

Stimulus–Transcription Coupling of TRPM3 Channels: A Signaling Pathway from the Plasma Membrane to the Nucleus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
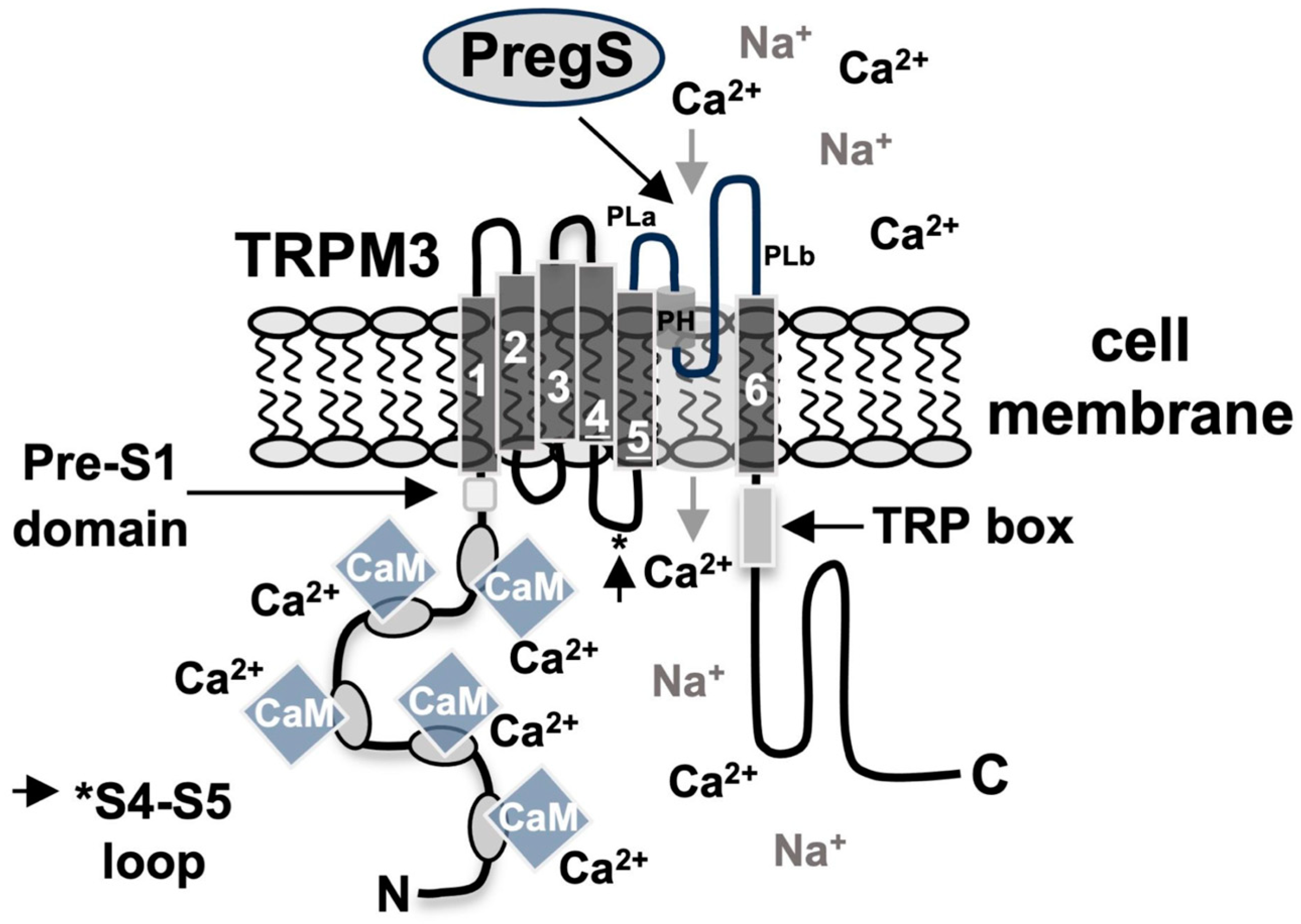
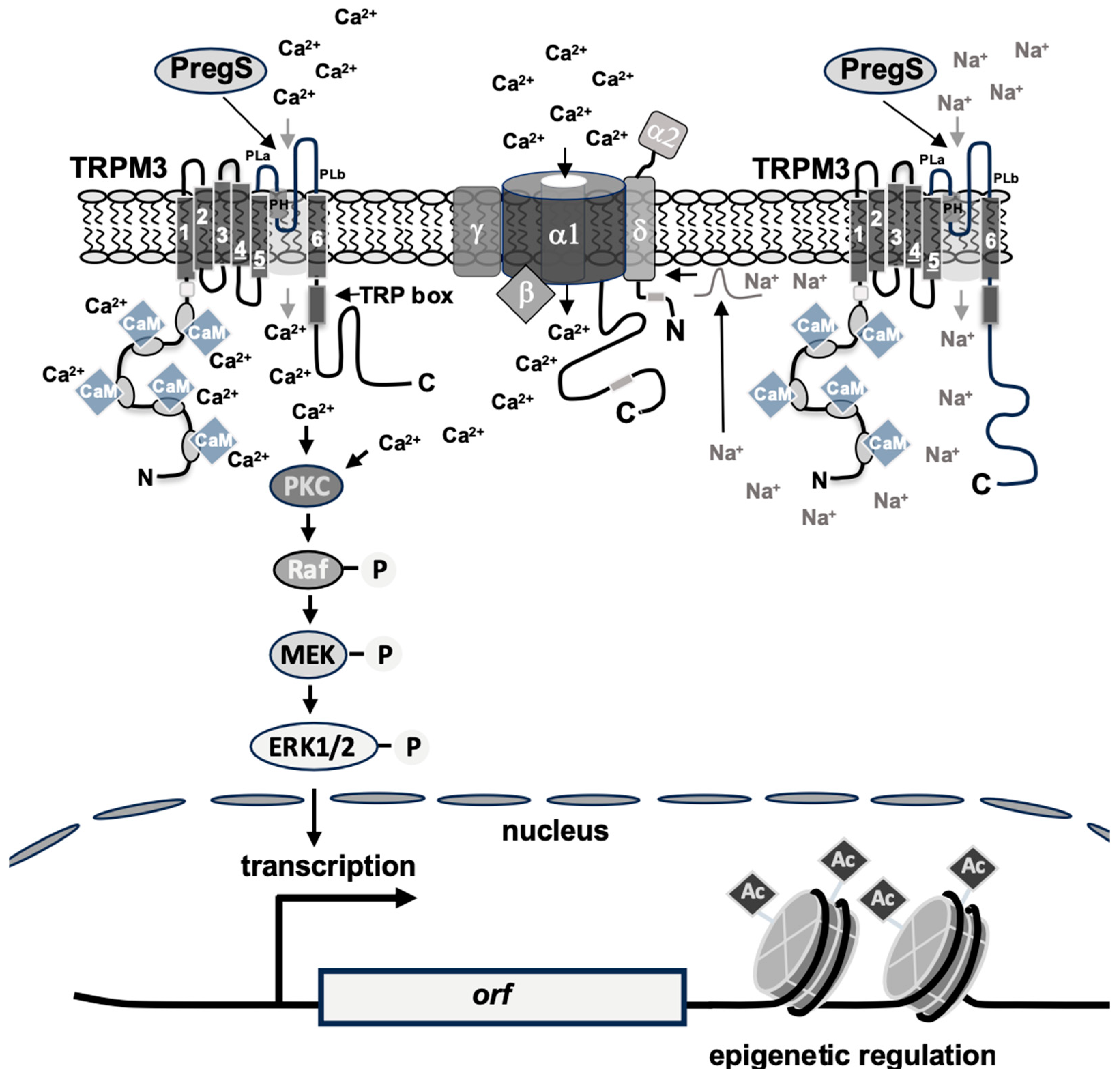
2. Structure and Function of TRPM3 Channels
3. TRPM3 Regulation by Intracellular Signaling Proteins, Lipids, and Ions
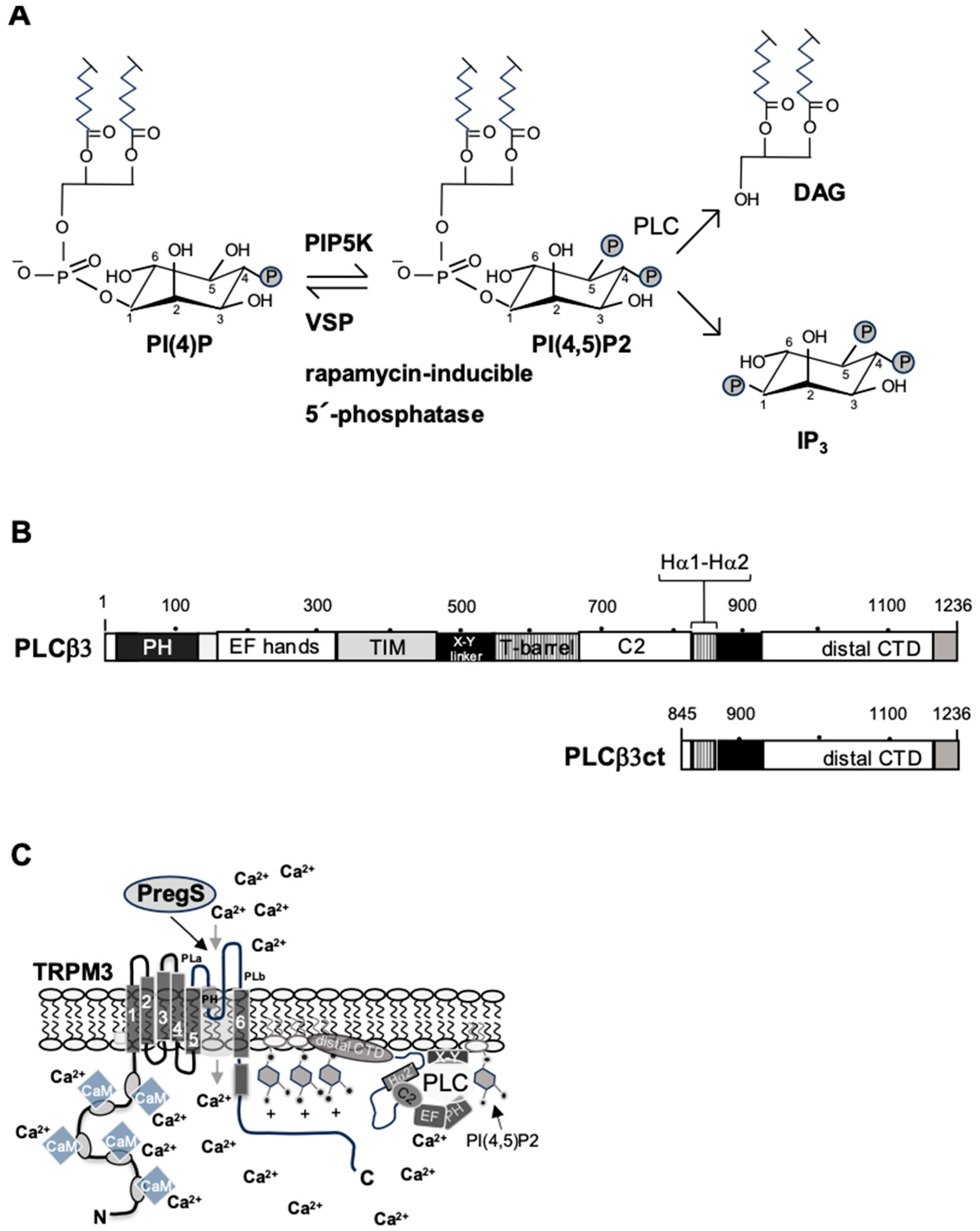
3.1. TRPM3-Induced Signaling Requires Phosphatidylinositol 4,5-Bisphosphate
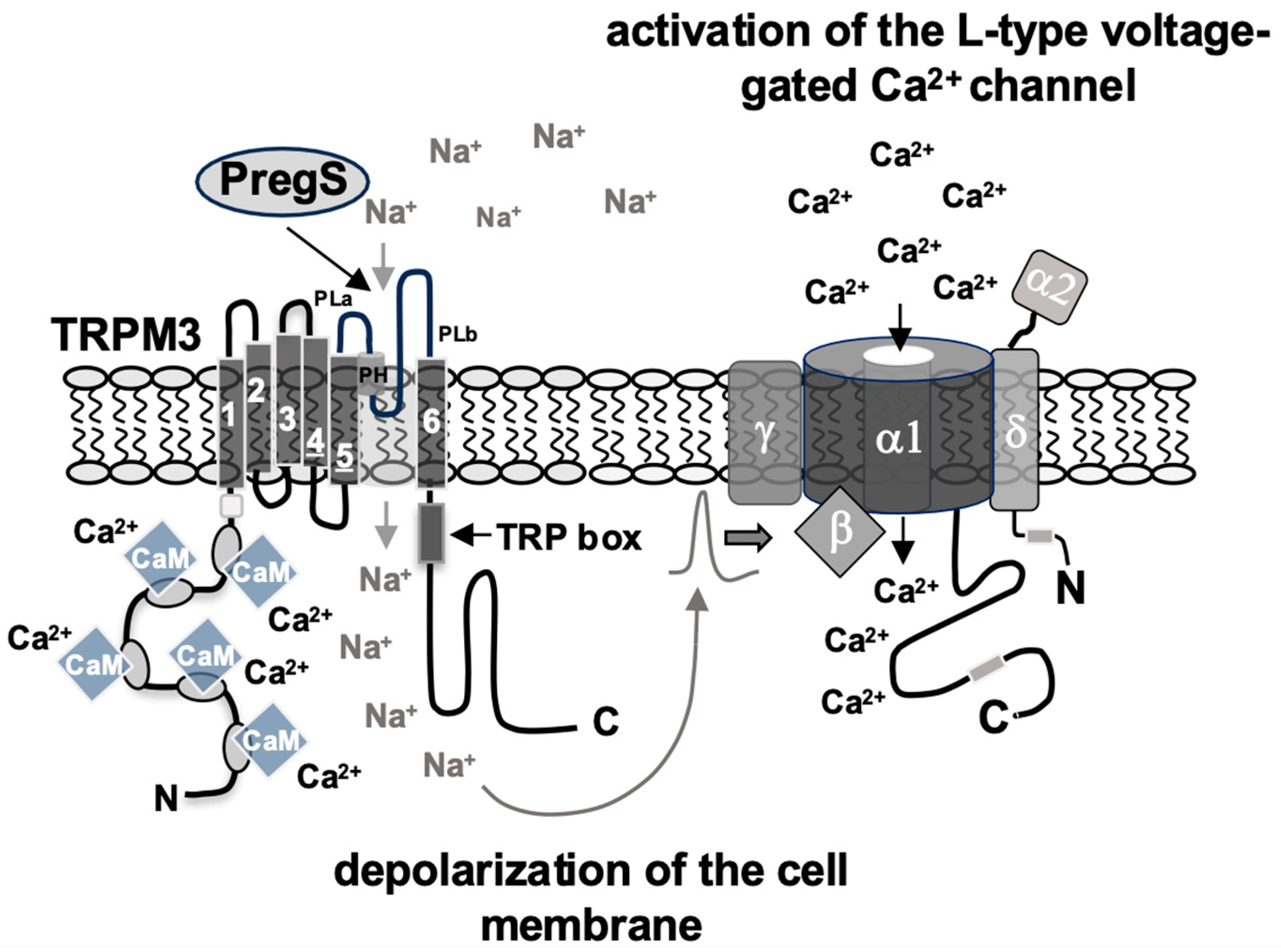
3.2. Inhibition of TRPM3 Signaling by the βγ Subunits of Trimeric G Proteins
3.3. Phospholipase C Negatively Affects TRPM3 Signaling
3.4. Calmodulin Is Required for TRPM3 Signaling
3.5. Zn2+ Ions Negatively Regulate TRPM3 Signaling
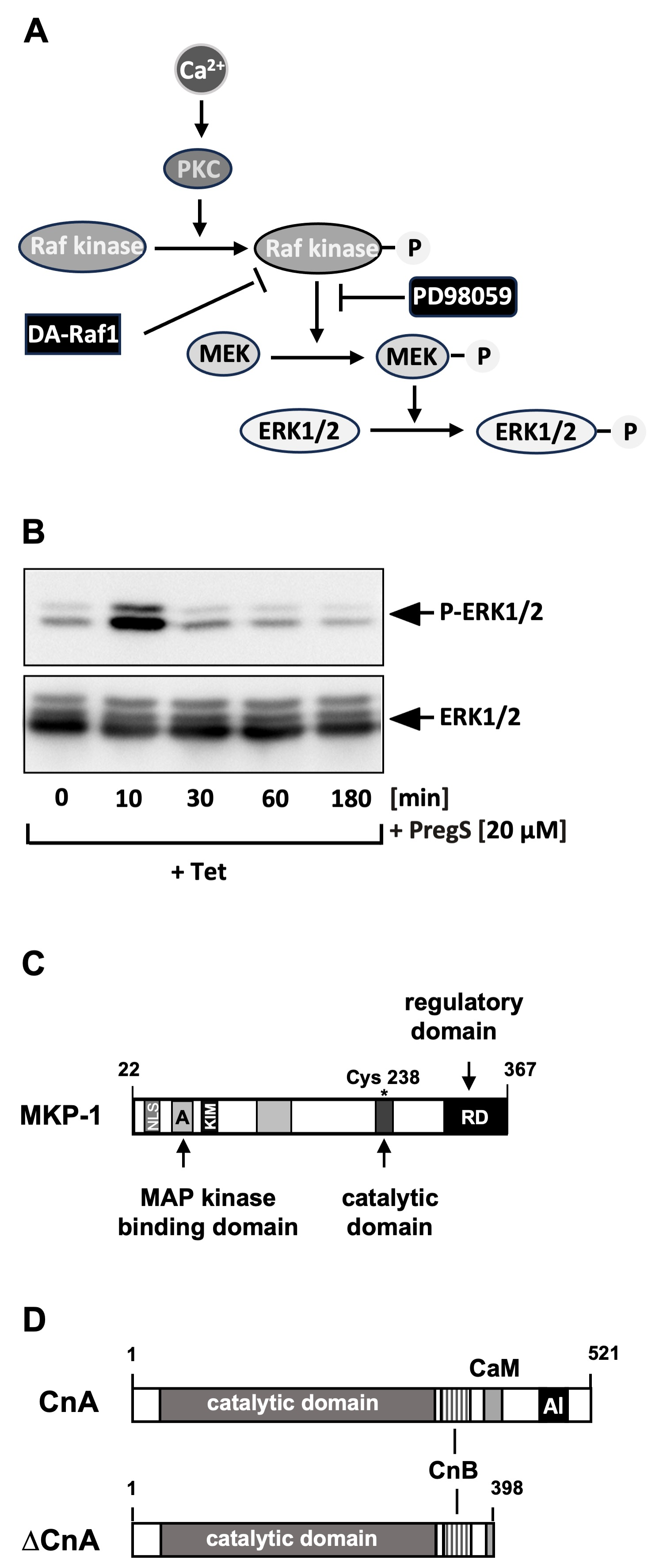
4. Stimulus-Responsive Protein Kinases Act as Signaling Transducers Within the TRPM3 Induced Signaling Cascade
5. Protein Phosphatases Act as Shut-Off Devices of the TRPM3-Induced Signaling Cascade
6. TRPM3 Stimulation Leads to the Activation of Stimulus-Responsive Transcription Factors
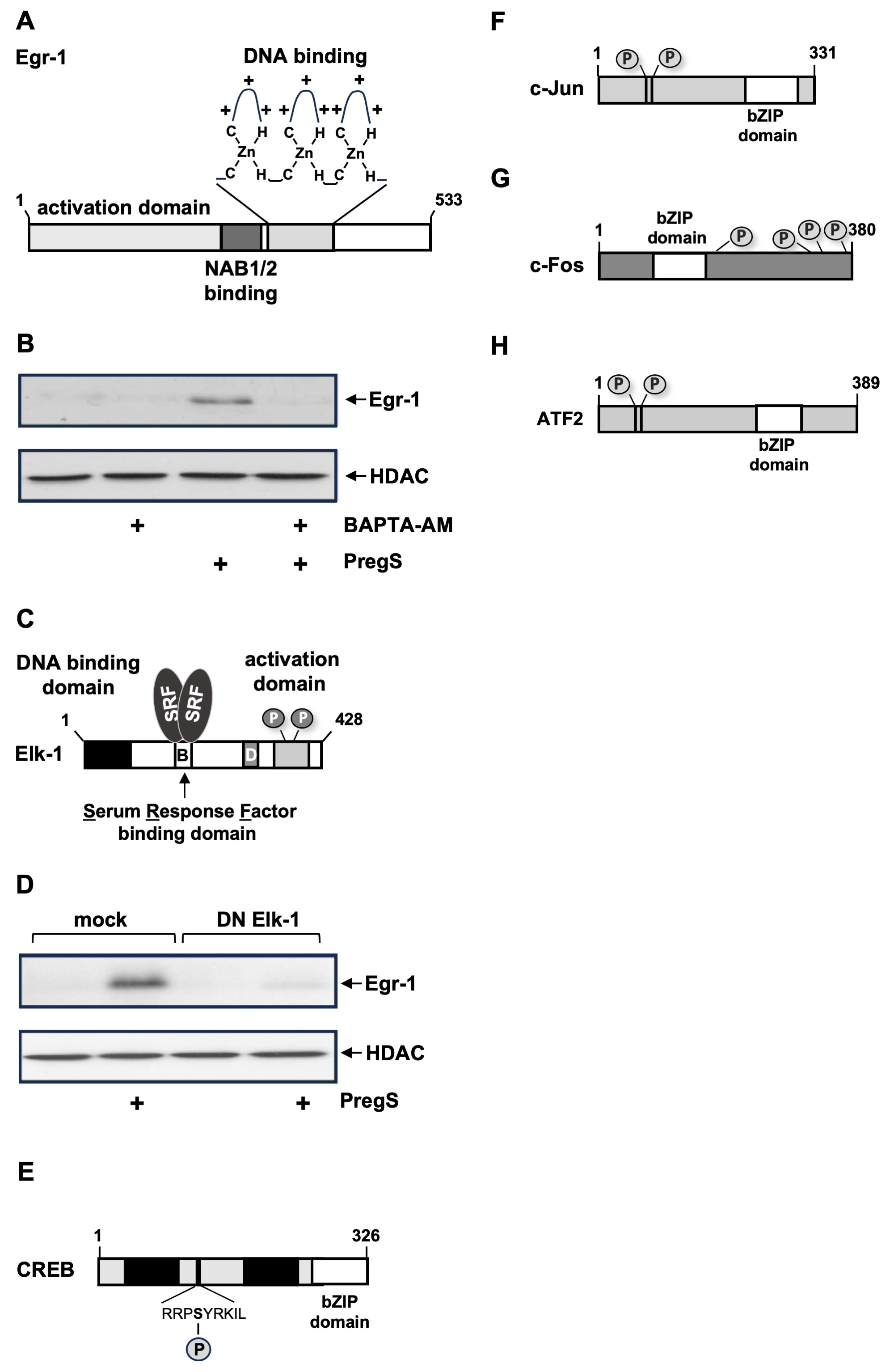
6.1. Egr-1, Elk-1
6.2. Basic Region Leucine Zipper (bZIP) Transcription Factors
6.3. Specificity of TRPM3-Induced Gene Transcription
7. Activation of Delayed-Response Genes After Stimulation of TRPM3 Channels
8. TRPM3-Induced Activation of Transcription Is Controlled by Epigenetic Regulators
9. Conclusions and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Samanta, A.; Hughes, T.E.T.; Moiseenkova-Bell, V.Y. Transient receptor potential (TRP) channels. Subcell. Biochem. 2018, 87, 141–165. [Google Scholar] [PubMed]
- Meotti, F.C.; Lemos de Andrade, E.; Calixto, J.B. TRP modulation by natural compounds. Handb. Exp. Pharmacol. 2014, 223, 1177–1238. [Google Scholar]
- Premkumar, L.S. Transient receptor potential channels a targets for phytochemicals. ACS Chem. Neurosci. 2014, 5, 1117–1130. [Google Scholar] [CrossRef]
- Nilius, B.; Szallasi, A. Transient receptor potential channels as drug targets: From the science of basic research to the art of medicine. Pharmacol. Rev. 2014, 66, 676–814. [Google Scholar]
- Zhou, Y.; Bennett, T.M.; Shiels, A. Mutation of the TRPM3 cation channel underlies progressive cataract development and lens calcification associated with pro-fibrotic and immune cell responses. FASEB J. 2021, 35, e21288. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.M.; Szallasi, A. Targeting nociceptive transient receptor potential channels to treat chronic pain: Current state of the field. Br. J. Pharmacol. 2018, 175, 2185–2203. [Google Scholar] [CrossRef]
- Koivisto, A.-P.; Voets, T.; Iadarola, M.J.; Szallasi, A. Targeting TRP channels for pain relief: A review of current evidence from bench to bedside. Curr. Opin. Pharmacol. 2024, 75, 102447. [Google Scholar] [CrossRef]
- Vriens, J.; Voets, T. Sensing the heat with TRPM3. Pflug. Arch. 2018, 470, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Held, K.; Tóth, B.I. TRPM3 in brain (patho)physiology. Front. Cell Dev. Biol. 2021, 9, 635659. [Google Scholar] [CrossRef]
- Kashio, M.; Tominaga, M. TRP channels in thermosensation. Curr. Opin. Neurobiol. 2022, 75, 102591. [Google Scholar] [CrossRef]
- Bousova, K.V.; Zouharova, M.; Jiraskova, K.; Vetyskova, V. Interaction of calmodulin with TRPM: An initiator of channel modification. Int. J. Mol. Sci. 2023, 24, 15162. [Google Scholar]
- Roelens, R.; Peigneur, A.N.F.; Voets, T.; Vriens, J. Neurodevelopmental disorders caused by variants in TRPM3. Biochim. Biophys. Acta–Mol. Cell Res. 2024, 1871, 119709. [Google Scholar] [CrossRef] [PubMed]
