

Integration of DNA Methylome and Transcriptome Analysis to Identify Novel Epigenetic Targets in the Acute Kidney Injury–Chronic Kidney Disease Transition

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Models and Experiment Design
2.2. Serum Creatinine and Blood Urea Nitrogen Tests
2.3. Histopathological Examination
2.4. Whole-Genome Bisulfite Sequencing (WGBS)
2.5. Data Processing and Differential DNA Methylation Analysis
2.6. Identification of Differentially Methylated Regions
2.7. DMR Annotation and Enrichment Analysis
2.8. RNA Sequencing and Data Processing
2.9. Detection of mRNA
2.10. Bisulfite Pyrosequencing
2.11. Statistical Analysis
3. Results
3.1. Establishment of AKI-CKD Animal Model
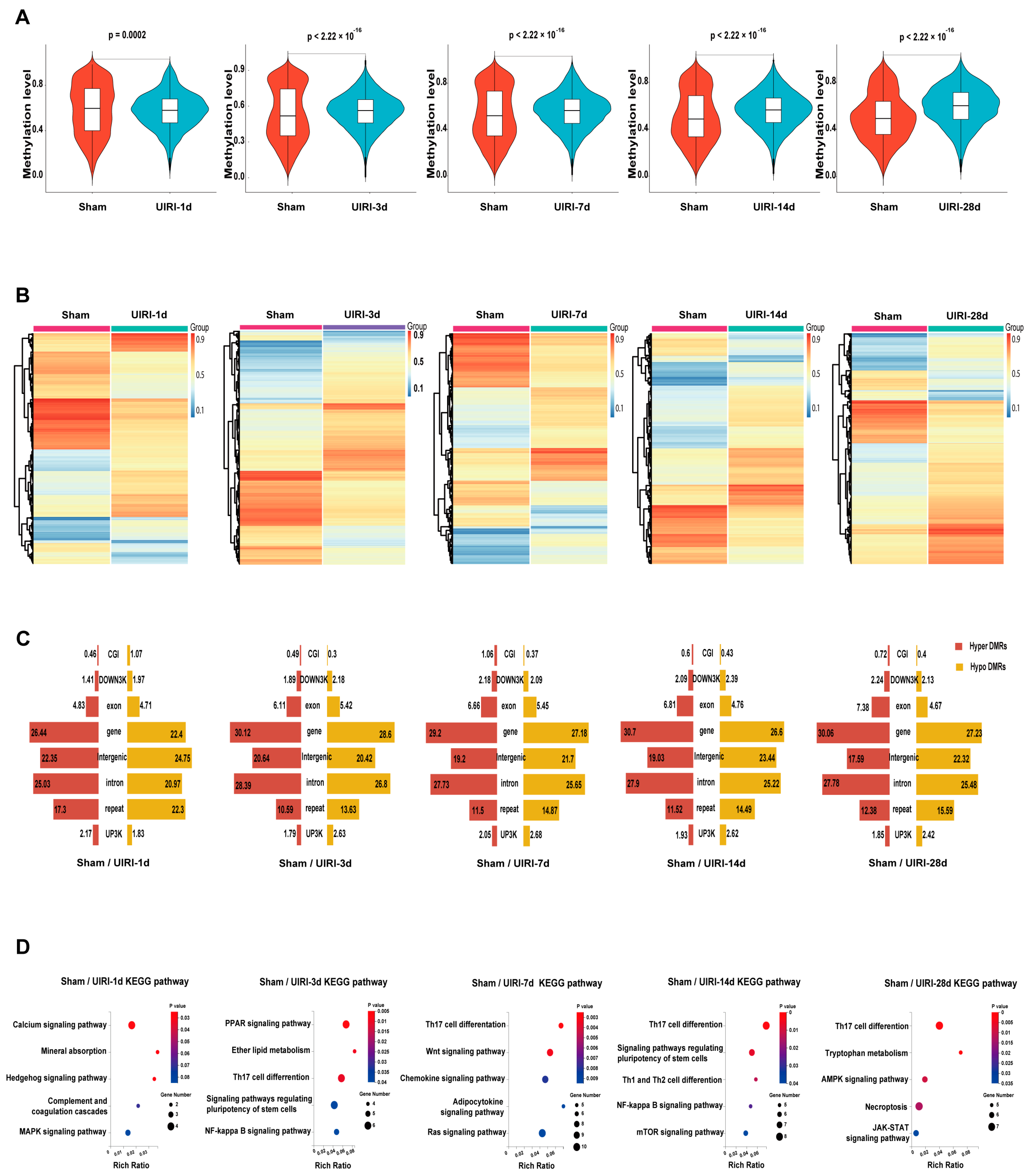
3.2. The Overall Landscape of DNA Methylation in the AKI-CKD Transition
3.3. DMR Landscape During AKI-CKD Progression
3.4. Integrated Analysis of DNA Methylome and Transcriptome to Identify Methylation-Driven Genes in AKI-CKD Transition
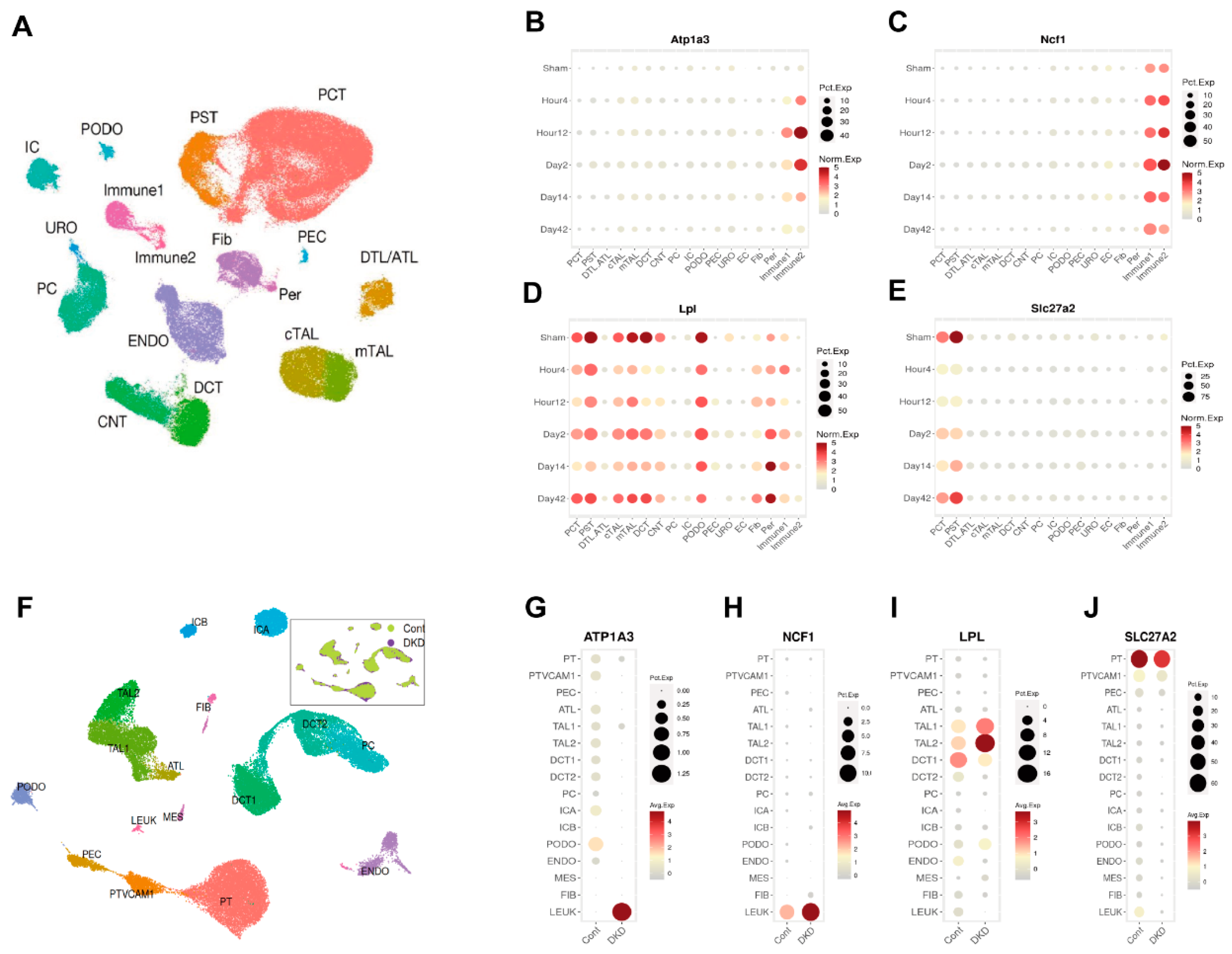
3.5. Identification of Atp1a3, Ncf1, Lpl, and Slc27a2 as Key Candidates in AKI-CKD Progression
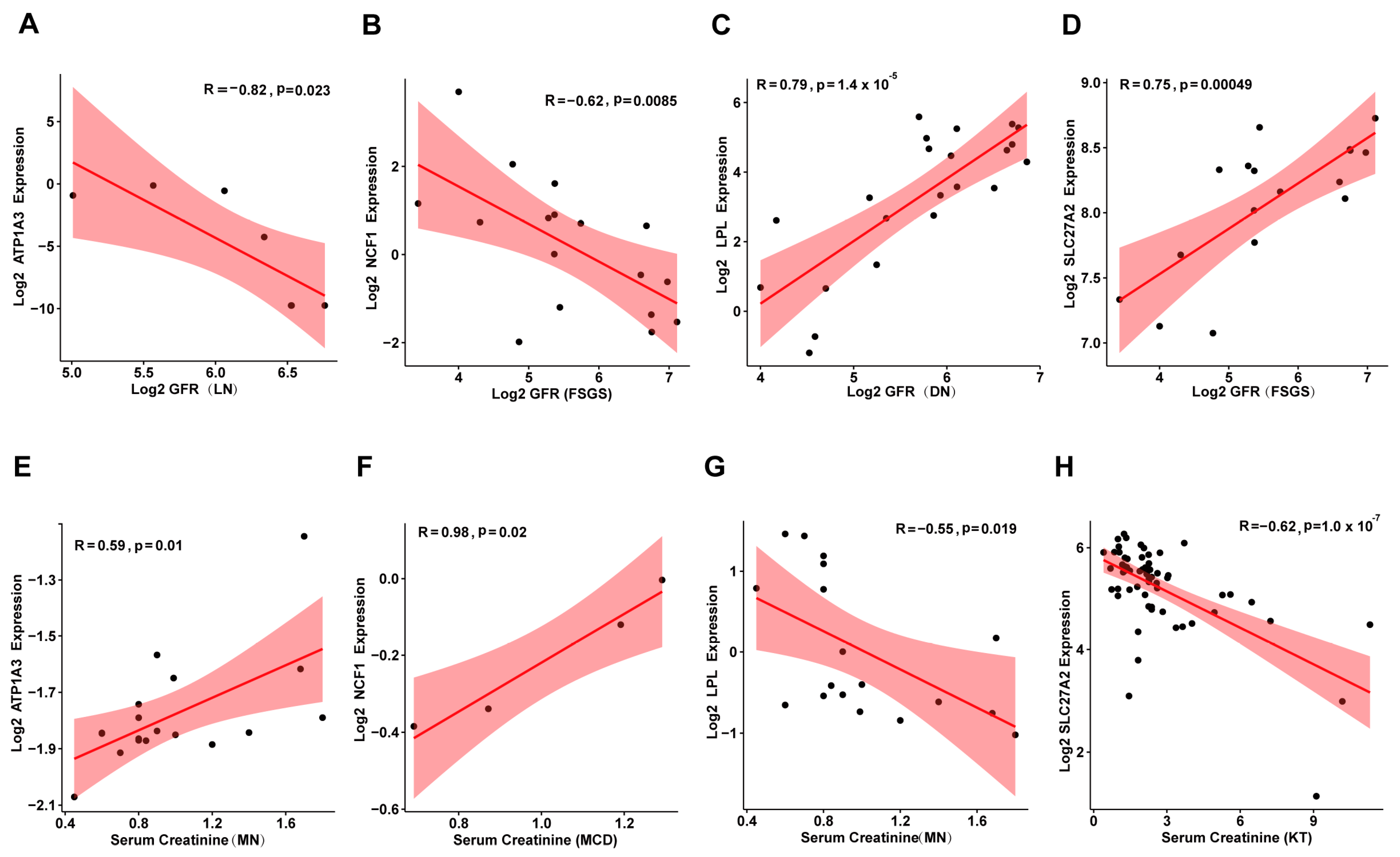
3.6. Correlation Between Candidate Genes and Kidney Disease Prognosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 5-Aza-dC | 5-Aza-2′-deoxycytidine |
| AKI | Acute Kidney Injury |
| ATL | Ascending Thin Limb |
| ATL | Ascending Thin Limb |
| Atp1a3 | ATPase Na+/K+ transporting subunit alpha 3 |
| BSP | Bisulfite Pyrosequencing |
| BUN | Blood Urea Nitrogen |
| cATL | Cortical Ascending Thin Limb |
| CG | Cytosine-Guanine dinucleotide |
| CHG | Cytosine-Homologous base-Guanine dinucleotide |
| CHH | Cytosine-Homologous base-Homologous base dinucleotide |
