.G.C.; Nani, J.V.; Oses, J.P.; Brietzke, E.; Hayashi, M.A.F. Neuroinflammation and glial cell activation in mental disorders. Brain Behav. Immun. Health 2020, 2, 100034. [Google Scholar] [CrossRef]
- Islam, A.; Kagawa, Y.; Miyazaki, H.; Shil, S.K.; Umaru, B.A.; Yasumoto, Y.; Yamamoto, Y.; Owada, Y. FABP7 Protects Astrocytes Against ROS Toxicity via Lipid Droplet Formation. Mol. Neurobiol. 2019, 56, 5763–5779. [Google Scholar] [CrossRef]
- Müller, N.; Schwarz, M.J. COX-2 inhibition in schizophrenia and major depression. Curr. Pharm. Des. 2008, 14, 1452–1465. [Google Scholar] [CrossRef]
- Südhof, T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008, 455, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Foster, J.B.; Lin, C.L. Glutamate transporter EAAT2: Regulation, function, and potential as a therapeutic target for neurological and psychiatric disease. Cell Mol. Life Sci. 2015, 72, 3489–3506. [Google Scholar] [CrossRef] [PubMed]
- Rygvold, T.W.; Hatlestad-Hall, C.; Elvsåshagen, T.; Moberget, T.; Andersson, S. Long-Term Potentiation-Like Visual Synaptic Plasticity Is Negatively Associated With Self-Reported Symptoms of Depression and Stress in Healthy Adults. Front. Hum. Neurosci. 2022, 16, 867675. [Google Scholar] [CrossRef]
- Hamilton, H.K.; Roach, B.J.; Cavus, I.; Teyler, T.J.; Clapp, W.C.; Ford, J.M.; Tarakci, E.; Krystal, J.H.; Mathalon, D.H. Impaired Potentiation of Theta Oscillations During a Visual Cortical Plasticity Paradigm in Individuals With Schizophrenia. Front. Psychiatry 2020, 11, 590567. [Google Scholar] [CrossRef]
- Hashimoto, A.; Nishikawa, T.; Hayashi, T.; Fujii, N.; Harada, K.; Oka, T.; Takahashi, K. The presence of free D-serine in rat brain. FEBS Lett. 1992, 296, 33–36. [Google Scholar] [CrossRef]
- Coyle, J.T.; Balu, D.; Wolosker, H. D-Serine, the Shape-Shifting NMDA Receptor Co-agonist. Neurochem. Res. 2020, 45, 1344–1353. [Google Scholar] [CrossRef]
- Allen, N.J.; Lyons, D.A. Glia as architects of central nervous system formation and function. Science 2018, 362, 181–185. [Google Scholar] [CrossRef]
- Pietiläinen, O.; Trehan, A.; Meyer, D.; Mitchell, J.; Tegtmeyer, M.; Valakh, V.; Gebre, H.; Chen, T.; Vartiainen, E.; Farhi, S.L.; et al. Astrocytic cell adhesion genes linked to schizophrenia correlate with synaptic programs in neurons. Cell Rep. 2023, 42, 111988. [Google Scholar] [CrossRef]
- Caldwell, A.L.M.; Sancho, L.; Deng, J.; Bosworth, A.; Miglietta, A.; Diedrich, J.K.; Shokhirev, M.N.; Allen, N.J. Aberrant astrocyte protein secretion contributes to altered neuronal development in multiple models of neurodevelopmental disorders. Nat. Neurosci. 2022, 25, 1163–1178. [Google Scholar] [CrossRef]
- Allen, M.; Huang, B.S.; Notaras, M.J.; Lodhi, A.; Barrio-Alonso, E.; Lituma, P.J.; Wolujewicz, P.; Witztum, J.; Longo, F.; Chen, M.; et al. Astrocytes derived from ASD individuals alter behavior and destabilize neuronal activity through aberrant Ca2+ signaling. Mol. Psychiatry 2022, 27, 2470–2484. [Google Scholar] [CrossRef]
- Wang, Q.; Kong, Y.; Wu, D.Y.; Liu, J.H.; Jie, W.; You, Q.L.; Huang, L.; Hu, J.; Chu, H.D.; Gao, F.; et al. Impaired calcium signaling in astrocytes modulates autism spectrum disorder-like behaviors in mice. Nat. Commun. 2021, 12, 3321. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Zhou, Z.; Han, B.; Xiang, X.; Huang, W.; Yao, H. Revisiting astrocytic calcium signaling in the brain. Fundam. Res. 2024, 4, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, M.; Perea, G.; Maglio, L.; Pastor, J.; García de Sola, R.; Araque, A. Astrocyte calcium signal and gliotransmission in human brain tissue. Cereb. Cortex 2013, 23, 1240–1246. [Google Scholar] [CrossRef]
- Fu, W.; Ruangkittisakul, A.; MacTavish, D.; Baker, G.B.; Ballanyi, K.; Jhamandas, J.H. Activity and metabolism-related Ca2+ and mitochondrial dynamics in co-cultured human fetal cortical neurons and astrocytes. Neuroscience 2013, 250, 520–535. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef]
- Sun, W.; McConnell, E.; Pare, J.F.; Xu, Q.; Chen, M.; Peng, W.; Lovatt, D.; Han, X.; Smith, Y.; Nedergaard, M. Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science 2013, 339, 197–200. [Google Scholar] [CrossRef]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.; Robitaille, R.; Volterra, A. Gliotransmitters travel in time and space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef]
- Rusakov, D.A.; Bard, L.; Stewart, M.G.; Henneberger, C. Diversity of astroglial functions alludes to subcellular specialisation. Trends Neurosci. 2014, 37, 228–242. [Google Scholar] [CrossRef]
- Volterra, A.; Liaudet, N.; Savtchouk, I. Astrocyte Ca²⁺ signalling: An unexpected complexity. Nat. Rev. Neurosci. 2014, 15, 327–335. [Google Scholar] [CrossRef]
- Reitman, M.E.; Tse, V.; Mi, X.; Willoughby, D.D.; Peinado, A.; Aivazidis, A.; Myagmar, B.E.; Simpson, P.C.; Bayraktar, O.A.; Yu, G.; et al. Norepinephrine links astrocytic activity to regulation of cortical state. Nat. Neurosci. 2023, 26, 579–593. [Google Scholar] [CrossRef]
- Pittolo, S.; Yokoyama, S.; Willoughby, D.D.; Taylor, C.R.; Reitman, M.E.; Tse, V.; Wu, Z.; Etchenique, R.; Li, Y.; Poskanzer, K.E. Dopamine activates astrocytes in prefrontal cortex via α1-adrenergic receptors. Cell Rep. 2022, 40, 111426. [Google Scholar] [CrossRef] [PubMed]
- Kellner, V.; Kersbergen, C.J.; Li, S.; Babola, T.A.; Saher, G.; Bergles, D.E. Dual metabotropic glutamate receptor signaling enables coordination of astrocyte and neuron activity in developing sensory domains. Neuron 2021, 109, 2545–2555.e7. [Google Scholar] [CrossRef] [PubMed]
- Corkrum, M.; Covelo, A.; Lines, J.; Bellocchio, L.; Pisansky, M.; Loke, K.; Quintana, R.; Rothwell, P.E.; Lujan, R.; Marsicano, G.; et al. Dopamine-Evoked Synaptic Regulation in the Nucleus Accumbens Requires Astrocyte Activity. Neuron 2020, 105, 1036–1047.e5. [Google Scholar] [CrossRef]
- Semyanov, A. Spatiotemporal pattern of calcium activity in astrocytic network. Cell Calcium 2019, 78, 15–25. [Google Scholar] [CrossRef]
- Bindocci, E.; Savtchouk, I.; Liaudet, N.; Becker, D.; Carriero, G.; Volterra, A. Three-dimensional Ca(2+) imaging advances understanding of astrocyte biology. Science 2017, 356, eaai8185. [Google Scholar] [CrossRef]