- Rohacs, T. Phosphoinositide regulation of TRP channels: A functional overview in the structural era. Annu. Rev. Physiol. 2024, 86, 329–355. [Google Scholar] [PubMed]
- Uchida, K. TRPM3, TRPM4, and TRPM5 as thermo-sensitive channels. J. Physiol. Sci. 2024, 74, 43. [Google Scholar]
- Thiel, G.; Rubil, S.; Lesch, A.; Guethlein, L.A.; Rössler, O.G. Transient receptor potential TRPM3 channels: Pharmacology, signaling, and biological functions. Pharmacol. Res. 2017, 124, 92–99. [Google Scholar]
- Vriens, J.; Held, K.; Janssens, A.; Tóth, B.I.; Kerselaers, S.; Nilius, B.; Vennekens, R.; Voets, T. Opening of an alternative ion permeation pathway in a nociceptor TRP channel. Nat. Chem. Biol. 2014, 10, 188–195. [Google Scholar] [CrossRef]
- Zhao, C.; MacKinnon, R. Structural and functional analyses of a GPCR-inhibited ion channel TRPM3. Neuron 2023, 111, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Park, C.-G.; Feng, S.; Guan, Z.; Lee, H.-J.; Zhang, F.; Sharma, K.; Borgnia, M.J.; Im, W.; Lee, S.-Y. Molecular basis of neurosteroid and anticonvulsant regulation of TRPM3. Nat. Struct. Mol. Biol. 2025. [Google Scholar] [CrossRef]
- Harteneck, C.; Klose, C.; Krautwurst, D. Synthetic modulators of TRP channel activity. Adv. Exp. Med. Biol. 2011, 704, 87–106. [Google Scholar]
- Thiel, G.; Backes, T.M.; Welck, J.; Steinhausen, S.; Fischer, A.-L.; Langfermann, D.S.; Ulrich, M.; Wissenbach, U.; Rössler, O.G. Pharmacological inhibition of TRPM8-induced gene transcription. Biochem. Pharmacol. 2019, 170, 113678. [Google Scholar]
- Hill, K.; McNulty, S.; Randall, A.D. Inhibition of TRPM2 channels by the antifungal agents clotrimazole and econazole. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2004, 370, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Meseguer, V.; Karashima, Y.; Talavera, K.; D’Hoedt, D.; Donovan-Rodriguez, T.; Viana, F.; Nilius, B.; Voets, T. Transient receptor potential channels in sensory neurons are targets of the antimycotic agent clotrimazole. J. Neurosci. 2008, 28, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Vriens, J.; Owsianik, G.; Hofmann, T.; Philipp, S.E.; Stab, J.; Chen, X.; Benoit, M.; Xue, F.; Janssens, A.; Kerselaers, S.; et al. TRPM3 is a nociceptor channel involved in the detection of noxious heat. Neuron 2011, 70, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Krügel, U.; Straub, I.; Beckmann, H.; Schaefer, M. Primidone inhibits TRPM3 and attenuates thermal nociception in vivo. Pain 2017, 158, 856–867. [Google Scholar] [CrossRef]
- Wagner, T.F.J.; Loch, S.; Lambert, S.; Straub, I.; Mannebach, S.; Mathar, I.; Düfer, M.; Lis, A.; Flockerzi, V.; Philipp, S.E.; et al. Transient receptor potential M3 channels are ionotropic steroid receptors in pancreatic beta cells. Nat. Cell Biol. 2008, 10, 1421–1430. [Google Scholar] [CrossRef]
- Lesch, A.; Rubil, S.; Thiel, G. Activation and inhibition of transient receptor potential TRPM3-induced gene transcription. Br. J. Pharmacol. 2014, 171, 2645–2658. [Google Scholar] [CrossRef]
- Held, K.; Kichko, T.; De Clercq, K.; Klaassen, H.; Van Bree, R.; Vanherck, J.-C.; Marchand, A.; Reeh, P.W.; Chaltin, P.; Voets, T.; et al. Activation of TRPM3 by a potent synthetic ligand reveals a role in peptide release. Proc. Natl. Acad. Sci. USA 2015, 112, E1363–E1372. [Google Scholar] [CrossRef]
- Rubil, S.; Thiel, G. Activation of gene transcription via CIM0216, a synthetic ligand of transient receptor potential melastatin-3 (TRPM3) channels. Channels 2017, 11, 79–83. [Google Scholar] [CrossRef]
