
Calcium-Sensing Receptor as a Novel Target for the Treatment of Idiopathic Pulmonary Fibrosis
 , , ,
, , ,  , ,
, ,  ,
,  ,
,  , , ,
, , ,  , and add
Show full author list
, and add
Show full author list
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. Human Studies
Flow Infusion Electrospray High-Resolution Mass Spectrometry (FIE-HRMS)
2.2. In Vitro Studies
2.2.1. Cell Culture
2.2.2. HLF Stimulation and Treatment
2.2.3. Immunofluorescence Staining
2.2.4. Quantitative Immunofluorescence Microscopy
2.2.5. Measurements of Intracellular Free Ionized Calcium Concentration ([Ca2+]i)
2.2.6. RNA Sequencing
2.2.7. Polyamine Secretion
Ornithine ELISA
Spermine ELISA
2.3. Statistics
3. Results
3.1. Naturally Occurring Polyamines Are Enriched in the Salivary Metabolome of IPF Patients
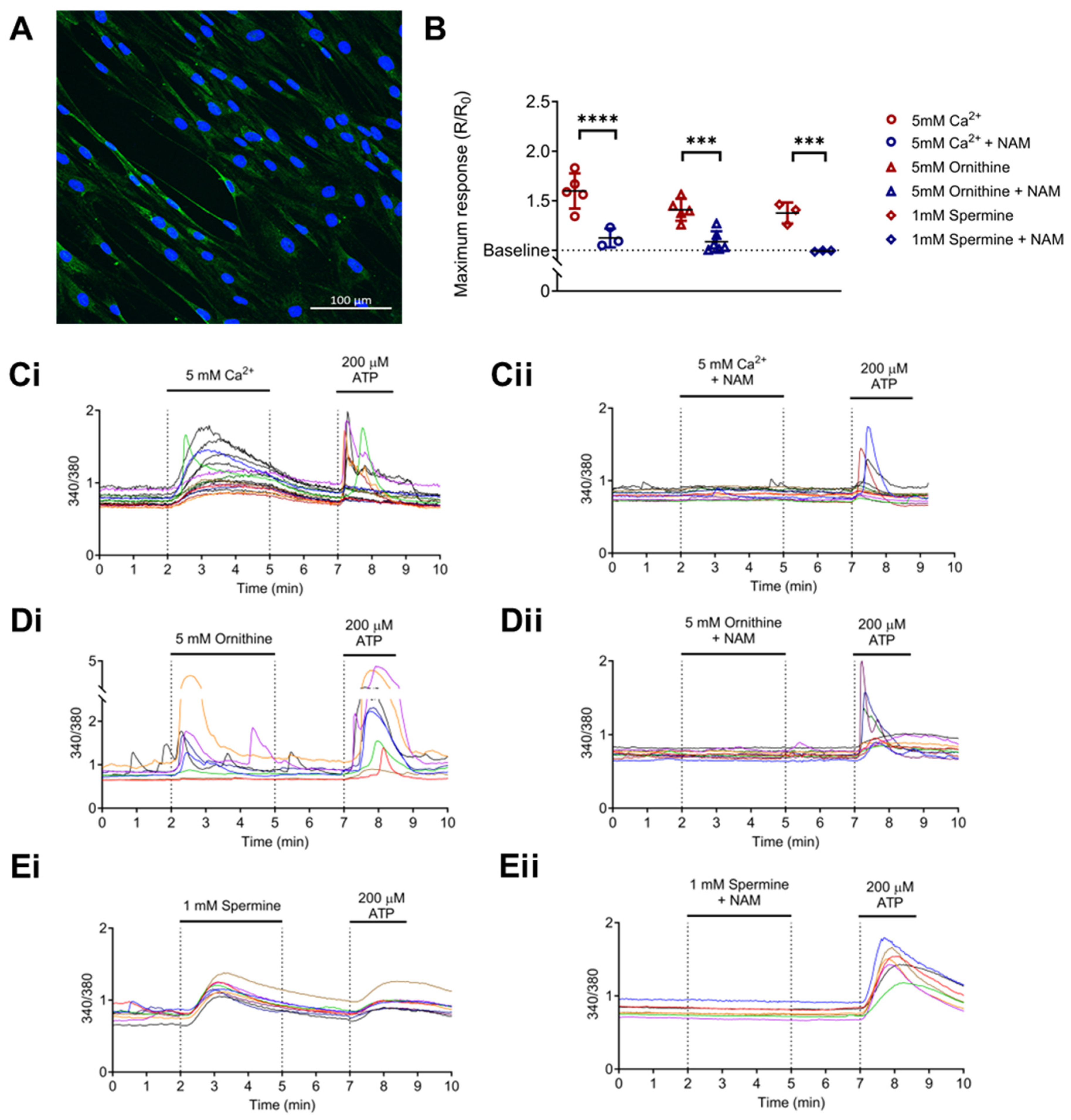
3.2. CaSR Is Functionally Expressed in Normal Human Lung Fibroblasts (NHLFs)
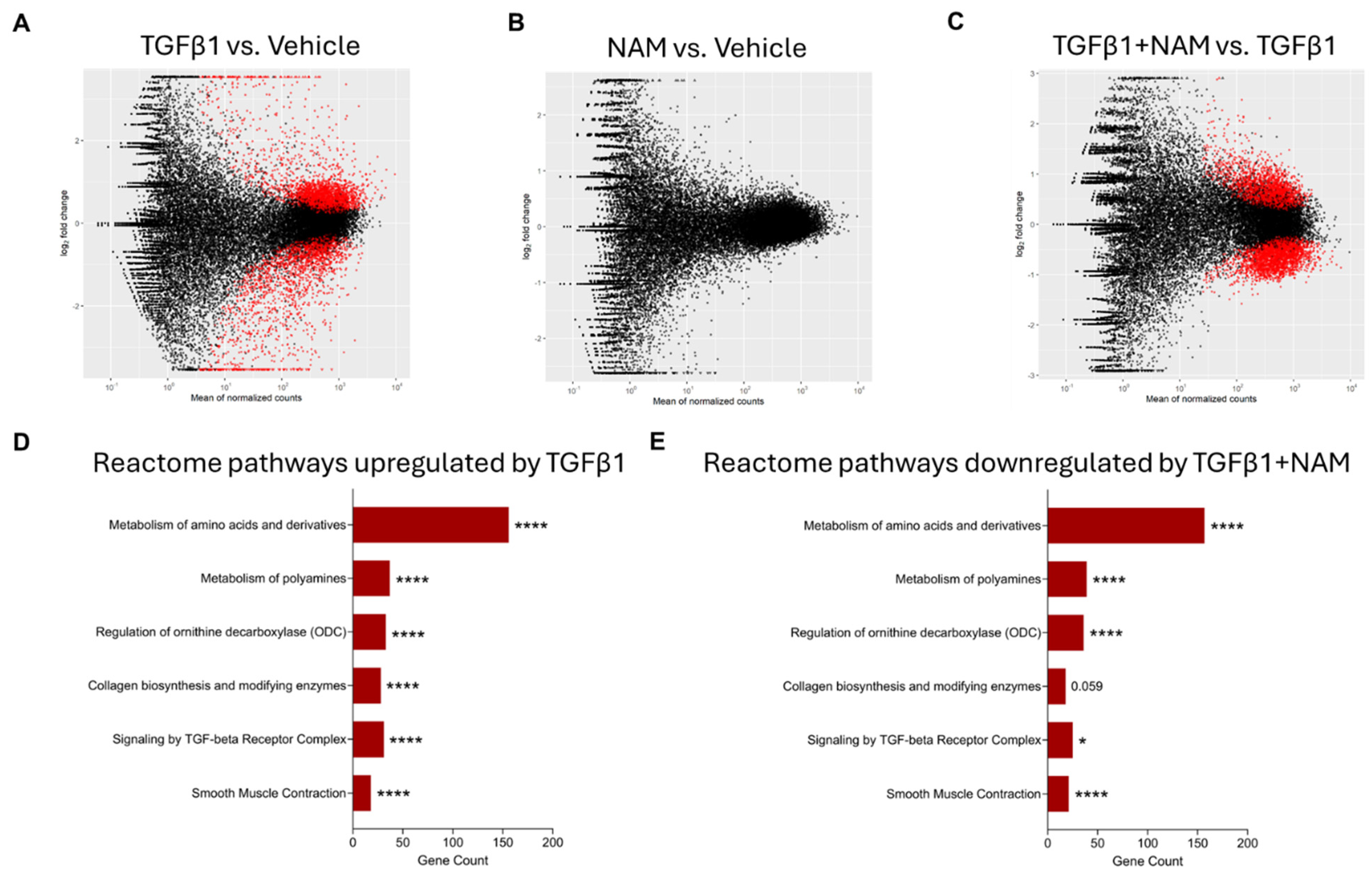
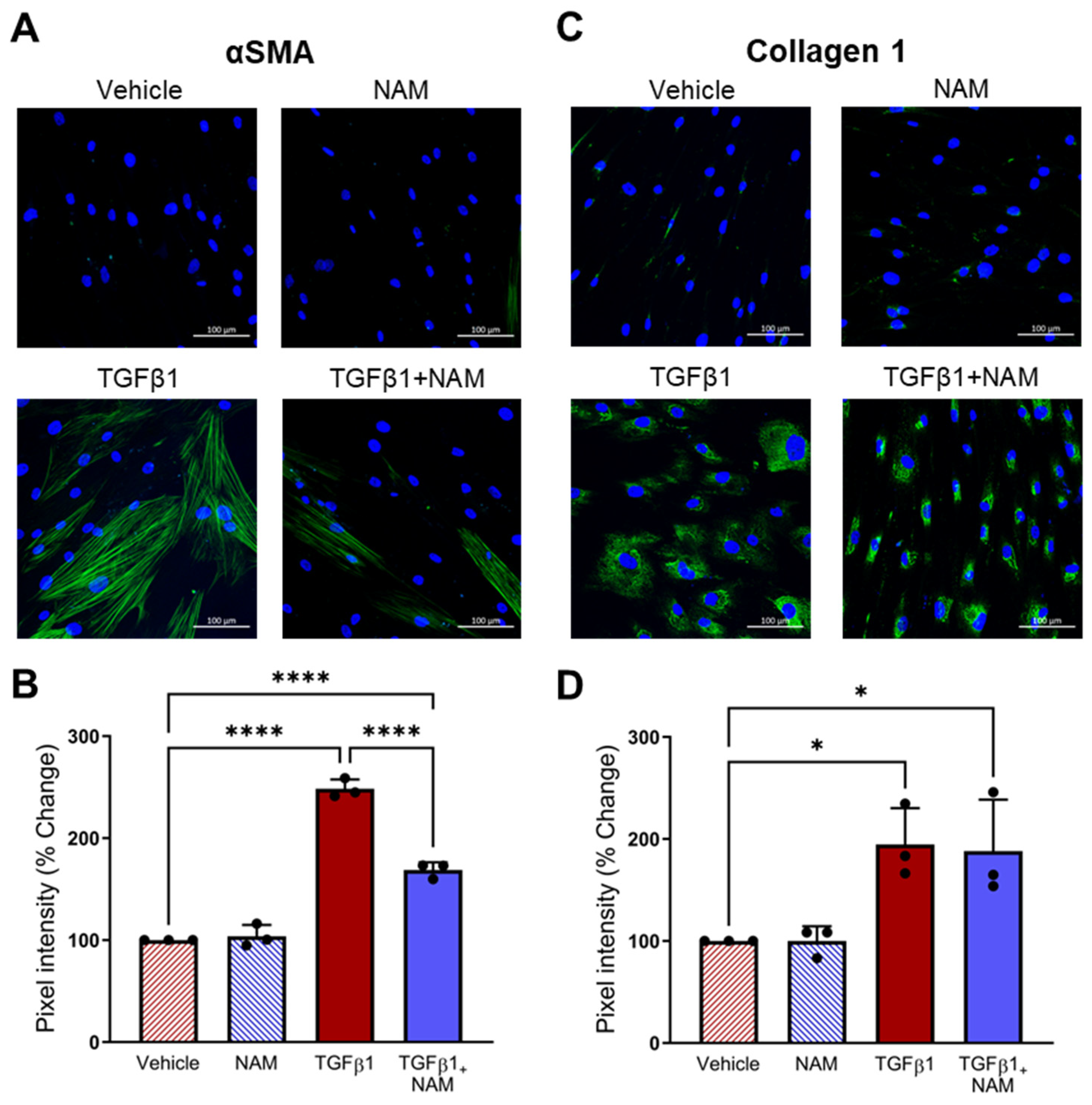
3.3. NHLFs Treated with TGFβ1 Display a Pro-Fibrotic Genetic Profile and Co-Treatment with CaSR NAM Alters TGFβ1-Mediated Changes
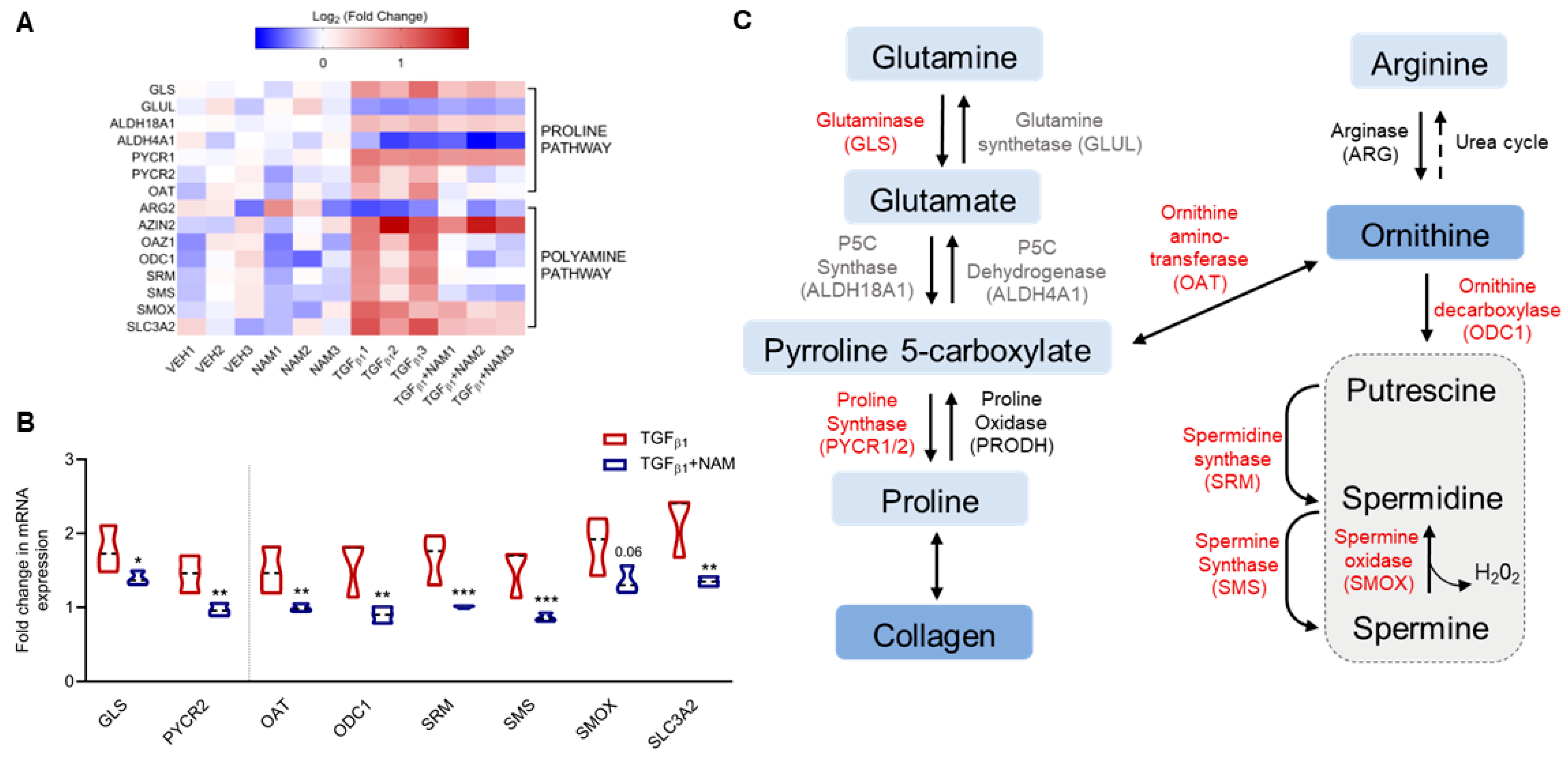
3.4. CaSR Regulates Polyamine Gene Expression in TGFβ1-Activated NHLF
3.5. CaSR Regulates Pro-Fibrotic Gene Expression in TGFβ1-Activated NHLF
3.6. Effects of TGFβ1 on CaSR Expression in Normal and IPF Primary Lung Fibroblasts
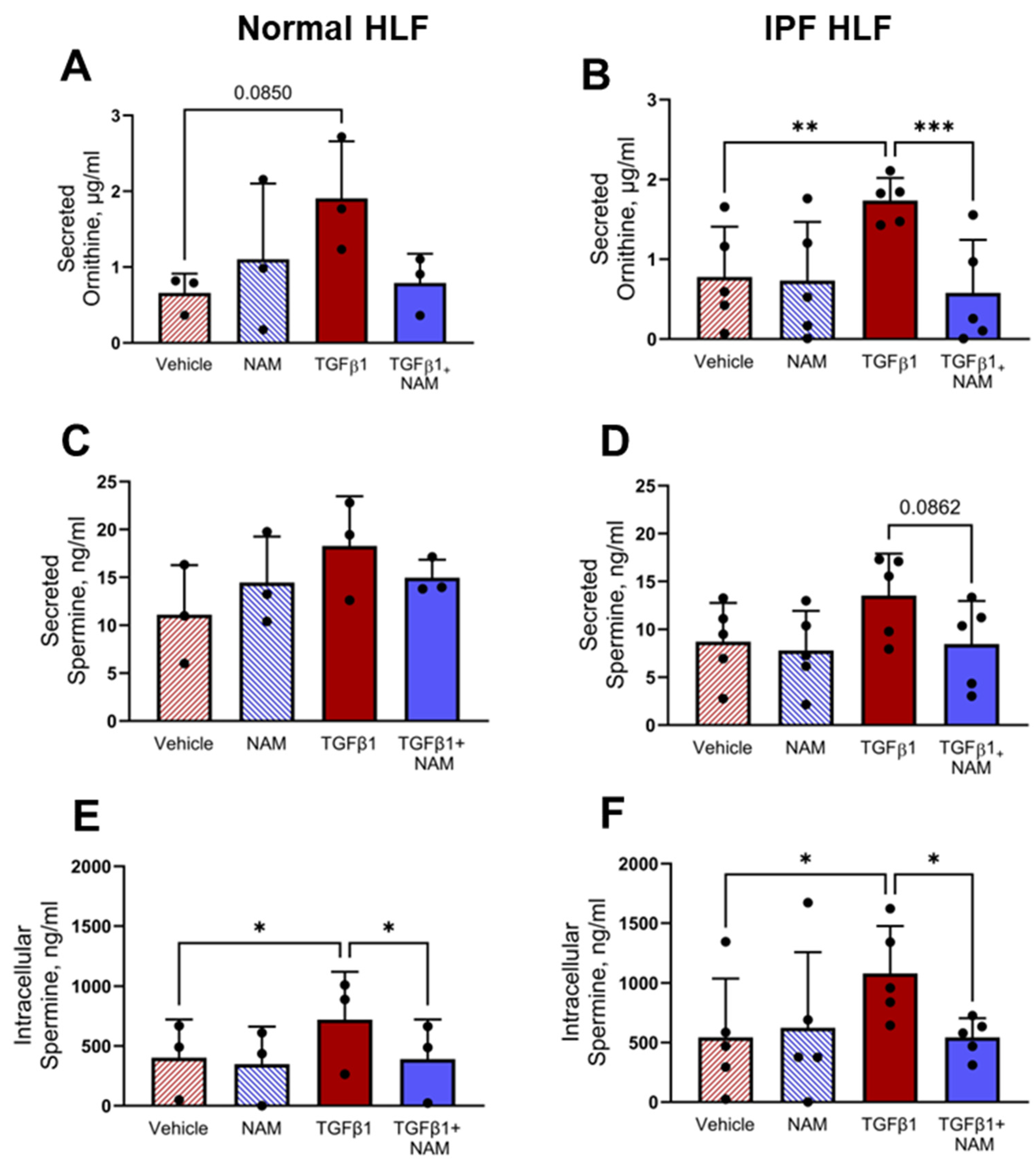
3.7. TGFβ1 Upregulates Polyamine Production in Normal and IPF HLF, and CaSR Antagonism Abrogates These Responses
3.8. CaSR Antagonism Reduces TGFβ1 Pro-Fibrotic Responses in IPF Lung Fibroblasts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adegunsoye, A.; Oldham, J.M.; Bellam, S.K.; Montner, S.; Churpek, M.M.; Noth, I.; Vij, R.; Strek, M.E.; Chung, J.H. Computed Tomography Honeycombing Identifies a Progressive Fibrotic Phenotype with Increased Mortality across Diverse Interstitial Lung Diseases. Ann. Am. Thorac. Soc. 2019, 16, 580–588. [Google Scholar] [CrossRef]
- Vancheri, C.; Failla, M.; Crimi, N.; Raghu, G. Idiopathic Pulmonary Fibrosis: A Disease with Similarities and Links to Cancer Biology. Eur. Respir. J. 2010, 35, 496–504. [Google Scholar] [CrossRef]