- De Pittà, M.; Goldberg, M.; Volman, V.; Berry, H.; Ben-Jacob, E. Glutamate regulation of calcium and IP3 oscillating and pulsating dynamics in astrocytes. J. Biol. Phys. 2009, 35, 383–411. [Google Scholar] [CrossRef]
- Fellin, T.; Pascual, O.; Gobbo, S.; Pozzan, T.; Haydon, P.G.; Carmignoto, G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 2004, 43, 729–743. [Google Scholar] [CrossRef]
- Petravicz, J.; Fiacco, T.A.; McCarthy, K.D. Loss of IP3 receptor-dependent Ca2+ increases in hippocampal astrocytes does not affect baseline CA1 pyramidal neuron synaptic activity. J. Neurosci. 2008, 28, 4967–4973. [Google Scholar] [CrossRef]
- Sherwood, M.W.; Arizono, M.; Hisatsune, C.; Bannai, H.; Ebisui, E.; Sherwood, J.L.; Panatier, A.; Oliet, S.H.; Mikoshiba, K. Astrocytic IP(3) Rs: Contribution to Ca(2+) signalling and hippocampal LTP. Glia 2017, 65, 502–513. [Google Scholar] [CrossRef]
- Oschmann, F.; Mergenthaler, K.; Jungnickel, E.; Obermayer, K. Spatial separation of two different pathways accounting for the generation of calcium signals in astrocytes. PLoS Comput. Biol. 2017, 13, e1005377. [Google Scholar] [CrossRef]
- Reyes, R.C.; Verkhratsky, A.; Parpura, V. Plasmalemmal Na+/Ca2+ exchanger modulates Ca2+-dependent exocytotic release of glutamate from rat cortical astrocytes. ASN Neuro 2012, 4, AN20110059. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, M.; Egawa, K.; Fukuda, A. Diverse Actions of Astrocytes in GABAergic Signaling. Int. J. Mol. Sci. 2019, 20, 2964. [Google Scholar] [CrossRef] [PubMed]
- Boddum, K.; Jensen, T.P.; Magloire, V.; Kristiansen, U.; Rusakov, D.A.; Pavlov, I.; Walker, M.C. Astrocytic GABA transporter activity modulates excitatory neurotransmission. Nat. Commun. 2016, 7, 13572. [Google Scholar] [CrossRef] [PubMed]
- Marcassa, G.; Dascenco, D.; Lorente-Echeverría, B.; Daaboul, D.; Vandensteen, J.; Leysen, E.; Baltussen, L.; Howden, A.J.M.; de Wit, J. Synaptic signatures and disease vulnerabilities of layer 5 pyramidal neurons. Nat. Commun. 2025, 16, 228. [Google Scholar] [CrossRef]
- Vaccarino, F.M.; Ganat, Y.; Zhang, Y.; Zheng, W. Stem cells in neurodevelopment and plasticity. Neuropsychopharmacology 2001, 25, 805–815. [Google Scholar] [CrossRef]
- Stiles, J.; Jernigan, T.L. The basics of brain development. Neuropsychol. Rev. 2010, 20, 327–348. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, J.; Wang, X.; Han, M.; Fei, Y.; Wang, J. Blood-Brain Barrier Disruption in Schizophrenia: Insights, Mechanisms, and Future Directions. Int. J. Mol. Sci. 2025, 26, 873. [Google Scholar] [CrossRef]
- Manu, D.R.; Slevin, M.; Barcutean, L.; Forro, T.; Boghitoiu, T.; Balasa, R. Astrocyte Involvement in Blood-Brain Barrier Function: A Critical Update Highlighting Novel, Complex, Neurovascular Interactions. Int. J. Mol. Sci. 2023, 24, 17146. [Google Scholar] [CrossRef]
- Xing, G.; Zhao, T.; Zhang, X.; Li, H.; Li, X.; Cui, P.; Li, M.; Li, D.; Zhang, N.; Jiang, W. Astrocytic Sonic Hedgehog Alleviates Intracerebral Hemorrhagic Brain Injury via Modulation of Blood-Brain Barrier Integrity. Front. Cell Neurosci. 2020, 14, 575690. [Google Scholar] [CrossRef]
- Najjar, S.; Pearlman, D.M. Neuroinflammation and white matter pathology in schizophrenia: Systematic review. Schizophr. Res. 2015, 161, 102–112. [Google Scholar] [CrossRef]
- Enwright Iii, J.F.; Huo, Z.; Arion, D.; Corradi, J.P.; Tseng, G.; Lewis, D.A. Transcriptome alterations of prefrontal cortical parvalbumin neurons in schizophrenia. Mol. Psychiatry 2018, 23, 1606–1613. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.; Hanley, N.; Campbell, M. Blood-brain barrier associated tight junction disruption is a hallmark feature of major psychiatric disorders. Transl. Psychiatry 2020, 10, 373. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, R.J.S.; Chouinard-Watkins, R.; Bazinet, R.P. Brain docosahexaenoic acid uptake and metabolism. Mol. Asp. Med. 2018, 64, 109–134. [Google Scholar] [CrossRef] [PubMed]
- Ahishali, B.; Kaya, M. Evaluation of Blood-Brain Barrier Integrity Using Vascular Permeability Markers: Evans Blue, Sodium Fluorescein, Albumin-Alexa Fluor Conjugates, and Horseradish Peroxidase. Methods Mol. Biol. 2021, 2367, 87–103. [Google Scholar] [CrossRef]
- Sharma, K.; Zhang, Y.; Paudel, K.R.; Kachelmeier, A.; Hansbro, P.M.; Shi, X. The Emerging Role of Pericyte-Derived Extracellular Vesicles in Vascular and Neurological Health. Cells 2022, 11, 3108. [Google Scholar] [CrossRef]
- Stanca, S.; Rossetti, M.; Bokulic Panichi, L.; Bongioanni, P. The Cellular Dysfunction of the Brain-Blood Barrier from Endothelial Cells to Astrocytes: The Pathway towards Neurotransmitter Impairment in Schizophrenia. Int. J. Mol. Sci. 2024, 25, 1250. [Google Scholar] [CrossRef]
- Webster, M.J.; Knable, M.B.; Johnston-Wilson, N.; Nagata, K.; Inagaki, M.; Yolken, R.H. Immunohistochemical localization of phosphorylated glial fibrillary acidic protein in the prefrontal cortex and hippocampus from patients with schizophrenia, bipolar disorder, and depression. Brain Behav. Immun. 2001, 15, 388–400. [Google Scholar] [CrossRef]
- Webster, M.J.; O’Grady, J.; Kleinman, J.E.; Weickert, C.S. Glial fibrillary acidic protein mRNA levels in the cingulate cortex of individuals with depression, bipolar disorder and schizophrenia. Neuroscience 2005, 133, 453–461. [Google Scholar] [CrossRef]
- Dalamaga, M.; Kounatidis, D.; Tsilingiris, D.; Vallianou, N.G.; Karampela, I.; Psallida, S.; Papavassiliou, A.G. The Role of Endocrine Disruptors Bisphenols and Phthalates in Obesity: Current Evidence, Perspectives and Controversies. Int. J. Mol. Sci. 2024, 25, 675. [Google Scholar] [CrossRef]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front. Cell Neurosci. 2021, 15, 661838. [Google Scholar] [CrossRef]
- Lee, C.Y.; Hyun, S.A.; Ko, M.Y.; Kim, H.R.; Rho, J.; Kim, K.K.; Kim, W.Y.; Ka, M. Maternal Bisphenol A (BPA) Exposure Alters Cerebral Cortical Morphogenesis and Synaptic Function in Mice. Cereb. Cortex 2021, 31, 5598–5612. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.H.; Wu, N.; Yuan, X.B. Toward a Better Understanding of Neuronal Migration Deficits in Autism Spectrum Disorders. Front. Cell Dev. Biol. 2019, 7, 205. [Google Scholar] [CrossRef] [PubMed]