- Jang, M.-K.; Mierke, D.F.; Russek, S.J.; Farb, D.H. A steroid modulatory domain on NR2B controls N-methyl-D-aspartate receptor proton sensitivity. Proc. Natl. Acad. Sci. USA 2004, 101, 8198–8203. [Google Scholar] [CrossRef]
- Chen, L.; Cai, W.; Chen, L.; Zhou, R.; Furuya, K.; Sokabe, M. Modulatory metaplasticity induced by pregnenolone sulfate in the rat hippocampus: A leftward shift in LTP/LTD-frequency curve. Hippocampus 2010, 20, 499–512. [Google Scholar] [CrossRef]
- Kobayashi, T.; Washiyama, K.; Ikeda, K. Pregnenolone sulfate potentiates the inwardly rectifying K channel Kir2.3. PLoS ONE 2009, 4, e6311. [Google Scholar]
- Kostakis, E.; Jang, M.K.; Russek, S.J.; Gibbs, T.T.; Farb, D.H. A steroid modulatory domain in NR2A collaborates with NR1 exon-5 to control NMDAR moduation by pregnenolone sulfate and protons. J. Neurochem. 2011, 119, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Cameron, K.; Bartle, E.; Roark, R.; Fanelli, D.; Pham, M.; Pollard, B.; Borkowski, B.; Rhoads, S.; Kim, J.; Rocha, M.; et al. Neurosteroid binding to the amino terminal and glutamate binding domains of inonotropic glutamate receptors. Steroids 2012, 77, 774–779. [Google Scholar]
- Mayer, S.I.; Müller, I.; Mannebach, S.; Endo, T.; Thiel, G. Signal transduction of pregnenolone sulfate in insulinoma cells. Activation of Egr-1 expression involving TRPM3, voltage-gated calcium channels, ERK, and ternary complex factors. J. Biol. Chem. 2011, 286, 10084–10096. [Google Scholar]
- Müller, I.; Rössler, O.G.; Thiel, G. Pregnenolone sulfate activates basic region leucine zipper transcription factors in insulinoma cells: Role of voltage-gated Ca2+ channels and transient receptor potential melastatin 3 channels. Mol. Pharmacol. 2011, 80, 1179–1189. [Google Scholar]
- Vangeel, L.; Benoit, M.; Miron, Y.; Miller, P.E.; De Clercq, K.; Chaltin, P.; Verfaillie, C.; Vriens, J.; Voets, T. Functional expression and pharmacological modulation of TRPM3 in human sensory neurons. Br. J. Pharmacol. 2020, 177, 2683–2695. [Google Scholar] [PubMed]
- Fajardo, O.; Meseguer, V.; Belmonte, C.; Viana, F. TRPA1 channels: Novel targets of 1,4-dihydropyridines. Channels 2008, 2, 429–438. [Google Scholar] [CrossRef]
- Klose, C.; Straub, I.; Riehle, M.; Ranta, F.; Krautwurst, D.; Ullrich, S.; Mayerhof, W.; Harteneck, C. Fenamates as TRP channel blocker: Mefenamic acid selectively blocks TRMP3. Br. J. Pharmacol. 2011, 162, 1757–1769. [Google Scholar]
- Straub, I.; Krügel, U.; Mohr, F.; Teichert, J.; Rizun, O.; Konrad, M.; Oberwinkler, J.; Schaefer, M. Flavanones that selectively inhibit TRPM3 attenuate thermal nociception in vivo. Mol. Pharmacol. 2013, 84, 736–750. [Google Scholar]
- Kumar, S.; Jin, F.; Park, S.J.; Choi, W.; Keuning, S.I.; Massimino, R.P.; Vu, S.; Lü, W.; Du, J. Convergent agonist and heat activation of nociceptor TRPM3. bioRxiv 2025. [Google Scholar] [CrossRef]
- Nilius, B.; Flockerzi, V. What do we really know and what do we need to know: Some controversies, perspectives, and surprises. Handb. Exp. Pharmacol. 2014, 223, 1239–1280. [Google Scholar] [PubMed]
- Lesch, A.; Hui, X.; Lipp, P.; Thiel, G. Transient receptor potential melastatin-3 (TRPM3)-induced activation of AP-1 requires Ca2+ ions and the transcription factors c-Jun, ATF2, and ternary complex factor. Mol. Pharmacol. 2015, 87, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Frühwald, J.; Camacho Londono, J.; Dembla, S.; Mannebach, S.; Lis, A.; Drews, A.; Wissenbach, U.; Oberwinkler, J.; Philipp, S.E. Alternative Splicing of a protein domain indispensable for function of transient receptor potential melastatin 3 (TRPM3) ion channels. J. Biol. Chem. 2012, 287, 36663–36672. [Google Scholar]
