1. Introduction
After undergoing meiosis, haploid round spermatids are differentiated into morphologically mature spermatozoa in a process known as spermiogenesis. During spermiogenesis, sperm undergo a series of molecular and morphological changes that include reorganization of the chromatin through the exchange of histones for protamine, formation of the flagellum, formation of the acrosome from the Golgi apparatus, degradation of proteins not needed for sperm function, and translation of spermatid and sperm-specific proteins. Among the post-meiotically expressed proteins, members of the testis-specific serine kinase (TSSK) family have been shown to be essential for spermiogenesis. The TSSK family is composed of 6 members (TSSK1 to -6), with TSSK5 being a pseudogene in humans and a putative nonfunctional protein in mice [
1,
2]. The other members, TSSK1, -2, -3, -4, and -6, are exclusively expressed in the testes during spermiogenesis [
1,
3,
4,
5,
6] and remain in mature sperm after their transit in the epididymis [
1], as reviewed in [
7].
The important roles in sperm production and function of the diverse TSSK family members is highlighted by the subfertile or infertile male phenotype of studied knockout (KO) animal models. Male mice lacking TSSK4 are subfertile, as KO animals were shown to produce half the number of the pups as their wild-type (WT) littermates [
8]. This subfertility phenotype was attributed to severe defects in sperm tail morphology with a concomitant decrease in sperm motility [
8]. TSSK6 male (but not female) KOs are sterile in vivo and infertile in vitro [
9,
10]. The sterility is due to a 50% reduction in sperm count and to reduced sperm motility, as well as to a high incidence rate (about 90%) of sperm head morphology defects in comparison to their WT littermates [
10]. Later, our group showed that sperm from TSSK6 KO animals are also unable to fertilize in vitro, even when the zona pellucida of the eggs is removed, indicating problems in sperm–egg fusion [
10]. More recently, our group used CRISPR/CAS9 to generate three independent TSSK3 KO lines. In the three lines,
Tssk3−/− male mice were sterile [
11]. Problems in the
Tssk3−/− male mice were mapped to spermiogenesis [
11]. The three lines of TSSK3 KO displayed a drastically reduced number of sperm combined with severe sperm head morphology defects in comparison to their heterozygous littermates [
11]. Consistent with our results, Nozawa and collaborators confirmed the same phenotype in an independent TSSK3 KO model [
12]. Interestingly, TSSK3 seems to be essential for the phosphorylation of proteins related to male fertility [
12].
Contrary to Tssk3, Tssk6, and Tssk4, three members of the TSSK family that have been independently knocked-out, two different groups eliminated Tssk1 and Tssk2 together, generating double-KO models [
13,
14]. The sequence similarity of TSSK1 and TSSK2, together with the close evolutionary origin [
7], led investigators to assume redundancy in the function of these kinases. However, only the generation of independent null mutant models would test the possible TSSK1 and TSSK2 divergence in protein function. This would be of great interest to understand TSSK1 and TSSK2’s autonomous function and to position these kinases as targets for the development of a male contraceptive independently from each other. In this work, the main goal was to use CRISPR/Cas9 technology to generate individual KOs for either Tssk1 or Tssk2 to evaluate the extent to which these two kinases can compensate for each other. Our results indicate that both TSSK1 and TSSK2 have a role in spermiogenesis that leads to problems in sperm morphology and function. TSSK1 and TSSK2 mutant lines were used with commercial antibodies against these proteins via Western blot and immunofluorescence. Anti-TSSK2 antibodies detected a band at the right molecular weight (MW ~40 KDa) in wild-type and heterozygous TSSK2 testis and sperm extracts but not in homozygous mutant lines. Therefore, both TSSK2 mutant lines were considered complete knockouts and named throughout the manuscript Tssk2
−/− or TSSK2 KOs. On the other hand, anti-TSSK1 antibodies were not validated; consequently, it is not clear whether the TSSK1 protein is completely gone or if the Crispr/Cas9-induced truncation represents a functional knockout without complete TSSK1 loss. Consequently, throughout this manuscript, we refer to the TSSK1 mutant line as Tssk1
M/M or simply as TSSK1 mutants. The similar sterile phenotypes of the two single KO models indicates that although these kinases have similar functions, there is no functional compensation between TSSK1 and TSSK2, as both are required for proper sperm function.
2. Materials and Methods
2.2. Generation of Tssk1 Mutant and Tssk2 Knockout Lines by CRISPR/Cas9
Tssk1 and Tssk2 KO alleles were engineered at the Animal Models Core Facility of the Institute for Applied Life Sciences (IALS) following the protocol approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Massachusetts Amherst. Briefly, adult female mice (8–10 weeks old) were superovulated with 7.5 IU of pregnant mare serum gonadotropin (PMSG), followed 48 h later by 7.5 IU of human chorionic gonadotropin (hCG), then paired with males of the same strain. At 20 h post-hCG injection, the females were euthanized for zygote collection. A volume of 5–10 pL containing gene-specific single-guide RNA (2 ng/µL) targeting Tssk1 (GCTGCCGTCCTCAAGCGACG) or Tssk2 (GGCAGATTTGACTTTTGCGT) and Cas9 mRNA (3.3 ng/µL) was microinjected into the cytoplasm of zygotes. The microinjected zygotes were cultured in KSOM medium at 37 °C in a humidified atmosphere of 5% CO2/5% O2 balanced with N2. After three days of in vitro culture, early blastocysts (E3.5) were transferred into CD-1 pseudo-pregnant female recipients (E2.5) using Nonsurgical Embryo Transfer. Pseudo-pregnancy was achieved by mating the CD-1 females with CD-1 vasectomized males 2.5 days before the embryo transfers. Only females with a visible vaginal plug were used as embryo recipients. The offspring were genotyped via PCR using gene-specific primers. All PCR amplicons were TA-cloned and verified by Sanger sequencing (Psomagen Inc., Cambridge, MA, USA) to identify founders carrying frameshift mutations. The founders were initially backcrossed with wild-type mice of the same strain to minimize potential off-target effects, followed by the intercrossing of heterozygous offspring to generate homozygous KO mice. The Tssk1 KO allele was generated and maintained on the C57BL/6J (JAX) background, while the Tssk2 KO alleles were generated and maintained on the B6D2F1 (C57BL/6J × DBA/2J, JAX) background, as hybrid females exhibit an improved superovulation response and higher mating success, facilitating more efficient KO generation.
