
Oxidant-Based Cytotoxic Agents During Aging: From Disturbed Energy Metabolism to Chronic Inflammation and Disease Progression


Abstract
1. Introduction
2. Key Concepts About Inflammation in Older Individuals
2.1. The Concept of Inflammaging
2.2. Molecular Patterns and Host-Derived Cytotoxic Agents in Inflammation
3. Peculiarities of Energy Metabolism and Oxidative Stress in Older Individuals
3.1. Energy Metabolism Under Normoxic Conditions
3.2. Deviations in Energy Metabolism in Older Individuals
3.3. Responses to Hypoxia and Oxidative Stress
3.4. Redox Regulation and Antioxidative Defense in Older Individuals
3.5. Activation of Proteolytic Systems in Older Individuals
3.6. Inflammaging and Necrotic Cell Death
4. Age-Related Alterations in Protection Against Oxidant-Based Cytotoxic Agents in Selected Disease Scenarios
4.1. Diseases of the Cardiovascular System
4.2. Diabetes Mellitus
4.3. Cancer
4.4. Neurodegenerative Diseases
5. Conclusions
Funding
Conflicts of Interest
References
- Medvedev, Z.A. An attempt at a rational classification of theories of aging. Biol. Rev. 1990, 65, 375–398. [Google Scholar] [CrossRef]
- Arnhold, J. Aging in complex multicellular organisms. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 231–247. [Google Scholar] [CrossRef]
- Arnhold, J. Cells and organisms as open systems. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 3–22. [Google Scholar] [CrossRef]
- Arnhold, J. Immune response and tissue damage. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 155–204. [Google Scholar] [CrossRef]
- Arnhold, J. Acute-phase proteins and additional protective systems. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 205–228. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; de Luca, M.; Ottaviani, E.; de Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Arnhold, J. Host-derived cytotoxic agents in chronic inflammation and disease progression. Int. J. Mol. Sci. 2023, 24, 3016. [Google Scholar] [CrossRef] [PubMed]
- Arnhold, J. Inflammation-associated cytotoxic agents in tumorigenesis. Cancers 2024, 16, 81. [Google Scholar] [CrossRef]
- Franceschi, C. Cell proliferation, cell death and aging. Aging Clin. Exp. Res. 1989, 1, 3–15. [Google Scholar] [CrossRef]
- Kirkwood, T.B.L.; Franceschi, C. Is ageing a complex as it would appear? New perspectives in gerontological research. Ann. N. Y. Acad. Sci. 1992, 663, 412–417. [Google Scholar] [CrossRef]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and antiinflammaging: A systemic perspective on aging and longevity emerged from studies on humans. Mech. Aging Dev. 2007, 128, 92–105. [Google Scholar] [CrossRef]
- Baggio, G.; Donazzan, S.; Monti, D.; Mari, D.; Martini, S.; Cabelli, C.; Dalla Vestra, M.; Previato, L.; Guido, M.; Pigozzo, S.; et al. Lipoprotein(a) and lipoprotein profile in healthy centenarians: A reappraisal of vascular risk factors. FASEB J. 1998, 12, 433–437. [Google Scholar] [CrossRef]
- Gangemi, S.; Basile, G.; Merendino, R.A.; Minciullo, P.L.; Novick, D.; Rubinstein, M.; Dinarello, C.A.; Lo Balbo, C.; Franceschi, C.; Basili, S.; et al. Increased circulating interleukin-18 levels in centenarians with no signs of vascular disease: Another paradox of longevity. Exp. Gerontol. 2003, 38, 669–672. [Google Scholar] [CrossRef]
- Mannucchi, P.M.; Mari, D.; Merati, G.; Peyvandi, F.; Tagliabue, L.; Sacchi, E.; Taioli, E.; Sansoni, P.; Bertolini, S.; Franceschi, C. Gene polymorphism predicting high plasma levels of coagulation and fibrinolysis proteins. A study in centenarians. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 755–759. [Google Scholar] [CrossRef]
- Coppola, R.; Mari, D.; Lattuada, A.; Franceschi, C. Von Willebrand factor in Italian centenarians. Haematologica 2003, 88, 39–43. [Google Scholar] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-related diseases. J. Gerontol. Biol. Sci. 2014, 69 (Suppl. S1), S4–S9. [Google Scholar] [CrossRef]
- Troiano, L.; Pini, G.; Petruzzi, E.; Ognibene, A.; Franceschi, C.; Monti, D.; Casotti, G.; Cilotti, A.; Forti, G. Evaluation of adrenal function in aging. J. Endocrinol. Investig. 1999, 22, 74–75. [Google Scholar]
- Carrieri, G.; Marzi, E.; Olivieri, F.; Marchigiani, F.; Cavallone, L.; Cardelli, M.; Giovanetti, S.; Stecconi, R.; Molendini, C.; Trapassi, C.; et al. The G/C915 polymorphism of transforming growth factor beta1 is associated with human longevity: A study in Italian centenarians. Aging Cell 2004, 3, 443–448. [Google Scholar] [CrossRef]
- Baylis, D.; Bartlett, D.B.; Patel, H.P.; Roberts, H.C. Understanding how we age: Insights into inflammaging. Longev. Heal. 2013, 2, 8. [Google Scholar] [CrossRef]
- Newman, A.B.; Sanders, J.L.; Kizer, J.R.; Boudreau, R.M.; Odden, M.C.; Zeki al Hazzouri, A.; Arnold, A.M. Trajectories of functions and biomarkers with age: The CHS all stars study. Int. J. Epidemiol. 2016, 45, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Zhang, X.; Zheng, S.; Khanabdali, S.; Kalionis, B.; Wu, J.; Wan, W.; Tai, X. An update of inflamm-aging: Mechanisms, prevention, and treatment. J. Immunol. Res. 2016, 2016, 8426874. [Google Scholar] [CrossRef]
- Dugan, B.; Conway, J.; Duggal, N.A. Inflammaging as a target for healthy ageing. Age Ageing 2023, 52, afac328. [Google Scholar] [CrossRef]
- Li, X.; Li, C.; Zhang, W.; Wang, Y.; Qian, P.; Huang, H. Inflammation and aging: Signaling pathways and intervention therapies. Signal Transd. Target. Ther. 2023, 8, 329. [Google Scholar] [CrossRef]
- Suresh, R.; Moser, D.M. Pattern recognition in innate immunity, host defense, and immunopathology. Adv. Physiol. Educ. 2013, 37, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef]
- Janeway, C.A.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells trigger inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Shi, Y.M.; Evans, J.E.; Rock, K.L. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 2003, 425, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Scheibner, K.A.; Lutz, M.A.; Boodoo, S.; Fenton, M.J.; Powell, J.D.; Horton, M.R. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J. Immunol. 2006, 177, 1272–1281. [Google Scholar] [CrossRef]
- Bours, M.J.; Swennon, E.L.; Di Virgilio, F.; Cronstein, B.N.; Dagnelie, P.C. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol. Ther. 2006, 112, 358–404. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliviera, M.F.; Graca-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of toll-like receptor 4. J. Biol. Chem. 2007, 282, 20221–20229. [Google Scholar] [CrossRef]
- Farkas, A.M.; Kilgore, T.M.; Lotze, M.T. Detecting DNA: Getting and begetting cancer. Curr. Opin. Investig. Drugs 2007, 8, 981–986. [Google Scholar]
- Pepys, M.B.; Baltz, M.I. Acute phase proteins with special reference to C-reactive protein and related proteins (pentraxins) and serum amyloid A protein. Adv. Immunol. 1983, 34, 141–212. [Google Scholar] [CrossRef]
- Vandivier, R.W.; Henson, P.M.; Douglas, I.S. Burying the death: The impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest 2006, 129, 1673–1682. [Google Scholar] [CrossRef] [PubMed]
- Pober, J.S.; Sessa, W.C. Inflammation and the blood microvascular system. Cold Spring Harb. Perspect. Biol. 2015, 7, 016345. [Google Scholar] [CrossRef]
- Young, B.; Gleeson, M.; Cripps, A.W. C-reactive protein. A critical review. Pathology 1991, 23, 118–124. [Google Scholar] [CrossRef]
- Gewurz, H.; Mold, C.; Siegel, J.; Fiedel, B. C-reactive protein and acute phase response. Adv. Intern. Med. 1982, 27, 345–372. [Google Scholar]
- Kopf, H.; de la Rosa, G.M.; Horward, O.M.; Chen, X. Rapamycin inhibits differentiation of Th17 cells and promotes generation of FoxP3+ T regulatory cells. Int. Immunopharmacol. 2007, 7, 1819–1824. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Prados, J.C.; Través, P.C.; Cuenca, J.; Rico, D.; Aragonés, J.; Martin-Sanz, P.; Casante, M.; Boscá, L. Substrate fate in activated macrophages; a comparison between innate, classic, and alternative activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blaigh, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749. [Google Scholar] [CrossRef]
- Morrison, T.; Watts, E.R.; Sadiku, P.; Walmsley, S.R. The emerging role for metabolism in fueling neutrophilic inflammation. Immunol. Rev. 2023, 314, 427–441. [Google Scholar] [CrossRef]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-beta regulation of immune response. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef]
- Li, M.O.; Flavell, R.A. Contextual regulation of inflammation: A duet of transforming growth factor-beta and interleukin-10. Immunity 2008, 28, 468–476. [Google Scholar] [CrossRef]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar] [CrossRef] [PubMed]
- Landén, N.X.; Li, D.; Ståhle, M. Transition from inflammation to proliferation: A critical step during wound healing. Cell Mol. Life Sci. 2016, 73, 3861–3885. [Google Scholar] [CrossRef] [PubMed]