- Fonfria, E.; Murdock, P.R.; Cusdin, F.S.; Benham, C.D.; Kesell, R.E.; McNulty, S. Tissue distribution profiles of the human TRPM cation channel family. J. Recept. Signal Transduct. 2006, 26, 159–178. [Google Scholar] [CrossRef]
- Vandewauw, I.; De Clercq, K.; Mulier, M.; Held, K.; Pinto, S.; Van Ranst, N.; Segal, A.; Voet, T.; Vennekens, R.; Zimmermann, K.; et al. A TRP channel trio mediates acute noxious heat sensing. Nature 2018, 555, 662–666. [Google Scholar] [PubMed]
- Cabanas, H.; Muraki, K.; Balinas, C.; Eaton-Fitch, N.; Staines, D.; Marshall-Gradisnik, S. Validation of impaired transient receptor potential melastatin 3 ion channel activity in natural killer cells from chronic fatigue syndrome/myalgic encephalomyelitis patients. Mol. Med. 2019, 25, 14. [Google Scholar]
- Wills, R.C.; Hammond, G.R.V. PI(4,5)P2: Signaling the plasma membrane. Biochem. J. 2022, 479, 2311–2325. [Google Scholar]
- Van den Bout, I.; Divecha, N. PIP5K-driven Ptd(4,5)P2 synthesis: Regulation and cellular functions. J. Cell Sci. 2009, 122, 3837–3850. [Google Scholar]
- Thiel, G.; Rössler, O.G. Expression of the C-terminal domain of phospholipase Cβ3 inhibits signaling via Gαq-coupled receptors and transient receptor potential channels. Int. J. Mol. Sci. 2022, 23, 9590. [Google Scholar] [CrossRef] [PubMed]
- Badheka, D.; Borbiro, I.; Rohacs, T. Transient receptor potential melastatin 3 is a phosphoinositide-dependent ion channel. J. Gen. Physiol. 2015, 146, 65–77. [Google Scholar]
- Tóth, B.I.; Konrad, M.; Ghosh, D.; Mohr, F.; Halaszovich, C.R.; Leitner, M.G.; Vriens, J.; Oberwinkler, J.; Voets, T. Regulation of the transient receptor potential channel TRPM3 by phosphoinositides. J. Gen. Physiol. 2015, 146, 51–63. [Google Scholar]
- Semenas, J.; Hedblom, A.; Miftakhova, R.R.; Sarwar, M.; Larsson, R.; Shcherbina, L.; Johansson, M.E.; Härkönen, P.; Sterner, O.; Persson, J.L. The role of PI3K/AKT-related PIP5K1α and the discovery of its selective inhibitor for treatment of advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2014, 111, E3689–E3698. [Google Scholar] [PubMed]
- Kunkl, M.; Porciello, N.; Mastrogiovanni, M.; Capuano, C.; Lucantoni, F.; Moretti, C.; Persson, J.L.; Galandrini, R.; Buzzetti, R.; Tuosto, L. ISA-2011B, a phosphatidylinositol 4-phosphate 5-kinase α inhibitor, impairs CD28-dependent costimulatory and pro-inflammatory signals in human T lymphocytes. Front. Immunol. 2017, 8, 502. [Google Scholar]
- Thiel, G.; Rössler, O.G. Signal transduction of transient receptor potential TRPM8 channels: Role of PIP5K, Gq-Proteins, and c-Jun. Molecules 2024, 29, 2602. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Carnevale, V.; Gabrielle, M.; Gianti, E.; Rohacs, T. Computational and functional studies of the PI(4,5)P2 binding site of the TRPM3 ion channel reveal interactions with other regulators. J. Biol. Chem. 2022, 298, 102547. [Google Scholar]
- Alkhatib, O.; da Costa, R.; Gentry, C.; Quallo, T.; Mannebach, S.; Weissgerber, P.; Freichel, M.; Philipp, S.E.; Bevan, S.; Andersson, D.A. Promiscuous G-protein-coupled receptor inhibition of transient receptor potential melastatin 3 ion channel by Gβγ subunits. J. Neurosci. 2019, 39, 7840–7852. [Google Scholar]