2.3. Genotyping and Sanger Sequencing
Genomic DNA was extracted from mice ear biopsies using DNA lysis buffer (50 mM Tris, 1 mM EDTA, 0.5% Tween 20) with incubation at 55 °C for 30 min. The samples were centrifuged for 5 min at full speed followed by supernatant (DNA) collection. Specific custom Tssk1 and Tssk2 primers were synthesized using Integrated DNA Technology. The following primers were used: Tssk1 F: ′5-GTGTGGCAGGGATGTAGAGG-3′; Tssk1 R: 5′-TCTTGCGATCGATGATCTTG-3′; Tssk2 F: 5′-GTGTGGGGAGGGGATGTAG-3′; Tssk2 R: 5′-TCCACAAAGTCAGTGGGTGT-3′. Conventional PCR was conducted using Taq polymerase. To distinguish wild-type, heterozygous, or homozygous mutants, PCR amplicons were analyzed on a 3.4% agarose gel running at 105 mAmps. TSSK1 wild-types were identified as a single band at 192 bp, heterozygous were identified as double bands at 192 bp and 184 bp, and homozygous mutants were identified as a single band at 184 bp. TSSK2 line 1 wild-types were identified as a single band at 210 bp, heterozygous were identified as two bands at 210 bp and 200 bp, and knockouts identified as a single band at 200 bp. TSSK2 line 2 wild-types were identified as a single band at 210 bp, heterozygous were identified as two bands at 210 bp and 205 bp, and knockouts were identified as a single band at 205 bp. DNA bands were excised and purified from agarose gel using a QIAquick PCR and Gel Cleanup Kit, as per the kit’s instructions. The DNA concentration and purity were determined using a Nanodrop Spectrophotometer (BioDrop, Cambridge, UK). The samples were prepared and sent to Azenta (Plainfield, NJ, USA) for sequencing, as instructed by the company.
2.4. In Vivo Fertility Test
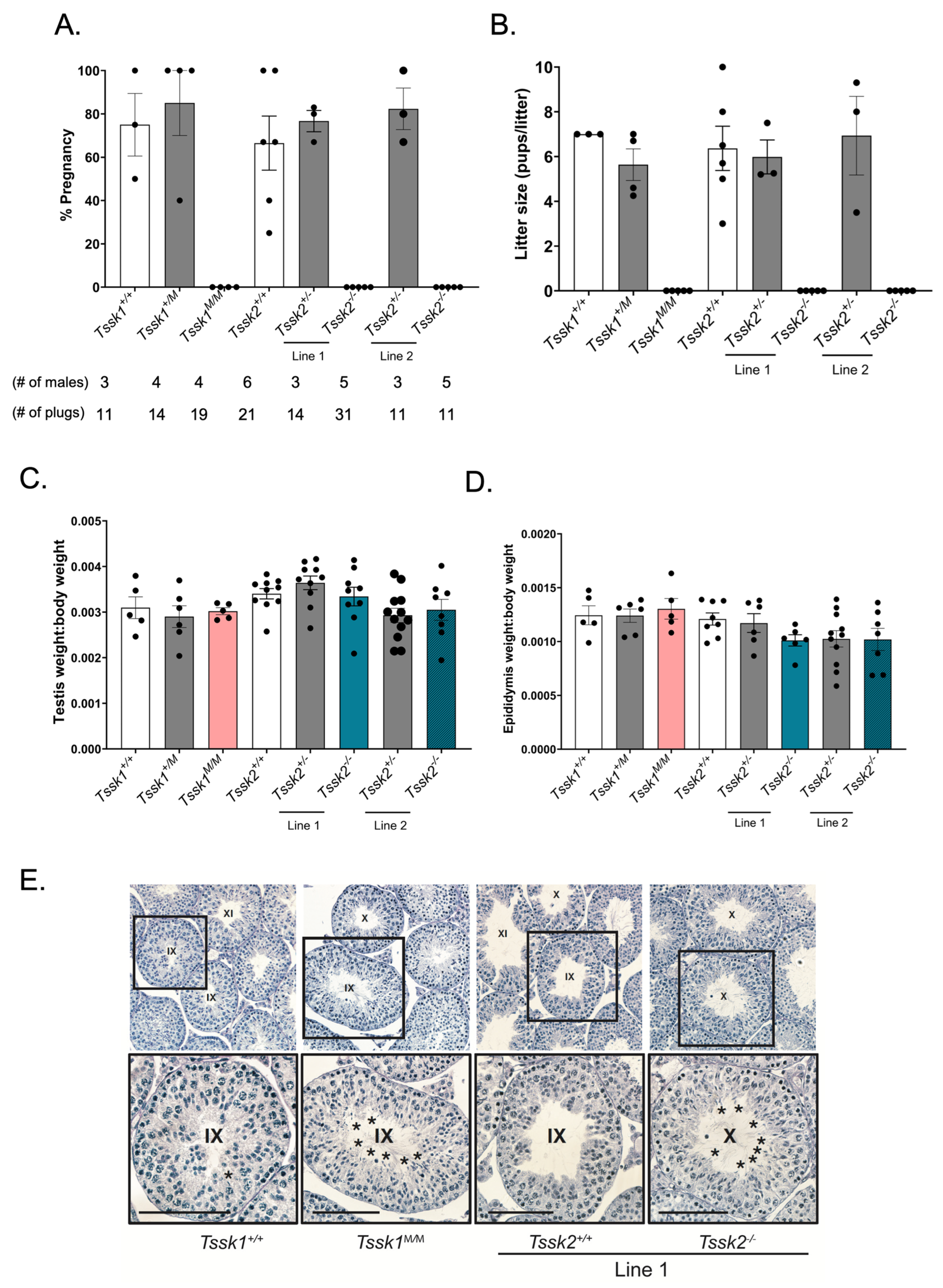
Two-month or older males of either Tssk1 or Tssk2 wild-type (+/+), heterozygous (+/m or +/−) and mutant (m/m or −/−) genotypes were mated with two wild-type C57BL/6J females in individual cages and their vaginal plugs were checked the following mornings. The plugged females were housed separately from the males and monitored for pregnancy and pups. The sexually active males were given one day of rest before housing them with additional females. Biological replicates were generated by using at least 3 different mice of each genotype. The pregnancy percentage was calculated as the number of pregnancies/number of plugs obtained multiplied by 100; the average percentage of pregnancies was determined for each genotype. The same test was conducted using female Tssk1 and Tssk2 mutant mice mated with C57BL/6J wild-type males.
2.5. Testes and Epididymis to Body Weight Analysis
The animal handling and euthanasia procedures were performed in accordance with the Animal Care and Use Committee (IACUC) guidelines of UMass-Amherst (protocol #3418). After the cervical dislocation of adult mice of each genotype of Tssk1 and Tssk2, the whole animal body was weighed on a scale, followed by weighing both the left and right dissected testicles or epididymis; all values are given in grams. The body to testis or body to epididymis weight ratio was calculated by the average of the left and right testes or epididymides over the body weight for each genotyped mouse.