- Somogyi, V.; Chaudhuri, N.; Torrisi, S.E.; Kahn, N.; Müller, V.; Kreuter, M. The Therapy of Idiopathic Pulmonary Fibrosis: What Is Next? Eur. Respir. Rev. 2019, 28, 190021. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G. Pharmacotherapy for Idiopathic Pulmonary Fibrosis: Current Landscape and Future Potential. Eur. Respir. Rev. 2017, 26, 170071. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Rochwerg, B.; Zhang, Y.; Garcia, C.A.C.; Azuma, A.; Behr, J.; Brozek, J.L.; Collard, H.R.; Cunningham, W.; Homma, S.; et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2015, 192, e3–e19. [Google Scholar] [CrossRef] [PubMed]
- Tarride, J.-E.; Hopkins, R.B.; Burke, N.; Guertin, J.R.; O’Reilly, D.; Fell, C.D.; Dion, G.; Kolb, M. Clinical and Economic Burden of Idiopathic Pulmonary Fibrosis in Quebec, Canada. Clin. Outcomes Res. 2018, 10, 127–137. [Google Scholar] [CrossRef]
- Van Manen, M.J.G.; Geelhoed, J.J.M.; Tak, N.C.; Wijsenbeek, M.S. Optimizing Quality of Life in Patients with Idiopathic Pulmonary Fibrosis. Ther. Adv. Respir. Dis. 2017, 11, 157–169. [Google Scholar] [CrossRef]
- Chianese, M.; Screm, G.; Salton, F.; Confalonieri, P.; Trotta, L.; Barbieri, M.; Ruggero, L.; Mari, M.; Reccardini, N.; Geri, P.; et al. Pirfenidone and Nintedanib in Pulmonary Fibrosis: Lights and Shadows. Pharmaceuticals 2024, 17, 709. [Google Scholar] [CrossRef]
- Kou, M.; Jiao, Y.; Li, Z.; Wei, B.; Li, Y.; Cai, Y.; Wei, W. Real-World Safety and Effectiveness of Pirfenidone and Nintedanib in the Treatment of Idiopathic Pulmonary Fibrosis: A Systematic Review and Meta-Analysis. Eur. J. Clin. Pharmacol. 2024, 80, 1445–1460. [Google Scholar] [CrossRef]
- Rajagopal, K.; Bryant, A.J.; Sahay, S.; Wareing, N.; Zhou, Y.; Pandit, L.M.; Karmouty-Quintana, H. Idiopathic Pulmonary Fibrosis and Pulmonary Hypertension: Heracles Meets the Hydra. Br. J. Pharmacol. 2021, 178, 172–186. [Google Scholar] [CrossRef]
- Haak, A.J.; Ducharme, M.T.; Diaz Espinosa, A.M.; Tschumperlin, D.J. Targeting GPCR Signaling for Idiopathic Pulmonary Fibrosis Therapies. Trends Pharmacol. Sci. 2020, 41, 172–182. [Google Scholar] [CrossRef]