- Badheka, D.; Yudin, Y.; Borbiro, I.; Hartle, C.M.; Yazici, A.; Mirshahi, T.; Rohacs, T. Inhibition of transient receptor potential melastatin 3 ion channels by G-protein βγ subunits. eLife 2017, 6, e26147. [Google Scholar] [CrossRef] [PubMed]
- Dembla, S.; Behrendt, M.; Mohr, F.; Goecke, C.; Sondermann, J.; Schneider, F.M.; Schmidt, M.; Stab, J.; Enzeroth, R.; Leitner, M.G.; et al. Ant-nociceptive action of peripheral mu-opioid receptor by G-beta-gamma protein-mediated inhibition of TRPM3 channels. eLife 2017, 6, e26280. [Google Scholar]
- Quallo, T.; Alkhatib, O.; Gentry, C.; Andersson, D.A.; Bevan, S. G protein βγ subunits inhibit TRPM3 ion channel in sensory neurons. eLife 2017, 6, e26138. [Google Scholar]
- Klasen, K.; Hollatz, D.; Zielke, S.; Gisselmann, G.; Hatt, H.; Wetzel, C.H. The TRPM8 ion channel comprises direct Gq protein-activating capacity. Pflügers Arch.–Eur. J. Physiol. 2012, 463, 779–797. [Google Scholar]
- Zhang, X.; Mak, S.; Li, L.; Parra, A.; Denlinger, B.; Belmonte, C.; McNaughton, P.A. Direct inhibition of the cold-activated TRPM8 ion channel by Gαq. Nat. Cell Biol. 2012, 14, 851–858. [Google Scholar]
- Behrendt, M.; Gruss, F.; Enzeroth, R.; Dembla, S.; Zhao, S.; Crassous, P.-A.; Mohr, F.; Nys, M.; Louros, N.; Gallardo, R.; et al. The structural basis for an on-off switch controlling Gβγ-mediated inhibition of TRPM3 channels. Proc. Nat. Acad. Sci. USA 2020, 117, 29090–29100. [Google Scholar]
- Wills, R.C.; Goulden, B.D.; Hammond, G.R.V. Genetically encoded lipid biosensors. Mol. Biol. Cell 2018, 29, 1526–1532. [Google Scholar] [PubMed]
- Stephens, L.; Jackson, T.R.; Hawkins, P.T. Activation of phosphatidylinositol 4,5-bisphosphate supply by agonists and non-hydrolysable GTP analogues. Biochem. J. 1993, 296, 481–488. [Google Scholar] [PubMed]
- Barneda, D.; Janardan, V.; Niewczas, I.; Collins, D.M.; Cosulich, S.; Clark, J.; Stephens, L.R.; Hawkins, P.T. Acyl chain selection couples the consumption and synthesis of phosphoinositides. EMBO J. 2022, 41, e110038. [Google Scholar]
- Falzone, M.E.; MacKinnon, R. The mechanism of Gαq regulation of PLCβ3-catalyzed PIP2 hydrolysis. Proc. Natl. Acad. Sci. USA 2023, 120, e2315011120. [Google Scholar]
- Falzone, M.E.; MacKinnon, R. Gbg activates PIP2 hydrolysis by recruiting and orienting PLCβ on the membrane surface. Proc. Natl. Acad. Sci. USA 2023, 120, e2301121120. [Google Scholar]
- Golebiewska, U.; Nyako, M.; Woturski, W.; Zaitseva, I.; McLaughlin, S. Diffusion coefficient of fluorescent phosphatidylinositol 4,5-bisphosphate in the plasma membrane of cells. Mol. Biol. Cell 2008, 19, 1663–1669. [Google Scholar]
- Holakovska, B.; Grycova, L.; Jirku, M.; Sulc, M.; Bumba, L.; Teisinger, J. Calmodulin and S100A1 protein interact with N terminus of TRPM3 channel. J. Biol. Chem. 2012, 287, 16645–16655. [Google Scholar]
- Przibilla, J.; Dembla, S.; Rizun, O.; Lis, A.; Jung, M.; Oberwinkler, J.; Beck, A.; Philipp, S.E. Ca2+-dependent regulation and binding of calmodulin to multiple sites of transient receptor potential melastatin 3 (TRPM3) ion channels. Cell Calcium 2018, 73, 40–52. [Google Scholar]
- Thiel, G.; Rössler, O.G. Calmodulin regulates transient receptor potential TRPM3 and TRPM8-induced gene transcription. Int. J. Mol. Sci. 2023, 24, 7902. [Google Scholar] [CrossRef]
- Peterson, B.Z.; DeMaria, C.D.; Yue, D.T. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron 1999, 22, 549–558. [Google Scholar]
- DeMaria, C.D.; Soong, T.W.; Alseikhan, B.A.; Alvania, R.S.; Yue, D.T. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature 2001, 411, 484–489. [Google Scholar] [PubMed]