2.6. Testis Fixing and Periodic Acid Solution (PAS) Staining
Whole testes from ≥2-month-old WT and mutant Tssk1 and Tssk2 line 1 male mice were collected and punctured 4–12 times with a 22 gauge needle before fixing in 4% paraformaldehyde in phosphate-buffered saline (PBS) overnight at 4 °C while rocking. After fixation, the tissues were rinsed twice with PBS and then washed 3 times with 1x PBS for 10 min each while rocking. Then, the tissues were washed and dehydrated with an alcohol gradient, cleared with xylene, and embedded in paraffin. The embedded samples were cut using a microtome at 4 microns and dried overnight on charged glass slides. The slides were deparaffinized in xylene and slowly rehydrated in a series of ethanol washes (100%, 95%, 80%, 70%, 50%) with tap or deionized (DI) water. The slides were PAS-stained following the manufacturer’s instructions. Briefly, the slides were immersed in PAS for 10 min and rinsed twice with deionized water before immersion in Schiff’s reagent for 20 min followed by a 5 min wash in running tap water. The samples were counterstained in Gill No. 3 hematoxylin solution for 4 min followed by a 2 min tap water rinse and 2 rinses in DI water. The samples were then dehydrated with alcohol and xylene and mounted with Permount. For the analysis, a minimum of three technical and biological replicates were performed.
2.7. Animal Usage, Sperm Collection, and Sperm Count
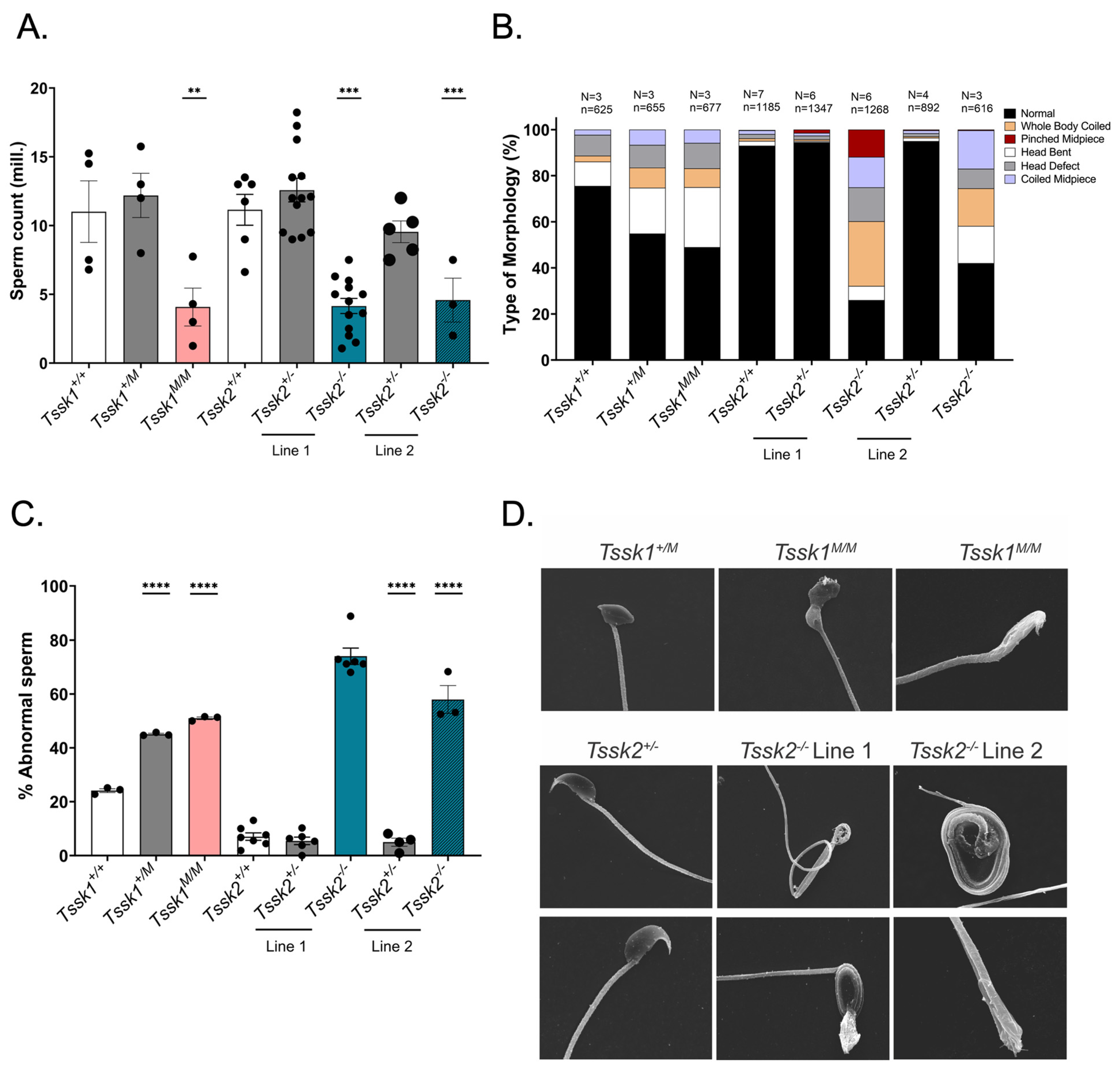
Following cervical dislocation, mature sperm were collected from epididymal cauda from ≥2-month-old male mice with 3–5 excisions, allowing the sperm to swim out of the tissue for 10 min in 1 mL modified noncapacitating Toyoda–Yokoyama–Hosi (mTYH) medium at 37 °C for 10 min. This medium consists of the following (concentrations are given in parenthesis): NaCl (119.3 mM), KCl (4.7 mM), CaCl2 × 2H2O (1.71 mM), KH2PO4 (1.2 mM), MgSO4 × 7H2O (1.2 mM), glucose (5.56 mM), Na pyruvate (0.51 mM), HEPES (20 mM), and gentamicin (10 mg/mL). After 10 min, the tissues were removed and the sperm samples were ready for usage. To establish sperm numbers and concentrations, 1:100 dilutions of the original sperm suspensions were made in H2O and counted in a hemocytometer. For sperm capacitation, the TYH medium was supplemented with 15 mM HCO3− and 5 mg/mL of bovine serum albumin (BSA).
2.8. Sperm Morphology Analysis
Sperm cells were collected from each genotyped mouse. After a 10 min swim-out step in mTYH media, the cells were fixed with 1 mL of 4% paraformaldehyde (PFA) for 10 min at room temperature on a rocker. After 2 washes with PBS, the cells were resuspended in 0.5–1.0 mL PBS, depending on the cell concentration. Next, 50 μL of cells was added to poly-l-lysine #1.5 Corning coverslips and air-dried. The coverslips were mounted on Global Diamond White Glass microscope slides with Vectashield and sealed with nail polish. The slides were imaged on an inverted microscope (Eclipse TE300; Nikon, Melville, NY, USA) with a 40× objective (NA 1.49; Nikon).
2.9. Scanning Electron Microscopy
After sperm collection, the cells were fixed with 2.5% v/v glutaraldehyde in 0.5 M sodium cacodylate pH 7.2 for 1 h at room temperature. The fixed cells were then washed and resuspended with 0.1 M cacodylate at pH 7.2. The samples were imaged at the Core Electron Microscopy Facility at University of Massachusetts Chan Medical School with an FEI/ThermoFisher (Waltham, MA, USA) Quanta 200 MK II Field Emission SEM.