- Marega, M.; Chen, C.; Bellusci, S. Cross-talk between inflammation and fibroblast growth factor 10 during organogenesis and pathogenesis: Lessons learnt from the lung and other organs. Front. Cell Dev. Biol. 2021, 9, 656883. [Google Scholar] [CrossRef]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef]
- Chandrasekharan, J.A.; Sharma-Walia, N. Lipoxins: Nature’s way to resolve inflammation. J. Inflamm. Res. 2015, 8, 181–192. [Google Scholar] [CrossRef]
- Sanchez-Pino, M.D.; Dean, M.J.; Ochoa, A.C. Myeloid-derived suppressor cells (MDSC): When good intentions go awry. Cell Immunol. 2021, 362, 104302. [Google Scholar]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S.; Locati, M.; Montavani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 2006, 177, 7303–7311. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Choi, J.; Gyamfi, J.; Jang, H.; Koo, J.S. The role of tumor-associated macrophage in breast cancer biology. Histol. Histopathol. 2018, 33, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Shi, W.; Xu, Y.; Xu, C.; Zhao, T.; Geng, B.; Yang, J.; Pan, J.; Hu, S.; Zhang, C.; et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle 2018, 17, 428–438. [Google Scholar] [CrossRef]
- Ratter, J.M.; Rooljackers, H.M.M.; Hoolveld, G.J.; Hijmans, A.G.M.; de Galan, B.E.; Tack, C.J.; Stienstra, R. In vitro and in vivo effects of lactate on metabolism and cytokine production of human primary PBMCs and monocytes. Front. Immunol. 2018, 9, 2564. [Google Scholar] [CrossRef]
- McCord, J.; Fridovich, I. Superoxide dismutase: An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1960, 224, 6049–6055. [Google Scholar]
- Chang, L.Y.; Slot, J.W.; Geuze, H.J.; Crapo, J.D. Molecular immunocytochemistry of the CuZn superoxide dismutase in rat hepatocytes. J. Cell Biol. 1988, 107, 2169–2179. [Google Scholar] [CrossRef] [PubMed]
- Weisiger, R.A.; Fridovich, I. Mitochondrial superoxide dismutase. Site of synthesis and intramolecular localization. J. Biol. Chem. 1973, 248, 4793–4796. [Google Scholar]
- Antonyuk, S.V.; Strange, R.W.; Marklund, S.L.; Hasnain, S.S. The structure of human extracellular copper-zinc superoxide dismutase at 1.7 Å resolution: Insights into heparin and collagen binding. J. Mol. Biol. 2009, 388, 310–326. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M.; Roveri, A. Phospholipid hydroperoxide glutathione peroxidase (PHGPx): More than an antioxidant enzyme? Biomed. Environm. Sci. 1997, 10, 327–332. [Google Scholar]
- Low, F.M.; Hampton, M.P.; Winterbourn, C.C. Prx2 and peroxide metabolism in the erythrocyte. Antioxid. Redox Signal. 2008, 10, 1621–1630. [Google Scholar] [CrossRef]
- Goyal, M.M.; Basak, A. Human catalase: Looking for complete identity. Protect. Cell 2010, 1, 888–897. [Google Scholar] [CrossRef]
- Gutteridge, J.M.C. Ferrous-salt-promoted damage to deoxyribose and benzoate. The increased effectiveness of hydroxyl-radical scavengers in the presence of EDTA. Biochem. J. 1987, 18, 37–49. [Google Scholar] [CrossRef]
- Zhao, N.; Zhang, A.-S.; Enns, C.A. Iron regulation by hepcidin. J. Clin. Investig. 2013, 123, 2337–2343. [Google Scholar] [CrossRef] [PubMed]
- Gkouvatsos, K.; Papanikolaou, G.; Pantopoulos, K. Regulation of iron transport and the role of transferrin. Biochim. Biophys. Acta 2011, 1820, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Gamella, E.; Buratti, P.; Cairo, G.; Recalcati, S. The transferrin receptor: The cellular iron gate. Metallomics 2017, 9, 1367–1375. [Google Scholar] [CrossRef]
- Massover, W.H. Ultrastructure of ferritin and apoferritin: A review. Micron 1993, 24, 389–437. [Google Scholar] [CrossRef]
- Prohaska, J.R. Role of copper transporters in copper homeostasis. Am. J. Clin. Nutr. 2008, 88, 826S–829S. [Google Scholar] [CrossRef]
- Sokolov, A.V.; Ageeva, K.V.; Pulina, M.O.; Cherkalina, O.S.; Samygina, V.R.; Vlasova, I.I.; Panasenko, O.M.; Zakharova, E.T.; Vasilyev, V.B. Ceruloplasmin and myeloperoxidase in complex affect the enzymatic properties of each other. Free Radic. Res. 2008, 42, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Chapman, A.L.P.; Mocatta, T.J.; Shiva, S.; Seidel, A.; Chen, B.; Khalilova, I.; Paumann-Page, M.E.; Jameson, G.N.L.; Winterbourn, C.C.; Kettle, A.J. Ceruloplasmin is an endogenous inhibitor of myeloperoxidase. J. Biol. Chem. 2013, 288, 6464–6477. [Google Scholar] [CrossRef]
- Samygina, V.R.; Sokolov, A.V.; Bourenkov, G.; Petoukhov, M.V.; Pulina, M.O.; Zakharova, E.T.; Vasilyev, V.B.; Bartunik, H.; Svergun, D.I. Ceruloplasmin: Macromolecular assemblies with iron-containing acute phase proteins. PLoS ONE 2013, 8, e67145. [Google Scholar] [CrossRef]
- Sokolov, A.V.; Kostevich, V.A.; Zakharova, E.T.; Samygina, V.R.; Panasenko, O.M.; Vasilyev, V.B. Interaction of ceruloplasmin with eosinophil peroxidase as compared to its interplay with myeloperoxidase: Reciprocal effect on enzymatic properties. Free Radic. Res. 2015, 49, 800–811. [Google Scholar] [CrossRef]
- Ashby, M.T.; Carlson, A.C.; Scott, M.J. Redox buffering of hypochlorous acid by thiocyanate in physiologic fluids. J. Am. Chem. Soc. 2004, 126, 15976–15977. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Beal, J.L.; Ashby, M.T. Thiocyanate is an efficient endogenous scavenger of the phagocytic killing agent hypobromous acid. Chem. Res. Toxicol. 2006, 19, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; Hawkins, C.L. The role of myeloperoxidase in biomolecule modification, chronic inflammation, and disease. Antioxid. Redox Signal. 2020, 32, 957–981. [Google Scholar] [CrossRef]
- Love, D.T.; Barrett, D.J.; White, M.Y.; Cordwell, S.J.; Davies, M.J.; Hawkins, C.L. Cellular targets of the myeloperoxidase-derived oxidant hypothiocyanous acid (HOSCN) and its role in the inhibition of glycolysis in macrophages. Free Radic. Biol. Med. 2016, 94, 88–98. [Google Scholar] [CrossRef]
- Frommherz, K.J.; Faller, B.; Bieth, J.G. Heparin strongly decreases the rate of inhibition of neutrophil elastase by α1-proteinase inhibitor. J. Biol. Chem. 1991, 266, 15356–15362. [Google Scholar] [CrossRef]
- Ermolieff, J.; Boudier, C.; Laine, A.; Meyer, B.; Bieth, J.G. Heparin protects cathepsin G against inhibition by protein proteinase inhibitors. J. Biol. Chem. 1994, 269, 29502–29508. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.C.; Ohlsson, K. Isolation, properties, and complete amino acid sequence of human secretory leukocyte protease inhibitor, a potent inhibitor of leukocyte elastase. Proc. Natl. Acad. Sci. USA 1986, 83, 6692–6696. [Google Scholar] [CrossRef]
- Williams, S.E.; Brown, T.I.; Roghanian, A.; Sallenave, J.M. SLPI and elafin: One glove, many fingers. Clin. Sci. 2006, 110, 21–35. [Google Scholar] [CrossRef]
- Verrier, T.; Solhonne, B.; Sallenave, J.M.; Garcia-Verdugo, I. The WAP protein Trappin-2/Elafin: A handyman in the regulation of inflammatory and immune responses. Int. J. Biochem. Cell Biol. 2012, 44, 1377–1380. [Google Scholar] [CrossRef]
- Duranton, J.; Adam, C.; Blieth, J.G. Kinetic mechanism of the inhibition of cathepsin G by α1-antichymotrypsin and α1-proteinase inhibitor. Biochemistry 1997, 37, 11239–11245. [Google Scholar] [CrossRef]
- Travis, J.; Bowen, J.; Baugh, R. Human α1-antichymotrypsin: Interaction with chymotrypsin-like proteinases. Biochemistry 1978, 26, 5651–5656. [Google Scholar] [CrossRef]
- Kalsheker, N.A. α1-Antichymotrypsin. Int. J. Biochem. Cell Biol. 1996, 28, 961–964. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.L. Extracellular ATP: Effects, sources and fate. Biochem. J. 1986, 233, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; Burnstock, G. The double life of ATP. Sci. Am. 2009, 301, 84–90. [Google Scholar] [CrossRef]
- Dou, L.; Chen, Y.F.; Cowan, P.J.; Chen, X.-P. Extracellular ATP signaling and clinical relevance. Clin. Immunol. 2018, 188, 67–73. [Google Scholar] [CrossRef]
- Flood, D.; Lee, E.S.; Taylor, C.T. Intracellular energy production and distribution in hypoxia. J. Biol. Chem. 2023, 299, 105103. [Google Scholar] [CrossRef]
- El Bacha, T.; Luz, M.R.M.P.; Da Poian, A.T. Dynamic adaptation of nutrient utilization in humans. Nat. Educ. 2010, 3, 8. [Google Scholar]
- Chandel, N.S. Amino acid metabolism. Cold Spring Harb. Perspect. Biol. 2021, 13, a040584. [Google Scholar] [CrossRef]
- Delgoffe, G.M.; Polizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, J.D. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 2011, 12, 295–303. [Google Scholar] [CrossRef]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T cell mechanism drives immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed]
- Ørtenblad, N.; Westerblad, H.; Nielsen, J. Muscle glycogen stores and fatigue. J. Physiol. 2013, 591, 4405–4413. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.E.; Richter, E.A. regulation of glucose and glycogen metabolism during and after exercise. J. Physiol. 2012, 590, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Laffel, L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 1999, 15, 412–416. [Google Scholar]
- Holeček, M. Origin and roles of alanine and glutamine in gluconeogenesis in the liver, kidneys, and small intestine under physiological and pathological conditions. Int. J. Mol. Sci. 2024, 25, 7037. [Google Scholar] [CrossRef]
