
Subtle Changes at the RBD/hACE2 Interface During SARS-CoV-2 Variant Evolution: A Molecular Dynamics Study
 , , ,
, , ,  and
and
Abstract

1. Introduction
2. Materials and Methods
2.1. Molecular Dynamics (MD) Simulations
2.2. PCA and RMSD Calculations
2.3. Pair Interactions (PairInt) Analyses
2.4. Computing Binding Free Energy Using MM/GBSA Model
2.5. Hydrophobic Interactions Analysis
2.6. Contact Principal Component Analysis (cPCA)
2.7. Cross-Correlation Analysis and Linear Mutual Information
3. Results
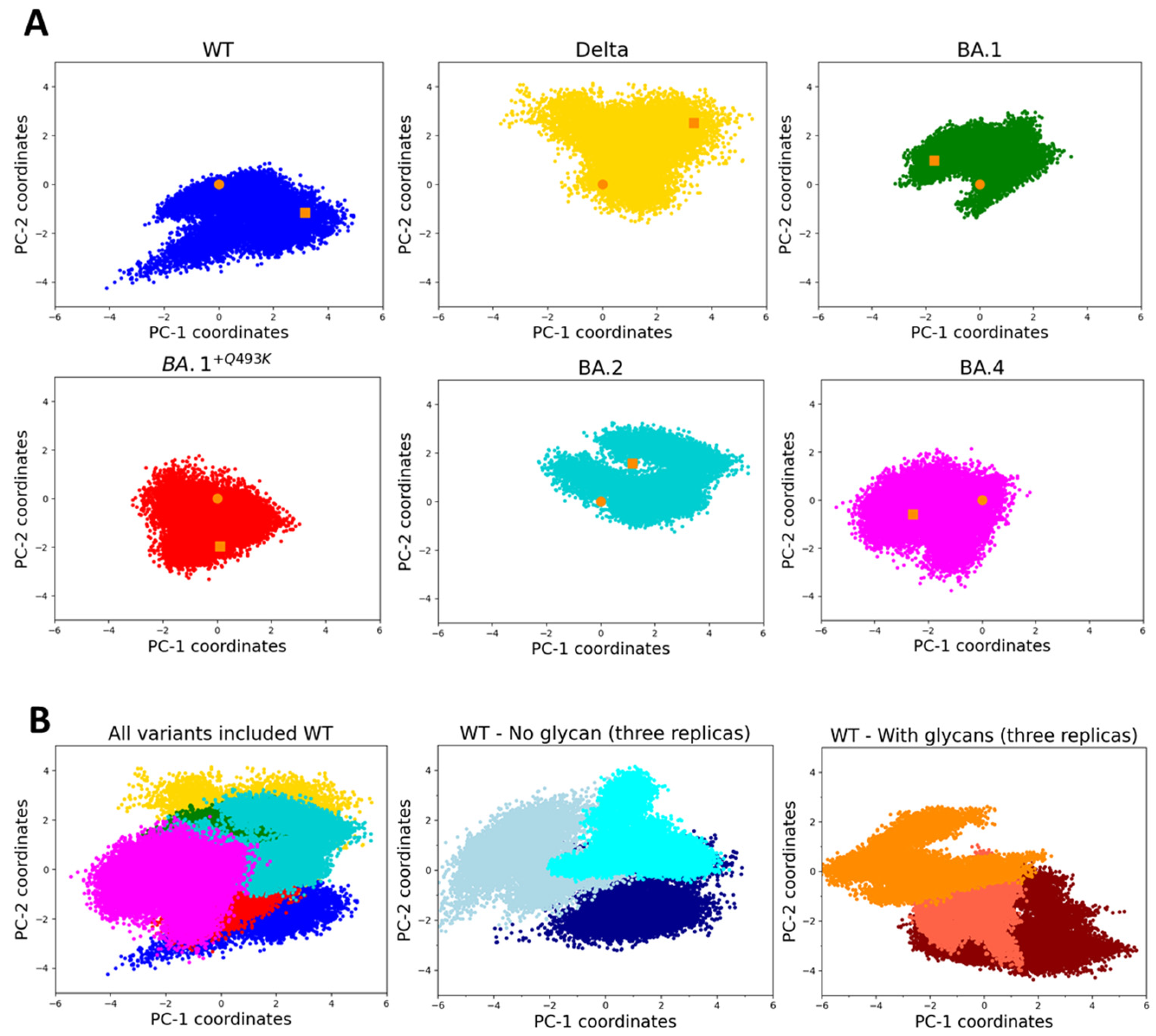
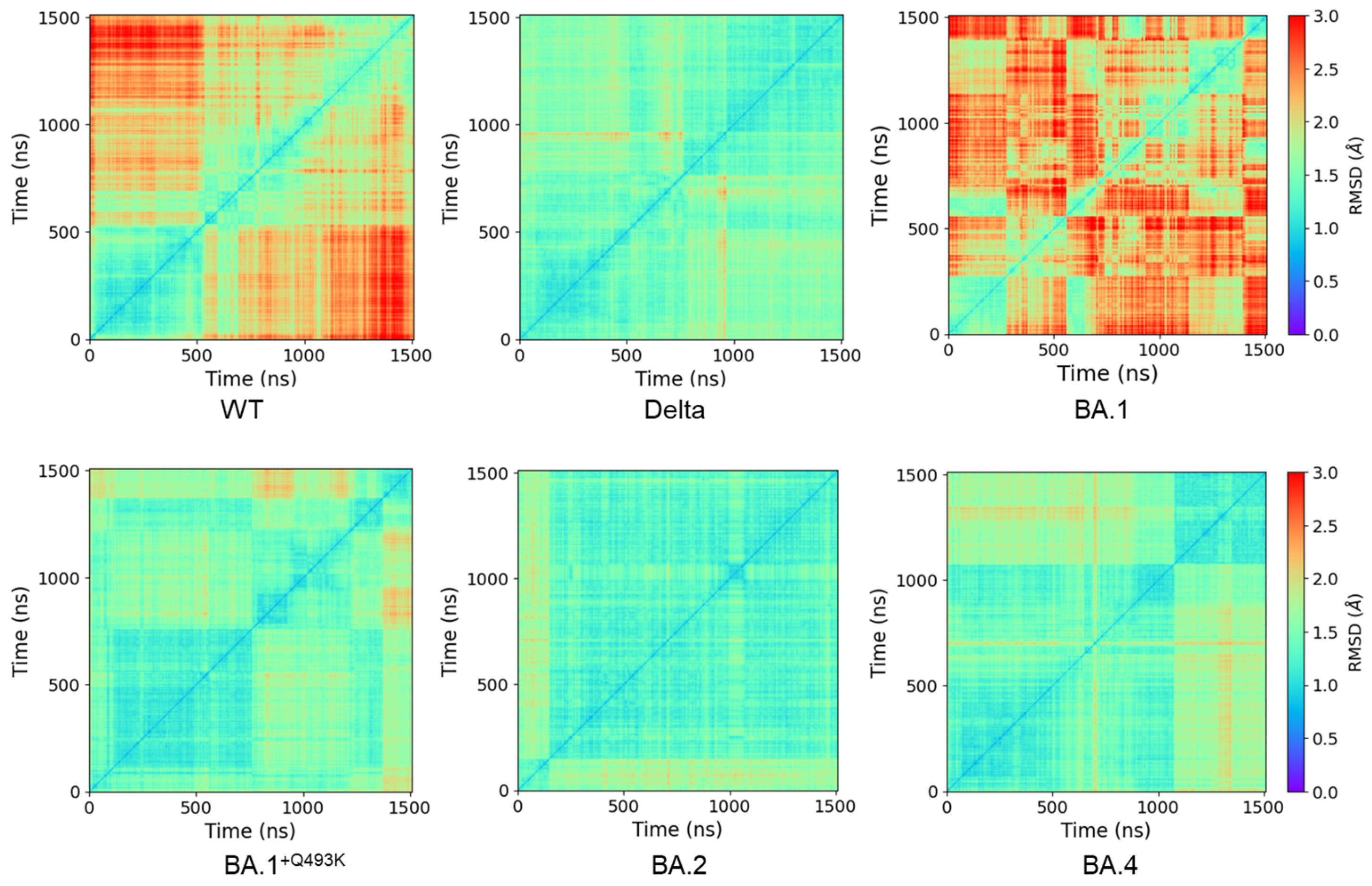
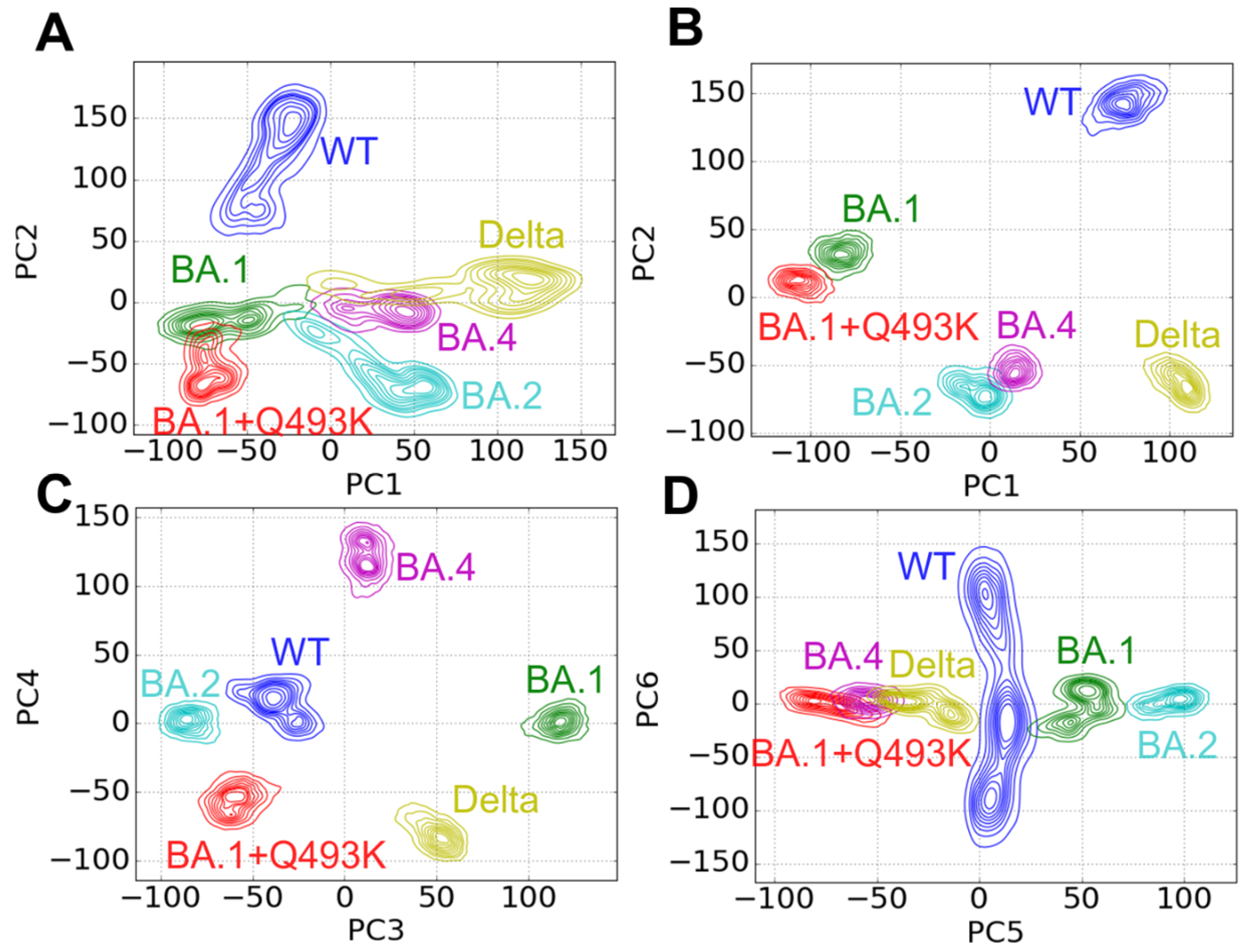
3.1. MD Simulations, Conformational Sampling and Dynamic Behaviors
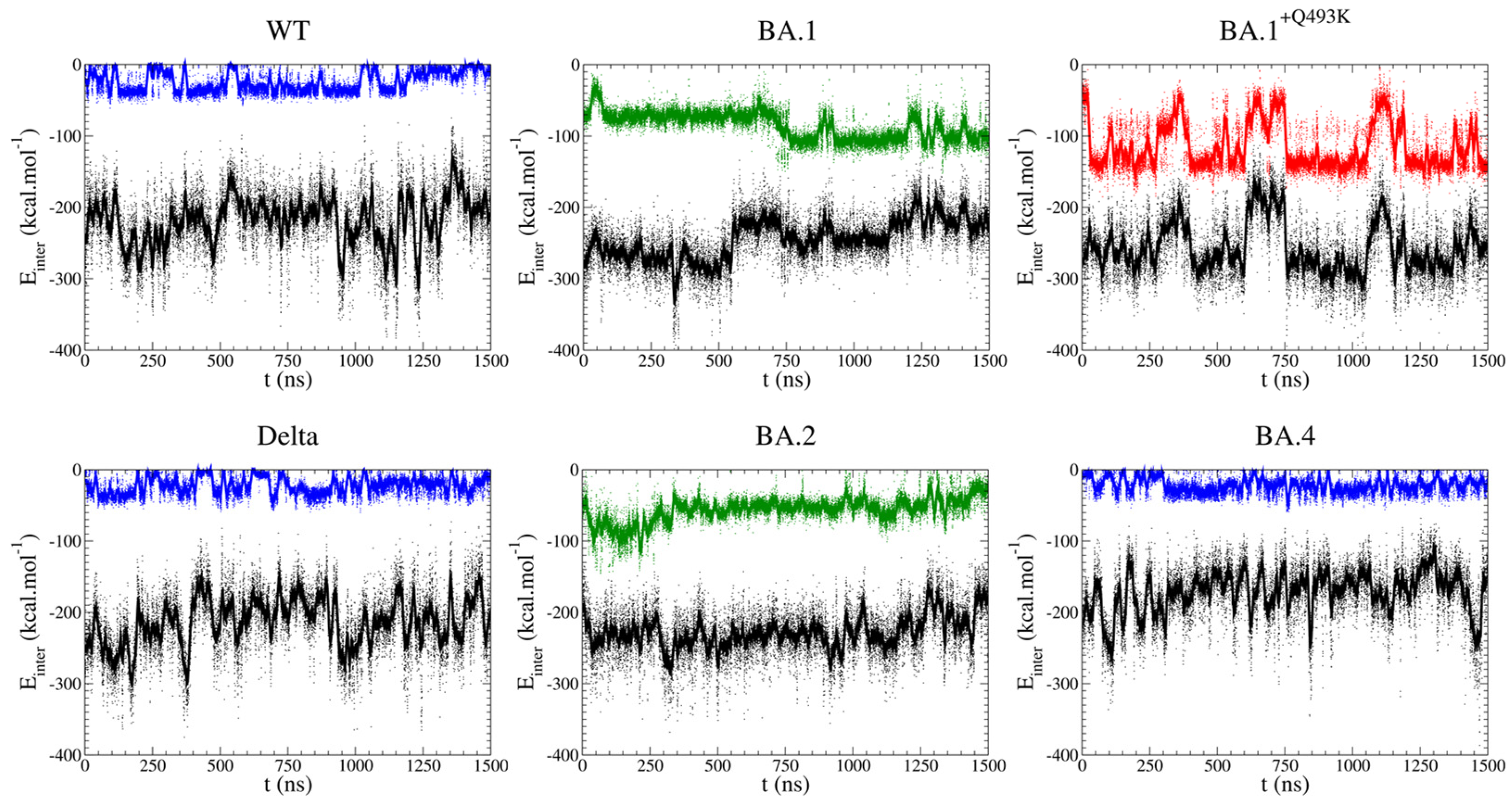
3.2. Pair Interactions Study
3.2.1. Focus on Q/K/R Spike Residue at Position 493
3.2.2. Residue–Residue PairInt Calculations
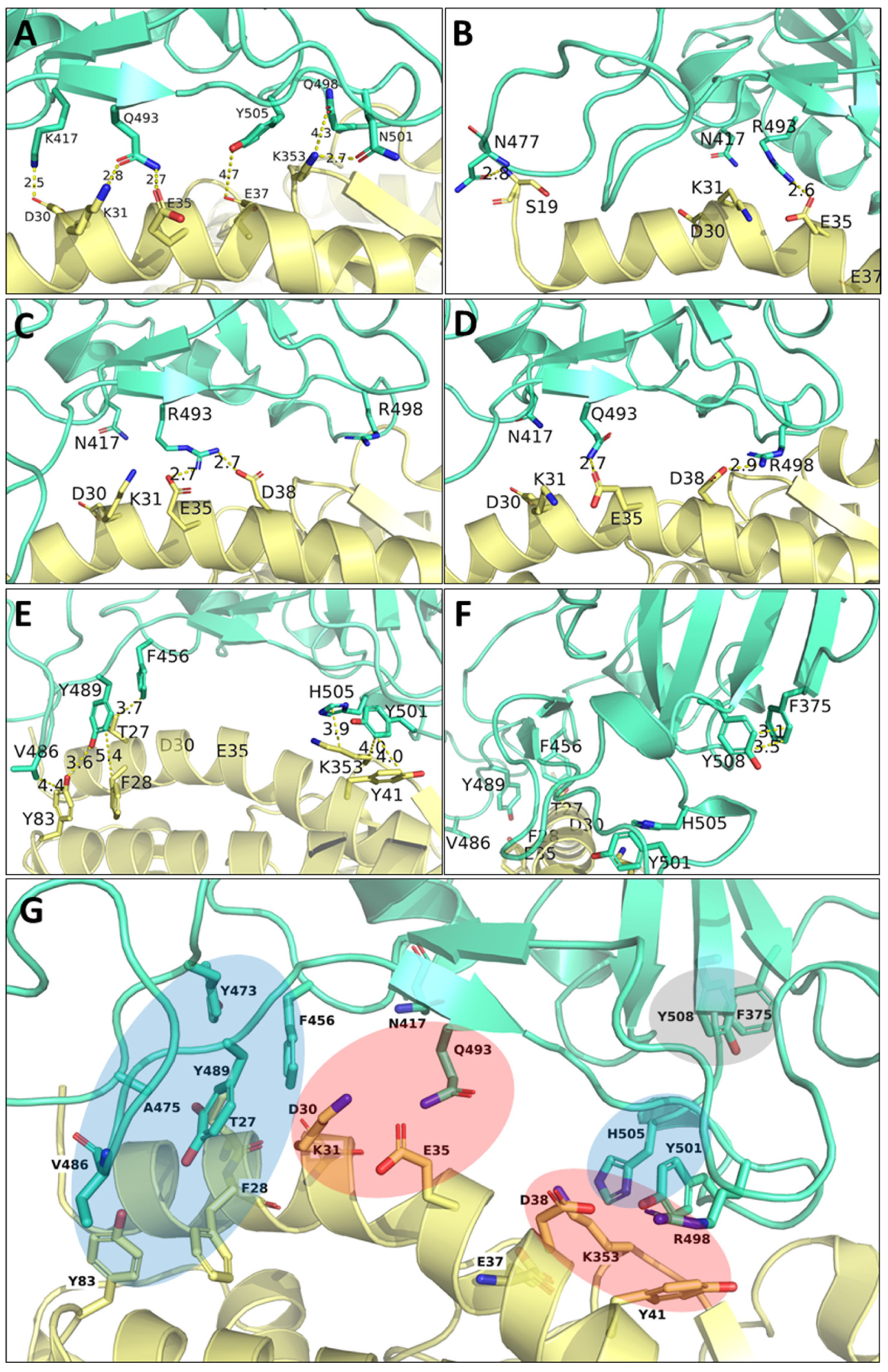
3.2.3. Identification of hACE2 Hot Spots for Spike Binding
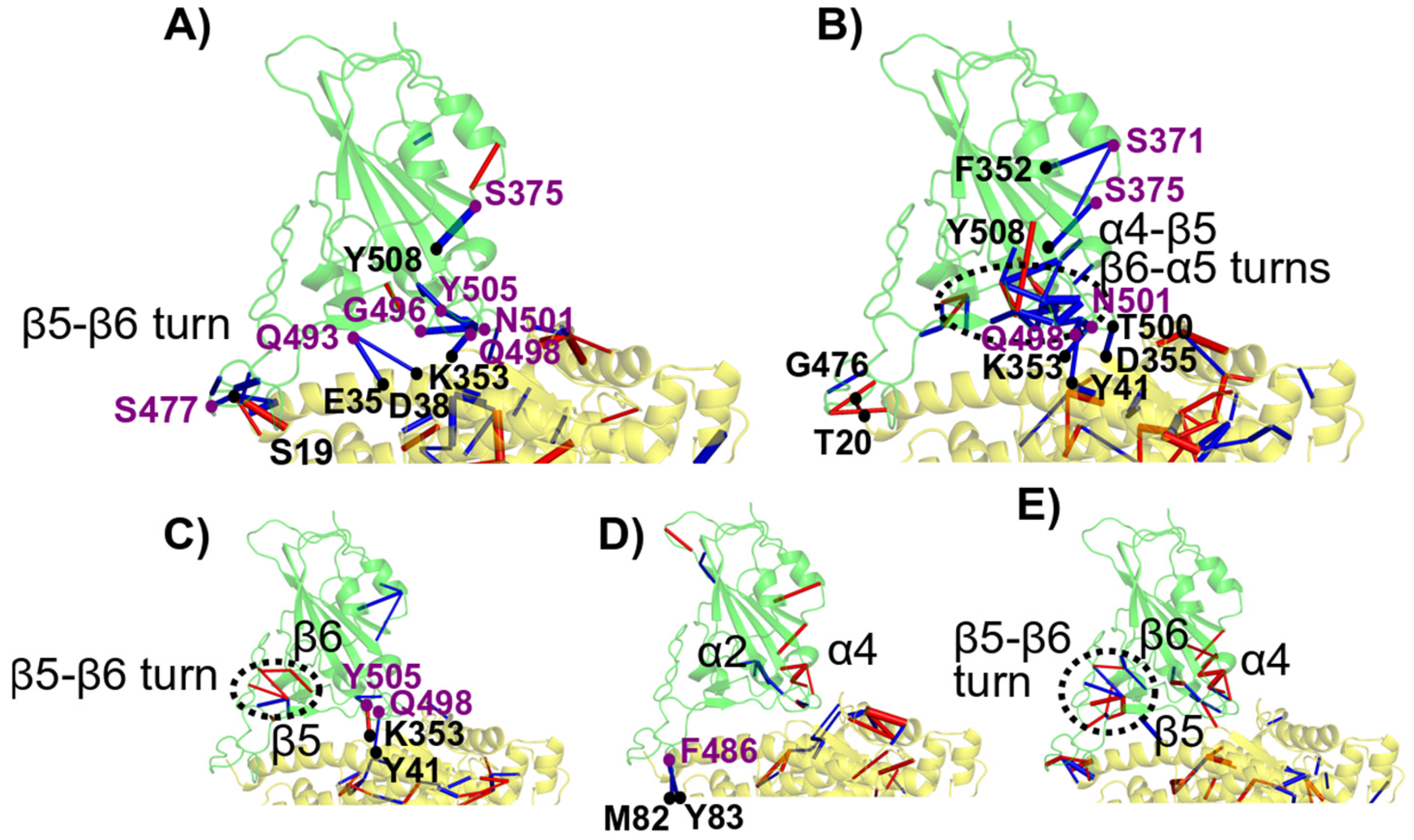
3.2.4. Identification of Distinct Regions of Spike RBD Involved in the Binding to hACE2
3.3. Hydrophobic Interactions
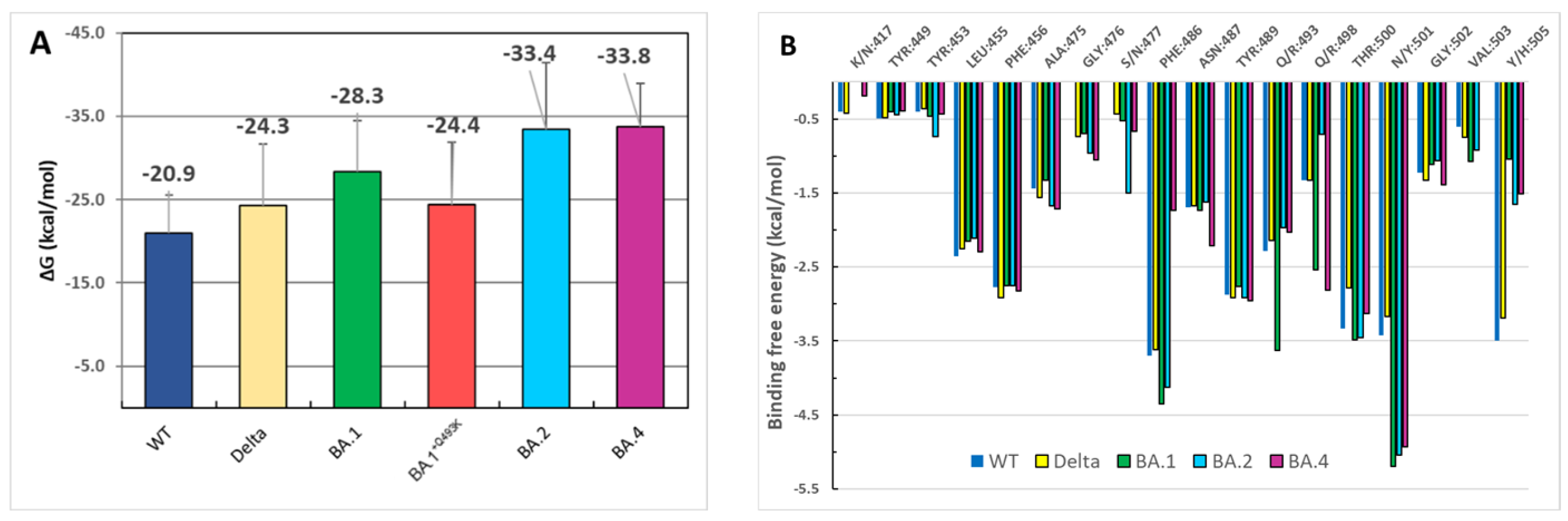
3.4. Binding Free Energies Evaluation
3.5. Contact Principal Component Analysis
3.6. Cross-Correlation Analysis
3.7. Visualization of the Main Contacts Identified by PairInt, MHP and cPCA Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chan, W.C.W.; Artzi, N.; Chen, C.; Chen, X.; Ho, D.; Hu, T.; Kataoka, K.; Liz-Marzán, L.M.; Oklu, R.; Parak, W.J. Noble Nanomedicine: Celebrating Groundbreaking mRNA Vaccine Innovations. ACS Nano 2023, 17, 19476–19477. [Google Scholar] [CrossRef]
- Graña, C.; Ghosn, L.; Evrenoglou, T.; Jarde, A.; Minozzi, S.; Bergman, H.; Buckley, B.S.; Probyn, K.; Villanueva, G.; Henschke, N.; et al. Efficacy and Safety of COVID-19 Vaccines. Cochrane Database Syst. Rev. 2022, 2023, CD015477. [Google Scholar] [CrossRef]
- Watson, O.J.; Barnsley, G.; Toor, J.; Hogan, A.B.; Winskill, P.; Ghani, A.C. Global Impact of the First Year of COVID-19 Vaccination: A Mathematical Modelling Study. Lancet Infect. Dis. 2022, 22, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Aschwanden, C. Five Reasons Why COVID Herd Immunity Is Probably Impossible. Nature 2021, 591, 520–522. [Google Scholar] [CrossRef]
- Phillips, N. The Coronavirus Is Here to Stay—Here’s What That Means. Nature 2021, 590, 382–384. [Google Scholar] [CrossRef]
- Karim, S.S.A.; Karim, Q.A. Omicron SARS-CoV-2 Variant: A New Chapter in the COVID-19 Pandemic. Lancet 2021, 398, 2126–2128. [Google Scholar] [CrossRef]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef]
- Dai, L.; Gao, G.F. Viral Targets for Vaccines against COVID-19. Nat. Rev. Immunol. 2021, 21, 73–82. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-Converting Enzyme 2 Is a Functional Receptor for the SARS Coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef]
- Ren, S.-Y.; Wang, W.-B.; Gao, R.-D.; Zhou, A.-M. Omicron Variant (B.1.1.529) of SARS-CoV-2: Mutation, Infectivity, Transmission, and Vaccine Resistance. World J. Clin. Cases 2022, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; De Silva, T.I.; Peacock, S.J.; Barclay, W.S.; De Silva, T.I.; COVID-19 Genomics UK Consortium; et al. SARS-CoV-2 Variant Biology: Immune Escape, Transmission and Fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Xie, C.; Bu, G.-L.; Zhong, L.-Y.; Zeng, M.-S. Molecular Characteristics, Immune Evasion, and Impact of SARS-CoV-2 Variants. Sig Transduct. Target. Ther. 2022, 7, 202. [Google Scholar] [CrossRef]