| CKD | Chronic Kidney Disease |
| CNT | Connecting Tubule |
| CpG | Cytosine-phosphate-Guanine |
| CTP1A | Carnitine palmitoyltransferase 1A |
| DCT | Distal Convoluted Tubule |
| DEGs | Differentially Expressed Genes |
| DKD | Diabetic Kidney Disease |
| DMRs | Differentially Methylated Regions |
| DTL | Descending Thin Limb |
| DTL | Descending Thin Limb |
| EC | Endothelial Cells |
| ENDO | Endothelial Cells |
| Fib | Fibroblasts |
| GFR | Glomerular Filtration Rate |
| GO | Gene Ontology |
| HDAC | Histone Deacetylase |
| IC | Intercalated Cells |
| ICA | Intercalated Cell Type A |
| ICB | Intercalated Cell Type B |
| Immune | Immune Cells |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LEUK | Leukocytes |
| Lpl | Lipoprotein lipase |
| mATL | Medullary Ascending Thin Limb |
| MES | Mesangial Cells |
| Ncf1 | Neutrophil cytosolic factor 1 |
| PAS | Periodic Acid-Schiff |
| PC | Principal Cells |
| PCT | Proximal Convoluted Tubule |
| PCT | Proximal Convoluted Tubule |
| PEC | Parietal Epithelial Cells |
| Per | Pericytes |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PODO | Podocytes |
| PPAR | Peroxisome Proliferator-Activated Receptor |
| PST | Proximal Straight Tubule |
| PST | Proximal Straight Tubule |
| PT | Proximal Tubule |
| PTVCAM1 | Proximal Tubule VCAM1+ Cells |
| SAHA | Suberoylanilide Hydroxamic Acid |
| Scr | Serum creatinine |
| Slc27a2 | Solute carrier family 27 member 2 |
| TAL1 | Thick Ascending Limb 1 |
| TAL2 | Thick Ascending Limb 2 |
| UIRI | Unilateral Renal Ischemia/Reperfusion Injury |
| URO | Urothelial Cells |
| VPA | Valproic Acid |
| WGBS | Whole Genome Bisulfite Sequencing |
References