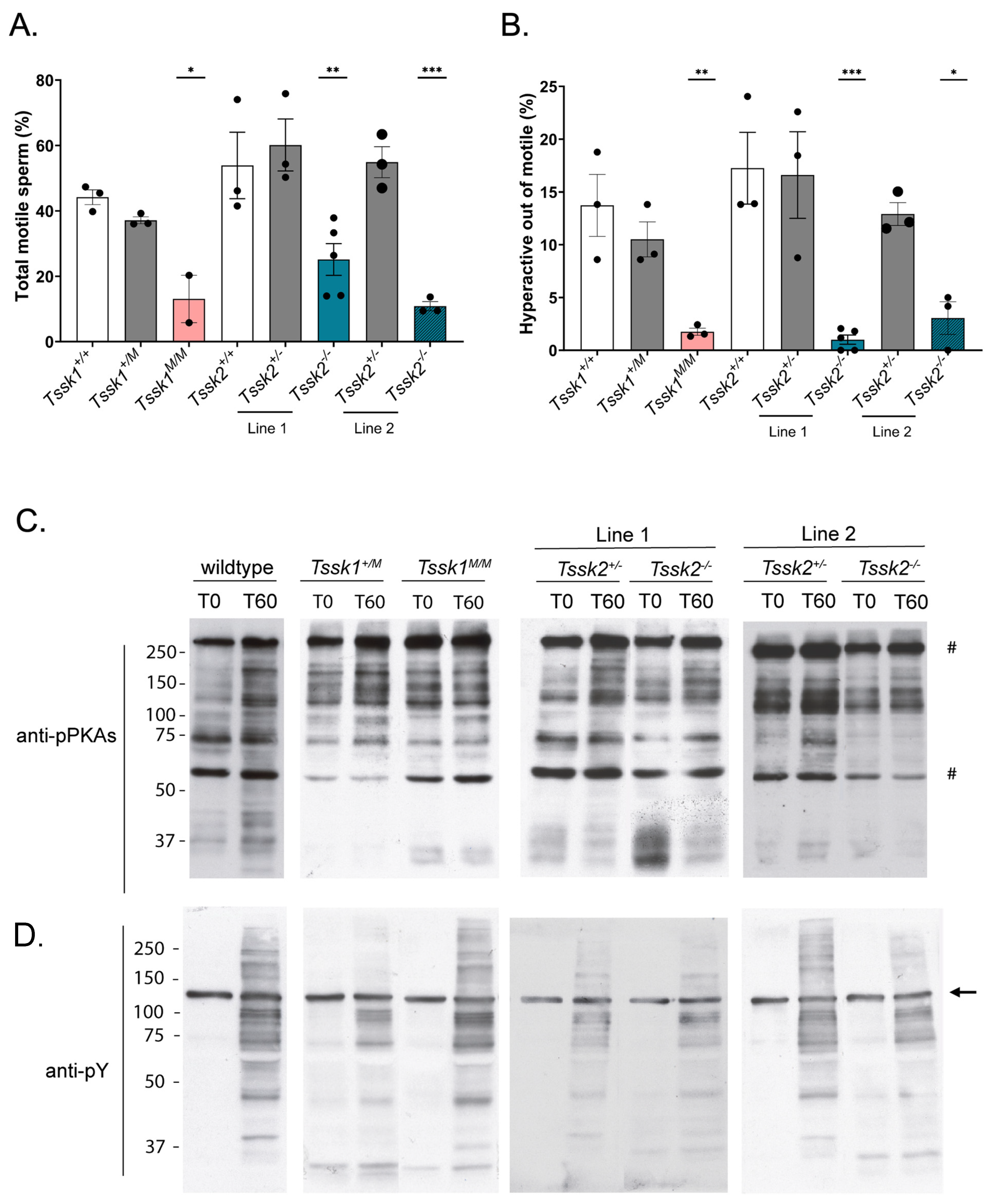
2.10. Computer-Assisted Sperm Analysis (CASA) Measurements
The sperm suspensions (30 μL; 2 × 106 sperm/mL) were loaded into a pre-warmed 4-chamber slide (depth 100 µm) and placed on a microscope stage at 37 °C. The sperm motility was examined using the CEROS computer-assisted sperm analysis (CASA) system (Hamilton Thorne Research, Beverly, MA, USA). The default settings include the following parameters: frames acquired: 90; frame rate: 60 Hz; minimum cell size: 4 pixels; static head size: 0.13–2.43; static head intensity: 0.10–1.52; static head elongation: 5–100. At least four microscopy fields corresponding to a minimum of 200 sperm were analyzed for each treatment in each experiment. The percentage of hyperactive sperm was calculated based on the total motile sperm, considering hyperactive sperm as those having the following parameters: curvilinear velocity (VCL) > 271.00 μm/s, linearity (LIN) < 50.00%, and amplitude of lateral head (ALH) > 3.50 µm.
2.11. Sperm Anti-Phospho PKA Substrates (pPKAs) and Anti Phosphotyrosine (pY) Western Blots
After sperm collection, the cells were exposed to capacitating mTYH for either 0 or 60 min at 37 °C. The cells were then centrifuged at 12.6 rpm for 2 min and washed with 0.6 mL of ice-cold PBS. The cells were then treated with 10 μL of 5× nonreducing Laemmli sample buffer [
15] boiled for 4 min at 100 °C, and centrifuged at max speed for 5 min, and then the supernatant was collected. After the addition of a 5% final concentration of β-mercapthoethanol, the protein lysates were boiled for an additional 5 min and loaded onto an 8% SDS–polyacrylamide gel for electrophoresis and electro-transferred to an Immobilon-P polyvinylidene difluoride (PVDF) membrane. The membrane was blocked with 5% fat-free milk in T-TBS for 1 h at room temperature and then treated with 1:10,000 polyclonal anti-pPKAs overnight at 4 °C in the dark. The following day, the PVDF membrane was washed 3× and incubated with IgG anti-rabbit (1:10,000) in 1% fat-free milk in T-TBS for 1 h at room temperature. After washing 3× in T-TBS, the membrane was treated with an ECL Prime Plus kit, and a signal for the pPKAs was detected. The PVDF membranes used for the pPKAs were then stripped at 55 °C for 20 min in 2% SDS, 0.74% β-mercaptoethanol, and 62.5 mM Tris at pH 6.5, followed by six washes for 5 min each in T-TBS prior to membrane blocking with 20% gelatin in T-PBS for 1 h at room temperature. They were then treated with anti-pY (1:10,000) in T-PBS and washed for 1 h at room temperature. Secondary IgG anti-mouse (1:10,000) was added, and after 30 min the membrane was washed 3× with T-PBS and the pY signal was detected using ECL Regular. A quantitative analysis was performed using ImageJ 1.47 V software (National Institutes of Health, Bethesda, MD, USA). The regions of interest (ROIs) used for quantification are indicated by a # on the left of the respective Western blot. The extent of hexokinase tyrosine phosphorylation does not change during capacitation [
16] and was used as the loading control. Therefore, the optical density of the bands was normalized to tyrosine-phosphorylated hexokinase.