- Furman, J.L.; Norris, C.M. Calcineurin and glial signaling: Neuroinflammation and beyond. J. Neuroinflammation 2014, 11, 158. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.E.; Imura, T.; Song, B.; Qi, J.; Ao, Y.; Nguyen, T.K.; Korsak, R.A.; Takeda, K.; Akira, S.; Sofroniew, M.V. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J. Neurosci. 2008, 28, 7231–7243. [Google Scholar] [CrossRef]
- Ceyzériat, K.; Abjean, L.; Carrillo-de Sauvage, M.A.; Ben Haim, L.; Escartin, C. The complex STATes of astrocyte reactivity: How are they controlled by the JAK-STAT3 pathway? Neuroscience 2016, 330, 205–218. [Google Scholar] [CrossRef]
- Roy Choudhury, G.; Ryou, M.G.; Poteet, E.; Wen, Y.; He, R.; Sun, F.; Yuan, F.; Jin, K.; Yang, S.H. Involvement of p38 MAPK in reactive astrogliosis induced by ischemic stroke. Brain Res. 2014, 1551, 45–58. [Google Scholar] [CrossRef]
- Simpson, I.A.; Chundu, K.R.; Davies-Hill, T.; Honer, W.G.; Davies, P. Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann. Neurol. 1994, 35, 546–551. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef]
- Pfrieger, F.W.; Ungerer, N. Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 2011, 50, 357–371. [Google Scholar] [CrossRef]
- Somjen, G.G. Ion regulation in the brain: Implications for pathophysiology. Neuroscientist 2002, 8, 254–267. [Google Scholar] [CrossRef]
- Silver, I.A.; Deas, J.; Erecińska, M. Ion homeostasis in brain cells: Differences in intracellular ion responses to energy limitation between cultured neurons and glial cells. Neuroscience 1997, 78, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.F.; Sytwu, H.K.; Lung, F.W. Human Aquaporin 4 Gene Polymorphisms and Haplotypes Are Associated With Serum S100B Level and Negative Symptoms of Schizophrenia in a Southern Chinese Han Population. Front. Psychiatry 2018, 9, 657. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.S.; Lung, F.W. Different impacts of aquaporin 4 and MAOA allele variation among olanzapine, risperidone, and paliperidone in schizophrenia. J. Clin. Psychopharmacol. 2012, 32, 394–397. [Google Scholar] [CrossRef]
- Amiry-Moghaddam, M.; Ottersen, O.P. The molecular basis of water transport in the brain. Nat. Rev. Neurosci. 2003, 4, 991–1001. [Google Scholar] [CrossRef]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef]
- Brandebura, A.N.; Paumier, A.; Onur, T.S.; Allen, N.J. Astrocyte contribution to dysfunction, risk and progression in neurodegenerative disorders. Nat. Rev. Neurosci. 2023, 24, 23–39. [Google Scholar] [CrossRef]
- Hoover, W.B.; Vertes, R.P. Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct. Funct. 2007, 212, 149–179. [Google Scholar] [CrossRef]
- Jin, J.; Maren, S. Prefrontal-Hippocampal Interactions in Memory and Emotion. Front. Syst. Neurosci. 2015, 9, 170. [Google Scholar] [CrossRef]
- Lander, S.S.; Chornyy, S.; Safory, H.; Gross, A.; Wolosker, H.; Gaisler-Salomon, I. Glutamate dehydrogenase deficiency disrupts glutamate homeostasis in hippocampus and prefrontal cortex and impairs recognition memory. Genes Brain Behav. 2020, 19, e12636. [Google Scholar] [CrossRef]
- Asraf, K.; Zaidan, H.; Natoor, B.; Gaisler-Salomon, I. Synergistic, long-term effects of glutamate dehydrogenase 1 deficiency and mild stress on cognitive function and mPFC gene and miRNA expression. Transl. Psychiatry 2023, 13, 248. [Google Scholar] [CrossRef] [PubMed]
- Dittlau, K.S.; Chandrasekaran, A.; Freude, K.; Van Den Bosch, L. Generation of Human Induced Pluripotent Stem Cell (hiPSC)-Derived Astrocytes for Amyotrophic Lateral Sclerosis and Other Neurodegenerative Disease Studies. Bio Protoc. 2024, 14, e4936. [Google Scholar] [CrossRef]
- Huang, J.; Li, C.; Shang, H. Astrocytes in Neurodegeneration: Inspiration From Genetics. Front. Neurosci. 2022, 16, 882316. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef]
- van Os, J.; Kapur, S. Schizophrenia. Lancet 2009, 374, 635–645. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Folsom, T.D. The neurodevelopmental hypothesis of schizophrenia, revisited. Schizophr. Bull. 2009, 35, 528–548. [Google Scholar] [CrossRef]
- Popovic, D.; Schmitt, A.; Kaurani, L.; Senner, F.; Papiol, S.; Malchow, B.; Fischer, A.; Schulze, T.G.; Koutsouleris, N.; Falkai, P. Childhood Trauma in Schizophrenia: Current Findings and Research Perspectives. Front. Neurosci. 2019, 13, 274. [Google Scholar] [CrossRef]
- van Winkel, R.; Stefanis, N.C.; Myin-Germeys, I. Psychosocial stress and psychosis. A review of the neurobiological mechanisms and the evidence for gene-stress interaction. Schizophr. Bull. 2008, 34, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Misiak, B.; Moustafa, A.A.; Kiejna, A.; Frydecka, D. Childhood traumatic events and types of auditory verbal hallucinations in first-episode schizophrenia patients. Compr. Psychiatry 2016, 66, 17–22. [Google Scholar] [CrossRef]
- Bernstein, H.G.; Steiner, J.; Guest, P.C.; Dobrowolny, H.; Bogerts, B. Glial cells as key players in schizophrenia pathology: Recent insights and concepts of therapy. Schizophr. Res. 2015, 161, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Scarante, F.F.; Ribeiro, M.A.; Almeida-Santos, A.F.; Guimarães, F.S.; Campos, A.C. Glial Cells and Their Contribution to the Mechanisms of Action of Cannabidiol in Neuropsychiatric Disorders. Front. Pharmacol. 2020, 11, 618065. [Google Scholar] [CrossRef]
- Wang, X.; Fu, J.; Wang, H.; Liu, C.; Zhang, Y.; Song, C.; Wang, C. Glia dysfunction in schizophrenia: Evidence of possible therapeutic effects of nervonic acid in a preclinical model. Psychopharmacology 2024, 241, 2271–2287. [Google Scholar] [CrossRef]
- Purves-Tyson, T.D.; Robinson, K.; Brown, A.M.; Boerrigter, D.; Cai, H.Q.; Weissleder, C.; Owens, S.J.; Rothmond, D.A.; Shannon Weickert, C. Increased Macrophages and C1qA, C3, C4 Transcripts in the Midbrain of People With Schizophrenia. Front. Immunol. 2020, 11, 2002. [Google Scholar] [CrossRef]
- Liu, S.H.; Du, Y.; Chen, L.; Cheng, Y. Glial Cell Abnormalities in Major Psychiatric Diseases: A Systematic Review of Postmortem Brain Studies. Mol. Neurobiol. 2022, 59, 1665–1692. [Google Scholar] [CrossRef]
- van Kesteren, C.F.; Gremmels, H.; de Witte, L.D.; Hol, E.M.; Van Gool, A.R.; Falkai, P.G.; Kahn, R.S.; Sommer, I.E. Immune involvement in the pathogenesis of schizophrenia: A meta-analysis on postmortem brain studies. Transl. Psychiatry 2017, 7, e1075. [Google Scholar] [CrossRef]