- Bratic, I.; Trifunovic, A. Mitochondrial energy metabolism and aging. Biochim. Biophys. Acta 2010, 1707, 961–967. [Google Scholar] [CrossRef]
- Santanasto, A.J.; Glynn, N.W.; Jubrias, S.A.; Conley, K.E.; Boudreau, R.M.; Amati, F.; Mackey, D.C.; Simonsick, E.M.; Strotmeyer, E.S.; Coen, P.M.; et al. Skeletal muscle mitochondrial function and fatigability in older adults. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 1379–1385. [Google Scholar] [CrossRef]
- Lima, T.; Li, T.Y.; Mottis, A.; Auwerx, J. Pleiotropic effects of mitochondria on aging. Nat. Aging 2022, 2, 199–213. [Google Scholar] [CrossRef]
- Miquel, J.; Economos, A.C.; Fleming, J.; Johnson, J.E., Jr. Mitochondrial role in cell aging. Exp. Gerontol. 1980, 15, 575–591. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial aging and age-related dysfunction of mitochondria. BioMed Res. Int. 2014, 2014, 238463. [Google Scholar] [CrossRef]
- Crane, J.D.; Devries, M.C.; Safdar, A.; Hamadeh, M.J.; Tarnopolsky, M.A. The effect of aging on human skeletal muscle mitochondrial and intramyocellular lipid ultrastructure. J. Gerontol. A 2010, 65, 119–128. [Google Scholar] [CrossRef]
- Marzetti, E.; Leeuwenburgh, C. Skeletal muscle apoptosis, sarcopenia and frailty at old age. Exp. Gerontol. 2006, 41, 1234–1238. [Google Scholar] [CrossRef] [PubMed]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.-S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD+/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef]
- Gomes, A.P.; Price, N.L.; Ling, A.J.Y.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef]
- Harrison, A.P.; Pierzynowski, S.G. Biological effects of 2-oxoglutarate with particular emphasis on the regulation of protein, mineral and lipid absorption/metabolism, muscle performance, kidney function, bone formation and cancerogenesis, all viewed from a healthy ageing perspective state of the art–review article. J. Physiol. Pharmacol. 2008, 59 (Suppl. S1), 91–106. [Google Scholar] [PubMed]
- Tian, Q.; Zhao, J.; Yang, Q.; Wang, B.; Deavila, J.M.; Zhu, M.-J.; Du, M. Dietary α-ketoglutarate promotes beige adipogenesis and prevents obesity in middle-aged mice. Aging Cell 2020, 19, e13059. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD+ homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic potential of NAD-boosting molecules: The in vivo evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef]
- Imai, S.-I.; Guarente, L. It takes two to tango: NAD+ and sirtuins in aging/longevity control. npj Aging Mech. Dis. 2016, 2, 16017. [Google Scholar] [CrossRef]
- Fasolino, M.; Liu, S.; Wang, Y.; Zhou, Z. Distinct cellular and molecular environments support aging-related DNA methylation changes in the substantia nigra. Epigenomics 2017, 9, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Deng, P.; Liu, Y.; Wu, Y.; Chen, Y.; Guo, Y.; Zhang, S.; Zheng, X.; Zhou, L.; Liu, W.; et al. Alpha-ketoglutarate ameliorates age-related osteoporosis via regulating histone methylations. Nat. Commun. 2020, 11, 5596. [Google Scholar] [CrossRef] [PubMed]
- Borkum, J.M. The tricarboxylic acid cycle as a central regulator of the rate of aging: Implications for metabolic interventions. Adv. Biol. 2023, 7, 2300095. [Google Scholar] [CrossRef]
- Gardner, P.R. Superoxide-driven aconitase Fe-S cycling. Biosci. Rep. 1997, 17, 33–42. [Google Scholar] [CrossRef]
- Gardner, P.R. Aconitase: Sensitive target and measure of superoxide. Meth. Enzymol. 2002, 349, 9–23. [Google Scholar] [CrossRef]
- Harmer, A.R.; Chisholm, D.J.; McKenna, M.J.; Hunter, S.K.; Ruell, P.A.; Naylor, J.M.; Maxwell, L.J.; Flack, J.R. Sprint training increases muscle oxidative metabolism during high-intensity exercise in patients with type 1 diabetes. Diabetes Care 2008, 31, 2097–2102. [Google Scholar] [CrossRef]
- Levy, B.; Gibot, S.; Franck, P.; Cravoisy, A.; Bollaert, P.-E. Relation between muscle Na+K+ATPase activity and raised lactate concentrations in septic shock: A prospective study. Lancet 2005, 365, 871–875. [Google Scholar] [CrossRef]
- Nalbandian, M.; Radak, Z.; Takeda, M. Lactate metabolism and satellite cell fate. Front. Physiol. 2020, 11, 610983. [Google Scholar] [CrossRef]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Bergman, B.C.; Horning, M.A.; Casazza, G.A.; Wolfel, E.E.; Butterfield, G.E.; Brooks, G.A. Endurance training increases gluconeogenesis during rest and exercise in men. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E244–E251. [Google Scholar] [CrossRef]
- Bergman, B.C.; Tsvetkova, T.; Lowes, B.; Wolfel, E.E. Myocardial glucose and lactate metabolism during rest and atrial pacing in humans. J. Physiol. 2009, 587, 2087–2099. [Google Scholar] [CrossRef]
- Glenn, T.C.; Martin, N.A.; Horning, M.A.; McArthur, D.L.; Hovda, D.A.; Vespa, P.; Brooks, G.A. Lactate: Brain fuel in human traumatic brain injury: A comparison with normal healthy control subjects. J. Neurotrauma 2015, 32, 820–832. [Google Scholar] [CrossRef] [PubMed]
- Suhara, T.; Hishiki, T.; Kasahara, M.; Hayakawa, N.; Oyaizu, T.; Nakanishi, T.; Kubo, A.; Morisaki, H.; Kaelin, W.G., Jr.; Suematsu, M.; et al. Inhibition of the oxygen sensor PHD2 in the liver improves survival in lactic acidosis by activating the Cori cycle. Proc. Natl. Acad. Sci. USA 2015, 112, 11642–11647. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, Y.; Zhang, B.; Lin, X.; Fu, X.; An, Y.; Zou, Y.; Wang, J.-X.; Wang, Z.; Yu, T. Lactate metabolism in human health and disease. Sign. Transd. Targ. Ther. 2022, 7, 305. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, B.; Mannelli, M.; Gamberi, T.; Fiaschi, T. The multiple roles of lactate in the skeletal muscle. Cells 2024, 13, 1177. [Google Scholar] [CrossRef]
- Colegio, O.R.; Shu, N.Q.; Shabo, A.I.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.E.; Philipps, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Goetze, K.; Walenta, S.; Ksiazkiewicz, M.; Kunz-Schughart, L.A.; Mueller-Klieser, W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol. 2011, 39, 453–463. [Google Scholar] [CrossRef]
- Ragni, M.; Fornelli, C.; Nisoli, E.; Penna, F. Amino acids in cancer and cachexia: An integrated view. Cancers 2022, 14, 5691. [Google Scholar] [CrossRef]
- Stalnecker, C.A.; Cluntun, A.A.; Cerione, R.A. Balancing redox stress: Anchorage-independent growth requires reductive carboxylation. Transl. Cancer Res. 2016, 5, S433–S437. [Google Scholar] [CrossRef]
- Yeo, E.-J. Hypoxia and aging. Exp. Mol. Med. 2019, 51, 67. [Google Scholar] [CrossRef]
- Taylor, C.T.; Scholz, C.C. The effect of HIF on metabolism and immunity. Nat. Rev. Nephrol. 2022, 18, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Giunta, S.; Xia, S. Hypoxia in aging and aging-related diseases: Mechanism and therapeutic strategies. Int. J. Mol. Sci. 2022, 23, 8165. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.M.; Lone, A.; Betts, D.H.; Cumming, R.C. Lactate preconditioning promotes a HIF-1α-mediated metabolic shift from OXPHOS to glycolysis in normal human diploid fibroblasts. Sci. Rep. 2020, 10, 8388. [Google Scholar] [CrossRef]
- Nishimura, K.; Fukuda, A.; Hisatake, K. Mechanisms of the metabolic shift during somatic cell reprogramming. Int. J. Mol. Sci. 2019, 20, 2254. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.; Recktenwald, M.; Hutt, E.; Fuller, S.; Briggs, M.; Goel, A.; Daringer, N. Targeting HIF-2α in the tumor microenvironment: Redefining the role of HIF-2α for solid cancer therapy. Cancers 2022, 14, 1259. [Google Scholar] [CrossRef]
- Löfstedt, T.; Fredlund, E.; Holmquist-Mengelbier, L.; Pietras, A.; Overnberger, M.; Pollinger, L.; Påhlman, S. Hypoxia inducible factor-2α in cancer. Cell Cycle 2007, 6, 919–926. [Google Scholar] [CrossRef]
- de Saedeleer, C.J.; Copetti, T.; Porporato, P.E.; Verrax, J.; Feron, O.; Sonveaux, P. Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. PLoS ONE 2012, 7, e46571. [Google Scholar] [CrossRef]
- Pires, B.R.B.; Mencalha, A.L.; Ferreira, G.M.; Panis, C.; Silva, R.C.M.C.; Abdelhay, E. The hypoxia-inducible factor-1 alpha signaling pathway and its relation to cancer and immunology. Am. J. Immunol. 2014, 10, 215–224. [Google Scholar] [CrossRef]
- Ivan, M.; Haberberger, T.; Gervasi, D.C.; Michelson, K.S.; Günzler, V.; Kondo, K.; Yang, H.; Sorokina, I.; Conawey, R.C.; Conawey, J.W.; et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc. Nat. Acad. Sci. USA 2002, 99, 13459–13464. [Google Scholar] [CrossRef]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide—Production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13, 39. [Google Scholar] [CrossRef] [PubMed]
- Cyran, A.M.; Zhitkovich, A. HIF1, HSF1, and NRF2: Oxidant-responsive trio raising cellular defenses and engaging immune system. Chem. Res. Toxicol. 2022, 35, 1690–1700. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [PubMed]
- Song, M.-Y.; Lee, D.-Y.; Chun, K.-S.; Kim, E.-H. The role of Nrf2/keap1 signaling pathway in cancer metabolism. Int. J. Mol. Sci. 2021, 22, 4376. [Google Scholar] [CrossRef]