2.12. In Vitro Fertilization (IVF)
Mouse cumulus–oocyte complexes (COCs) were collected from 8–10-week-old superovulated CD1 females (Charles River Laboratories, Wilmington, MA, USA). For superovulation, the females were first injected with 7.5–10 IU of pregnant mare serum gonadotropin (PMSG) (Lee BioSolutions, cat # 493-10, Maryland Heights, MO, USA) followed by 7.5–10 IU of human chorionic gonadotrophin (hCG) (Sigma, cat # CG5, St. Louis, MO, USA) 48 h later. Thirteen hours post-hCG injection, the COCs were collected in TL-HEPES medium (NaCl (114 mM), KCl (3.22 mM), NaHCO3 (2.02 mM), NaH2PO4 (0.348 mM), HEPES (10.1 mM), lactic acid (sodium salt) (10 mM), CaCl2.2H2O (1.71 mM), MgCl2.7H2O (1.2 mM), gentamicin (10 mg/mL)), with the addition of bovine serum albumin (5 mg/mL) on the day of experiment, washed in mTYH (see above for composition), and placed in the insemination droplet. Tssk1+/+, Tssk1+/M, Tssk1M/M, Tssk2+/+, Tssk2+/−, and Tssk2−/− from line 1 and line 2 male mice sperm were collected as described previously, and approximately 100,000 sperm were added to a 90 µL insemination droplet. After 4 h, the oocytes were washed in mTYH and allowed to culture for 20 h. The following day, fertilization was determined via the visualization of cleavage into a 2-cell stage embryo.
2.13. Intracytoplasmic Sperm Injection (ICSI)
ICSI was performed as previously reported by [
17] using the described setup and micromanipulators (Narishige, Japan). Sperm from
Tssk1+/+,
Tssk1+/M,
Tssk1M/M,
Tssk2+/+,
Tssk2+/−, and
Tssk2−/− from line 1 and line 2 male mice (≥2 months old) were collected from the cauda epididymis in mTYH and washed three times in the same media; then, heads were separated from tails via sonication (XL2020; Heat Systems Inc., Phoenix, AZ, USA) for 5 sec at 4 °C. The sperm lysate was washed in mTYH and diluted with 12% polyvinylpyrrolidone (PVP) to a final PVP concentration of 6%. A piezo micropipette driving unit was used to deliver the sperm into the ooplasm (Primetech, Ibaraki, Japan); a few piezo-pulses were applied to puncture the eggs’ plasma membrane following penetration of the zona pellucida. After ICSI, the eggs were cultured in KSOM to evaluate the 2-cell development at 36.5 °C in a humidified atmosphere containing 5% CO
2.
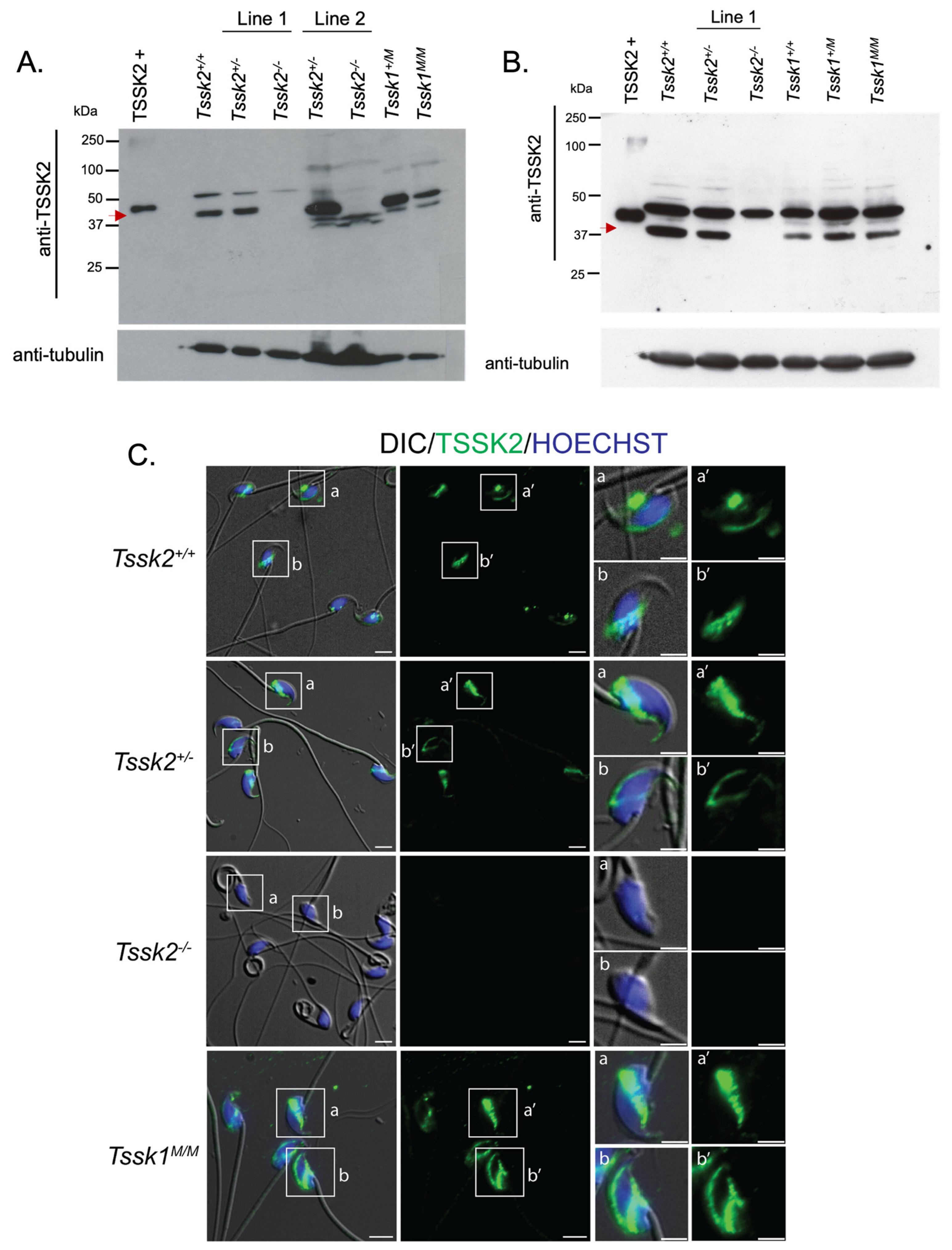
2.14. TSSK2 Protein Detection via Western Blots
2.14.1. Testes
Testis protein extracts were obtained [
16] from
Tssk1+/+,
Tssk1+/M,
Tssk1M/M,
Tssk2+/+,
Tssk2+/−, and
Tssk2−/− from line 1 and line 2 male mice. Briefly, both the left and right testes were dissected from ≥2-month-old mice and the tunica albuginea was removed in Dulbecco’s modified Eagle’s medium (DMEM). The seminiferous tubes were cut into small fragments and transferred into a disaggregation cartridge. The fragments were manually processed in the cartridge for 5 min and passed through 40 μm cell strainers twice. The samples were then processed again manually in the disaggregation cartridge for 5 min. Once germ cells from the testes were obtained, the protein was extracted using the same method as for the sperm cells.