- Esshili, A.; Manitz, M.P.; Freund, N.; Juckel, G. Induction of inducible nitric oxide synthase expression in activated microglia and astrocytes following pre- and postnatal immune challenge in an animal model of schizophrenia. Eur. Neuropsychopharmacol. 2020, 35, 100–110. [Google Scholar] [CrossRef]
- Francisco, R.D.; Fernando, V.; Norma, E.; Madai, M.E.; Marcelo, B. Glial changes in schizophrenia: Genetic and epigenetic approach. Indian. J. Psychiatry 2022, 64, 3–12. [Google Scholar] [CrossRef]
- Yeung, R.K.; Xiang, Z.H.; Tsang, S.Y.; Li, R.; Ho, T.Y.C.; Li, Q.; Hui, C.K.; Sham, P.C.; Qiao, M.Q.; Xue, H. Gabrb2-knockout mice displayed schizophrenia-like and comorbid phenotypes with interneuron-astrocyte-microglia dysregulation. Transl. Psychiatry 2018, 8, 128. [Google Scholar] [CrossRef] [PubMed]
- de Bartolomeis, A.; Tomasetti, C. Calcium-dependent networks in dopamine-glutamate interaction: The role of postsynaptic scaffolding proteins. Mol. Neurobiol. 2012, 46, 275–296. [Google Scholar] [CrossRef] [PubMed]
- Bayés, À.; Collins, M.O.; Reig-Viader, R.; Gou, G.; Goulding, D.; Izquierdo, A.; Choudhary, J.S.; Emes, R.D.; Grant, S.G. Evolution of complexity in the zebrafish synapse proteome. Nat. Commun. 2017, 8, 14613. [Google Scholar] [CrossRef] [PubMed]
- Dosemeci, A.; Weinberg, R.J.; Reese, T.S.; Tao-Cheng, J.H. The Postsynaptic Density: There Is More than Meets the Eye. Front. Synaptic Neurosci. 2016, 8, 23. [Google Scholar] [CrossRef]
- Berdenis van Berlekom, A.; Muflihah, C.H.; Snijders, G.; MacGillavry, H.D.; Middeldorp, J.; Hol, E.M.; Kahn, R.S.; de Witte, L.D. Synapse Pathology in Schizophrenia: A Meta-analysis of Postsynaptic Elements in Postmortem Brain Studies. Schizophr. Bull. 2020, 46, 374–386. [Google Scholar] [CrossRef]
- Osimo, E.F.; Beck, K.; Reis Marques, T.; Howes, O.D. Synaptic loss in schizophrenia: A meta-analysis and systematic review of synaptic protein and mRNA measures. Mol. Psychiatry 2019, 24, 549–561. [Google Scholar] [CrossRef]
- Kaizuka, T.; Takumi, T. Postsynaptic density proteins and their involvement in neurodevelopmental disorders. J. Biochem. 2018, 163, 447–455. [Google Scholar] [CrossRef]
- de Bartolomeis, A.; Aloj, L.; Ambesi-Impiombato, A.; Bravi, D.; Caracò, C.; Muscettola, G.; Barone, P. Acute administration of antipsychotics modulates Homer striatal gene expression differentially. Brain Res. Mol. Brain Res. 2002, 98, 124–129. [Google Scholar] [CrossRef]
- Buonaguro, E.F.; Iasevoli, F.; Marmo, F.; Eramo, A.; Latte, G.; Avagliano, C.; Tomasetti, C.; de Bartolomeis, A. Re-arrangements of gene transcripts at glutamatergic synapses after prolonged treatments with antipsychotics: A putative link with synaptic remodeling. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 76, 29–41. [Google Scholar] [CrossRef]
- Iasevoli, F.; Tomasetti, C.; Marmo, F.; Bravi, D.; Arnt, J.; de Bartolomeis, A. Divergent acute and chronic modulation of glutamatergic postsynaptic density genes expression by the antipsychotics haloperidol and sertindole. Psychopharmacology 2010, 212, 329–344. [Google Scholar] [CrossRef]
- de Bartolomeis, A.; Iasevoli, F.; Marmo, F.; Buonaguro, E.F.; Eramo, A.; Rossi, R.; Avvisati, L.; Latte, G.; Tomasetti, C. Progressive recruitment of cortical and striatal regions by inducible postsynaptic density transcripts after increasing doses of antipsychotics with different receptor profiles: Insights for psychosis treatment. Eur. Neuropsychopharmacol. 2015, 25, 566–582. [Google Scholar] [CrossRef] [PubMed]
- Buonaguro, E.F.; Tomasetti, C.; Chiodini, P.; Marmo, F.; Latte, G.; Rossi, R.; Avvisati, L.; Iasevoli, F.; de Bartolomeis, A. Postsynaptic density protein transcripts are differentially modulated by minocycline alone or in add-on to haloperidol: Implications for treatment resistant schizophrenia. J. Psychopharmacol. 2017, 31, 406–417. [Google Scholar] [CrossRef] [PubMed]
- de Bartolomeis, A.; Iasevoli, F.; Marmo, F.; Buonaguro, E.F.; Avvisati, L.; Latte, G.; Tomasetti, C. Nicotine and caffeine modulate haloperidol-induced changes in postsynaptic density transcripts expression: Translational insights in psychosis therapy and treatment resistance. Eur. Neuropsychopharmacol. 2018, 28, 538–559. [Google Scholar] [CrossRef] [PubMed]
- de Bartolomeis, A.; Avagliano, C.; Vellucci, L.; D’Ambrosio, L.; Manchia, M.; D’Urso, G.; Buonaguro, E.F.; Iasevoli, F. Translating preclinical findings in clinically relevant new antipsychotic targets: Focus on the glutamatergic postsynaptic density. Implications for treatment resistant schizophrenia. Neurosci. Biobehav. Rev. 2019, 107, 795–827. [Google Scholar] [CrossRef]
- Pierre, W.C.; Londono, I.; Quiniou, C.; Chemtob, S.; Lodygensky, G.A. Modulatory effect of IL-1 inhibition following lipopolysaccharide-induced neuroinflammation in neonatal microglia and astrocytes. Int. J. Dev. Neurosci. 2022, 82, 243–260. [Google Scholar] [CrossRef]
- Banker, G.; Churchill, L.; Cotman, C.W. Proteins of the postsynaptic density. J. Cell Biol. 1974, 63, 456–465. [Google Scholar] [CrossRef]
- Granger, A.J.; Nicoll, R.A. Expression mechanisms underlying long-term potentiation: A postsynaptic view, 10 years on. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130136. [Google Scholar] [CrossRef]
- Berretta, S. Extracellular matrix abnormalities in schizophrenia. Neuropharmacology 2012, 62, 1584–1597. [Google Scholar] [CrossRef]
- Chelini, G.; Pantazopoulos, H.; Durning, P.; Berretta, S. The tetrapartite synapse: A key concept in the pathophysiology of schizophrenia. Eur. Psychiatry 2018, 50, 60–69. [Google Scholar] [CrossRef]
- Ventura, R.; Harris, K.M. Three-dimensional relationships between hippocampal synapses and astrocytes. J. Neurosci. 1999, 19, 6897–6906. [Google Scholar] [CrossRef]
- Witcher, M.R.; Kirov, S.A.; Harris, K.M. Plasticity of perisynaptic astroglia during synaptogenesis in the mature rat hippocampus. Glia 2007, 55, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Halassa, M.M.; Haydon, P.G. Integrated brain circuits: Astrocytic networks modulate neuronal activity and behavior. Annu. Rev. Physiol. 2010, 72, 335–355. [Google Scholar] [CrossRef] [PubMed]