- Mehta, R.L.; Cerdá, J.; Burdmann, E.A.; Tonelli, M.; García-García, G.; Jha, V.; Susantitaphong, P.; Rocco, M.; Vanholder, R.; Sever, M.S.; et al. International Society of Nephrology’s 0by25 initiative for acute kidney injury (zero preventable deaths by 2025): A human rights case for nephrology. Lancet 2015, 385, 2616–2643. [Google Scholar] [CrossRef]
- Chertow, G.M.; Burdick, E.; Honour, M.; Bonventre, J.V.; Bates, D.W. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J. Am. Soc. Nephrol. 2005, 16, 3365–3370. [Google Scholar] [CrossRef]
- Wald, R.; Quinn, R.R.; Luo, J.; Li, P.; Scales, D.C.; Mamdani, M.M.; Ray, J.G. Chronic dialysis and death among survivors of acute kidney injury requiring dialysis. JAMA 2009, 302, 1179–1185. [Google Scholar] [CrossRef] [PubMed]
- Lo, L.J.; Go, A.S.; Chertow, G.M.; McCulloch, C.E.; Fan, D.; Ordoñez, J.D.; Hsu, C.Y. Dialysis-requiring acute renal failure increases the risk of progressive chronic kidney disease. Kidney Int. 2009, 76, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Amdur, R.L.; Chawla, L.S.; Amodeo, S.; Kimmel, P.L.; Palant, C.E. Outcomes following diagnosis of acute renal failure in U.S. veterans: Focus on acute tubular necrosis. Kidney Int. 2009, 76, 1089–1097. [Google Scholar] [CrossRef]
- Global, regional, and national burden of chronic kidney disease, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [CrossRef] [PubMed]
- Zhang, L.; Chen, F.; Dong, J.; Wang, R.; Bi, G.; Xu, D.; Zhang, Y.; Deng, Y.; Lin, W.; Yang, Z.; et al. HDAC3 aberration-incurred GPX4 suppression drives renal ferroptosis and AKI-CKD progression. Redox Biol. 2023, 68, 102939. [Google Scholar] [CrossRef]