- Meister, A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988, 263, 17205–17208. [Google Scholar]
- Mustacich, D.; Powis, G. Thioredoxin reductase. Biochem. J. 2000, 346, 1–8. [Google Scholar]
- Holmgren, A.; Johansson, C.; Berndt, C.; Lönn, M.E.; Hudemann, C.; Lillig, C.H. Thiol redox control via thioredoxin and glutaredoxin systems. Biochem. Soc. Trans. 2005, 33, 1375–1377. [Google Scholar] [CrossRef]
- Riganti, C.; Gazzano, E.; Polimeni, M.; Aldieri, E.; Ghigo, D. The pentose phosphate pathway: An antioxidant defense and a crossroad in tumor cell fate. Free Radic. Biol. Med. 2012, 53, 421–436. [Google Scholar] [CrossRef]
- Chen, L.; Xing, X.; Zhang, P.; Chen, L.; Pei, H. Homeostatic regulation of NAD(H) and NADP(H) in cells. Genes Dis. 2024, 11, 101146. [Google Scholar] [CrossRef]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amigo, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef]
- Bai, P.; Cantó, C.; Oudart, H.; Brunyánszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Pirinen, E.; Cantó, C.; Jo, Y.S.; Morato, L.; Zhang, H.; Menzies, K.J.; Williams, E.G.; Mouchiraud, L.; Moullan, N.; Hagberg, C.; et al. Pharmacological Inhibition of poly(ADP-ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab. 2014, 19, 1034–1041. [Google Scholar] [CrossRef]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef]
- Cantó, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Arnhold, J. Oxidation and reduction of biological material. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 55–97. [Google Scholar] [CrossRef]
- Diaz-Del Cerro, E.; de Toda, I.M.; Félix, J.; Baca, A.; De la Fuente, M. Components of the glutathione cycle as markers of biological age: An approach to clinical application in aging. Antioxidants 2023, 12, 1529. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.A.; Naryshkin, S.; Schneider, D.L.; Mills, B.J.; Lindemann, R.D. Low blood glutathione levels in healthy aging adults. J. Lab. Clin. Med. 1992, 120, 720–725. [Google Scholar]
- Loguercio, C.; Taranto, D.; Vitale, L.M.; Beneduce, F.; Del Vecchio Blanco, C. Effect of liver cirrhosis and age on the glutathione concentration in the plasma, erythrocytes, and gastric mucosa of man. Free Radic. Biol. Med. 1996, 20, 483–488. [Google Scholar] [CrossRef]
- de Toda, M.; Vida, C.; Garrido, A.; De la Fuente, M. Redox parameters as markers of the rate of aging and predictors of life span. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2020, 75, 613–620. [Google Scholar] [CrossRef]
- Espinoza, S.E.; Guo, H.; Fedarko, N.; DeZern, A.; Fried, L.P.; Xue, Q.-L.; Leng, S.; Beamer, B.; Walston, J.D. Glutathione peroxidase enzyme activity in aging. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2008, 63, 505–509. [Google Scholar] [CrossRef]
- Belrose, J.C.; Xie, Y.F.; Gierszewski, L.J.; MacDonald, J.F.; Jackson, M.F. Loss of glutathione homeostasis associated with neuronal senescence facilitates TRPM2 channel activation in cultured hippocampal pyramidal neurons. Mol. Brain 2012, 5, 11. [Google Scholar] [CrossRef]
- Iskusnykh, I.Y.; Zakharova, A.A.; Pathak, D. Glutathione in brain disorders and aging. Molecules 2022, 27, 324. [Google Scholar] [CrossRef]
- Yang, B.; Lin, Y.; Huang, Y.; Shen, Y.-Q.; Chen, Q. Thioredoxin (Trx): A redox target and modulator of cellular senescence and aging-related diseases. Redox Biol. 2024, 70, 103032. [Google Scholar] [CrossRef]
- Lee, S.; Kim, S.M.; Lee, R.T. Thioredoxin and thioredoxin target proteins: From molecular mechanisms to functional significance. Antioxid. Redox Signal. 2013, 18, 1165–1207. [Google Scholar] [CrossRef]
- Pan, M.; Zhang, F.; Qu, K.; Liu, C.; Zhang, J. TXNIP: A double-edged sword in disease and therapeutic outlook. Oxid. Med. Cell. Longev. 2022, 2022, 7805115. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Suh, H.-W.; Jeon, Y.H.; Hwang, E.; Nguyen, L.T.; Yeom, J.; Lee, S.-G.; Lee, C.; Kim, K.J.; Kang, B.S.; et al. The structural basis for the negative regulation of thioredoxin by thioredoxin-interacting protein. Nat. Commun. 2014, 5, 2958. [Google Scholar] [CrossRef] [PubMed]
- Ismael, S.; Nasochi, S.; Li, L.; Aslam, K.S.; Khan, M.M.; El-Remessy, A.B.; McDonald, M.P.; Liao, F.-F.; Ishrat, T. Thioredoxin interacting protein regulates age-associated neuroinflammation. Neurobiol. Dis. 2021, 156, 105399. [Google Scholar] [CrossRef]
- Choi, E.-H.; Park, S.-J. TXNIP: A key protein in the cellular stress response pathway and a potential therapeutic target. Exp. Mol. Med. 2023, 55, 1348–1356. [Google Scholar] [CrossRef]
- Alhawiti, N.M.; Al Mahri, S.; Aziz, M.A.; Malik, S.S.; Mohammad, S. TXNIP in metabolic regulation: Physiological role and therapeutic outlook. Curr. Drug Targets 2017, 18, 1095–1103. [Google Scholar] [CrossRef]
- Wu, N.; Zheng, B.; Shaywitz, A.; Dagon, Y.; Tower, C.; Bellinger, G.; Shen, C.H.; Wen, J.; Asara, J.; McGraw, T.E.; et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell 2013, 49, 1167–1175. [Google Scholar] [CrossRef]
- Waldhart, A.N.; Dykstra, H.; Peck, A.S.; Boguslawski, E.A.; Madaj, Z.B.; Wen, J.; Veldkamp, K.; Hollowell, M.; Zheng, B.; Cantley, L.C.; et al. Phosphorylation of TXNIP by AKT mediates acute influx of glucose in response to insulin. Cell. Rep. 2017, 19, 2005–2013. [Google Scholar] [CrossRef]
- Chen, K.S.; DeLuca, H.F. Isolation and characterization of a novel cDNA from HL-60 cells treated with 1,25-dihydroxyvitamin D-3. Biochim. Biophys. Acta 1994, 1219, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Oberacker, T.; Bajorat, J.; Ziola, S.; Schroeder, A.; Röth, D.; Kastl, L.; Edgar, B.A.; Wagner, W.; Gülow, K.; Krammer, P.H. Enhanced expression of thioredoxin-interacting-protein regulates oxidative DNA damage and aging. FEBS Lett. 2018, 592, 2297–2307. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Saxena, G.; Mungrue, I.N.; Lusis, A.J.; Shalev, A. Thioredoxin-interacting protein: A critical link between glucose toxicity and beta-cell apoptosis. Diabetes 2008, 57, 938–944. [Google Scholar] [CrossRef]
- Gokulakrishnan, K.; Mohanavalli, K.T.; Monickaraj, F.; Mohan, V.; Balasubramanyam, M. Subclinical inflammation/oxidation as revealed by altered gene expression profiles in subjects with impaired glucose tolerance and Type 2 diabetes patients. Mol. Cell. Biochem. 2009, 324, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ismael, S.; Nasoohi, S.; Sakata, K.; Liao, F.-F.; McDonald, M.P.; Ishrat, T. Thioredoxin-interacting protein (TXNIP) associated NLRP3 inflammasome activation in human Alzheimer’s disease Brain. J. Alzheimer’s Dis. 2019, 68, 255–265. [Google Scholar] [CrossRef]
- Su, C.-J.; Feng, Y.; Liu, T.-T.; Liu, X.; Bao, J.-J.; Shi, A.-M.; Hu, D.-M.; Liu, T.; Yu, Y.-L. Thioredoxin-interacting protein induced alpha-synuclein accumulation via inhibition of autophagic flux: Implications for Parkinson’s disease. CNS Neurosci. Ther. 2017, 23, 717–723. [Google Scholar] [CrossRef]
- Ismael, S.; Wajidunnisa; Sakata, K.; McDonald, M.P.; Liao, F.-F.; Ishrat, T. ER stress associated TXNIP-NLRP3 inflammasome activation in hippocampus of human Alzheimer’s disease. Neurochem. Int. 2021, 148, 105104. [Google Scholar] [CrossRef]
- Tsubaki, H.; Mendsaikhan, A.; Buyandelger, U.; Tooyama, I.; Walker, D.G. Localization of thioredoxin-interacting protein in aging and Alzheimer’s disease brains. NeuroSci 2022, 3, 166–185. [Google Scholar] [CrossRef]
- Kim, S.Y.; Suh, H.-W.; Chung, J.W.; Yoon, S.-R.; Choi, I. Diverse functions of VDUP1 in cell proliferation, differentiation, and diseases. Cell. Mol. Immunol. 2007, 4, 345–351. [Google Scholar]
- Yu, F.-Y.; Chai, T.F.; He, H.; Hagen, T.; Luo, Y. Thioredoxin-interacting protein (Txnip) gene expression. Sensing oxidative phosphorylation status and glycolytic rate. J. Biol. Chem. 2010, 285, 25822–25830. [Google Scholar] [CrossRef] [PubMed]
- Fabrizio, P.; Liou, L.-L.; Moy, V.N.; Diaspro, A.; Valentine, J.S.; Gralla, R.B.; Longo, V.D. SOD2 functions downstream of Sch9 to extend longevity in yeast. Genetics 2013, 163, 35–46. [Google Scholar] [CrossRef]
- Harris, N.; Costa, V.; Maclean, M.; Mollapour, M.; Moradas-Ferreira, P.; Piper, P.W. MnSOD overexpression extends the yeast chronological (G0) life span but acts independently of Sir2p histone deacetylase to shorten the replicative life span of dividing cells. Free Radic. Biol. Med. 2003, 34, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, J.M.; Hekimi, S. Superoxide dismutase is dispensable for normal animal lifespan. Proc. Natl. Acad. Sci. USA 2012, 109, 5785–5790. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Yuan, J.-Q.; Lv, Y.-B.; Gao, X.; Yin, Z.-X.; Kraus, V.B.; Luo, J.-S.; Chei, C.-L.; Matchar, D.B.; Zeng, Y.; et al. Associations between superoxide dismutase, malondialdehyde and all-cause mortality in older adults: A community-based cohort study. BMC Genet. 2019, 19, 104. [Google Scholar] [CrossRef]
- Huie, R.E.; Padmaja, S. Reactions of NO and O2•−. Free Radic. Res. Commun. 1993, 18, 195–199. [Google Scholar] [CrossRef]