- Charles, A.C.; Merrill, J.E.; Dirksen, E.R.; Sanderson, M.J. Intercellular signaling in glial cells: Calcium waves and oscillations in response to mechanical stimulation and glutamate. Neuron 1991, 6, 983–992. [Google Scholar] [CrossRef]
- Harada, K.; Kamiya, T.; Tsuboi, T. Gliotransmitter Release from Astrocytes: Functional, Developmental, and Pathological Implications in the Brain. Front. Neurosci. 2015, 9, 499. [Google Scholar] [CrossRef]
- Rivera, A.; Vanzulli, I.; Butt, A.M. A Central Role for ATP Signalling in Glial Interactions in the CNS. Curr. Drug Targets 2016, 17, 1829–1833. [Google Scholar] [CrossRef]
- Syková, E.; Nicholson, C. Diffusion in brain extracellular space. Physiol. Rev. 2008, 88, 1277–1340. [Google Scholar] [CrossRef]
- Evers, M.R.; Salmen, B.; Bukalo, O.; Rollenhagen, A.; Bösl, M.R.; Morellini, F.; Bartsch, U.; Dityatev, A.; Schachner, M. Impairment of L-type Ca2+ channel-dependent forms of hippocampal synaptic plasticity in mice deficient in the extracellular matrix glycoprotein tenascin-C. J. Neurosci. 2002, 22, 7177–7194. [Google Scholar] [CrossRef]
- Dityatev, A.; Schachner, M.; Sonderegger, P. The dual role of the extracellular matrix in synaptic plasticity and homeostasis. Nat. Rev. Neurosci. 2010, 11, 735–746. [Google Scholar] [CrossRef]
- Pantazopoulos, H.; Woo, T.U.; Lim, M.P.; Lange, N.; Berretta, S. Extracellular matrix-glial abnormalities in the amygdala and entorhinal cortex of subjects diagnosed with schizophrenia. Arch. Gen. Psychiatry 2010, 67, 155–166. [Google Scholar] [CrossRef]
- Pantazopoulos, H.; Markota, M.; Jaquet, F.; Ghosh, D.; Wallin, A.; Santos, A.; Caterson, B.; Berretta, S. Aggrecan and chondroitin-6-sulfate abnormalities in schizophrenia and bipolar disorder: A postmortem study on the amygdala. Transl. Psychiatry 2015, 5, e496. [Google Scholar] [CrossRef]
- Koskuvi, M.; Lehtonen, Š.; Trontti, K.; Keuters, M.; Wu, Y.C.; Koivisto, H.; Ludwig, A.; Plotnikova, L.; Virtanen, P.L.J.; Räsänen, N.; et al. Contribution of astrocytes to familial risk and clinical manifestation of schizophrenia. Glia 2022, 70, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Mohan, V.; Wade, S.D.; Sullivan, C.S.; Kasten, M.R.; Sweetman, C.; Stewart, R.; Truong, Y.; Schachner, M.; Manis, P.B.; Maness, P.F. Close Homolog of L1 Regulates Dendritic Spine Density in the Mouse Cerebral Cortex Through Semaphorin 3B. J. Neurosci. 2019, 39, 6233–6250. [Google Scholar] [CrossRef] [PubMed]
- Boksa, P. Abnormal synaptic pruning in schizophrenia: Urban myth or reality? J. Psychiatry Neurosci. 2012, 37, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Keshavan, M.; Lizano, P.; Prasad, K. The synaptic pruning hypothesis of schizophrenia: Promises and challenges. World Psychiatry 2020, 19, 110–111. [Google Scholar] [CrossRef]
- Sakai, J. Core Concept: How synaptic pruning shapes neural wiring during development and, possibly, in disease. Proc. Natl. Acad. Sci. USA 2020, 117, 16096–16099. [Google Scholar] [CrossRef]
- Sellgren, C.M.; Gracias, J.; Watmuff, B.; Biag, J.D.; Thanos, J.M.; Whittredge, P.B.; Fu, T.; Worringer, K.; Brown, H.E.; Wang, J.; et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat. Neurosci. 2019, 22, 374–385. [Google Scholar] [CrossRef]
- Bosworth, A.P.; Allen, N.J. The diverse actions of astrocytes during synaptic development. Curr. Opin. Neurobiol. 2017, 47, 38–43. [Google Scholar] [CrossRef]
- Chung, W.S.; Clarke, L.E.; Wang, G.X.; Stafford, B.K.; Sher, A.; Chakraborty, C.; Joung, J.; Foo, L.C.; Thompson, A.; Chen, C.; et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 2013, 504, 394–400. [Google Scholar] [CrossRef]
- Iino, M.; Goto, K.; Kakegawa, W.; Okado, H.; Sudo, M.; Ishiuchi, S.; Miwa, A.; Takayasu, Y.; Saito, I.; Tsuzuki, K.; et al. Glia-synapse interaction through Ca2+-permeable AMPA receptors in Bergmann glia. Science 2001, 292, 926–929. [Google Scholar] [CrossRef]
- Vainchtein, I.D.; Chin, G.; Cho, F.S.; Kelley, K.W.; Miller, J.G.; Chien, E.C.; Liddelow, S.A.; Nguyen, P.T.; Nakao-Inoue, H.; Dorman, L.C.; et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 2018, 359, 1269–1273. [Google Scholar] [CrossRef]
- Yang, J.; Yang, H.; Liu, Y.; Li, X.; Qin, L.; Lou, H.; Duan, S.; Wang, H. Astrocytes contribute to synapse elimination via type 2 inositol 1,4,5-trisphosphate receptor-dependent release of ATP. eLife 2016, 5, e15043. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.M.; Beach, M.G.; Porpiglia, E.; Sheehan, A.E.; Watts, R.J.; Freeman, M.R. The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron 2006, 50, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Ziegenfuss, J.S.; Biswas, R.; Avery, M.A.; Hong, K.; Sheehan, A.E.; Yeung, Y.G.; Stanley, E.R.; Freeman, M.R. Draper-dependent glial phagocytic activity is mediated by Src and Syk family kinase signalling. Nature 2008, 453, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Oliet, S.H.; Mothet, J.P. Regulation of N-methyl-D-aspartate receptors by astrocytic D-serine. Neuroscience 2009, 158, 275–283. [Google Scholar] [CrossRef]
- Fossat, P.; Turpin, F.R.; Sacchi, S.; Dulong, J.; Shi, T.; Rivet, J.M.; Sweedler, J.V.; Pollegioni, L.; Millan, M.J.; Oliet, S.H.; et al. Glial D-serine gates NMDA receptors at excitatory synapses in prefrontal cortex. Cereb. Cortex 2012, 22, 595–606. [Google Scholar] [CrossRef]
- Henneberger, C.; Papouin, T.; Oliet, S.H.; Rusakov, D.A. Long-term potentiation depends on release of D-serine from astrocytes. Nature 2010, 463, 232–236. [Google Scholar] [CrossRef]
- Mothet, J.P.; Rouaud, E.; Sinet, P.M.; Potier, B.; Jouvenceau, A.; Dutar, P.; Videau, C.; Epelbaum, J.; Billard, J.M. A critical role for the glial-derived neuromodulator D-serine in the age-related deficits of cellular mechanisms of learning and memory. Aging Cell 2006, 5, 267–274. [Google Scholar] [CrossRef]
- Panatier, A.; Theodosis, D.T.; Mothet, J.P.; Touquet, B.; Pollegioni, L.; Poulain, D.A.; Oliet, S.H. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell 2006, 125, 775–784. [Google Scholar] [CrossRef]
- Diniz, L.P.; Almeida, J.C.; Tortelli, V.; Vargas Lopes, C.; Setti-Perdigão, P.; Stipursky, J.; Kahn, S.A.; Romão, L.F.; de Miranda, J.; Alves-Leon, S.V.; et al. Astrocyte-induced synaptogenesis is mediated by transforming growth factor β signaling through modulation of D-serine levels in cerebral cortex neurons. J. Biol. Chem. 2012, 287, 41432–41445. [Google Scholar] [CrossRef]