- Mikolasch, T.A.; Garthwaite, H.S.; Porter, J.C. Update in Diagnosis and Management of Interstitial Lung Disease. Clin. Med. 2017, 17, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Hewlett, J.C.; Kropski, J.A.; Blackwell, T.S. Idiopathic Pulmonary Fibrosis: Epithelial-Mesenchymal Interactions and Emerging Therapeutic Targets. Matrix Biol. 2018, 71–72, 112. [Google Scholar] [CrossRef]
- Pardo, A.; Selman, M. The Interplay of the Genetic Architecture, Aging, and Environmental Factors in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 64, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Roque, W.; Romero, F. Cellular Metabolomics of Pulmonary Fibrosis, from Amino Acids to Lipids. Am. J. Physiol. Physiol. 2021, 320, C689–C695. [Google Scholar] [CrossRef] [PubMed]
- Bernard, K.; Logsdon, N.J.; Benavides, G.A.; Sanders, Y.; Zhang, J.; Darley-Usmar, V.M.; Thannickal, V.J. Glutaminolysis Is Required for Transforming Growth Factor-Β1–Induced Myofibroblast Differentiation and Activation. J. Biol. Chem. 2018, 293, 1218–1228. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; O’Leary, E.M.; Witt, L.J.; Tian, Y.; Gökalp, G.A.; Meliton, A.Y.; Dulin, N.O.; Mutlu, G.M. Glutamine Metabolism Is Required for Collagen Protein Synthesis in Lung Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2019, 61, 597–606. [Google Scholar] [CrossRef]
- Zhao, Y.D.; Yin, L.; Archer, S.; Lu, C.; Zhao, G.; Yao, Y.; Wu, L.; Hsin, M.; Waddell, T.K.; Keshavjee, S.; et al. Metabolic Heterogeneity of Idiopathic Pulmonary Fibrosis: A Metabolomic Study. BMJ Open Respir. Res. 2017, 4, e000183. [Google Scholar] [CrossRef]
- Kitowska, K.; Zakrzewicz, D.; Königshoff, M.; Chrobak, I.; Grimminger, F.; Seeger, W.; Bulau, P.; Eickelberg, O. Functional Role and Species-Specific Contribution of Arginases in Pulmonary Fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L34–L45. [Google Scholar] [CrossRef]
- Endo, M.; Oyadomari, S.; Terasaki, Y.; Takeya, M.; Suga, M.; Mori, M.; Gotoh, T. Induction of Arginase I and II in Bleomycin-Induced Fibrosis of Mouse Lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 285, L313–L321. [Google Scholar] [CrossRef]