- Chen, D.-Y.; Chin, C.V.; Kenney, D.; Tavares, A.H.; Khan, N.; Conway, H.L.; Liu, G.; Choudhary, M.C.; Gertje, H.P.; O’Connell, A.K.; et al. Spike and Nsp6 Are Key Determinants of SARS-CoV-2 Omicron BA.1 Attenuation. Nature 2023, 615, 143–150. [Google Scholar] [CrossRef]
- Planas, D.; Saunders, N.; Maes, P.; Guivel-Benhassine, F.; Planchais, C.; Buchrieser, J.; Bolland, W.-H.; Porrot, F.; Staropoli, I.; Lemoine, F.; et al. Considerable Escape of SARS-CoV-2 Omicron to Antibody Neutralization. Nature 2022, 602, 671–675. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 Variants, Spike Mutations and Immune Escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Goldberg, E.E.; Lin, Q.; Romero-Severson, E.O.; Ke, R. Swift and Extensive Omicron Outbreak in China after Sudden Exit from ‘Zero-COVID’ Policy. Nat. Commun. 2023, 14, 3888. [Google Scholar] [CrossRef]
- Grewal, R.; Nguyen, L.; Buchan, S.A.; Wilson, S.E.; Nasreen, S.; Austin, P.C.; Brown, K.A.; Fell, D.B.; Gubbay, J.B.; Schwartz, K.L.; et al. Effectiveness of mRNA COVID-19 Vaccine Booster Doses against Omicron Severe Outcomes. Nat. Commun. 2023, 14, 1273. [Google Scholar] [CrossRef]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X.; et al. BA.2.12.1, BA.4 and BA.5 Escape Antibodies Elicited by Omicron Infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef]
- Shrestha, L.B.; Foster, C.; Rawlinson, W.; Tedla, N.; Bull, R.A. Evolution of the SARS-CoV-2 Omicron Variants BA.1 to BA.5: Implications for Immune Escape and Transmission. Rev. Med. Virol. 2022, 32, e2381. [Google Scholar] [CrossRef]
- Wolter, N.; Jassat, W.; Walaza, S.; Welch, R.; Moultrie, H.; Groome, M.J.; Amoako, D.G.; Everatt, J.; Bhiman, J.N.; Scheepers, C.; et al. Clinical Severity of SARS-CoV-2 Omicron BA.4 and BA.5 Lineages Compared to BA.1 and Delta in South Africa. Nat. Commun. 2022, 13, 5860. [Google Scholar] [CrossRef] [PubMed]
- Parsons, R.J.; Acharya, P. Evolution of the SARS-CoV-2 Omicron Spike. Cell Rep. 2023, 42, 113444. [Google Scholar] [CrossRef] [PubMed]
- Triveri, A.; Casali, E.; Frasnetti, E.; Doria, F.; Frigerio, F.; Cinquini, F.; Pavoni, S.; Moroni, E.; Marchetti, F.; Serapian, S.A.; et al. Conformational Behavior of SARS-Cov-2 Spike Protein Variants: Evolutionary Jumps in Sequence Reverberate in Structural Dynamic Differences. J. Chem. Theory Comput. 2023, 19, 2120–2134. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Zardecki, C.; Di Costanzo, L.; Duarte, J.M.; Hudson, B.P.; Persikova, I.; Segura, J.; Shao, C.; Voigt, M.; Westbrook, J.D.; et al. RCSB Protein Data Bank: Enabling Biomedical Research and Drug Discovery. Protein Sci. 2020, 29, 52–65. [Google Scholar] [CrossRef]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C.; et al. Receptor Binding and Complex Structures of Human ACE2 to Spike RBD from Omicron and Delta SARS-CoV-2. Cell 2022, 185, 630–640.e10. [Google Scholar] [CrossRef]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 Omicron Variant: Antibody Evasion and Cryo-EM Structure of Spike Protein-ACE2 Complex. Science 2022, 375, 760–764. [Google Scholar] [CrossRef]