- Packard, M.; Mathew, D.; Budnik, V. Wnts and TGF beta in synaptogenesis: Old friends signalling at new places. Nat. Rev. Neurosci. 2003, 4, 113–120. [Google Scholar] [CrossRef]
- Morita, Y.; Ujike, H.; Tanaka, Y.; Otani, K.; Kishimoto, M.; Morio, A.; Kotaka, T.; Okahisa, Y.; Matsushita, M.; Morikawa, A.; et al. A genetic variant of the serine racemase gene is associated with schizophrenia. Biol. Psychiatry 2007, 61, 1200–1203. [Google Scholar] [CrossRef] [PubMed]
- Boks, M.P.; Rietkerk, T.; van de Beek, M.H.; Sommer, I.E.; de Koning, T.J.; Kahn, R.S. Reviewing the role of the genes G72 and DAAO in glutamate neurotransmission in schizophrenia. Eur. Neuropsychopharmacol. 2007, 17, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Müller, D.J.; Zai, C.C.; Shinkai, T.; Strauss, J.; Kennedy, J.L. Association between the DAOA/G72 gene and bipolar disorder and meta-analyses in bipolar disorder and schizophrenia. Bipolar Disord. 2011, 13, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Burnet, P.W.; Hutchinson, L.; von Hesling, M.; Gilbert, E.J.; Brandon, N.J.; Rutter, A.R.; Hutson, P.H.; Harrison, P.J. Expression of D-serine and glycine transporters in the prefrontal cortex and cerebellum in schizophrenia. Schizophr. Res. 2008, 102, 283–294. [Google Scholar] [CrossRef]
- Bendikov, I.; Nadri, C.; Amar, S.; Panizzutti, R.; De Miranda, J.; Wolosker, H.; Agam, G. A CSF and postmortem brain study of D-serine metabolic parameters in schizophrenia. Schizophr. Res. 2007, 90, 41–51. [Google Scholar] [CrossRef]
- Hashimoto, K.; Fukushima, T.; Shimizu, E.; Komatsu, N.; Watanabe, H.; Shinoda, N.; Nakazato, M.; Kumakiri, C.; Okada, S.; Hasegawa, H.; et al. Decreased serum levels of D-serine in patients with schizophrenia: Evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch. Gen. Psychiatry 2003, 60, 572–576. [Google Scholar] [CrossRef]
- Garofalo, M.; De Simone, G.; Motta, Z.; Nuzzo, T.; De Grandis, E.; Bruno, C.; Boeri, S.; Riccio, M.P.; Pastore, L.; Bravaccio, C.; et al. Decreased free D-aspartate levels in the blood serum of patients with schizophrenia. Front. Psychiatry 2024, 15, 1408175. [Google Scholar] [CrossRef]
- De Rosa, A.; Fontana, A.; Nuzzo, T.; Garofalo, M.; Di Maio, A.; Punzo, D.; Copetti, M.; Bertolino, A.; Errico, F.; Rampino, A.; et al. Machine Learning algorithm unveils glutamatergic alterations in the post-mortem schizophrenia brain. Schizophrenia 2022, 8, 8. [Google Scholar] [CrossRef]
- de Bartolomeis, A.; Vellucci, L.; Austin, M.C.; De Simone, G.; Barone, A. Rational and Translational Implications of D-Amino Acids for Treatment-Resistant Schizophrenia: From Neurobiology to the Clinics. Biomolecules 2022, 12, 909. [Google Scholar] [CrossRef]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef]
- Youdim, M.B.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Aras, R.; Christian, W.V.; Rappold, P.M.; Hatwar, M.; Panza, J.; Jackson-Lewis, V.; Javitch, J.A.; Ballatori, N.; Przedborski, S.; et al. The organic cation transporter-3 is a pivotal modulator of neurodegeneration in the nigrostriatal dopaminergic pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 8043–8048. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.; Naganuma, F.; Iida, T.; Nakamura, T.; Harada, R.; Mohsen, A.S.; Kasajima, A.; Sasano, H.; Yanai, K. Molecular mechanism of histamine clearance by primary human astrocytes. Glia 2013, 61, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Zehnder, T.; Pucci, L.; Cali, C.; Bondiolotti, B.M.; Perez, A.M.; Dallerac, G.; Déglon, N.; Giros, B.; Magara, F.; et al. Astrocytic VMAT2 in the developing prefrontal cortex is required for normal grooming behavior in mice. bioRxiv 2021. [Google Scholar] [CrossRef]
- Paterlini, M.; Zakharenko, S.S.; Lai, W.S.; Qin, J.; Zhang, H.; Mukai, J.; Westphal, K.G.; Olivier, B.; Sulzer, D.; Pavlidis, P.; et al. Transcriptional and behavioral interaction between 22q11.2 orthologs modulates schizophrenia-related phenotypes in mice. Nat. Neurosci. 2005, 8, 1586–1594. [Google Scholar] [CrossRef]
- Bezerra, T.O.; Roque, A.C. Dopamine facilitates the response to glutamatergic inputs in astrocyte cell models. PLoS Comput. Biol. 2024, 20, e1012688. [Google Scholar] [CrossRef]
- Lee, H.G.; Wheeler, M.A.; Quintana, F.J. Function and therapeutic value of astrocytes in neurological diseases. Nat. Rev. Drug Discov. 2022, 21, 339–358. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef]
- Khakh, B.S.; Deneen, B. The Emerging Nature of Astrocyte Diversity. Annu. Rev. Neurosci. 2019, 42, 187–207. [Google Scholar] [CrossRef]
- Hochstim, C.; Deneen, B.; Lukaszewicz, A.; Zhou, Q.; Anderson, D.J. Identification of positionally distinct astrocyte subtypes whose identities are specified by a homeodomain code. Cell 2008, 133, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; Kelley, K.W.; Tsai, H.H.; Redmond, S.A.; Chang, S.M.; Madireddy, L.; Chan, J.R.; Baranzini, S.E.; Ullian, E.M.; Rowitch, D.H. Astrocyte-encoded positional cues maintain sensorimotor circuit integrity. Nature 2014, 509, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.H.; Li, H.; Fuentealba, L.C.; Molofsky, A.V.; Taveira-Marques, R.; Zhuang, H.; Tenney, A.; Murnen, A.T.; Fancy, S.P.; Merkle, F.; et al. Regional astrocyte allocation regulates CNS synaptogenesis and repair. Science 2012, 337, 358–362. [Google Scholar] [CrossRef]
- Skene, N.G.; Roy, M.; Grant, S.G. A genomic lifespan program that reorganises the young adult brain is targeted in schizophrenia. eLife 2017, 6, e17915. [Google Scholar] [CrossRef]
- Li, R.; Ma, X.; Wang, G.; Yang, J.; Wang, C. Why sex differences in schizophrenia? J. Transl. Neurosci. 2016, 1, 37–42. [Google Scholar]
- Dietz, A.G.; Goldman, S.A.; Nedergaard, M. Glial cells in schizophrenia: A unified hypothesis. Lancet Psychiatry 2020, 7, 272–281. [Google Scholar] [CrossRef]
- Khandaker, G.M.; Cousins, L.; Deakin, J.; Lennox, B.R.; Yolken, R.; Jones, P.B. Inflammation and immunity in schizophrenia: Implications for pathophysiology and treatment. Lancet Psychiatry 2015, 2, 258–270. [Google Scholar] [CrossRef]