- Komuves, L.; Oda, Y.; Tu, C.L.; Chang, W.H.; Ho-Pao, C.L.; Mauro, T.; Bikle, D.D. Epidermal Expression of the Full-Length Extracellular Calcium-Sensing Receptor Is Required for Normal Keratinocyte Differentiation. J. Cell Physiol. 2002, 192, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Schepelmann, M.; Yarova, P.L.; Lopez-Fernandez, I.; Davies, T.S.; Brennan, S.C.; Edwards, P.J.; Aggarwal, A.; Graça, J.; Rietdorf, K.; Matchkov, V.; et al. The Vascular Ca2+-Sensing Receptor Regulates Blood Vessel Tone and Blood Pressure. Am. J. Physiol. Cell Physiol. 2016, 310, C193–C204. [Google Scholar] [CrossRef] [PubMed]
- Sundararaman, S.S.; van der Vorst, E.P.C. Calcium-Sensing Receptor (Casr), Its Impact on Inflammation and the Consequences on Cardiovascular Health. Int. J. Mol. Sci. 2021, 22, 2478. [Google Scholar] [CrossRef] [PubMed]
- Yarova, P.L.; Stewart, A.L.; Sathish, V.; Britt, R.D.; Thompson, M.A.; P Lowe, A.P.; Freeman, M.; Aravamudan, B.; Kita, H.; Brennan, S.C.; et al. Calcium-Sensing Receptor Antagonists Abrogate Airway Hyperresponsiveness and Inflammation in Allergic Asthma. Sci. Transl. Med. 2015, 7, 284ra60. [Google Scholar] [CrossRef]
- Brennan, S.C.; Wilkinson, W.J.; Tseng, H.-E.; Finney, B.; Monk, B.; Dibble, H.; Quilliam, S.; Warburton, D.; Galietta, L.J.; Kemp, P.J.; et al. The Extracellular Calcium-Sensing Receptor Regulates Human Fetal Lung Development via CFTR. Sci. Rep. 2016, 6, 21975. [Google Scholar] [CrossRef]
- Finney, B.A.; Del Moral, P.M.; Wilkinson, W.J.; Cayzac, S.; Cole, M.; Warburton, D.; Kemp, P.J.; Riccardi, D. Regulation of Mouse Lung Development by the Extracellular Calcium-Sensing Receptor, CaR. J. Physiol. 2008, 586, 6007–6019. [Google Scholar] [CrossRef]
- Riccardi, D.; Brennan, S.C.; Chang, W. The Extracellular Calcium-Sensing Receptor, CaSR, in Fetal Development. Best. Pr. Res. Clin. Endocrinol. Metab. 2013, 27, 443–453. [Google Scholar] [CrossRef]
- Brown, E.M.; MacLeod, R.J. Extracellular Calcium Sensing and Extracellular Calcium Signaling. Physiol. Rev. 2001, 81, 239–297. [Google Scholar] [CrossRef]