- Zhang, J.; Cai, Y.; Lavine, C.L.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; Mayer, M.L.; Rits-Volloch, S.; et al. Structural and Functional Impact by SARS-CoV-2 Omicron Spike Mutations. Cell Rep. 2022, 39, 110729. [Google Scholar] [CrossRef]
- Gobeil, S.M.-C.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Stalls, V.; Kopp, M.F.; Manne, K.; Li, D.; Wiehe, K.; et al. Effect of Natural Mutations of SARS-CoV-2 on Spike Structure, Conformation, and Antigenicity. Science 2021, 373, eabi6226. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Chiu, W.; Dai, W.; Flatt, J.W.; Hudson, B.P.; Kaelber, J.T.; Khare, S.D.; Kulczyk, A.W.; Lawson, C.L.; et al. Electron Microscopy Holdings of the Protein Data Bank: The Impact of the Resolution Revolution, New Validation Tools, and Implications for the Future. Biophys. Rev. 2022, 14, 1281–1301. [Google Scholar] [CrossRef]
- Henderson, R. From Electron Crystallography to Single Particle CryoEM (Nobel Lecture). Angew. Chem. Int. Ed. 2018, 57, 10804–10825. [Google Scholar] [CrossRef]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular Simulation: A Computational Microscope for Molecular Biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef] [PubMed]
- Dadonaite, B.; Brown, J.; McMahon, T.E.; Farrell, A.G.; Figgins, M.D.; Asarnow, D.; Stewart, C.; Lee, J.; Logue, J.; Bedford, T.; et al. Spike Deep Mutational Scanning Helps Predict Success of SARS-CoV-2 Clades. Nature 2024, 631, 617–626. [Google Scholar] [CrossRef]
- Rath, S.L.; Padhi, A.K.; Mandal, N. Scanning the RBD-ACE2 Molecular Interactions in Omicron Variant. Biochem. Biophys. Res. Commun. 2022, 592, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Maurya, S.; Mahale, A.; Rath, S.L.; Tripathi, T.; Padhi, A.K. Delineating the Structure–Dynamics–Binding Differences among BA.1, BA.4/5, and BF.7 SARS-CoV-2 Variants through Atomistic Simulations: Correlation with Structural and Epidemiological Features. ACS Omega 2023, 8, 37852–37863. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Waris, H.; Rafique, M.; Suleman, M.; Mohammad, A.; Ali, S.S.; Khan, T.; Waheed, Y.; Liao, C.; Wei, D.-Q. The Omicron (B.1.1.529) Variant of SARS-CoV-2 Binds to the hACE2 Receptor More Strongly and Escapes the Antibody Response: Insights from Structural and Simulation Data. Int. J. Biol. Macromol. 2022, 200, 438–448. [Google Scholar] [CrossRef]
- Wrobel, A.G. Mechanism and Evolution of Human ACE2 Binding by SARS-CoV-2 Spike. Curr. Opin. Struct. Biol. 2023, 81, 102619. [Google Scholar] [CrossRef]
- Maity, S.; Acharya, A. Many Roles of Carbohydrates: A Computational Spotlight on the Coronavirus S Protein Binding. ACS Appl. Bio Mater. 2023, 7, 646–656. [Google Scholar] [CrossRef]
- Gheeraert, A.; Vuillon, L.; Chaloin, L.; Moncorgé, O.; Very, T.; Perez, S.; Leroux, V.; Chauvot de Beauchêne, I.; Mias-Lucquin, D.; Devignes, M.-D.; et al. Singular Interface Dynamics of the SARS-CoV-2 Delta Variant Explained with Contact Perturbation Analysis. J. Chem. Inf. Model. 2022, 62, 3107–3122. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert. Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Nutalai, R.; Zhou, D.; Tuekprakhon, A.; Ginn, H.M.; Supasa, P.; Liu, C.; Huo, J.; Mentzer, A.J.; Duyvesteyn, H.M.E.; Dijokaite-Guraliuc, A.; et al. Potent Cross-Reactive Antibodies Following Omicron Breakthrough in Vaccinees. Cell 2022, 185, 2116–2131.e18. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Tsybovsky, Y.; Gorman, J.; Rapp, M.; Cerutti, G.; Chuang, G.-Y.; Katsamba, P.S.; Sampson, J.M.; Schön, A.; Bimela, J.; et al. Cryo-EM Structures of SARS-CoV-2 Spike without and with ACE2 Reveal a pH-Dependent Switch to Mediate Endosomal Positioning of Receptor-Binding Domains. Cell Host Microbe 2020, 28, 867–879.e5. [Google Scholar] [CrossRef]
- Zahradník, J.; Marciano, S.; Shemesh, M.; Zoler, E.; Harari, D.; Chiaravalli, J.; Meyer, B.; Rudich, Y.; Li, C.; Marton, I.; et al. SARS-CoV-2 Variant Prediction and Antiviral Drug Design Are Enabled by RBD in Vitro Evolution. Nat. Microbiol. 2021, 6, 1188–1198. [Google Scholar] [CrossRef]
- Liu, N.; Zheng, L.; Xu, J.; Wang, J.; Hu, C.; Lan, J.; Zhang, X.; Zhang, J.; Xu, K.; Cheng, H.; et al. Reduced Graphene Oxide Membrane as Supporting Film for High-Resolution Cryo-EM. Biophys. Rep. 2021, 7, 227–238. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, C.; Zhang, C.; Wang, Y.; Hong, Q.; Xu, S.; Li, Z.; Yang, Y.; Huang, Z.; Cong, Y. Structural Basis for SARS-CoV-2 Delta Variant Recognition of ACE2 Receptor and Broadly Neutralizing Antibodies. Nat. Commun. 2022, 13, 871. [Google Scholar] [CrossRef]
- Hong, Q.; Han, W.; Li, J.; Xu, S.; Wang, Y.; Xu, C.; Li, Z.; Wang, Y.; Zhang, C.; Huang, Z.; et al. Molecular Basis of Receptor Binding and Antibody Neutralization of Omicron. Nature 2022, 604, 546–552. [Google Scholar] [CrossRef]
- Li, L.; Liao, H.; Meng, Y.; Li, W.; Han, P.; Liu, K.; Wang, Q.; Li, D.; Zhang, Y.; Wang, L.; et al. Structural Basis of Human ACE2 Higher Binding Affinity to Currently Circulating Omicron SARS-CoV-2 Sub-Variants BA.2 and BA.1.1. Cell 2022, 185, 2952–2960.e10. [Google Scholar] [CrossRef]
- Kimura, I.; Yamasoba, D.; Tamura, T.; Nao, N.; Suzuki, T.; Oda, Y.; Mitoma, S.; Ito, J.; Nasser, H.; Zahradnik, J.; et al. Virological Characteristics of the SARS-CoV-2 Omicron BA.2 Subvariants, Including BA.4 and BA.5. Cell 2022, 185, 3992–4007.e16. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Newby, M.L.; Fogarty, C.A.; Allen, J.D.; Butler, J.; Fadda, E.; Crispin, M. Variations within the Glycan Shield of SARS-CoV-2 Impact Viral Spike Dynamics. J. Mol. Biol. 2023, 435, 167928. [Google Scholar] [CrossRef]
- Park, S.-J.; Lee, J.; Qi, Y.; Kern, N.R.; Lee, H.S.; Jo, S.; Joung, I.; Joo, K.; Lee, J.; Im, W. CHARMM-GUI Glycan Modeler for Modeling and Simulation of Carbohydrates and Glycoconjugates. Glycobiology 2019, 29, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Krieger, J.M.; Zhang, Y.; Kaya, C.; Kaynak, B.; Mikulska-Ruminska, K.; Doruker, P.; Li, H.; Bahar, I. ProDy 2.0: Increased Scale and Scope after 10 Years of Protein Dynamics Modelling with Python. Bioinformatics 2021, 37, 3657–3659. [Google Scholar] [CrossRef] [PubMed]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Dotson, S.L.; Domanski, J.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the 15th Python in Science Conference, Austin, TX, USA, 11–17 July 2016; pp. 98–105. [Google Scholar]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef]