- Szabo, A.; Akkouh, I.A.; Vandenberghe, M.; Osete, J.R.; Hughes, T.; Heine, V.; Smeland, O.B.; Glover, J.C.; Andreassen, O.A.; Djurovic, S. A human iPSC-astroglia neurodevelopmental model reveals divergent transcriptomic patterns in schizophrenia. Transl. Psychiatry 2021, 11, 554. [Google Scholar] [CrossRef]
- Mena, A.; Ruiz-Salas, J.C.; Puentes, A.; Dorado, I.; Ruiz-Veguilla, M.; De la Casa, L.G. Reduced Prepulse Inhibition as a Biomarker of Schizophrenia. Front. Behav. Neurosci. 2016, 10, 202. [Google Scholar] [CrossRef]
- Baev, A.Y.; Vinokurov, A.Y.; Novikova, I.N.; Dremin, V.V.; Potapova, E.V.; Abramov, A.Y. Interaction of Mitochondrial Calcium and ROS in Neurodegeneration. Cells 2022, 11, 706. [Google Scholar] [CrossRef]
- Chavda, V.; Singh, K.; Patel, V.; Mishra, M.; Mishra, A.K. Neuronal Glial Crosstalk: Specific and Shared Mechanisms in Alzheimer’s Disease. Brain Sci. 2022, 12, 75. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, R.A.L. Reactive gliosis in Alzheimer’s disease: A crucial role for cognitive impairment and memory loss. Metab. Brain Dis. 2022, 37, 851–857. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Marsh, S.E.; Stevens, B. Microglia and Astrocytes in Disease: Dynamic Duo or Partners in Crime? Trends Immunol. 2020, 41, 820–835. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrogliosis. Cold Spring Harb. Perspect. Biol. 2014, 7, a020420. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol. 2020, 41, 758–770. [Google Scholar] [CrossRef]
- Burnet, P.W.; Anderson, P.N.; Chen, L.; Nikiforova, N.; Harrison, P.J.; Wood, M.J. D-amino acid oxidase knockdown in the mouse cerebellum reduces NR2A mRNA. Mol. Cell Neurosci. 2011, 46, 167–175. [Google Scholar] [CrossRef]
- Papouin, T.; Ladépêche, L.; Ruel, J.; Sacchi, S.; Labasque, M.; Hanini, M.; Groc, L.; Pollegioni, L.; Mothet, J.P.; Oliet, S.H. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell 2012, 150, 633–646. [Google Scholar] [CrossRef]
- van Zundert, B.; Yoshii, A.; Constantine-Paton, M. Receptor compartmentalization and trafficking at glutamate synapses: A developmental proposal. Trends Neurosci. 2004, 27, 428–437. [Google Scholar] [CrossRef]
- Yashiro, K.; Philpot, B.D. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology 2008, 55, 1081–1094. [Google Scholar] [CrossRef]
- Allen, N.J.; Bennett, M.L.; Foo, L.C.; Wang, G.X.; Chakraborty, C.; Smith, S.J.; Barres, B.A. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 2012, 486, 410–414. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Figueiredo, E.C.; Calì, C.; Petrelli, F.; Bezzi, P. Emerging evidence for astrocyte dysfunction in schizophrenia. Glia 2022, 70, 1585–1604. [Google Scholar] [CrossRef] [PubMed]
- Farhy-Tselnicker, I.; van Casteren, A.C.M.; Lee, A.; Chang, V.T.; Aricescu, A.R.; Allen, N.J. Astrocyte-Secreted Glypican 4 Regulates Release of Neuronal Pentraxin 1 from Axons to Induce Functional Synapse Formation. Neuron 2017, 96, 428–445.e13. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Woo, J.; Cristobal, C.D.; Choi, J.M.; Wang, C.Y.; Ye, Q.; Smith, J.A.; Ung, K.; Liu, G.; Cortes, D.; et al. Regional heterogeneity of astrocyte morphogenesis dictated by the formin protein, Daam2, modifies circuit function. EMBO Rep. 2021, 22, e53200. [Google Scholar] [CrossRef]
- Clarke, L.E.; Barres, B.A. Emerging roles of astrocytes in neural circuit development. Nat. Rev. Neurosci. 2013, 14, 311–321. [Google Scholar] [CrossRef]
- Schmitz, I.; da Silva, A.; Bobermin, L.D.; Gonçalves, C.A.; Steiner, J.; Quincozes-Santos, A. The Janus face of antipsychotics in glial cells: Focus on glioprotection. Exp. Biol. Med. 2023, 248, 2120–2130. [Google Scholar] [CrossRef]
- de Bartolomeis, A.; Barone, A.; Vellucci, L.; Mazza, B.; Austin, M.C.; Iasevoli, F.; Ciccarelli, M. Linking Inflammation, Aberrant Glutamate-Dopamine Interaction, and Post-synaptic Changes: Translational Relevance for Schizophrenia and Antipsychotic Treatment: A Systematic Review. Mol. Neurobiol. 2022, 59, 6460–6501. [Google Scholar] [CrossRef]
- Heider, J.; Vogel, S.; Volkmer, H.; Breitmeyer, R. Human iPSC-Derived Glia as a Tool for Neuropsychiatric Research and Drug Development. Int. J. Mol. Sci. 2021, 22, 10254. [Google Scholar] [CrossRef]
- Khan, Z.U.; Koulen, P.; Rubinstein, M.; Grandy, D.K.; Goldman-Rakic, P.S. An astroglia-linked dopamine D2-receptor action in prefrontal cortex. Proc. Natl. Acad. Sci. USA 2001, 98, 1964–1969. [Google Scholar] [CrossRef]
- Molofsky, A.V.; Krencik, R.; Ullian, E.M.; Tsai, H.H.; Deneen, B.; Richardson, W.D.; Barres, B.A.; Rowitch, D.H. Astrocytes and disease: A neurodevelopmental perspective. Genes Dev. 2012, 26, 891–907. [Google Scholar] [CrossRef]
- Howes, O.; McCutcheon, R.; Stone, J. Glutamate and dopamine in schizophrenia: An update for the 21st century. J. Psychopharmacol. 2015, 29, 97–115. [Google Scholar] [CrossRef] [PubMed]
- Quincozes-Santos, A.; Santos, C.L.; de Souza Almeida, R.R.; da Silva, A.; Thomaz, N.K.; Costa, N.L.F.; Weber, F.B.; Schmitz, I.; Medeiros, L.S.; Medeiros, L.; et al. Gliotoxicity and Glioprotection: The Dual Role of Glial Cells. Mol. Neurobiol. 2021, 58, 6577–6592. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Okada, M. Effects of Atypical Antipsychotics, Clozapine, Quetiapine and Brexpiprazole on Astroglial Transmission Associated with Connexin43. Int. J. Mol. Sci. 2021, 22, 5623. [Google Scholar] [CrossRef] [PubMed]
- Akkouh, I.A.; Hribkova, H.; Grabiec, M.; Budinska, E.; Szabo, A.; Kasparek, T.; Andreassen, O.A.; Sun, Y.M.; Djurovic, S. Derivation and Molecular Characterization of a Morphological Subpopulation of Human iPSC Astrocytes Reveal a Potential Role in Schizophrenia and Clozapine Response. Schizophr. Bull. 2022, 48, 190–198. [Google Scholar] [CrossRef]
- De Simone, G.; Mazza, B.; Vellucci, L.; Barone, A.; Ciccarelli, M.; de Bartolomeis, A. Schizophrenia Synaptic Pathology and Antipsychotic Treatment in the Framework of Oxidative and Mitochondrial Dysfunction: Translational Highlights for the Clinics and Treatment. Antioxidants 2023, 12, 975. [Google Scholar] [CrossRef]
- Thaakur, S.R.; Jyothi, B. Effect of spirulina maxima on the haloperidol induced tardive dyskinesia and oxidative stress in rats. J. Neural Transm. 2007, 114, 1217–1225. [Google Scholar] [CrossRef]
- Chu, C.S.; Lin, Y.S.; Liang, W.Z. The Impact of the Antipsychotic Medication Chlorpromazine on Cytotoxicity through Ca(2+) Signaling Pathway in Glial Cell Models. Neurotox. Res. 2022, 40, 791–802. [Google Scholar] [CrossRef]