- Erickson, M.G.; Alseikhan, B.A.; Peterson, B.Z.; Yue, D.T. Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells. Neuron 2001, 31, 973–985. [Google Scholar]
- Pitt, G.S.; Zühlke, R.D.; Hudmon, A.; Schulman, H.; Reuter, H.; Tsien, R.W. Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type Ca2+ channels. J. Biol. Chem. 2001, 276, 30794–30802. [Google Scholar] [PubMed]
- Adams, P.J.; Ben-Johny, M.; Dick, I.E.; Inoue, T.; Yue, D.T. Apocalmodulin itself promotes ion channel opening and Ca2+ regulation. Cell 2014, 159, 608–622. [Google Scholar] [PubMed]
- Chemin, J.; Taiakina, V.; Monteil, A.; Piazza, M.; Guan, W.; Stephens, R.F.; Kitmitto, A.; Pang, Z.P.; Dolphin, A.C.; Perez-Reyes, E.; et al. Calmodulin regulates Cav3 T-type channels at their gating brake. J. Biol. Chem. 2017, 292, 20010–20031. [Google Scholar]
- Fanger, C.M.; Ghanshani, S.; Logsdon, N.J.; Rauer, H.; Kalman, K.; Zhou, J.; Beckingham, K.; Chandy, K.G.; Cahalan, M.D.; Aiyar, J. Calmodulin mediates calcium-dependent activation of the intermediate conductance KCa channel, IKCα1. J. Biol. Chem. 1999, 274, 5746–5754. [Google Scholar]
- Yang, T.; Colecraft, H.M. Calmodulin regulation of TMEM16A and 16B Ca2+-activated chloride channels. Channels 2016, 10, 38–44. [Google Scholar]
- Wagner, T.F.J.; Drews, A.; Loch, S.; Mohr, F.; Philipp, S.E.; Lambert, S.; Oberwinkler, J. TRPM3 channels provide a regulated influx pathway for zinc in pancreatic beta cells. Pflügers Arch.–Eur. J. Physiol. 2010, 460, 755–765. [Google Scholar]
- Loviscach, L.; Backes, T.M.; Langfermann, D.S.; Ulrich, M.; Thiel, G. Zn2+ ions inhibit gene transcription following stimulation of the Ca2+ channels Cav1.2 and TRPM3. Metallomics 2020, 12, 1735–1747. [Google Scholar] [PubMed]
- Bellomo, E.; Massarotti, A.; Hogstrand, C.; Maret, W. Zinc ions modulate protein tyrosine phosphatase 1B activity. Metallomics 2014, 6, 1229–1239. [Google Scholar] [PubMed]
- Gore, A.; Moran, A.; Hershfinkel, M.; Sekler, I. Inhibitory mechanism of store-operated Ca2+ channels by zinc. J. Biol. Chem. 2004, 279, 11106–11111. [Google Scholar]
- Yuan, H.; Hansen, K.B.; Zhang, J.; Pierson, T.M.; Markello, T.C.; Fuentes Fajardo, K.V.; Holloman, C.M.; Golas, G.; Adams, D.R.; Boerkoel, C.F.; et al. Functional analysis of a de novo GRIN2A missense mutation associated with early-onset epileptic encephalopathy. Nat. Commun. 2014, 5, 3251. [Google Scholar]
- Schulze, A.; Nicke, B.; Warne, P.H.; Tomlinson, S.; Downward, J. The transcriptional response to Raf activation is almost completely dependent on mitogen-activated protein kinase kinase activity and shows a major autocrine component. Mol. Biol. Cell 2004, 15, 3450–3463. [Google Scholar]
- Wellbock, C.; Karasarides, M.; Marais, R. The Raf proteins take centre stage. Nat. Mol. Cell Biol. 2004, 5, 875–885. [Google Scholar]
- Lesch, A.; Rössler, O.G.; Thiel, G. Extracellular signal-regulated protein kinase, c-Jun N-terminal protein kinase, and calcineurin regulate transient receptor potential M3 (TRPM3) induced activation of AP-1. J. Cell. Biochem. 2017, 118, 2409–2419. [Google Scholar]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK signalling: A master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar]
- Arnette, D.; Gibson, T.B.; Lawrence, M.C.; January, B.; Khoo, S.; McGlynn, K.; Vanderbilt, C.A.; Cobb, M.H. Regulation of ERK1 and ERK2 by glucose and peptide hormones in pancreatic β cells. J. Biol. Chem. 2003, 278, 32517–32525. [Google Scholar]