- Quinn, S.J.; Ye, C.-P.; Diaz, R.; Kifor, O.; Bai, M.; Vassilev, P.; Brown, E. The Ca2+-sensing receptor: A target for polyamines. Am. J. Physiol. 1997, 273, C1315–C1323. [Google Scholar] [CrossRef] [PubMed]
- Hoet, P.H.M.; Nemery, B. Polyamines in the Lung: Polyamine Uptake and Polyamine-Linked Pathological or Toxicological Conditions. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 278, L417–L433. [Google Scholar] [CrossRef]
- Davies, S.L.; Gibbons, C.E.; Vizard, T.; Ward, D.T. Ca2+ -Sensing Receptor Induces Rho Kinase-Mediated Actin Stress Fiber Assembly and Altered Cell Morphology, but Not in Response to Aromatic Amino Acids. Am. J. Physiol. Cell Physiol. 2006, 290, C1543–C1551. [Google Scholar] [CrossRef] [PubMed]
- Rybchyn, M.S.; Slater, M.; Conigrave, A.D.; Mason, R.S. An Akt-Dependent Increase in Canonical Wnt Signaling and a Decrease in Sclerostin Protein Levels Are Involved in Strontium Ranelate-Induced Osteogenic Effects in Human Osteoblasts. J. Biol. Chem. 2011, 286, 23771–23779. [Google Scholar] [CrossRef]
- Zhao, W.; Zhang, Z.; Zheng, L.; You, C.; Chi, H.; Zhang, T.; Xu, G. Calcium-Sensing Receptor Activating ERK1/2 and PI3K-Akt Pathways to Induce the Proliferation of Osteosarcoma Cells. Clin. Exp. Pharmacol. Physiol. 2020, 47, 517–519. [Google Scholar] [CrossRef]
- Yamamura, A.; Guo, Q.; Yamamura, H.; Zimnicka, A.M.; Pohl, N.M.; Smith, K.A.; Fernandez, R.A.; Zeifman, A.; Makino, A.; Dong, H.; et al. Enhanced Ca(2+)-Sensing Receptor Function in Idiopathic Pulmonary Arterial Hypertension. Circ. Res. 2012, 111, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Yarova, P.; Davies, C.; Price, S.A.; Huang, Q.; Graca, J.A.; Maleki-Toyserkani, S.; Lowe, A.P.P.; Kidd, E.J.; Ford, W.R.; Broadley, K.J.; et al. Inhaled Calcilytics: Effects on Airway Inflammation and Remodeling. Respir. Drug Deliv. 2016, 1, 1–12. [Google Scholar]
- Buendía-Roldán, I.; Mejía, M.; Navarro, C.; Selman, M. Idiopathic Pulmonary Fibrosis: Clinical Behavior and Aging Associated Comorbidities. Respir. Med. 2017, 129, 46–52. [Google Scholar] [CrossRef]