2.14.2. Sperm
After mature sperm collection from TSSK2 wild-type, het, and KO mice, the cells were washed with ice-cold PBS. The cell pellets were treated with 1x Laemmli buffer [
15], 1x protease inhibitor, and 2 mM of dithiothreitol and incubated on ice for 30 min, with vortexing every 5 min. The cells were then sonicated for 3 sections (10 sec pulse and 30 sec pause) on ice. The cells were centrifuged at 15,000×
g for 20 min at 4 °C and 95 μL of supernatant was collected. After the addition of 5% β-mercapthoethanol, the samples were boiled for 5 min at 100 °C, then 30 μg of protein was loaded onto an 8% SDS–polyacrylamide gel for electrophoresis and electro-transferred to a PVDF membrane. The membrane was blocked with 5% milk in T-PBS for 1 h at room temperature and then treated with anti-TSSK2 (clone 1E12) at a concentration of 2 μg/mL overnight at 4 °C in the dark. The following day, the membrane was washed 3× with T-PBS and incubated with secondary IgG anti-mouse (1:10,000) in 1% milk in T-PBS for one hour at room temperature. The membrane was then washed 3× with T-PBS and the TSSK2 signal was detected with an ECL Prime Plus kit. Equal loading was determined with blot stripping and reprobing was performed with monoclonal α anti-tubulin (E7).
2.15. TSSK2 Immunofluorescence
2.15.1. Sperm
TSSK2 wild-type, het, or KO epididymal caudal sperm were collected from ≥2-month-old mice and fixed in 1 mL 4% PFA in 1x PBS for 10 min at room temperature. After fixing, the cells were centrifuged at 150× g for 5 min and washed 2x with 1 mL PBS. The cells were resuspended in 0.1 mL–1 mL PBS, depending on the sperm concentration. Then, 50 μL of cells was added to poly-l-lysine #1.5 Corning coverslips and air dried at room temperature. The cells were permeabilized with 0.5% v/v Triton X-100 in PBS for 5 min at room temperature and then washed 3× with 0.01% Tween-PBS (T-PBS) for 5 min each time. The cells were blocked with 10% BSA in T-PBS for 1 h at room temperature. After the hour blocking, anti-TSSK2 (clone 1E12) was added at a 20 μg/mL concentration in 1% BSA in T-PBS and incubated at 4 °C overnight in the dark. The next day, the coverslips were washed 3× with T-PBS and treated with anti-mouse IgG AlexaFluoro 488 (1:1000), peanut agglutinin (1:100), and Hoechst (1:100) for one hour at room temperature. The cells were then washed 5× with 1x T-PBS for 5 min in the dark at room temperature and mounted on Global Diamond White Glass microscope slides with Vectashield. The slides were imaged on an inverted microscope (Eclipse TE300; Nikon) with epifluorescence and DIC images were taken with a 60x PlanApo/DIC objective (NA 1.49; Nikon).
2.15.2. Testes
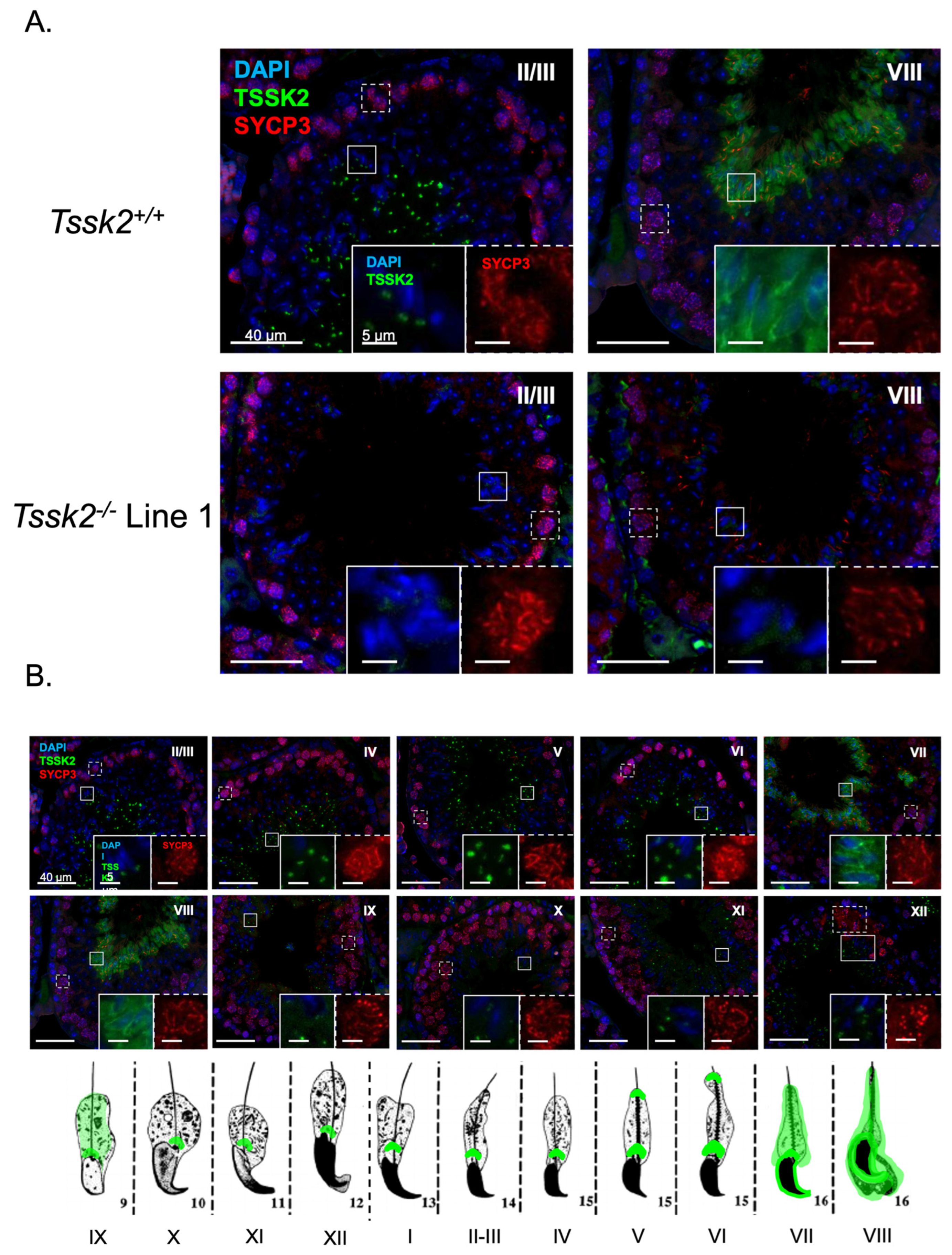
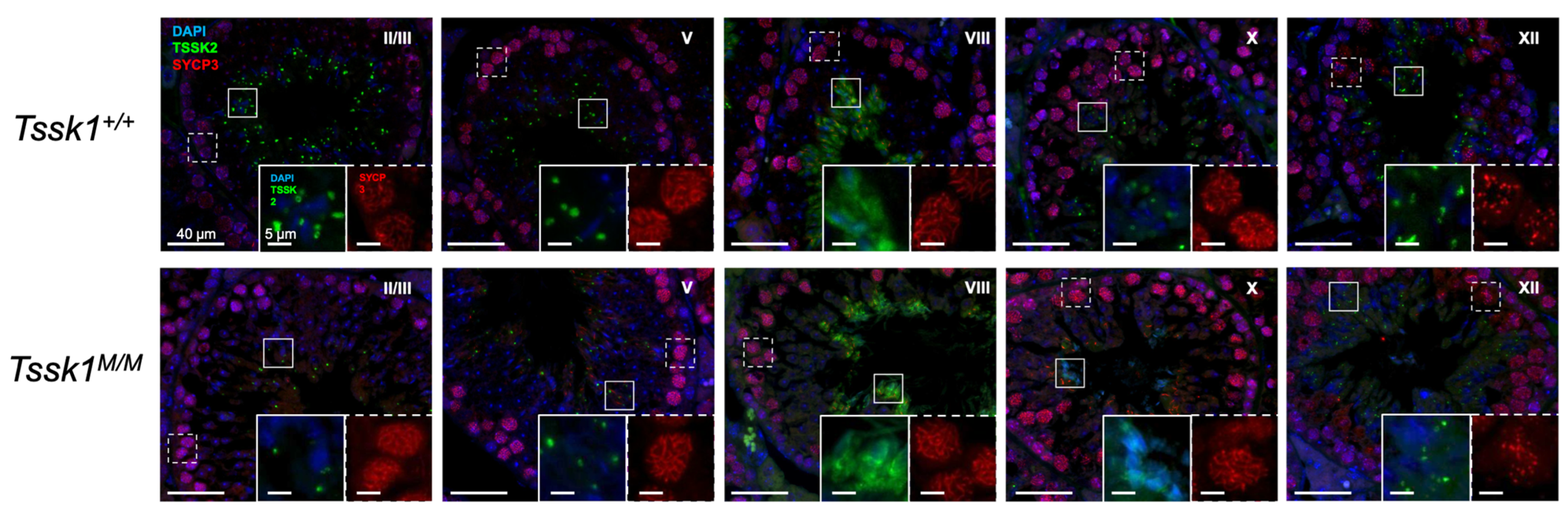
The testes were dissected from adult mice and fixed overnight in 4% PFA. The tissue was rinsed in PBS and dehydrated in increasing concentrations of ethanol before embedding in paraffin wax and sectioning to 4 μm. Antigen retrieval was performed as previously described [