- Theobald, D.L. Rapid Calculation of RMSDs Using a Quaternion-Based Characteristic Polynomial. Acta Crystallogr. A 2005, 61, 478–480. [Google Scholar] [CrossRef]
- Asses, Y.; Leroux, V.; Tairi-Kellou, S.; Dono, R.; Maina, F.; Maigret, B. Analysis of C-Met Kinase Domain Complexes: A New Specific Catalytic Site Receptor Model for Defining Binding Modes of ATP-Competitive Ligands. Chem. Biol. Drug Des. 2009, 74, 560–570. [Google Scholar] [CrossRef]
- Patané, S.; Pietrancosta, N.; Hassani, H.; Leroux, V.; Maigret, B.; Kraus, J.-L.; Dono, R.; Maina, F. A New Met Inhibitory-Scaffold Identified by a Focused Forward Chemical Biological Screen. Biochem. Biophys. Res. Commun. 2008, 375, 184–189. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Li, Y.; Cong, Y.; Feng, G.; Zhong, S.; Zhang, J.Z.H.; Sun, H.; Duan, L. The Impact of Interior Dielectric Constant and Entropic Change on HIV-1 Complex Binding Free Energy Prediction. Struct. Dyn. 2018, 5, 064101. [Google Scholar] [CrossRef] [PubMed]
- Pyrkov, T.V.; Chugunov, A.O.; Krylov, N.A.; Nolde, D.E.; Efremov, R.G. PLATINUM: A Web Tool for Analysis of Hydrophobic/Hydrophilic Organization of Biomolecular Complexes. Bioinformatics 2009, 25, 1201–1202. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. Prediction of Hydrophobic (Lipophilic) Properties of Small Organic Molecules Using Fragmental Methods: An Analysis of ALOGP and CLOGP Methods. J. Phys. Chem. A 1998, 102, 3762–3772. [Google Scholar] [CrossRef]
- Mias-Lucquin, D.; Chéron, A.; Le Rumeur, E.; Hubert, J.-F.; Delalande, O. Fine Mapping of Hydrophobic Contacts Reassesses the Organization of the First Three Dystrophin Coiled-Coil Repeats. Protein Sci. 2019, 28, 561–570. [Google Scholar] [CrossRef]
- Altis, A.; Otten, M.; Nguyen, P.H.; Hegger, R.; Stock, G. Construction of the Free Energy Landscape of Biomolecules via Dihedral Angle Principal Component Analysis. J. Chem. Phys. 2008, 128, 245102. [Google Scholar] [CrossRef]
- Lange, O.F.; Grubmüller, H. Generalized Correlation for Biomolecular Dynamics. Proteins 2006, 62, 1053–1061. [Google Scholar] [CrossRef]
- Tekpinar, M.; Neron, B.; Delarue, M. Extracting Dynamical Correlations and Identifying Key Residues for Allosteric Communication in Proteins by Correlationplus. J. Chem. Inf. Model. 2021, 61, 4832–4838. [Google Scholar] [CrossRef]
- Clarke, D.; Sethi, A.; Li, S.; Kumar, S.; Chang, R.W.F.; Chen, J.; Gerstein, M. Identifying Allosteric Hotspots with Dynamics: Application to Inter- and Intra-Species Conservation. Structure 2016, 24, 826–837. [Google Scholar] [CrossRef]
- Rivalta, I.; Sultan, M.M.; Lee, N.-S.; Manley, G.A.; Loria, J.P.; Batista, V.S. Allosteric Pathways in Imidazole Glycerol Phosphate Synthase. Proc. Natl. Acad. Sci. USA 2012, 109, E1428–E1436. [Google Scholar] [CrossRef]
- Sethi, A.; Eargle, J.; Black, A.A.; Luthey-Schulten, Z. Dynamical Networks in tRNA:Protein Complexes. Proc. Natl. Acad. Sci. USA 2009, 106, 6620–6625. [Google Scholar] [CrossRef]
- Negre, C.F.A.; Morzan, U.N.; Hendrickson, H.P.; Pal, R.; Lisi, G.P.; Loria, J.P.; Rivalta, I.; Ho, J.; Batista, V.S. Eigenvector Centrality for Characterization of Protein Allosteric Pathways. Proc. Natl. Acad. Sci. USA 2018, 115, E12201–E12208. [Google Scholar] [CrossRef] [PubMed]