- Xiao, X.; Wang, W.; Guo, C.; Wu, J.; Zhang, S.; Shi, H.; Kwon, S.; Chen, J.; Dong, Z. Hypermethylation leads to the loss of HOXA5, resulting in JAG1 expression and NOTCH signaling contributing to kidney fibrosis. Kidney Int. 2024, 106, 98–114. [Google Scholar] [CrossRef]
- Chou, Y.H.; Pan, S.Y.; Shao, Y.H.; Shih, H.M.; Wei, S.Y.; Lai, C.F.; Chiang, W.C.; Schrimpf, C.; Yang, K.C.; Lai, L.C.; et al. Methylation in pericytes after acute injury promotes chronic kidney disease. J. Clin. Invest. 2020, 130, 4845–4857. [Google Scholar] [CrossRef]
- Guo, C.; Dong, G.; Liang, X.; Dong, Z. Epigenetic regulation in AKI and kidney repair: Mechanisms and therapeutic implications. Nat. Rev. Nephrol. 2019, 15, 220–239. [Google Scholar] [CrossRef]
- Tampe, B.; Tampe, D.; Müller, C.A.; Sugimoto, H.; LeBleu, V.; Xu, X.; Müller, G.A.; Zeisberg, E.M.; Kalluri, R.; Zeisberg, M. Tet3-mediated hydroxymethylation of epigenetically silenced genes contributes to bone morphogenic protein 7-induced reversal of kidney fibrosis. J. Am. Soc. Nephrol. 2014, 25, 905–912. [Google Scholar] [CrossRef]
- Tampe, B.; Steinle, U.; Tampe, D.; Carstens, J.L.; Korsten, P.; Zeisberg, E.M.; Müller, G.A.; Kalluri, R.; Zeisberg, M. Low-dose hydralazine prevents fibrosis in a murine model of acute kidney injury-to-chronic kidney disease progression. Kidney Int. 2017, 91, 157–176. [Google Scholar] [CrossRef] [PubMed]
- Bechtel, W.; McGoohan, S.; Zeisberg, E.M.; Müller, G.A.; Kalbacher, H.; Salant, D.J.; Müller, C.A.; Kalluri, R.; Zeisberg, M. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat. Med. 2010, 16, 544–550. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Catoni, M.; Tsang, J.M.; Greco, A.P.; Zabet, N.R. DMRcaller: A versatile R/Bioconductor package for detection and visualization of differentially methylated regions in CpG and non-CpG contexts. Nucleic Acids Res. 2018, 46, e114. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.K.; Kanehisa, M. Systematic survey for novel types of prokaryotic retroelements based on gene neighborhood and protein architecture. Mol. Biol. Evol. 2008, 25, 1395–1404. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Pei, L.; Xiao, X.; Wei, Q.; Chen, J.K.; Ding, H.F.; Huang, S.; Fan, G.; Shi, H.; Dong, Z. DNA methylation protects against cisplatin-induced kidney injury by regulating specific genes, including interferon regulatory factor 8. Kidney Int. 2017, 92, 1194–1205. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Tan, L.; Xue, Z.; Cang, J.; Wang, H. Reduction of DNA hydroxymethylation in the mouse kidney insulted by ischemia reperfusion. Biochem. Biophys. Res. Commun. 2012, 422, 697–702. [Google Scholar] [CrossRef]
- Zhao, Y.; Ding, C.; Xue, W.; Ding, X.; Zheng, J.; Gao, Y.; Xia, X.; Li, S.; Liu, J.; Han, F.; et al. Genome-wide DNA methylation analysis in renal ischemia reperfusion injury. Gene 2017, 610, 32–43. [Google Scholar] [CrossRef]
- Lan, Y.L.; Zou, S.; Qin, B.; Zhu, X. Analysis of the sodium pump subunit ATP1A3 in glioma patients: Potential value in prognostic prediction and immunotherapy. Int. Immunopharmacol. 2024, 133, 112045. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.C.; Fang, F.; Zhou, J.; Koulajian, K.; Yang, S.; Lam, L.; Reich, H.N.; John, R.; Herzenberg, A.M.; Giacca, A.; et al. Deletion of p47phox attenuates the progression of diabetic nephropathy and reduces the severity of diabetes in the Akita mouse. Diabetologia 2012, 55, 2522–2532. [Google Scholar] [CrossRef]