- Ruffenach, G.; Hong, J.; Vaillancourt, M.; Medzikovic, L.; Eghbali, M. Pulmonary Hypertension Secondary to Pulmonary Fibrosis: Clinical Data, Histopathology and Molecular Insights. Respir. Res. 2020, 21, 303. [Google Scholar] [CrossRef]
- de Araújo, R.P.; Bertoni, N.; Seneda, A.L.; Felix, T.F.; Carvalho, M.; Lewis, K.E.; Hasimoto, É.N.; Beckmann, M.; Drigo, S.A.; Reis, P.P.; et al. Defining Metabolic Rewiring in Lung Squamous Cell Carcinoma. Metabolites 2019, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, U.; Llamazares Prada, M.; Pohl, S.T.; Richter, M.; Tamas, R.; Schuler, M.; Keller, C.; Mijosek, V.; Muley, T.; Schneider, M.A.; et al. High-resolution Transcriptomic and Epigenetic Profiling Identifies Novel Regulators of COPD. EMBO J. 2023, 42, e111272. [Google Scholar] [CrossRef]
- Llamazares-Prada, M.; Espinet, E.; Mijošek, V.; Schwartz, U.; Lutsik, P.; Tamas, R.; Richter, M.; Behrendt, A.; Pohl, S.T.; Benz, N.P.; et al. Versatile Workflow for Cell Type-Resolved Transcriptional and Epigenetic Profiles from Cryopreserved Human Lung. JCI Insight 2021, 6, e140443. [Google Scholar] [CrossRef]
- Andrews, S. Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 2 November 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic. Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and Accurate Filtering of Ribosomal RNAs in Metatranscriptomic Data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Varet, H.; Brillet-Guéguen, L.; Coppée, J.Y.; Dillies, M.A. SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Yu, G. Thirteen Years of ClusterProfiler. Innovation 2024, 5, 100722. [Google Scholar] [CrossRef]
- Xu, S.; Hu, E.; Cai, Y.; Xie, Z.; Luo, X.; Zhan, L.; Tang, W.; Wang, Q.; Liu, B.; Wang, R.; et al. Using ClusterProfiler to Characterize Multiomics Data. Nat. Protoc. 2024, 19, 3292–3320. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. ClusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; He, Q.Y. ReactomePA: An R/Bioconductor Package for Reactome Pathway Analysis and Visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar] [CrossRef]
- Kolde, R. Pheatmap: Pretty Heatmaps. Available online: https://github.com/raivokolde/pheatmap (accessed on 13 February 2025).