- Kissner, R.; Nauser, T.; Bugnon, P.; Lye, P.G.; Koppenol, W.H. Formation and properties of peroxynitrite as studied by laser flash photolysis, high-pressure stopped-flow technique, and pulse radiolysis. Chem. Res. Toxicol. 1997, 10, 1285–1292. [Google Scholar] [CrossRef]
- Nandi, A.; Yan, L.-L.; Jana, C.K.; Das, N. Role of catalase in oxidative stress- and age-associated degenerative diseases. Oxid. Med. Cell. Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef]
- Wu, M.; Deng, C.; Lo, T.-H.; Chan, K.-Y.; Li, X.; Wong, C.-M. Peroxiredoxin, senescence, and cancer. Cells 2022, 11, 1772. [Google Scholar] [CrossRef]
- Wonsey, D.R.; Zeller, K.I.; Dang, C.V. The c-Myc target gene PRDX3 is required for mitochondrial homeostasis and neoplastic transformation. Prod. Natl. Acad. Sci. USA 2002, 99, 6649–6655. [Google Scholar] [CrossRef]
- Huh, J.Y.; Kim, Y.; Jeong, J.; Park, J.; Kim, I.; Huh, K.H.; Kim, Y.S.; Woo, H.A.; Rhee, S.G.; Lee, K.-J.; et al. Peroxiredoxin 3 is a key molecule regulating adipocyte oxidative stress, mitochondrial biogenesis, and adipokine expression. Antioxid. Redox Signal. 2012, 16, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto-Imoto, H.; Minami, S.; Shioda, T.; Yamashita, Y.; Sakai, S.; Maeda, S.; Yamamoto, T.; Oki, S.; Takashima, M.; Yamamura, T.; et al. Age-associated decline of mondoA drives cellular senescence through impaired autophagy and mitochondrial homeostasis. Cell Rep. 2022, 38, 110444. [Google Scholar] [CrossRef]
- Ahmed, W.; Lingner, J. PRDX1 and MTH1 cooperate to prevent ROS-mediated inhibition of telomerase. Genes Dev. 2018, 32, 658–669. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.; Lingner, J. PRDX1 counteracts catastrophic telomeric cleavage events that are triggered by DNA repair activities post oxidative damage. Cell Rep. 2020, 33, 108347. [Google Scholar] [CrossRef] [PubMed]
- Aeby, E.; Ahmed, W.; Redon, S.; Simanis, V.; Lingner, J. Peroxiredoxin 1 protects telomeres from oxidative damage and preserves telomeric DNA for extension by telomerase. Cell Rep. 2016, 17, 3107–3114. [Google Scholar] [CrossRef]
- Chrichton, R.R.; Ward, R.J. Iron homeostasis. Met. Ions Biol. Syst. 1998, 35, 633–665. [Google Scholar]
- Ponka, P. Cellular iron metabolism. Kidney Int. 1999, 55, S2–S11. [Google Scholar] [CrossRef]
- Cankurtaran, M.; Yavuz, B.B.; Halil, M.; Ulger, Z.; Haznedaroglu, I.C.; Anogul, S. increased ferritin levels could reflect ongoing aging-associated inflammation and may obscure underlying iron deficiency in the geriatric population. Eur. Geriat. Med. 2012, 3, 277–280. [Google Scholar] [CrossRef]
- Kirsipuu, T.; Zadorožnaja, A.; Smirnova, J.; Friedemann, M.; Plitz, T.; Tougu, V.; Palumaa, P. Copper(II)-binding equilibria in human blood. Sci. Rep. 2020, 10, 5686. [Google Scholar] [CrossRef]
- Cabrera, A.; Alonzo, E.; Sauble, E.; Chu, Y.L.; Nguyen, D.; Linder, M.C.; Sato, D.S.; Mason, A.Z. Copper binding components of blood plasma and organs, and their responses to influx of large doses of (65)Cu, in the mouse. Biometals 2008, 21, 525–543. [Google Scholar] [CrossRef]
- Osaki, S. Kinetic studies of ferrous ion oxidation with crystalline human ferroxidase (ceruloplasmin). J. Biol. Chem. 1996, 241, 5053–5059. [Google Scholar]
- Patel, B.N.; Dunn, R.J.; Jeong, S.Y.; Zhu, Q.; Julien, J.-P.; David, S. Ceruloplasmin regulates iron levels in the CNS and prevents free radical injury. J. Neurosci. 2002, 22, 6578–6586. [Google Scholar] [CrossRef] [PubMed]
- Kono, S.; Yoshida, K.; Tomosugi, N.; Terada, T.; Hamaya, Y.; Kanaoka, S.; Miyajima, H. Biological effects of mutant ceruloplasmin on hepcidin-mediated internalization of ferroportin. Biochim. Biophys. Acta 2010, 1802, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Kono, S. Aceruloplasminemia: An update. Int. Rev. Neurobiol. 2013, 110, 125–151. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A red carpet for iron metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef]
- Cheli, V.T.; Sekhar, M.; Santiago Conzález, D.A.; Angeliu, C.G.; Denaroso, G.E.; Smith, Z.; Wang, C.; Paez, P.M. The expression of ceruloplasmin in astrocytes is essential for postnatal myelination and myelin maintenance in the adult brain. Glia 2023, 71, 2323–2342. [Google Scholar] [CrossRef]
- Harris, Z.L.; Takahashi, Y.; Miyajima, H.; Serizawa, M.; MacGillivray, R.T.; Gitlin, J.D. Aceruloplasminemia: Molecular characterization of this disorder of iron metabolism. Proc. Natl. Acad. Sci. USA 1995, 92, 2539–2543. [Google Scholar] [CrossRef]
- Morita, H.; Ikeda, S.; Yamamoto, K.; Morita, S.; Yoshida, K.; Nomoto, S.; Kato, M.; Yanagisawa, N. Hereditary ceruloplasmin deficiency with hemosiderosis: A clinicopathological study of a Japanese family. Ann. Neurol. 1995, 37, 646–656. [Google Scholar] [CrossRef]
- Yoshida, K.; Furihata, K.; Takeda, S.; Nakamura, A.; Yamamoto, K.; Morita, H.; Hiyamuta, S.; Ikeda, S.; Shimizu, N.; Yanagisawa, N. A mutation in the ceruloplasmin gene is associated with systemic hemosiderosis in humans. Nat. Genet. 1995, 9, 267–272. [Google Scholar] [CrossRef]
- Jeong, S.Y.; David, S. Age-related changes in iron homeostasis and cell death in the cerebellum of ceruloplasmin-deficient mice. J. Neurosci. 2006, 26, 9810–9819. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Chen, X.-b.; Hong, Y.-c.; Zhu, H.; He, Q.-j.; Yang, B.; Ying, M.-d.; Cao, J. Identification of PRDX6 as a regulator of ferroptosis. Acta Pharmacol. Sin. 2019, 40, 1334–1342. [Google Scholar] [CrossRef]
- Torres-Velarde, J.M.; Allen, K.N.; Salvador-Pascual, A.; Leija, R.G.; Luang, D.; Moreno-Santillán, D.D.; Ensminger, D.C.; Vázquez-Medina, J.P. peroxiredoxin 6 suppresses ferroptosis in lung endothelial cells. Free Radic. Biol. Med. 2024, 218, 82–93. [Google Scholar] [CrossRef]
- Sauret, J.M.; Marinides, G.; Wang, G.K. Rhabdomyolysis. Am. Fam. Phys. 2002, 65, 907–912. [Google Scholar]
- Rother, R.P.; Bell, L.; Hillman, P.; Gladwin, M.T. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin. J. Am. Med. Assoc. 2005, 293, 1653–1662. [Google Scholar] [CrossRef]
- Kristiansen, M.; Graversen, J.H.; Jacobsen, C.; Sonne, O.; Hoffmann, H.J.; Law, S.K.; Moestrup, S.K. Identification of the haemoglobin scavenger receptor. Nature 2001, 409, 198–201. [Google Scholar] [CrossRef]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Tolosano, E. Haptoglobin and hemopexin in heme detoxification and iron recycling. In Acute Phase Proteins—Regulation and Functions of Acute Phase Proteins; Veas, F., Ed.; Intech: Rijeka, Croatia, 2011; pp. 261–288. [Google Scholar] [CrossRef]
- Bunn, H.F.; Jandl, J.H. Exchange of heme among hemoglobins and hemoglobin and albumin. J. Biol. Chem. 1968, 243, 465–475. [Google Scholar]
- Hvidberg, V.; Maniecki, M.B.; Jacobson, C.; Højrup, P.; Møller, H.J.; Moestrup, S.K. Identification of the receptor scavenging hemopexin-heme complexes. Blood 2005, 106, 2572–2579. [Google Scholar] [CrossRef]
- Pan, M.; Wang, P.; Zheng, C.; Zhang, H.Y.; Lin, S.; Shao, B.; Zhuge, Q.; Jin, K. Aging systemic milieu impairs outcome after ischemic stroke in rats. Aging Dis. 2017, 8, 519–530. [Google Scholar] [CrossRef]
- Sarpong-Kumankomah, S.; Knox, K.B.; Kelly, M.E.; Hunter, G.; Popescu, B.; Nichol, H.; Kopciuk, K.; Ntanda, H.; Galler, J. Quantification of human plasma metalloproteins in multiple sclerosis, ischemic stroke and healthy controls reveals an association of haptoglobin-hemoglobin complexes with age. PLoS ONE 2022, 17, e0262160. [Google Scholar] [CrossRef]
- Schipper, H.M.; Song, W.; Tavitan, A.; Cressatti, M. The sinister face of heme oxygenase-1 in brain aging and disease. Progr. Neurobiol. 2019, 172, 40–70. [Google Scholar] [CrossRef]
- Wei, D.; Qu, C.; Zhao, N.; Li, S.; Pu, N.; Song, Z.; Tao, Y. The significance of precisely regulating heme oxygenase-1 expression: Another avenue for treating age-related ocular disease? Ageing Res. Rev. 2024, 97, 102308. [Google Scholar] [CrossRef]
- Picard, E.; Ranchon-Cole, I.; Jonet, L.; Beaumont, C.; Behar-Cohen, F.; Courtois, Y.; Jeanny, J.-C. Light-induced retinal degeneration correlates with changes in iron metabolism gene expression, ferritin level, and aging. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1261–1274. [Google Scholar] [CrossRef]
- Chen, C.; Yang, K.; He, D.; Yang, B.; Tao, L.; Chen, J.; Wu, Y. Induction of ferroptosis by HO-1 contributes to retinal degeneration in mice with defective clearance of all-trans-retinal. Free Radic. Biol. Med. 2023, 194, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Miyamura, N.; Ogawa, T.; Boylan, S.; Morse, L.S.; Handa, J.T.; Hjelmeland, L.M. Topographic and age-dependent expression of heme oxygenase-1 and catalase in the human retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1562–1565. [Google Scholar] [CrossRef]
- Ma, C.; Pi, C.; Yang, Y.; Lin, L.; Shi, Y.; Li, Y.; Li, Y.; He, X. Nampt expression decreases age-related senescence in rat bone marrow mesenchymal stem cells by targeting Sirt1. PLoS ONE 2017, 12, e0170930. [Google Scholar] [CrossRef]