- Tekpinar, M.; Yildirim, A. Impact of Dimerization and N3 Binding on Molecular Dynamics of SARS-CoV and SARS-CoV-2 Main Proteases. J. Biomol. Struct. Dyn. 2022, 40, 6243–6254. [Google Scholar] [CrossRef] [PubMed]
- David, C.C.; Jacobs, D.J. Principal Component Analysis: A Method for Determining the Essential Dynamics of Proteins. Methods Mol. Biol. 2014, 1084, 193–226. [Google Scholar] [CrossRef] [PubMed]
- Leist, S.R.; Dinnon, K.H.; Schäfer, A.; Tse, L.V.; Okuda, K.; Hou, Y.J.; West, A.; Edwards, C.E.; Sanders, W.; Fritch, E.J.; et al. A Mouse-Adapted SARS-CoV-2 Induces Acute Lung Injury and Mortality in Standard Laboratory Mice. Cell 2020, 183, 1070–1085.e12. [Google Scholar] [CrossRef]
- Mandal, N.; Padhi, A.K.; Rath, S.L. Molecular Insights into the Differential Dynamics of SARS-CoV-2 Variants of Concern. J. Mol. Graph. Model. 2022, 114, 108194. [Google Scholar] [CrossRef]
- Socher, E.; Conrad, M.; Heger, L.; Paulsen, F.; Sticht, H.; Zunke, F.; Arnold, P. Computational Decomposition Reveals Reshaping of the SARS-CoV-2-ACE2 Interface among Viral Variants Expressing the N501Y Mutation. J. Cell Biochem. 2021, 122, 1863–1872. [Google Scholar] [CrossRef]
- Abduljalil, J.M.; Elghareib, A.M.; Samir, A.; Ezat, A.A.; Elfiky, A.A. How Helpful Were Molecular Dynamics Simulations in Shaping Our Understanding of SARS-CoV-2 Spike Protein Dynamics? Int. J. Biol. Macromol. 2023, 242, 125153. [Google Scholar] [CrossRef]
- Lupala, C.S.; Ye, Y.; Chen, H.; Su, X.-D.; Liu, H. Mutations on RBD of SARS-CoV-2 Omicron Variant Result in Stronger Binding to Human ACE2 Receptor. Biochem. Biophys. Res. Commun. 2022, 590, 34–41. [Google Scholar] [CrossRef]
- Omotuyi, O.; Olubiyi, O.; Nash, O.; Afolabi, E.; Oyinloye, B.; Fatumo, S.; Femi-Oyewo, M.; Bogoro, S. SARS-CoV-2 Omicron Spike Glycoprotein Receptor Binding Domain Exhibits Super-Binder Ability with ACE2 but Not Convalescent Monoclonal Antibody. Comput. Biol. Med. 2022, 142, 105226. [Google Scholar] [CrossRef]
- Shah, M.; Woo, H.G. Omicron: A Heavily Mutated SARS-CoV-2 Variant Exhibits Stronger Binding to ACE2 and Potently Escapes Approved COVID-19 Therapeutic Antibodies. Front. Immunol. 2021, 12, 830527. [Google Scholar] [CrossRef]
- Koley, T.; Kumar, M.; Goswami, A.; Ethayathulla, A.S.; Hariprasad, G. Structural Modeling of Omicron Spike Protein and Its Complex with Human ACE-2 Receptor: Molecular Basis for High Transmissibility of the Virus. Biochem. Biophys. Res. Commun. 2022, 592, 51–53. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhou, L.; Mo, M.; Liu, T.; Wu, C.; Gong, C.; Lu, K.; Gong, L.; Zhu, W.; Xu, Z. SARS-CoV-2 Omicron RBD Shows Weaker Binding Affinity than the Currently Dominant Delta Variant to Human ACE2. Signal Transduct. Target. Ther. 2022, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Panthi, B.; Chandra, A. All-Atom Simulations of Human ACE2-Spike Protein RBD Complexes for SARS-CoV-2 and Some of Its Variants: Nature of Interactions and Free Energy Diagrams for Dissociation of the Protein Complexes. J. Phys. Chem. B 2022, 126, 5375–5389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, S.; Wu, B.; Yang, Q.; Chen, A.; Li, Y.; Zhang, Y.; Pan, T.; Zhang, H.; He, X. SARS-CoV-2 Omicron Strain Exhibits Potent Capabilities for Immune Evasion and Viral Entrance. Sig Transduct. Target. Ther. 2021, 6, 430. [Google Scholar] [CrossRef]