- R Core Team R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 22 November 2021).
- Janssen, L.J.; Mukherjee, S.; Ask, K. Calcium Homeostasis and Ionic Mechanisms in Pulmonary Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2015, 53, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Transforming Growth Factor–β in Tissue Fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Mutlu, G.M. Metabolic Requirements of Pulmonary Fibrosis: Role of Fibroblast Metabolism. FEBS J. 2021, 288, 6331–6352. [Google Scholar] [CrossRef]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.K.; Jang, H.G.; Jha, A.K.; et al. Functional Genomics Reveal That the Serine Synthesis Pathway Is Essential in Breast Cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Fear, M.W.; Suk Choi, Y.; Wood, F.M.; Allahham, A.; Mutsaers, S.E.; Prêle, C.M. The Extracellular Matrix and Mechanotransduction in Pulmonary Fibrosis. Int. J. Biochem. Cell Biol. 2020, 126, 105802. [Google Scholar] [CrossRef]
- D’Urso, M.; Kurniawan, N.A. Mechanical and Physical Regulation of Fibroblast-Myofibroblast Transition: From Cellular Mechanoresponse to Tissue Pathology. Front. Bioeng. Biotechnol. 2020, 8, 609653. [Google Scholar] [CrossRef]
- Zimmermann, N.; Rothenberg, M.E. The Arginine-Arginase Balance in Asthma and Lung Inflammation. Eur. J. Pharmacol. 2006, 533, 253–262. [Google Scholar] [CrossRef]
- Hasan, C.M.; Pottenger, S.; Green, A.E.; Cox, A.A.; White, J.S.; Jones, T.; Winstanley, C.; Kadioglu, A.; Wright, M.H.; Neill, D.R.; et al. Pseudomonas Aeruginosa Utilizes the Host-Derived Polyamine Spermidine to Facilitate Antimicrobial Tolerance. JCI Insight 2022, 7, e158879. [Google Scholar] [CrossRef]
- Grasemann, H.; Dhaliwal, R.; Ivanovska, J.; Kantores, C.; McNamara, P.J.; Scott, J.A.; Belik, J.; Jankov, R.P. Arginase Inhibition Prevents Bleomycin-Induced Pulmonary Hypertension, Vascular Remodeling, and Collagen Deposition in Neonatal Rat Lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L503–L510. [Google Scholar] [CrossRef]
- He, Y.Y.; Yan, Y.; Jiang, X.; Zhao, J.H.; Wang, Z.; Wu, T.; Wang, Y.; Guo, S.S.; Ye, J.; Lian, T.Y.; et al. Spermine Promotes Pulmonary Vascular Remodelling and Its Synthase Is a Therapeutic Target for Pulmonary Arterial Hypertension. Eur. Respir. J. 2020, 56, 2000522. [Google Scholar] [CrossRef] [PubMed]
- Firpo, M.R.; Mounce, B.C. Diverse Functions of Polyamines in Virus Infection. Biomolecules 2020, 10, 628. [Google Scholar] [CrossRef] [PubMed]
- Summer, R.; Todd, J.L.; Neely, M.L.; Lobo, L.J.; Namen, A.; Newby, L.K.; Shafazand, S.; Suliman, S.; Hesslinger, C.; Keller, S.; et al. Circulating Metabolic Profile in Idiopathic Pulmonary Fibrosis: Data from the IPF-PRO Registry. Respir. Res. 2024, 25, 58. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, A. Advances in G Protein-Coupled Receptor Allostery: From Function to Structure. Mol. Pharmacol. 2014, 86, 463–478. [Google Scholar] [CrossRef]
- Minois, N.; Carmona-Gutierrez, D.; Madeo, F. Polyamines in Aging and Disease. Aging 2011, 3, 716–732. [Google Scholar] [CrossRef]
- Zhu, L.; Xiao, R.; Zhang, X.; Lang, Y.; Liu, F.; Yu, Z.; Zhang, J.; Su, Y.; Lu, Y.; Wang, T.; et al. Spermine on Endothelial Extracellular Vesicles Mediates Smoking-Induced Pulmonary Hypertension Partially through Calcium-Sensing Receptor. Arter. Thromb. Vasc. Biol. 2019, 39, 482–495. [Google Scholar] [CrossRef]
- Zhou, Y.; Frey, T.K.; Yang, J.J. Viral Calciomics: Interplays between Ca2+ and Virus. Cell Calcium 2009, 46, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, M.L.; Campilongo, R.; Casalino, M.; Micheli, G.; Colonna, B.; Prosseda, G. Polyamines: Emerging Players in Bacteria–Host Interactions. Int. J. Med. Microbiol. 2013, 303, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Sheng, G.; Chen, P.; Wei, Y.; Yue, H.; Chu, J.; Zhao, J.; Wang, Y.; Zhang, W.; Zhang, H.L. Viral Infection Increases the Risk of Idiopathic Pulmonary Fibrosis: A Meta-Analysis. Chest 2020, 157, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Pulkkinen, V.; Salmenkivi, K.; Kinnula, V.L.; Sutinen, E.; Halme, M.; Hodgson, U.; Lehto, J.; Jääskelinen, A.; Piiparinen, H.; Kere, J.; et al. A Novel Screening Method Detects Herpesviral DNA in the Idiopathic Pulmonary Fibrosis Lung. Ann. Med. 2012, 44, 178–186. [Google Scholar] [CrossRef]