- Wu, Y.; Williams, E.G.; Dubuis, S.; Mottis, A.; Jovaisaite, V.; Houton, S.M.; Argmann, C.A.; Faridi, P.; Wolski, W.; Kutalik, Z.; et al. Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population. Cell 2014, 158, 1415–1430. [Google Scholar] [CrossRef]
- Migliavacca, E.; Tay, S.K.H.; Patel, H.P.; Sonntag, T.; Civiletto, G.; McFarlane, C.; Forrester, T.; Barton, S.J.; Leow, M.K.; Antoun, E.; et al. Mitochondrial oxidative capacity and NAD+ biosynthesis are reduced in human sarcopenia across ethnicities. Nat. Commun. 2019, 10, 5808. [Google Scholar] [CrossRef]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef]
- Beck, J.S.; Mufson, E.J.; Counts, S.E. Evidence for mitochondrial UPR gene activation in familial and sporadic Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 610–614. [Google Scholar] [CrossRef]
- Eisner, V.; Picard, M.; Hajnóczky, G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 2018, 20, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, O.; Ishihara, T.; Maeda, M.; Matsunaga, Y.; Tsukamoto, S.; Kawano, N.; Miyado, K.; Shitara, H.; Yokota, S.; Nomura, M.; et al. Mitochondrial fission factor Drp1 maintains oocyte quality via dynamic rearrangement of multiple organelles. Curr. Biol. 2014, 24, 2451–2458. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, Y.; Zhang, Z.; Roda, R.; Fukaya, M.; Wakabayashi, J.; Wakabayashi, N.; Kensler, T.W.; Reddy, H.; Iijima, M.; Sesaki, H. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J. Cell Biol. 2012, 197, 535–551. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–429. [Google Scholar] [CrossRef]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structure, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef]
- López-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Murshid, A.; Prince, T. The shock of aging: Molecular chaperones and the heat shock response in longevity and aging--a mini-review. Gerontology 2009, 55, 550–558. [Google Scholar] [CrossRef]
- Löw, P. The role of ubiquitin-proteasome system in aging. Gen. Comp. Endocrinol. 2011, 172, 39–43. [Google Scholar] [CrossRef]
- Saez, I.; Vilchez, D. The mechanistic links between proteasome activity, aging and age-related diseases. Curr. Genom. 2014, 15, 38–51. [Google Scholar] [CrossRef]
- Frankowska, N.; Lisowska, K.; Witkowski, J.M. Proteolysis dysfunction in the process of aging and age-related diseases. Front. Aging 2022, 3, 927630. [Google Scholar] [CrossRef]
- Ponnappan, U.; Zhong, M.; Trebilcock, G.U. Decreased proteasome-mediated degradation in T cells from the elderly: A role in immune senescence. Cell. Immunol. 1999, 192, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Chondrogianni, N.; Stratford, F.L.L.; Trougakos, I.P.; Friguet, B.; Rivett, A.J.; Gonos, E.S. Central role of the proteasome in senescence and survival of human fibroblasts. Induction of a senescence-like phenotype upon its inhibition and resistance to stress upon its activation. J. Biol. Chem. 2003, 278, 28026–28037. [Google Scholar] [CrossRef] [PubMed]
- Dahlmann, B. Role of proteasomes in disease. BMC Biochem. 2007, 8 (Suppl. S1), S3. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Kobayashi, S. Choose delicately and reuse adequately: The newly revealed process of autophagy. Biol. Pharmaceut. Bull. 2015, 38, 1098–1103. [Google Scholar] [CrossRef]
- Collins, T.J.; Berridge, M.J.; Lipp, P.; Bootman, M.D. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 2002, 21, 1616–1627. [Google Scholar] [CrossRef]
- Hoshino, A.; Mita, Y.; Okawa, Y.; Ariyoshi, M.; Iwai-Kanai, E.; Ueyama, T.; Ikeda, K.; Ogata, T.; Matoba, S. Cytosolic p53 inhibits parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 2013, 4, 2308. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Prescott, A.R.; Allen, G.F.G.; Tamjar, J.; Munson, M.J.; Thomson, C.; Muqit, M.M.K.; Ganley, I.G. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 2016, 214, 333–345. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- García-Prat, L.; Martinez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodriguez-Ubreva, J.; Rebello, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.Y.; Blum, A.; Liu, J.; Finkel, T. The role of mitochondria in aging. J. Clin. Investig. 2018, 128, 3662–3670. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.; Cevenini, E.; Nasi, M.; de Biasi, S.; Salvioli, S.; Monti, D.; Benatti, S.; Gibellini, L.; Cotichini, R.; Stazi, M.A.; et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur. J. Immunol. 2014, 44, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narenda, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Penna, F.; Ballarò, R.; Beltrá, M.; de Lucia, S.; Costelli, P. Modulating metabolism to improve cancer-imduced muscle wasting. Oxid. Med. Cell. Longev. 2018, 2018, 7153610. [Google Scholar] [CrossRef]
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef]
- Bye, A.; Sjøblom, B.; Wentzel-Larsen, T.; Grønberg, B.H.; Baracos, V.E.; Hjemstad, M.J.; Aass, N.; Bremnes, R.M.; Fløtten, Ø.; Jordhøy, M. Muscle mass and association to quality of life in non-small cell lung cancer patients. J. Cachexia Sarcopenia Muscle 2017, 8, 759–767. [Google Scholar] [CrossRef]
- Nipp, R.D.; Fuchs, G.; El-Jawahri, A.; Mario, J.; Troschel, E.M.; Greer, J.A.; Gallgher, E.R.; Jackson, V.A.; Kambadakone, A.; Hong, T.S.; et al. Sarcopenia is associated with quality of life and depression in patients with advanced cancer. Oncologist 2017, 23, 97–104. [Google Scholar] [CrossRef]
- Morley, J.E.; Anker, S.D.; Evans, W.J. Cachexia and aging: An update based on the fourth international cachexia meeting. J. Nutr. Health Aging 2009, 13, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Garcia, J.M. Sarcopenia, cachexia and aging: Diagnosis, mechanisms and therapeutic options. Gerontology 2014, 60, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Roubenoff, R. The pathophysiology of wasting in elderly. J. Nutr. 1999, 129, 256S–259S. [Google Scholar]
- De la Fuente, M.; Miquel, J. An update of the oxidation-inflammation theory of aging: The involvement of the immune system in oxi-inflamm-aging. Curr. Pharm. Des. 2009, 15, 3003–3026. [Google Scholar] [CrossRef]
- Martinez de Toda, I.; Ceprian, N.; Diaz-Del Cerro, E.; De la Fuente, M. The role of immune cells in oxi-inflamm-aging. Cells 2021, 10, 2974. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Bientinesi, E.; Monti, D. Immunosenescence and inflammaging in the aging process: Age-related diseases or longevity? Ageing Res. Rev. 2021, 71, 101422. [Google Scholar] [CrossRef]
- Mohammed, S.; Thadathil, N.; Selvarani, R.; Nicklas, E.H.; Wang, D.; Miller, B.F.; Richardson, A.; Deepa, S.S. Necroptosis contributes to chronic inflammation and fibrosis in aging liver. Aging Cell 2021, 20, e13512. [Google Scholar] [CrossRef]
- Miao, E.A.; Leaf, I.A.; Treuting, P.M.; Mao, D.P.; Dors, M.; Sarkar, A.; Warren, S.E.; Wewers, M.D.; Aderem, A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 2010, 11, 1136–1142. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirios, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Ardehali, H.; Min, J.; Wang, F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat. Rev. Cardiol. 2023, 20, 7–23. [Google Scholar] [CrossRef]
- Pittet, D.; Wenzel, R.P. Nosocomial bloodstream infections. Secular trends in rates, mortality, and contribution to total hospital deaths. Arch. Intern. Med. 1995, 155, 1177–1184. [Google Scholar] [CrossRef]
- Llewelyn, M.J.; Cohen, J. Tracking the microbes in sepsis: Advancements in treatment bring challenges for microbial epidemiology. Clin. Infect. Dis. 2007, 44, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Kollef, K.E.; Schramm, G.E.; Wills, A.R.; Reichley, R.M.; Mirek, S.T.; Kollef, M.H. Predictors of 30-day mortality and hospital costs in patients with ventilator-associated pneumonia attributed to potentially antibiotic-resistant gram-negative bacteria. Chest 2008, 134, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Babu, M.; Menon, V.P.; Devi, P.U. Prevalence of antimicrobial resistant pathogens in severe sepsis and septic shock patients. J. Young Pharm. 2018, 10, 358–361. [Google Scholar] [CrossRef]
- Lin, G.-L.; McGinley, J.P.; Drysdale, S.B.; Pollard, A.J. Epidemiology and immune pathogenesis of viral sepsis. Front. Immunol. 2018, 9, 2147. [Google Scholar] [CrossRef]
- Podnos, Y.D.; Jiminez, J.C.; Wilson, S.E. Intraabdominal sepsis in elderly persons. Clin. Infect. Dis. 2002, 35, 62–68. [Google Scholar] [CrossRef]
- Williams, M.D.; Braun, L.A.; Cooper, I.M.; Johnston, J.; Weiss, R.V.; Qualy, R.I.; Linde-Zwirble, W. Hospitalized cancer patients with severe sepsis: Analysis of incidence, mortality, and associated costs of care. Crit. Care 2004, 8, R291–R298. [Google Scholar] [CrossRef]
- Steinberg, D.; Parthasarathy, S.; Carew, T.E.; Khoo, J.C.; Witztum, J.L. Beyond cholesterol: Modification of low-density lipoprotein that increases its atherogenicity. N. Engl. J. Med. 1989, 320, 915–924. [Google Scholar] [CrossRef]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Wick, G.; Knoflach, M.; Xu, Q. Autoimmune and inflammatory mechanisms in atherosclerosis. Annu. Rev. Atheroscl. 2004, 22, 361–403. [Google Scholar] [CrossRef]