18]. Briefly, the slides were boiled in Tris-EDTA (10 mM Tris-HCl, 1 mM EDTA, and 0.05% Tween (pH 9.0)) for 30 min on low. The slides were blocked in 4% goat serum for 1 h followed by primary antibodies (anti-TSSK2 1:250; anti-SYCP3, 1:1000) overnight at room temperature. The slides were washed in 0.1% Triton PBS then incubated with goat anti-rabbit (1:1000) and goat anti-mouse (1:1000) secondary antibodies for 1 h at room temperature. The slides were washed in 0.1% Triton PBS, followed by 1x PBS, and mounted using DAPI Fluoromount-G. Once dry, they were stored at 4 °C with light protection until imaging. The slides were visualized on a custom-built microscope (Zeiss, Jena, Germany) with fluorescent and bright-field capabilities. The provided images are representative of three or more biological samples. The signal intensity was matched across slides by matching the background (interstitial) signal intensity. The developmental stages were determined according to the parameters set forth by [
19,
20].
2.16. Statistics
Statistical analyses were performed using Prism 8.4.3 software (by GraphPad). Data are expressed as the mean ± SEM. All data were verified to accomplish the parametric assumptions of homogeneity of variances and normality. Variables were compared via a one-way analysis of variance, and the statistical significance between groups was determined using Tukey’s post-hoc test. Statistical analyses for optical densitometries were performed using two-tailed t-tests ranked for parametric data and a Wilcoxon matched pair ranked test for nonparametric data. Significance was considered as * p < 0.05, ** p < 0.01, *** p < 0.0005, or **** p < 0.0001.
4. Discussion
Spermatogenesis encompasses a series of events that culminate in the postmeiotic differentiation of haploid spermatids. This last step is known as spermiogenesis and involves the conversion of round spermatids into elongated spermatozoa. Spermiogenesis includes the formation of the sperm acrosome and the flagellum, redistribution of the mitochondria to the sperm tail, synthesis of new proteins, and elimination of others through the activation of degradation and secretory pathways. Importantly, spermiogenesis induces chromatin compaction through the exchange of histones for protamines. All of these changes in the spermatid architecture rely on finely controlled signaling pathways; among them, post-translational modifications, such as phosphorylation, play essential roles. The importance of phosphorylation pathways is highlighted by a growing number of protein kinases that are abundantly expressed in the testis and have been shown to be critical for spermiogenesis [
7,
22]. Among these kinases, the testis-specific serine kinase (TSSK) family has six members named numerically from 1 to 6. The focus of this manuscript is on TSSK1 and TSSK2.
Tssk1 was discovered in 1994 using a degenerate primer strategy [
3], and three years later, the same group identified TSSK2 using
32P-labeled
Tssk1 as the probe [
4] to screen a mouse testis cDNA library. In the mouse,
Tssk1 and
Tssk2 are encoded in chromosome 16 and separated by only 3 kb. In humans,
TSSK2 is found in the DiGeorge syndrome minimal region in chromosome 22. This region in chromosome 22 is syntenic to the mouse chromosome 16; however, in higher primates,
TSSK1 in the syntenic chromosome 22 location is a pseudogene. Interestingly, a new
TSSK member with high homology to the mouse
Tssk1 is found in a nonsyntenic region in chromosome 5. This gene was named
TSSK1b and is hypothesized to have appeared in higher primates through a retro-transposition event [
13]. Importantly, in humans,
TSSK1b is also mainly expressed in the testes. At the same time of the
TSSK2 discovery, Ziemiecki’s group reported that both TSSK1 and TSSK2 phosphorylate another testis-specific protein, and they named it testis-specific kinase substrate (TSKS) [
4].