- Hsu, S.S.; Liang, W.Z. Ca2+ signaling as a mechanism of haloperidol-induced cytotoxicity in human astrocytes and assessing the protective role of a Ca2+ chelator. Naunyn Schmiedebergs Arch. Pharmacol. 2020, 393, 2117–2127. [Google Scholar] [CrossRef]
- Smythies, J. Redox mechanisms at the glutamate synapse and their significance: A review. Eur. J. Pharmacol. 1999, 370, 1–7. [Google Scholar] [CrossRef]
- Pillai, A.; Parikh, V.; Terry, A.V., Jr.; Mahadik, S.P. Long-term antipsychotic treatments and crossover studies in rats: Differential effects of typical and atypical agents on the expression of antioxidant enzymes and membrane lipid peroxidation in rat brain. J. Psychiatr. Res. 2007, 41, 372–386. [Google Scholar] [CrossRef]
- Krogmann, A.; Peters, L.; von Hardenberg, L.; Bödeker, K.; Nöhles, V.B.; Correll, C.U. Keeping up with the therapeutic advances in schizophrenia: A review of novel and emerging pharmacological entities. CNS Spectr. 2019, 24, 38–69. [Google Scholar] [CrossRef] [PubMed]
- Dutheil, S.; Watson, L.S.; Davis, R.E.; Snyder, G.L. Lumateperone Normalizes Pathological Levels of Acute Inflammation through Important Pathways Known to Be Involved in Mood Regulation. J. Neurosci. 2023, 43, 863–877. [Google Scholar] [CrossRef] [PubMed]
- Power, S.K.; Venkatesan, S.; Lambe, E.K. Xanomeline restores endogenous nicotinic acetylcholine receptor signaling in mouse prefrontal cortex. Neuropsychopharmacology 2023, 48, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Vallejo-Illarramendi, A.; Torres-Ramos, M.; Melone, M.; Conti, F.; Matute, C. Clozapine reduces GLT-1 expression and glutamate uptake in astrocyte cultures. Glia 2005, 50, 276–279. [Google Scholar] [CrossRef]
- Melone, M.; Bragina, L.; Conti, F. Clozapine-induced reduction of glutamate transport in the frontal cortex is not mediated by GLAST and EAAC1. Mol. Psychiatry 2003, 8, 12–13. [Google Scholar] [CrossRef]
- Kim, R.; Healey, K.L.; Sepulveda-Orengo, M.T.; Reissner, K.J. Astroglial correlates of neuropsychiatric disease: From astrocytopathy to astrogliosis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 87, 126–146. [Google Scholar] [CrossRef]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef]
- Deneen, B.; Ho, R.; Lukaszewicz, A.; Hochstim, C.J.; Gronostajski, R.M.; Anderson, D.J. The transcription factor NFIA controls the onset of gliogenesis in the developing spinal cord. Neuron 2006, 52, 953–968. [Google Scholar] [CrossRef]
- He, F.; Ge, W.; Martinowich, K.; Becker-Catania, S.; Coskun, V.; Zhu, W.; Wu, H.; Castro, D.; Guillemot, F.; Fan, G.; et al. A positive autoregulatory loop of Jak-STAT signaling controls the onset of astrogliogenesis. Nat. Neurosci. 2005, 8, 616–625. [Google Scholar] [CrossRef]
- Kang, P.; Lee, H.K.; Glasgow, S.M.; Finley, M.; Donti, T.; Gaber, Z.B.; Graham, B.H.; Foster, A.E.; Novitch, B.G.; Gronostajski, R.M.; et al. Sox9 and NFIA coordinate a transcriptional regulatory cascade during the initiation of gliogenesis. Neuron 2012, 74, 79–94. [Google Scholar] [CrossRef]
- Bragina, L.; Melone, M.; Fattorini, G.; Torres-Ramos, M.; Vallejo-Illarramendi, A.; Matute, C.; Conti, F. GLT-1 down-regulation induced by clozapine in rat frontal cortex is associated with synaptophysin up-regulation. J. Neurochem. 2006, 99, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Crews, L.; Masliah, E. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum. Mol. Genet. 2010, 19, R12–R20. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Price, A.C.; Kaiser, A.; Shaikh, A.Y.; Liu, Z. Mechanisms for neuronal degeneration in amyotrophic lateral sclerosis and in models of motor neuron death (Review). Int. J. Mol. Med. 2000, 5, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Gozlan, E.; Lewit-Cohen, Y.; Frenkel, D. Sex Differences in Astrocyte Activity. Cells 2024, 13, 1724. [Google Scholar] [CrossRef]
- Breeze, B.; Connell, E.; Wileman, T.; Muller, M.; Vauzour, D.; Pontifex, M.G. Menopause and Alzheimer’s disease susceptibility: Exploring the potential mechanisms. Brain Res. 2024, 1844, 149170. [Google Scholar] [CrossRef]
- Cheng, Y.J.; Lin, C.H.; Lane, H.Y. From Menopause to Neurodegeneration-Molecular Basis and Potential Therapy. Int. J. Mol. Sci. 2021, 22, 8654. [Google Scholar] [CrossRef]
- Kimura, M.; Oda, Y.; Hirose, Y.; Kimura, H.; Yoshino, K.; Niitsu, T.; Kanahara, N.; Shirayama, Y.; Hashimoto, K.; Iyo, M. Upregulation of heat-shock protein HSP-70 and glutamate transporter-1/glutamine synthetase in the striatum and hippocampus in haloperidol-induced dopamine-supersensitivity-state rats. Pharmacol. Biochem. Behav. 2021, 211, 173288. [Google Scholar] [CrossRef]
- De Souza, I.E.; McBean, G.J.; Meredith, G.E. Chronic haloperidol treatment impairs glutamate transport in the rat striatum. Eur. J. Pharmacol. 1999, 382, 139–142. [Google Scholar] [CrossRef]
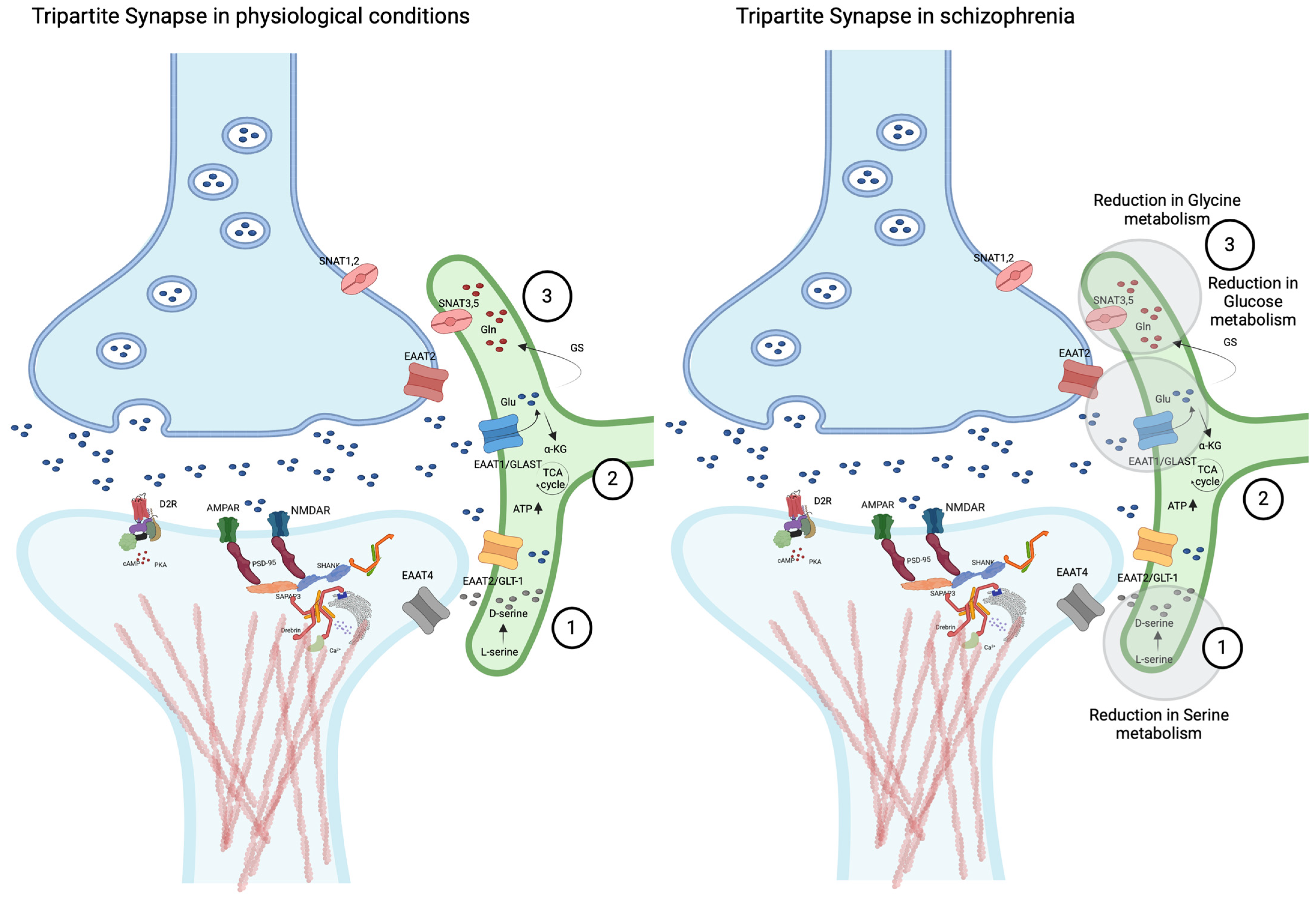
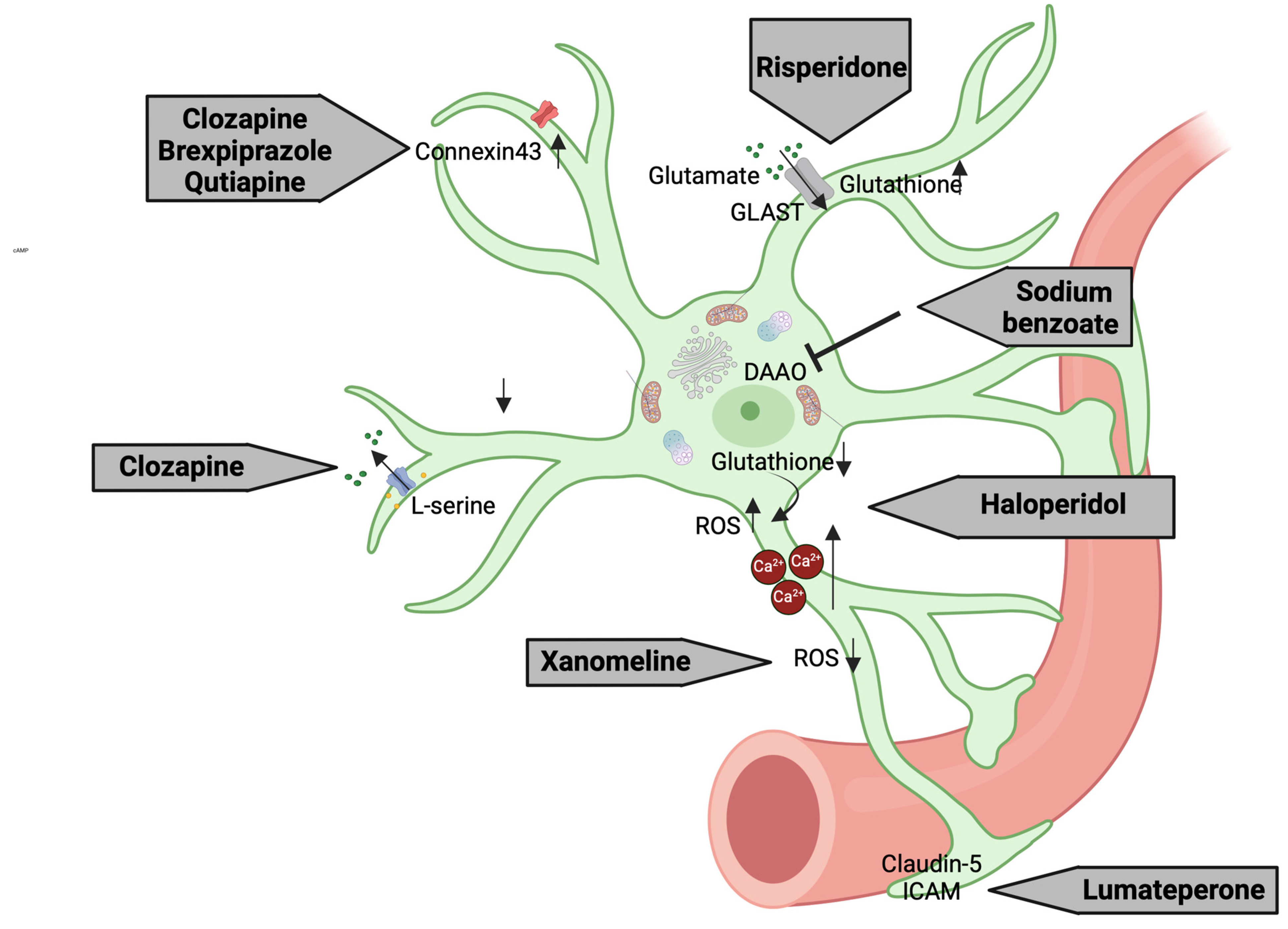
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vellucci, L.; Mazza, B.; Barone, A.; Nasti, A.; De Simone, G.; Iasevoli, F.; de Bartolomeis, A. The Role of Astrocytes in the Molecular Pathophysiology of Schizophrenia: Between Neurodevelopment and Neurodegeneration. Biomolecules 2025, 15, 615. https://doi.org/10.3390/biom15050615
Vellucci L, Mazza B, Barone A, Nasti A, De Simone G, Iasevoli F, de Bartolomeis A. The Role of Astrocytes in the Molecular Pathophysiology of Schizophrenia: Between Neurodevelopment and Neurodegeneration. Biomolecules. 2025; 15(5):615. https://doi.org/10.3390/biom15050615
Chicago/Turabian StyleVellucci, Licia, Benedetta Mazza, Annarita Barone, Anita Nasti, Giuseppe De Simone, Felice Iasevoli, and Andrea de Bartolomeis. 2025. "The Role of Astrocytes in the Molecular Pathophysiology of Schizophrenia: Between Neurodevelopment and Neurodegeneration" Biomolecules 15, no. 5: 615. https://doi.org/10.3390/biom15050615
APA StyleVellucci, L., Mazza, B., Barone, A., Nasti, A., De Simone, G., Iasevoli, F., & de Bartolomeis, A. (2025). The Role of Astrocytes in the Molecular Pathophysiology of Schizophrenia: Between Neurodevelopment and Neurodegeneration. Biomolecules, 15(5), 615. https://doi.org/10.3390/biom15050615