- Tabas, I.; Garcia-Gardeña, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 2015, 209, 13–22. [Google Scholar] [CrossRef]
- Bergheanu, S.C.; Bodde, M.C.; Jukema, J.W. Pathophysiology and treatment of atherosclerosis. Current view and future perspective on lipoprotein modification treatment. Neth. Heart 2017, 25, 231–242. [Google Scholar] [CrossRef]
- Gisterå, A.; Hansson, G. The immunology of atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis. Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef]
- Head, T.; Daunert, S.; Goldschmidt-Clermont, P.J. The aging risk and atherosclerosis: A fresh look at arterial homeostasis. Front. Genet. 2017, 8, 216. [Google Scholar] [CrossRef]
- Vecoli, C.; Borghini, A.; Andreassi, M.G. The molecular biomarkers of vascular aging and atherosclerosis: Telomere length and mitochondrial DNA4977 common deletion. Mut. Res. 2020, 784, 108309. [Google Scholar] [CrossRef]
- Wong, J.J.; Hong, R.; Teo, L.L.Y.; Tan, R.-S.; Koh, A.S. Atherosclerotic cardiovascular disease in aging and the role of advanced cardiovascular imaging. Cardiovasc. Health 2024, 1, 11. [Google Scholar] [CrossRef]
- Daugherty, A.; Dunn, J.L.; Rateri, D.L.; Heinecke, J.W. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J. Clin. Investig. 1994, 94, 437–444. [Google Scholar] [CrossRef]
- Hazen, S.L.; Heinecke, J.W. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J. Clin. Investig. 1997, 99, 2075–2081. [Google Scholar] [CrossRef] [PubMed]
- Malle, E.; Marsche, G.; Arnhold, J.; Davies, M.J. Modification of low-density lipoprotein by myeloperoxidase-derived oxidants and reagent hypochlorous acid. Biochim. Biophys. Acta 2006, 1761, 392–415. [Google Scholar] [CrossRef] [PubMed]
- Balla, G.; Jacob, H.S.; Eaton, J.W.; Belcher, J.D.; Vercelotti, G.M. Hemin: A possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscl. Thromb. 1991, 11, 1700–1711. [Google Scholar] [CrossRef]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercelotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887. [Google Scholar] [CrossRef]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon III, R.O.; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Eich, R.F.; Li, T.; Lemon, D.D.; Doherty, D.H.; Curry, S.R.; Aitken, J.F.; Mathews, A.J.; Johnson, K.A.; Smith, R.D.; Philips, G.N., Jr.; et al. Mechanisms of NO-induced oxidation of myoglobin and hemoglobin. Biochemistry 1996, 35, 6076–6983. [Google Scholar] [CrossRef]
- Olson, J.S.; Foley, E.W.; Rogge, C.; Tsai, A.L.; Doyle, M.P.; Lemon, D.D. NO scavenging and the hypertensive effect of hemoglobin-based blood substitutes. Free Radic. Biol. Med. 2004, 36, 685–697. [Google Scholar] [CrossRef]
- Flögel, U.; Fago, A.; Rassaf, T. Keeping the heart in balance: The functional interactions of myoglobin with nitrogen oxides. J. Exp. Biol. 2010, 213, 2726–2733. [Google Scholar] [CrossRef]
- Eiserich, J.P.; Baldus, S.; Brennan, M.-L.; Ma, W.; Zhang, C.; Tousson, A.; Castro, L.; Lusis, A.J.; Nauseef, W.M.; White, C.R.; et al. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science 2002, 196, 2391–2394. [Google Scholar] [CrossRef]
- Ariesen, M.J.; Claus, S.P.; Rinkel, G.J.; Algra, A. Risk factors for intracerebral hemorrhage in the general population: A systematic view. Stroke 2003, 34, 2060–2065. [Google Scholar] [CrossRef]
- Jani, B.; Rajkumar, C. Ageing and vascular ageing. Postgrad. Med. J. 2006, 82, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Dastur, C.K.; Yu, W. Current management of spontaneous intracerebral haemorrhage. Stroke Vasc. Neurol. 2017, 2, e000047. [Google Scholar] [CrossRef]
- Flemmig, J.; Schlorke, D.; Kühne, F.-W.; Arnhold, J. Inhibition of heme-induced hemolysis of red blood cells by the chlorite-based drug WF10. Free Rad. Res. 2016, 50, 1386–1395. [Google Scholar] [CrossRef]
- Deuel, J.W.; Schaer, C.A.; Boretti, F.S.; Opitz, L.; Garcia-Rubio, I.; Baek, J.H.; Spahn, D.R.; Buehler, P.W.; Schaer, D.J. Hemoglobinuria-related acute kidney injury is driven by intrarenal oxidative reactions triggering a heme toxicity response. Cell Death Dis. 2016, 7, e2064. [Google Scholar] [CrossRef] [PubMed]
- Rosendaal, F.R.; van Hylckama Vlieg, A.; Doggen, C.J.M. Venous thrombosis in the elderly. J. Thromb. Haemost. 2007, 5 (Suppl. S1), 310–317. [Google Scholar] [CrossRef]
- Engbers, M.J.; van Hylckama Vlieg, A.; Rosendaal, F.R. Venous thrombosis in the elderly: Incidence, risk factors and risk groups. J. Thromb. Haemost. 2010, 8, 2105–2112. [Google Scholar] [CrossRef]
- Akrivou, D.; Perlepe, G.; Kirgou, P.; Gourgoulianis, K.I.; Malli, F. Pathophysiological aspects of aging in venous thromboembolism: An update. Medicina 2022, 58, 1078. [Google Scholar] [CrossRef]
- Sinclair, A.; Dunning, T.; Rodriguez-Mañas, L. Diabetes in older people: New insights and remaining challenges. Lancet Diabet. Endocrinol. 2015, 3, 275–285. [Google Scholar] [CrossRef]
- Arnhold, J. Cell and tissue destruction in selected disorders. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 249–287. [Google Scholar] [CrossRef]
- Potenza, M.A.; Gagliardi, S.; Nacci, C.; Carratu, M.R.; Montagnani, M. Endothelial dysfunction in diabetes: From mechanisms to therapeutic targets. Curr. Med. Chem. 2009, 16, 94–112. [Google Scholar] [CrossRef]
- Hirose, A.; Tanikawa, T.; Mori, H.; Okada, Y.; Tanaka, Y. Advanced glycation end products increase endothelial permeability through RAGE/Rho signaling pathway. FEBS Lett. 2010, 584, 61–66. [Google Scholar] [CrossRef]
- Koenig, R.J.; Peterson, C.M.; Jones, R.L.; Saudek, C.; Lehrmann, M.; Cerami, A. Correlation of glucose regulation and hemoglobin A1c in diabetes mellitus. N. Engl. J. Med. 1976, 295, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Bunn, H.F.; Gabbay, K.H.; Gallop, P.M. The glycosylation of hemoglobin: Relevance to diabetes mellitus. Science 1978, 200, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Ku, Y.-H.; Ho, J.-X.; Kim, Y.-K.; Suh, J.-S.; Singh, M. Progressive impairment of erythrocyte deformability as indicator of microangiopathy in type 2 diabetes mellitus. Clin. Hemorheol. Microcirc. 2007, 36, 253–261. [Google Scholar] [PubMed]
- Babu, N.; Singh, M. Influence of hyperglycemia on aggregation, deformability and shape parameters of erythrocytes. Clin. Hemorheol. Microcirc. 2004, 31, 273–280. [Google Scholar]
- Kung, C.-M.; Tseng, Z.-I.; Wang, H.-L. Erythrocyte fragility with level of glycosylated hemoglobin in type 2 diabetic patients. Clin. Hemorheol. Microcirc. 2009, 43, 345–351. [Google Scholar] [CrossRef]
- Agarwal, R.; Smart, T.; Nobre-Cordosa, J.; Richards, C.; Bhatnagar, R.; Tufail, A.; Shima, D.; Jones, P.H.; Pavesio, C. Assessment of red blood cell deformability in type 2 diabetes mellitus and diabetic retinopathy by dual optical tweezers stretching technique. Sci. Rep. 2016, 6, 15873. [Google Scholar] [CrossRef]
- Ernst, E.; Matrai, A. Altered red and white blood cell rheology in type II diabetes. Diabetes 1986, 35, 1412–1415. [Google Scholar] [CrossRef]
- Popov, D. Endothelial cell dysfunction in hyperglycemia: Phenotypic change, intracellular signaling modification, ultrastructural alteration, and potential clinical outcomes. Int. J. Diabetes Mellitus 2010, 2, 189–195. [Google Scholar] [CrossRef]
- Popov, D. Towards understanding the mechanisms of impeded vascular function in the diabetic kidney. In Cellular Dysfunction in Atherosclerosis and Diabetes—Reports from Bench to Bedside; Simionescu, M., Sima, A., Popov, D., Eds.; Romanian Academy’s Publishing House: Bucharest, Romania, 2004; pp. 244–259. [Google Scholar]
- Dokken, B.B. The pathophysiology of cardiovascular disease and diabetes: Beyond blood pressure and lipids. Diabetes Spectr. 2008, 21, 160–165. [Google Scholar] [CrossRef]
- Xiang, Y.; Cheng, J.; Wang, D.; Hu, X.; Xie, Y.; Stitham, J.; Atteva, G.; Du, J.; Tang, W.H.; Lee, S.H.; et al. Hyperglycemia repression of miR-24 coordinately upregulates endothelial cell expression and secretion of von Willebrand factor. Blood 2015, 125, 3377–3387. [Google Scholar] [CrossRef]
- Whincup, P.H.; Danesh, J.; Walker, M.; Lennon, L.; Thomson, A.; Appleby, P.; Rumley, A.; Lowe, G.D. Von Willebrand factor and coronary heart disease: Prospective study and meta-analysis. Eur. Heart J. 2002, 23, 1764–1770. [Google Scholar] [CrossRef]
- Frankel, D.S.; Meigs, J.B.; Massaro, J.M.; Wilson, P.W.; O’Donnell, C.J.; D’Agostino, R.B.; Tofler, G.H. Von Willebrand factor, type 2 diabetes mellitus, and risk of cardiovascular disease: The Framingham offspring study. Circulation 2008, 118, 2533–2539. [Google Scholar] [CrossRef]
- Nakamura, K.; Smyth, M.J. Targeting cancer-related inflammation in the era of immunotherapy. Immunol. Cell Biol. 2017, 95, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Smyth, M.J. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell Mol. Immunol. 2020, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The tumor microenvironment: A milieu hindering and obstructing antitumor immune response. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef]