Taking advantage of the proximity between the
Tssk1 and
Tssk2 genes in the mouse, two groups generated double-knockout mouse models lacking both genes using a homologous recombination in ES cells [
13,
14]. One of these groups was not able to generate progeny lacking both genes, and it was concluded that these knockout mice were haploinsufficient [
14]; the second group was able to generate homozygous mutant mice [
13]. This last group showed that the double
Tssk1−/−/Tssk2−/− mice have a male sterility phenotype without presenting defects in females. This manuscript highlighted the relevance of these kinases for reproduction; however, the work was silent regarding the individual TSSK1 and TSSK2 roles. Here, we took advantage of CRISPR/Cas9 methodology to generate mutant mice that eliminated the functional expression of either TSSK1 or TSSK2 and evaluated their phenotype. We found that individual mutations in both genes generated male sterility without affecting female reproduction. In both mutant models, the overall testis and epididymis anatomy was not altered but the number of spermatozoa and their morphology were significantly affected. Moreover, a higher percentage of the
Tssk2 mutant spermatozoa had motility problems, while both
Tssk1 and
Tssk2 homozygous mutants were not able to undergo hyperactivation. Given these phenotypic characteristics, it was not surprising to find that the sperm from these mice were not able to fertilize metaphase II-arrested eggs in vitro; interestingly, they were able to generate two-cell embryos when injected by ICSI, suggesting that chromatin condensation and decondensation were not affected.
We also used these mouse models as controls for specific antibodies. However, we were only able to validate anti-TSSK2 antibodies, which recognize specific Western blot bands in sperm and testis extracts from
Tssk2+/+ and
Tssk2+/− but not in extracts from
Tssk2−/− mouse models. Therefore, follow-up experiments were only conducted with the validated anti-TSSK2 antibody. Using this antibody, we showed that TSSK2 is present in sperm and testis extracts from
Tssk1−/− mutants. This result indicated that TSSK2 is still expressed in mice lacking the functional expression of TSSK1. We also used the anti-TSSK2 antibody to localize TSSK2 in the sperm and testes. Consistent with previous results [
1], TSSK2 has a post-acrosomal localization like the one observed for TSSK6 [
9]. In addition to this localization, TSSK2 was found in the anterior head. In the testes, TSSK2 is observed in post-meiotic spermatids, with variable localization based on spermatid developmental state. Consistent with previous reports [
13], this refined localization pattern suggests dynamic regulation of TSSK2 localization during spermatid development, specifically during spermatid elongation, and begs the question of why TSSK2 has such a distinct localization pattern. Based on the wide range of mutant sperm phenotypes, it is tempting to postulate that TSSK2 may play a dual role in regulating sperm head morphology and sperm tail formation, in particular the midpiece. These suppositions are also consistent with the localization pattern of TSSK2 in mature sperm. A second question arising from these findings is whether the two TSSK2 foci share distinct or similar protein substrates. Based on the previous localization of TSKS [
13], overlapping substrates seems highly likely. Regardless, directly addressing these questions will require detailed localization, protein binding, and substrate identification studies of TSSK2.
Our results indicate that both TSSK1 and TSSK2 are essential for male reproduction. These findings add to the growing body of evidence indicating the critical role of kinases and phosphorylation in spermiogenesis. As an example, the recent work by Pasand et al. indicates that a methamphetamine-induced increase in inflammatory cell populations decreases TSSK protein expression [
23]. In addition to TSSK1 and TSSK2, among the relevant spermatid kinases required for male reproduction, we can mention TSSK3 [
11,
12], TSSK6 [
10], HIPK4 [
24], and STK33 [
25]. In addition, mice lacking TSSK4 are sub-fertile [
8]. As previously published [
7], the TSSK family is evolutionarily conserved. Recent studies in Drosophila melanogaster (D.m.) showed that the relevance of this kinase family for male reproduction is also conserved. Contrary to mammals, in D.m., only one
dTssk gene (aka CG14305) is found; this gene has between 37 and 46% shared identity with mammalian TSSKs, with the highest homology to TSSK1 and TSSK2. The elimination of this gene from the D.m. genome using CRISPR/Cas9 resulted in a sterile fly phenotype [
24]. Flies lacking the
dTssk gene present severe spermiogenesis defects, including impairment of the histone-to-protamine transition and morphological defects. As part of this work, the authors crossed
dTssk−/− flies with transgenic flies expressing human
TSSK1B,
TSSK2,
TSSK3,
TSSK4, or
TSSK6 under the control of the endogenous
dTssk promoter. Each of the individual human TSSKs partially restored spermiogenesis defects but none of them was able to rescue the sterility phenotype. Interestingly, in
dTssk−/− flies,
TSSK1B (the TSSK1 human homologue) but not
TSSK2 rescued nucleus morphology, as well as defects in histone-to-protamine transition [
26]. The differential ability of each human TSSK to rescue spermiogenesis defects in
dTssk−/− flies suggests that during evolution, the dTSSK function was divided between multiple TSSKs.
From a clinical perspective, a deletion of ~8-Mb in the 5q22.2q23.1 locus harboring the TSSK1B gene causes asthenoteratozoospermia [
27]. The same group performed Sanger sequencing of the coding regions and exon borders of TSSK1B in a cohort of 100 male patients with unresolved infertility causes. They discovered missense mutations in 10% of the cases that were associated with sperm abnormalities, including two previously unreported TSSK1B mutations [
28]. In 2013, a study found that 5 out of 372 infertile Chinese men who had oligospermia or azoospermia contained some mutation in the
Tssk4 gene. The study found 4 single nucleotide changes within the gene: 2 within the 5′ and 3′ untranslated region (UTR) and two in exon 3 [
29]. These clinical reports, together with loss-of-function studies in flies and mice, including those in this paper, strongly suggest that small molecules blocking TSSK activity will induce reversible sterility in men and other species. Reproductive control is a key aspect of human health. It is estimated that about 50% of pregnancies worldwide are unintended, and this has a direct impact, primarily in women of reproductive age and society at large [
30]. Currently, the burden of family planning relies on women, leaving men with limited options [
31]. In recent years, the efforts to develop safe and effective male-directed contraceptives have significantly increased.
Ideally, male contraceptive targets should exclusively affect reproductive processes. Among the different strategies, hormonal approaches such as exposure to testosterone induces suppression of spermatogenesis via the inhibition of gonadotropin release [
32,
33]. More recently, efforts have been made to find nonhormonal compounds that target reproductive molecules. These compounds have higher safety and efficacy requirements than other small molecules targeting diseases. As is also the case for women’s contraceptives, the small molecules that block male reproduction should have negligible or no side effects and ideally should be completely reversible. The targets for nonhormonal male contraceptives should have some relevant properties: (1) they should be expressed exclusively or almost exclusively in reproductive cells and tissues, including testis, epididymis, or germ cells or spermatozoa; (2) they should be amenable for finding drugs that inhibit their function; (3) they should be essential for male reproduction. TSSKs have all of these properties; they are testis-specific proteins and are druggable, and loss-of-function models showed that they are indispensable for male reproduction without affecting female reproduction or other tissues. In this regard, the work from our group has produced recombinant TSSK2 [
34] and identified potent TSSK1 and TSSK2 inhibitors having pyrrolopyrimidine and pyrimidine cores [
35]. Some of these compounds have IC50s in low nanomolar concentrations; however, their selectivity is still poor. These findings warrant the use of molecular medicine strategies to achieve higher selectivity.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}