- Kim, S.; Liu, Y.; Lei, Z.; Dicker, J.; Cao, Y.; Zhang, X.F.; Im, W. Differential Interactions between Human ACE2 and Spike RBD of SARS-CoV-2 Variants of Concern. J. Chem. Theory Comput. 2021, 17, 7972–7979. [Google Scholar] [CrossRef]
- Kim, S.; Liu, Y.; Ziarnik, M.; Seo, S.; Cao, Y.; Zhang, X.F.; Im, W. Binding of Human ACE2 and RBD of Omicron Enhanced by Unique Interaction Patterns among SARS-CoV-2 Variants of Concern. J. Comput. Chem. 2023, 44, 594–601. [Google Scholar] [CrossRef]
- Meng, B.; Abdullahi, A.; Ferreira, I.A.T.M.; Goonawardane, N.; Saito, A.; Kimura, I.; Yamasoba, D.; Gerber, P.P.; Fatihi, S.; Rathore, S.; et al. Altered TMPRSS2 Usage by SARS-CoV-2 Omicron Impacts Infectivity and Fusogenicity. Nature 2022, 603, 706–714. [Google Scholar] [CrossRef]
- Cameroni, E.; Bowen, J.E.; Rosen, L.E.; Saliba, C.; Zepeda, S.K.; Culap, K.; Pinto, D.; VanBlargan, L.A.; De Marco, A.; di Iulio, J.; et al. Broadly Neutralizing Antibodies Overcome SARS-CoV-2 Omicron Antigenic Shift. Nature 2022, 602, 664–670. [Google Scholar] [CrossRef]
- Chowdhury, R.; Boorla, V.S.; Maranas, C.D. Computational Biophysical Characterization of the SARS-CoV-2 Spike Protein Binding with the ACE2 Receptor and Implications for Infectivity. Comput. Struct. Biotechnol. J. 2020, 18, 2573–2582. [Google Scholar] [CrossRef]
- Guo, W.; Xie, Y.; E Lopez-Hernandez, A.; Sun, S.; Li, L. Electrostatic Features for Nucleocapsid Proteins of SARS-CoV and SARS-CoV-2. Math. Biosci. Eng. 2021, 18, 2372–2383. [Google Scholar] [CrossRef]
- Charlier, L.; Louet, M.; Chaloin, L.; Fuchs, P.; Martinez, J.; Muriaux, D.; Favard, C.; Floquet, N. Coarse-Grained Simulations of the HIV-1 Matrix Protein Anchoring: Revisiting Its Assembly on Membrane Domains. Biophys. J. 2014, 106, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Azzaz, F.; Chahinian, H.; Yahi, N. Electrostatic Surface Potential as a Key Parameter in Virus Transmission and Evolution: How to Manage Future Virus Pandemics in the Post-COVID-19 Era. Viruses 2023, 15, 284. [Google Scholar] [CrossRef] [PubMed]
- Cele, S.; Jackson, L.; Khoury, D.S.; Khan, K.; Moyo-Gwete, T.; Tegally, H.; San, J.E.; Cromer, D.; Scheepers, C.; Amoako, D.G.; et al. Omicron Extensively but Incompletely Escapes Pfizer BNT162b2 Neutralization. Nature 2022, 602, 654–656. [Google Scholar] [CrossRef]
- Dejnirattisai, W.; Huo, J.; Zhou, D.; Zahradník, J.; Supasa, P.; Liu, C.; Duyvesteyn, H.M.E.; Ginn, H.M.; Mentzer, A.J.; Tuekprakhon, A.; et al. SARS-CoV-2 Omicron-B.1.1.529 Leads to Widespread Escape from Neutralizing Antibody Responses. Cell 2022, 185, 467–484.e15. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, Y.; Iketani, S.; Nair, M.S.; Li, Z.; Mohri, H.; Wang, M.; Yu, J.; Bowen, A.D.; Chang, J.Y.; et al. Antibody Evasion by SARS-CoV-2 Omicron Subvariants BA.2.12.1, BA.4 and BA.5. Nature 2022, 608, 603–608. [Google Scholar] [CrossRef]
- Qu, P.; Evans, J.P.; Faraone, J.N.; Zheng, Y.-M.; Carlin, C.; Anghelina, M.; Stevens, P.; Fernandez, S.; Jones, D.; Lozanski, G.; et al. Enhanced Neutralization Resistance of SARS-CoV-2 Omicron Subvariants BQ.1, BQ.1.1, BA.4.6, BF.7, and BA.2.75.2. Cell Host Microbe 2023, 31, 9–17.e3. [Google Scholar] [CrossRef]