- Barnestein, R.; Galland, L.; Kalfeist, L.; Ghiringhelli, F.; Ladoire, S.; Limagne, E. Immunosuppressive tumor microenvironment modulation by chemotherapies and targeted therapies to enhance immunotherapy effectiveness. OncoImmunology 2022, 11, 2120676. [Google Scholar] [CrossRef]
- Tie, Y.; Tang, F.; Wei, Y.-Q.; Wie, X.-W. Immunosuppressive cells in cancer: Mechanisms and potential therapeutic targets. J. Hematol. Oncol. 2022, 15, 61. [Google Scholar] [CrossRef]
- Othman, N.; Jamal, R.; Abu, N. Cancer-derived exosomes as effectors of key inflammation-related players. Front. Immunol. 2019, 10, 2103. [Google Scholar] [CrossRef]
- He, Z.; Zhang, S. Tumor-associated macrophages and their functional transformation in the hypoxic tumor microenvironment. Front. Immunol. 2021, 12, 741305. [Google Scholar] [CrossRef]
- Pastorevka, S.; Ratcliffe, P.J.; Pastorek, J. Molecular mechanisms of carbonic anhydrase IX-mediated pH regulation under hypoxia. J. Compil. 2008, 101 (Suppl. S4), 8–15. [Google Scholar] [CrossRef]
- Kaluz, S.; Kaluzová, M.; Liao, S.-Y.; Lerman, M.; Stanbridge, E.J. Transcriptional control of the tumor- and hypoxia-marker carbonic anhydrase 9: A one transcription factor (HIF-1) show? Biochim. Biophys. Acta 2009, 1795, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Becker, H.M. Carbonic anhydrase IX and acid transport in cancer. Br. J. Cancer 2020, 122, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Jäättelä, M.; Liu, B. pH gradient reversal fuels cancer progression. Int. J. Biochem. Cell Biol. 2020, 125, 105796. [Google Scholar] [CrossRef]
- Park, Y.; Jeong, J.; Seong, S.; Kim, W. In silico evaluation of natural compounds for an acidic extracellular environment in human breast cancer. Cells 2021, 10, 2673. [Google Scholar] [CrossRef]
- Logozzi, M.; Spugnini, E.; Mizzoni, D.; Di Raimo, R.; Fais, S. Extracellular acidity and increased exosome release as key phenotypes of malignant tumors. Cancer Metastasis Rev. 2019, 38, 93–101. [Google Scholar] [CrossRef]
- Riek, R.; Eisenberg, D.S. The activities of amyloids from a structural perspective. Nature 2016, 539, 227–235. [Google Scholar] [CrossRef]
- Walti, M.A.; Ravotti, F.; Arai, H.; Glabe, C.G.; Wall, J.S.; Bockmann, A.; Guntert, P.; Meier, B.H.; Riek, R. Atomic-resolution structure of a disease-relevant Abeta(1-42) amyloid fibril. Proc. Natl. Acad. Sci. USA 2016, 113, E4976–E4984. [Google Scholar] [CrossRef]
- Fezoui, Y.; Teplow, D.B. Kinetic studies of amyloid-bate protein fibril assembly. Differential effects of alpha-helix stabilization. J. Biol. Chem. 2002, 277, 36948–36954. [Google Scholar] [CrossRef]
- Uversky, V.N. Protein misfolding in lipid-mimetic environments. Adv. Exp. Med. Biol. 2015, 855, 33–66. [Google Scholar] [CrossRef]
- Prasad, H.; Rao, R. Endosomal acid-base homeostasis in neurodegenerative diseases. Rev. Physiol. Biochem. Pharmacol. 2020, 185, 195–231. [Google Scholar] [CrossRef]
- Decker, Y.; Németh, E.; Schomburg, R.; Chemla, A.; Fülöp, L.; Menger, M.D.; Liu, Y.; Fassbender, K. Decreased pH in the aging brain and Alzheimer’s disease. Neurobiol. Aging 2021, 101, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Hagihara, H.; Miyakawa, T. Decreased brain pH correlated with progression of Alzheimer disease neuropathology: A systematic review and meta-analyses of postmortem studies. Int. J. Neuropsychopharmacol. 2024, 27, pyae047. [Google Scholar] [CrossRef]
- Levi, S.; Finazzi, D. Neurodegeneration with brain iron accumulation: Update on pathogenic mechanisms. Front. Pharmacol. 2014, 5, 99. [Google Scholar] [CrossRef]
- Mena, N.P.; Urrutia, P.J.; Lourido, F.; Carrasco, C.M.; Núñez, M.T. Mitochondrial iron homeostasis and its dysfunction in neurodegenerative disorders. Mitochondrion 2015, 21, 92–105. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.; et al. Ferroptosis: A regulated death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- De Bie, P.; Muller, P.; Wijmenga, C.; Klomp, L.W.J. Molecular pathogenesis of Wilson and Menkes disease: Correlation of mutations with molecular defects and disease phenotypes. J. Med. Genet. 2007, 44, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R.; Pasqualetti, P.; Dal Forno, G.; Moffa, F.; Cassetta, E.; Lupoi, D.; Vernieri, F.; Rossi, L.; Baldassini, M.; Rossini, P.M. Excess of serum copper not related to ceruloplasmin in Alzheimer disease. Neurology 2005, 64, 1040–1046. [Google Scholar] [CrossRef]
- Squitti, R.; Bressi, F.; Pasqualetti, P.; Bonomini, C.; Ghidoni, R.; Binetti, G.; Cassetta, E.; Moffa, F.; Ventriglia, M.; Vernieri, F.; et al. Longitudinal prognostic value of serum “free” copper in patients with Alzheimer disease. Neurology 2009, 72, 50–55. [Google Scholar] [CrossRef]
- Righy, C.; Bozza, M.T.; Oliveira, M.F.; Bozza, F.A. Molecular, cellular and clinical aspects of intracerebral hemorrhage: Are the enemies within? Curr. Neuropharmacol. 2016, 14, 392–402. [Google Scholar] [CrossRef]
- Chiabrando, D.; Fiorito, V.; Petrillo, S.; Tolosano, E. Unraveling the role of heme in neurodegeneration. Front. Neurosci. 2018, 12, 712. [Google Scholar] [CrossRef]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercelotti, G.M. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014, 13, 377–390. [Google Scholar] [CrossRef]
- Chiziane, E.; Telemann, H.; Krueger, M.; Adler, J.; Arnhold, J.; Alia, A.; Flemmig, J. Free heme and amyloid-β: A fatal liaison in Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 61, 963–984. [Google Scholar] [CrossRef]
- Song, I.-U.; Kim, Y.-D.; Chung, S.-W.; Cho, H.J. Association between serum haptoglobin and the pathogenesis of Alzheimer’s disease. Intern. Med. 2015, 54, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, A.; Clark, M.; So, P.-W. The aging of iron man. Front. Aging Neurosci. 2018, 10, 65. [Google Scholar] [CrossRef] [PubMed]
- Moshage, H. Cytokines and the hepatic acute phase response. J. Pathol. 1997, 181, 74–80. [Google Scholar]
- Lin, T.; Maita, D.; Thundivalappil, S.R.; Riley, F.E.; Hambsch, J.; van Marter, L.J.; Christou, H.A.; Berra, L.; Fagan, S.; Christiani, D.C.; et al. Hemopexin in severe inflammation and infection: Mouse models and human diseases. Crit. Care 2015, 19, 166. [Google Scholar] [CrossRef]
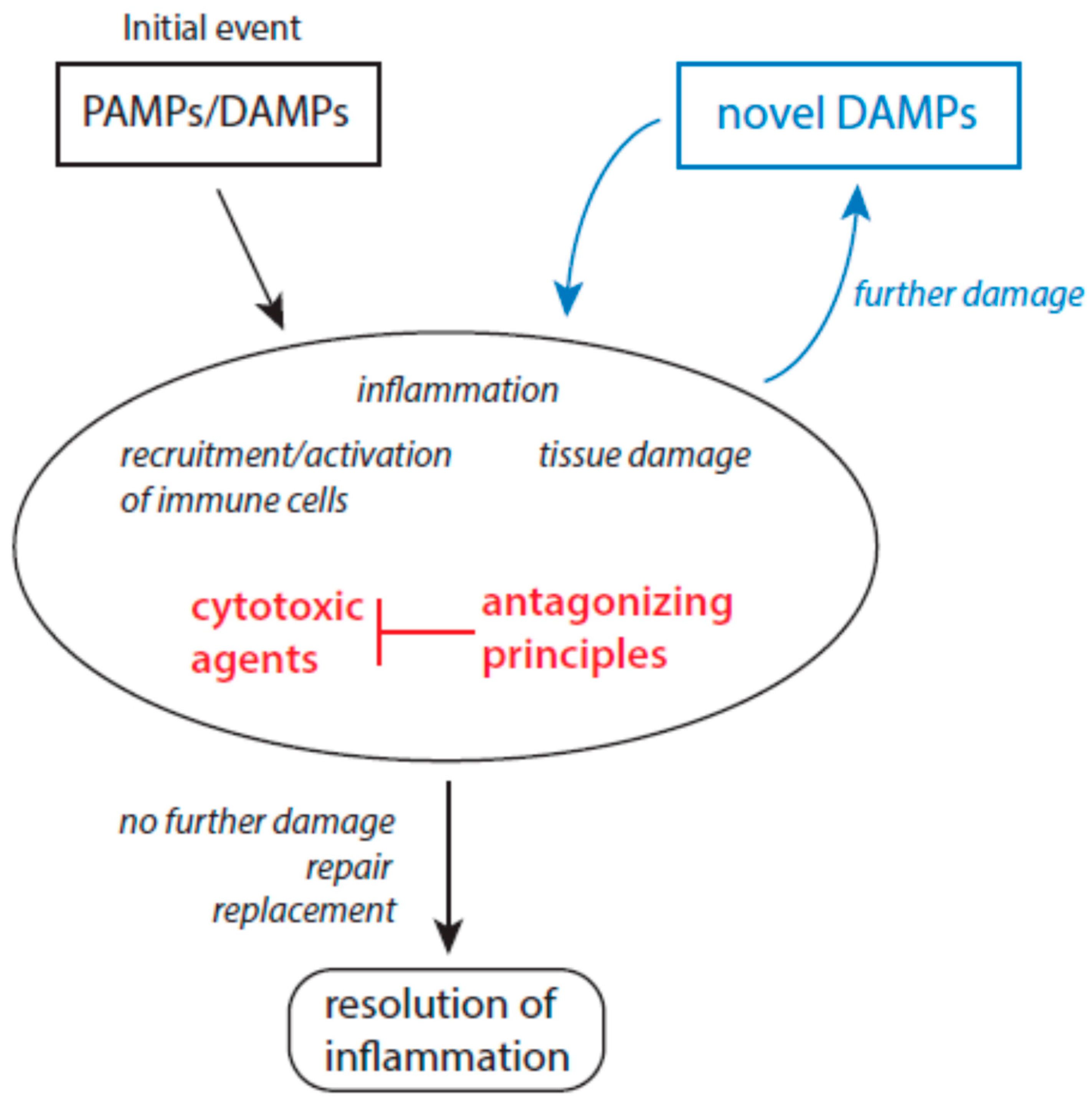
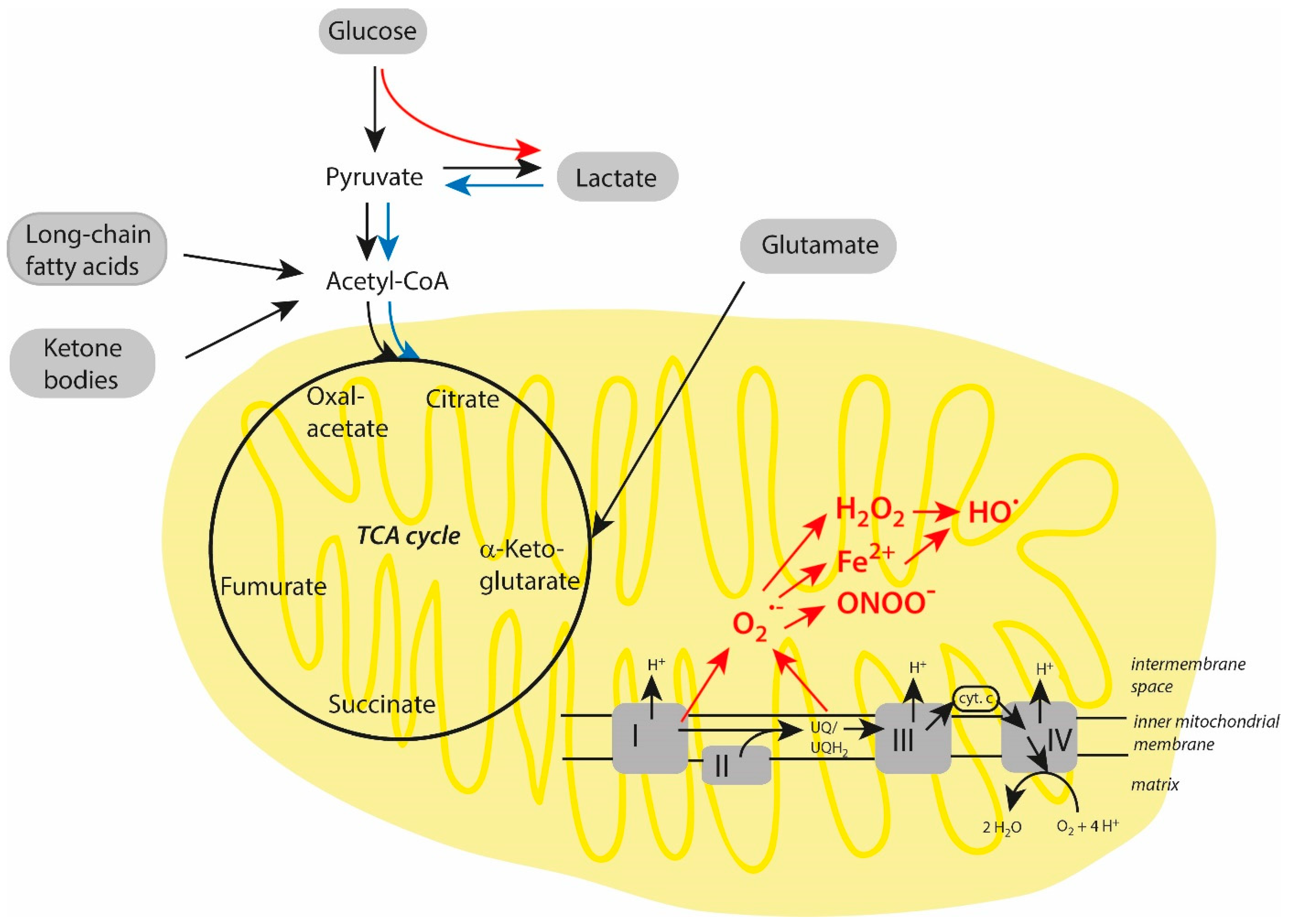
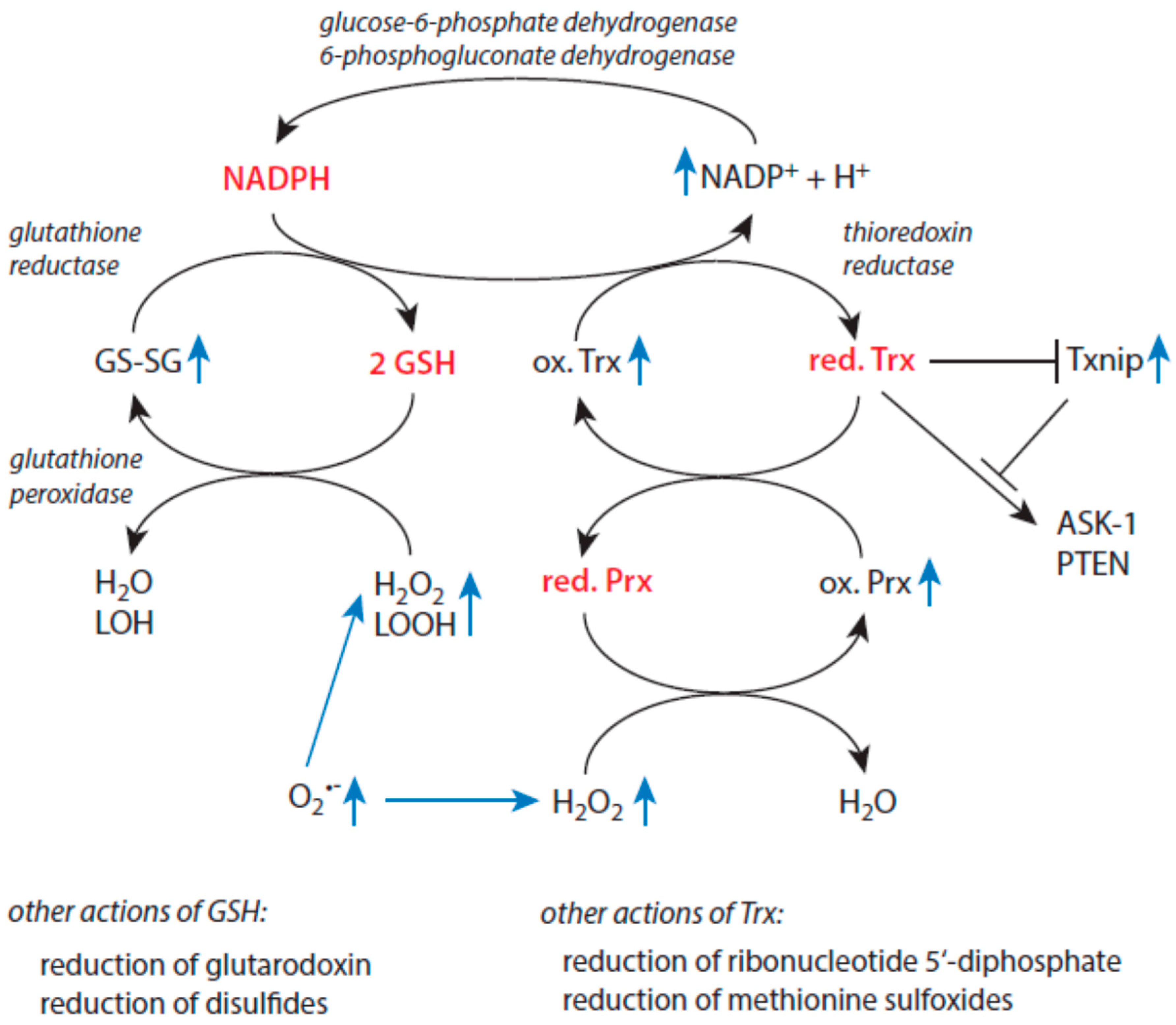

{kind=link}
{kind=link}
{kind=link}
| Predominant Features or Properties | Initiation and Propagation of Inflammation | Resolution of Inflammation |
|---|---|---|
| Immune response | Recruitment and activation of immune cells | Deactivation of immune cells, immunosuppression |
| Presence of molecular patterns | Yes | No |
| Main physiological processes | Elimination of pathogens, removal of damaged cells and cell debris | Repair processes, synthesis of novel extracellular matrix |
| Typical mediators | IL-1, IL-6, IL-15, IL-8, TNF-α | TGF-β, IL-10, VEGF, lipoxins |
| Presence of CRP and SAA | Enhanced values | Decreasing values |
| Macrophage subtypes | M1 type | M2 type |
| Major pathways for ATP production in macrophages | Glycolysis | Oxidative phosphorylation |
| Presence of MDSCs | Low | Enhanced |
| Cytotoxic Agent | Antagonizing Principles | References |
|---|---|---|
| Superoxide anion radicals | Superoxide dismutases | [58,59,60,61] |
| Hydrogen peroxide | Catalase, peroxiredoxins, glutathione peroxidases | [62,63,64] |
| Hydroxyl radicals | Limited protection by carbohydrates | [65] |
| Free transition metal ions | Proper control over all aspects of iron and copper ion metabolism | [66,67,68,69,70] |
| Myeloperoxidase | Ceruloplasmin | [71,72,73,74] |
| Hypochlorous acid, hypobromous acid | SCN−, taurine, glutathione, ascorbate | [75,76,77] |
| Hypothiocyanite | Glutathione | [78] |
| Elastase | α1-antitrypsin (A1AT), secretory leukocyte protease inhibitor (SLPI), elafin, serpin B1, α2-macroglobulin | [79,80,81,82,83] |
| Cathepsin G | A1AT, SLPI, α1-antichymotrypsin | [79,80,81,84,85,86] |
| Proteinase 3 | A1AT, elafin | [79,80,82,83] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnhold, J. Oxidant-Based Cytotoxic Agents During Aging: From Disturbed Energy Metabolism to Chronic Inflammation and Disease Progression. Biomolecules 2025, 15, 547. https://doi.org/10.3390/biom15040547
Arnhold J. Oxidant-Based Cytotoxic Agents During Aging: From Disturbed Energy Metabolism to Chronic Inflammation and Disease Progression. Biomolecules. 2025; 15(4):547. https://doi.org/10.3390/biom15040547
Chicago/Turabian StyleArnhold, Jürgen. 2025. "Oxidant-Based Cytotoxic Agents During Aging: From Disturbed Energy Metabolism to Chronic Inflammation and Disease Progression" Biomolecules 15, no. 4: 547. https://doi.org/10.3390/biom15040547
APA StyleArnhold, J. (2025). Oxidant-Based Cytotoxic Agents During Aging: From Disturbed Energy Metabolism to Chronic Inflammation and Disease Progression. Biomolecules, 15(4), 547. https://doi.org/10.3390/biom15040547