- Ram, A.; Murphy, D.; DeCuzzi, N.; Patankar, M.; Hu, J.; Pargett, M.; Albeck, J.G. A guide to ERK dynamics, part 2: Downstream decoding. Biochem. J. 2023, 480, 1909–1928. [Google Scholar]
- Zhou, Y.; Bennett, T.M.; Ruzycki, P.A.; Guo, Z.; Cao, Y.-Q.; Shahidullah, M.; Delamere, N.A.; Shiels, A. A cataract-causing mutation in the TRPM3 cation channel disrupts calcium dynamics in the lens. Cells 2024, 13, 257. [Google Scholar] [CrossRef] [PubMed]
- Tanoue, T.; Moriguchi, T.; Nishida, E. Molecular cloning and characterization of a novel dual specificity phosphatase, MKP-5. J. Biol. Chem. 1999, 274, 19949–19956. [Google Scholar]
- Shapiro, P.S.; Ahn, N.G. Feedback regulation of Raf-1 and mitogen-activated protein kinase (MAP) kinase kinase 1 and 2 by MAP kinase phosphatase-1 (MKP-1). J. Biol. Chem. 1998, 273, 1788–1793. [Google Scholar] [PubMed]
- Slack, D.N.; Seternes, O.-M.; Gabrielsen, M.; Keyse, S.M. Distinct binding determinants for ERK2/p38α and JNK MAP kinases mediate catalytic activation and substrate selectivity of MAP kinase phosphatase-1. J. Biol. Chem. 2001, 276, 16491–16500. [Google Scholar] [CrossRef] [PubMed]
- Rubil, S.; Lesch, A.; Mukaida, N.; Thiel, G. Stimulation of transient receptor potential M3 (TRPM3) increases interleukin-8 gene promoter activity involving AP-1 and extracellular signal-regulated protein kinase. Cytokine 2018, 103, 133–141. [Google Scholar]
- Thiel, G.; Schmidt, T.; Rössler, O.G. Ca2+ microdomains, calcineurin and the regulation of gene transcription. Cells 2021, 10, 875. [Google Scholar] [CrossRef]
- Wang, H.; Du, Y.; Xiang, B.; Lin, W.; Li, X.; Wie, Q. A renewed model of CNA regulation involving its C-terminal regulatory domain and CaM. Biochemistry 2008, 47, 4461–4468. [Google Scholar]
- Rumi-Masante, J.; Rusinga, F.I.; Lester, T.E.; Dunlap, T.B.; Williams, T.D.; Dunker, A.K.; Weiss, D.D.; Creamer, T.P. Structural basis for activation of calcineurin by calmodulin. J. Mol. Biol. 2012, 415, 307–317. [Google Scholar]
- Sugimoto, T.; Stewart, S.; Guan, K.-L. The calcium/calmodulin-dependent protein phosphatase calcineurin is the major Elk-1 phosphatase. J. Biol. Chem. 1997, 272, 29415–29418. [Google Scholar]
- Tian, J.; Karin, M. Stimulation of Elk1 transcriptional activity by mitogen-activated protein kinases is negatively regulated by protein phosphatase 2B (calcineurin). J. Biol. Chem. 1999, 274, 15173–15180. [Google Scholar] [CrossRef]
- Thiel, G.; Backes, T.M.; Guethlein, L.A.; Rössler, O.G. Critical protein–protein interactions determine the biological activity of Elk-1, a master regulator of stimulus-induced gene transcription. Molecules 2021, 26, 6125. [Google Scholar] [CrossRef]
- Rubil, S.; Thiel, G. Stimulation of TRPM3 channels increases the transcriptional activation potential of Elk-1 involving cytosolic Ca2+, extracellular signal-regulated protein kinase, and calcineurin. Eur. J. Pharmacol. 2019, 884, 225–230. [Google Scholar]
- Rubil, S.; Rössler, O.G.; Thiel, G. CREB, AP-1, ternary complex factors and MAP kinases connect transient receptor potential melastatin-3 (TRPM3) channel stimulation with increased c-Fos expression. Br. J. Pharmacol. 2016, 173, 305–318. [Google Scholar]
- Müller, I.; Rössler, O.G.; Wittig, C.; Menger, M.D.; Thiel, G. Critical role of Egr transcription factors in regulating insulin biosynthesis, blood glucose homeostasis and islet size. Endocrinology 2012, 53, 3040–3053. [Google Scholar]
- Lesch, A.; Backes, T.M.; Langfermann, D.S.; Rössler, O.G.; Laschke, M.W.; Thiel, G. Ternary complex factor regulates pancreatic islet size and blood glucose homeostasis in transgenic mice. Pharmacol. Res. 2020, 159, 104983. [Google Scholar] [PubMed]