- Egan, J.J.; Woodcock, A.A.; Stewart, J.P. Viruses and Idiopathic Pulmonary Fibrosis. Eur. Respir. J. 1997, 10, 1433–1437. [Google Scholar] [CrossRef]
- Bharadwaj, S.; Singh, M.; Kirtipal, N.; Kang, S.G. SARS-CoV-2 and Glutamine: SARS-CoV-2 Triggered Pathogenesis via Metabolic Reprograming of Glutamine in Host Cells. Front. Mol. Biosci. 2021, 7, 627842. [Google Scholar] [CrossRef]
- Xu, T.; Liu, C.; Ning, X.; Gao, Z.; Li, A.; Wang, S.; Leng, L.; Kong, P.; Liu, P.; Zhang, S.; et al. Causal Relationship between Circulating Glutamine Levels and Idiopathic Pulmonary Fibrosis: A Two-Sample Mendelian Randomization Study. BMC Pulm. Med. 2024, 24, 451. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, J.; Sun, H.; Zhang, Y.; Zou, D. New Insights into Fibrosis from the ECM Degradation Perspective: The Macrophage-MMP-ECM Interaction. Cell Biosci. 2022, 12, 117. [Google Scholar] [CrossRef]
- Upagupta, C.; Shimbori, C.; Alsilmi, R.; Kolb, M. Matrix Abnormalities in Pulmonary Fibrosis. Eur. Respir. Rev. 2018, 27, 180033. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Eickelberg, O. The Impact of TGF-β on Lung Fibrosis: From Targeting to Biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Pardo, A.; Cabrera, S.; Maldonado, M.; Selman, M. Role of Matrix Metalloproteinases in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Respir. Res. 2016, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Pardo, A.; Selman, M.; Kaminski, N. Approaching the Degradome in Idiopathic Pulmonary Fibrosis. Int. J. Biochem. Cell Biol. 2008, 40, 1141–1155. [Google Scholar] [CrossRef]
- Pardo, A.; Selman, M. Matrix Metalloproteases in Aberrant Fibrotic Tissue Remodeling. Proc. Am. Thorac. Soc. 2006, 3, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Manicone, A.M.; McGuire, J.K. Matrix Metalloproteinases as Modulators of Inflammation. Semin. Cell Dev. Biol. 2008, 19, 34–41. [Google Scholar] [CrossRef]
- Limb, G.A.; Matter, K.; Murphy, G.; Cambrey, A.D.; Bishop, P.N.; Morris, G.E.; Khaw, P.T. Matrix Metalloproteinase-1 Associates with Intracellular Organelles and Confers Resistance to Lamin A/C Degradation during Apoptosis. Am. J. Pathol. 2005, 166, 1555–1563. [Google Scholar] [CrossRef]
- Selman, M.; Ruiz, V.; Cabrera, S.; Segura, L.; Ramírez, R.; Barrios, R.; Pardo, A. TIMP-1, -2, -3, and -4 in Idiopathic Pulmonary Fibrosis. A Prevailing Nondegradative Lung Microenvironment? Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L562–L574. [Google Scholar] [CrossRef]
- Bergeron, S.; Lemieux, E.; Durand, V.; Cagnol, S.; Carrier, J.C.; Lussier, J.G.; Boucher, M.J.; Rivard, N. The Serine Protease Inhibitor SerpinE2 Is a Novel Target of ERK Signaling Involved in Human Colorectal Tumorigenesis. Mol. Cancer 2010, 9, 271. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wolffs, K.; Li, R.; Mansfield, B.; Pass, D.A.; Bruce, R.T.; Huang, P.; de Araújo, R.P.; Haddadi, B.S.; Mur, L.A.J.; Dally, J.; et al. Calcium-Sensing Receptor as a Novel Target for the Treatment of Idiopathic Pulmonary Fibrosis. Biomolecules 2025, 15, 509. https://doi.org/10.3390/biom15040509
Wolffs K, Li R, Mansfield B, Pass DA, Bruce RT, Huang P, de Araújo RP, Haddadi BS, Mur LAJ, Dally J, et al. Calcium-Sensing Receptor as a Novel Target for the Treatment of Idiopathic Pulmonary Fibrosis. Biomolecules. 2025; 15(4):509. https://doi.org/10.3390/biom15040509
Chicago/Turabian StyleWolffs, Kasope, Renjiao Li, Bethan Mansfield, Daniel A. Pass, Richard T. Bruce, Ping Huang, Rachel Paes de Araújo, Bahareh Sadat Haddadi, Luis A. J. Mur, Jordanna Dally, and et al. 2025. "Calcium-Sensing Receptor as a Novel Target for the Treatment of Idiopathic Pulmonary Fibrosis" Biomolecules 15, no. 4: 509. https://doi.org/10.3390/biom15040509
APA StyleWolffs, K., Li, R., Mansfield, B., Pass, D. A., Bruce, R. T., Huang, P., de Araújo, R. P., Haddadi, B. S., Mur, L. A. J., Dally, J., Moseley, R., Ecker, R., Karmouty-Quintana, H., Lewis, K. E., Simpson, A. J., Ward, J. P. T., Corrigan, C. J., Jurkowska, R. Z., Hope-Gill, B. D., ... Yarova, P. L. (2025). Calcium-Sensing Receptor as a Novel Target for the Treatment of Idiopathic Pulmonary Fibrosis. Biomolecules, 15(4), 509. https://doi.org/10.3390/biom15040509