- Planas, D.; Bruel, T.; Staropoli, I.; Guivel-Benhassine, F.; Porrot, F.; Maes, P.; Grzelak, L.; Prot, M.; Mougari, S.; Planchais, C.; et al. Resistance of Omicron Subvariants BA.2.75.2, BA.4.6, and BQ.1.1 to Neutralizing Antibodies. Nat. Commun. 2023, 14, 824. [Google Scholar] [CrossRef]
- Wang, Q.; Iketani, S.; Li, Z.; Liu, L.; Guo, Y.; Huang, Y.; Bowen, A.D.; Liu, M.; Wang, M.; Yu, J.; et al. Alarming Antibody Evasion Properties of Rising SARS-CoV-2 BQ and XBB Subvariants. Cell 2023, 186, 279–286.e8. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, Y.; Liu, L.; Schwanz, L.T.; Li, Z.; Nair, M.S.; Ho, J.; Zhang, R.M.; Iketani, S.; Yu, J.; et al. Antigenicity and Receptor Affinity of SARS-CoV-2 BA.2.86 Spike. Nature 2023, 624, 639–644. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variants | PDB IDs | Reference | Method | Resolution (Å) |
|---|---|---|---|---|
| WT | 6M0J | [41] | X-ray | 2.5 |
| 7KMB | [43] | Cryo-EM | 3.4 | |
| 7BH9 | [44] | Cryo-EM | 2.9 | |
| 6LZG | [10] | X-ray | 2.5 | |
| 7DQA | [45] | Cryo-EM | 2.8 | |
| Delta | 7W9I | [46] | Cryo-EM | 3.4 |
| 7V8B | (Yang et al., unpublished data) | Cryo-EM | 3.5 | |
| 7WBQ | [25] | X-ray | 3.3 | |
| Omicron | 7WBP | [25] | X-ray | 3.0 |
| BA.1 | 7WBL | [25] | Cryo-EM | 3.4 |
| 7T9L | [26] | Cryo-EM | 2.7 | |
| 7WK6 | [47] | Cryo-EM | 3.7 | |
| BA.2 | 7XB0 7XAZ | [48] | X-ray X-ray | 2.9 3.0 |
| 7ZF7 | [42] | X-ray | 3.46 | |
| BA.4 | 7XNQ 7XWA | [19] [49] | Cryo-EM X-ray | 3.52 3.36 |
| MD Simulation | Within RBD | Within hACE2 | Between RBD and hACE2 | Total |
|---|---|---|---|---|
| WT | 0.64 ± 0.14 | 0.59 ± 0.12 | 0.55 ± 0.12 | 0.58 ± 0.12 |
| Delta | 0.71 ± 0.13 | 0.53 ± 0.13 | 0.54 ± 0.11 | 0.54 ± 0.13 |
| BA.1+Q493R | 0.61 ± 0.15 | 0.53 ± 0.13 | 0.50 ± 0.12 | 0.52 ± 0.13 |
| BA.1 | 0.61 ± 0.14 | 0.54 ± 0.13 | 0.51 ± 0.10 | 0.53 ± 0.12 |
| BA.2 | 0.67 ± 0.13 | 0.59 ± 0.13 | 0.58 ± 0.12 | 0.59 ± 0.13 |
| BA.4 | 0.66 ± 0.15 | 0.57 ± 0.12 | 0.56 ± 0.12 | 0.57 ± 0.13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gheeraert, A.; Leroux, V.; Mias-Lucquin, D.; Karami, Y.; Vuillon, L.; Chauvot de Beauchêne, I.; Devignes, M.-D.; Rivalta, I.; Maigret, B.; Chaloin, L. Subtle Changes at the RBD/hACE2 Interface During SARS-CoV-2 Variant Evolution: A Molecular Dynamics Study. Biomolecules 2025, 15, 541. https://doi.org/10.3390/biom15040541
Gheeraert A, Leroux V, Mias-Lucquin D, Karami Y, Vuillon L, Chauvot de Beauchêne I, Devignes M-D, Rivalta I, Maigret B, Chaloin L. Subtle Changes at the RBD/hACE2 Interface During SARS-CoV-2 Variant Evolution: A Molecular Dynamics Study. Biomolecules. 2025; 15(4):541. https://doi.org/10.3390/biom15040541
Chicago/Turabian StyleGheeraert, Aria, Vincent Leroux, Dominique Mias-Lucquin, Yasaman Karami, Laurent Vuillon, Isaure Chauvot de Beauchêne, Marie-Dominique Devignes, Ivan Rivalta, Bernard Maigret, and Laurent Chaloin. 2025. "Subtle Changes at the RBD/hACE2 Interface During SARS-CoV-2 Variant Evolution: A Molecular Dynamics Study" Biomolecules 15, no. 4: 541. https://doi.org/10.3390/biom15040541
APA StyleGheeraert, A., Leroux, V., Mias-Lucquin, D., Karami, Y., Vuillon, L., Chauvot de Beauchêne, I., Devignes, M.-D., Rivalta, I., Maigret, B., & Chaloin, L. (2025). Subtle Changes at the RBD/hACE2 Interface During SARS-CoV-2 Variant Evolution: A Molecular Dynamics Study. Biomolecules, 15(4), 541. https://doi.org/10.3390/biom15040541