- Newman, J.R.S.; Keating, A.E. Comprehensive identification of human bZIP interactions with coiled-coil arrays. Science 2003, 300, 2097–2101. [Google Scholar]
- Thiel, G.; Rössler, O.G. TRPM3-induced gene transcription is under epigenetic control. Pharmaceuticals 2022, 15, 846. [Google Scholar] [CrossRef]
- Leppä, S.; Saffrich, R.; Ansorge, W.; Bohmann, D. Differential regulation of c-Jun by ERK and JNK during PC12 cell differentiation. EMBO J. 1998, 17, 4404–4413. [Google Scholar]
- Morton, S.; Davis, R.J.; McLaren, A.; Cohen, P. A reinvestigation of the multisite phosphorylation of the transcription factor c-Jun. EMBO J. 2003, 22, 3876–3886. [Google Scholar]
- Nakanishi, M.; Hata, K.; Nagayama, T.; Sakurai, T.; Nishisho, T.; Wakabayashi, H.; Hiraga, T.; Ebisu, S.; Yoneda, T. Acid activation of Trpv1 leads to an up-regulation of calcitonin gene-related peptide expression in dorsal root ganglion neurons via the CamK-CREB cascade: A potential mechanism of inflammatory pain. Mol. Biol. Cell 2010, 21, 2568–2577. [Google Scholar]
- Nitsch, S.; Zorro Shahidian, L.; Schneider, R. Histone acetylation and chromatin dynamics: Concepts, challenges, and links to metabolism. EMBO Rep. 2021, 22, e52774. [Google Scholar] [PubMed]
- Vo, N.; Goodman, R.H. CREB-binding protein and p300 in transcriptional regulation. J. Biol. Chem. 2001, 276, 13505–13508. [Google Scholar] [PubMed]
- Kwok, R.P.; Lundblad, J.R.; Chrivia, J.C.; Richards, J.P.; Bächinger, H.P.; Brennan, R.G.; Roberts, S.G.; Green, M.R.; Goodman, R.H. Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature 1994, 370, 223–226. [Google Scholar] [PubMed]
- Silverman, E.S.; Du, J.; Williams, A.J.; Wadgaonkar, R.; Drazen, J.M.; Collins, T. cAMP-response-element-binding-protein-binding protein (CBP) and p300 are transcriptional co-activators of early growth response factor-1 (Egr-1). Biochem. J. 1998, 336, 183–189. [Google Scholar] [PubMed]
- Miller, R.S.; Wolfe, A.; He, L.; Radovick, S.; Wondisford, F.E. CREB binding protein (CBP) activation is required for luteinizing hormone beta expression and normal fertility in mice. Mol. Cell. Biol. 2012, 32, 2349–2358. [Google Scholar]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar]
- Devaiah, B.; Case-Borden, C.; Gegonne, A.; Hsu, C.H.; Chen, Q.; Meerzaman, D.; Dey, A.; Ozato, K.; Singer, S.S. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nat. Struct. Mol. Biol. 2016, 23, 540–548. [Google Scholar]
- Wang, N.; Wu, R.; Tang, D.; Kang, R. The BET family in immunity and disease. Signal Transduct. Target. Ther. 2021, 6, 23. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thiel, G.; Rössler, O.G. Stimulus–Transcription Coupling of TRPM3 Channels: A Signaling Pathway from the Plasma Membrane to the Nucleus. Biomolecules 2025, 15, 521. https://doi.org/10.3390/biom15040521
Thiel G, Rössler OG. Stimulus–Transcription Coupling of TRPM3 Channels: A Signaling Pathway from the Plasma Membrane to the Nucleus. Biomolecules. 2025; 15(4):521. https://doi.org/10.3390/biom15040521
Chicago/Turabian StyleThiel, Gerald, and Oliver G. Rössler. 2025. "Stimulus–Transcription Coupling of TRPM3 Channels: A Signaling Pathway from the Plasma Membrane to the Nucleus" Biomolecules 15, no. 4: 521. https://doi.org/10.3390/biom15040521
APA StyleThiel, G., & Rössler, O. G. (2025). Stimulus–Transcription Coupling of TRPM3 Channels: A Signaling Pathway from the Plasma Membrane to the Nucleus. Biomolecules, 15(4), 521. https://doi.org/10.3390/biom15040521