- Huang, F.; Ren, X.; Yuan, B.; Yang, W.; Xu, L.; Zhang, J.; Zhang, H.; Geng, M.; Li, X.; Zhang, F.; et al. Systemic Mutation of Ncf1 Ameliorates Obstruction-Induced Renal Fibrosis While Macrophage-Rescued NCF1 Further Alleviates Renal Fibrosis. Antioxid. Redox Signal 2023. [Google Scholar] [CrossRef]
- Geng, L.; Zhao, J.; Deng, Y.; Molano, I.; Xu, X.; Xu, L.; Ruiz, P.; Li, Q.; Feng, X.; Zhang, M.; et al. Human SLE variant NCF1-R90H promotes kidney damage and murine lupus through enhanced Tfh2 responses induced by defective efferocytosis of macrophages. Ann. Rheum. Dis. 2022, 81, 255–267. [Google Scholar] [CrossRef]
- Wu, Y.; Cheng, S.; Gu, H.; Yang, K.; Xu, Z.; Meng, X.; Wang, Y.; Jiang, Y.; Li, H.; Zhou, Y.; et al. Variants within the LPL gene confer susceptility to diabetic kidney disease and rapid decline in kidney function in Chinese patients with type 2 diabetes. Diabetes Obes. Metab. 2023, 25, 3012–3019. [Google Scholar] [CrossRef]
- Wu, H.; Xu, H.; Lei, S.; Yang, Z.; Yang, S.; Du, J.; Zhou, Y.; Liu, Y.; Yang, Y.; Hu, Z. Loss-of-Function Homozygous Variant in LPL Causes Type I Hyperlipoproteinemia and Renal Lipidosis. Kidney Int. Rep. 2023, 8, 2428–2438. [Google Scholar] [CrossRef]
- Yu, H.; Tang, H.; Wang, M.; Xu, Q.; Yu, J.; Ge, H.; Qiang, L.; Tang, W.; Gu, H.F. Effects of total flavones of Abelmoschus manihot (L.) on the treatment of diabetic nephropathy via the activation of solute carriers in renal tubular epithelial cells. Biomed. Pharmacother. 2023, 169, 115899. [Google Scholar] [CrossRef]
- Khan, S.; Gaivin, R.; Abramovich, C.; Boylan, M.; Calles, J.; Schelling, J.R. Fatty acid transport protein-2 regulates glycemic control and diabetic kidney disease progression. JCI Insight 2020, 5, e136845. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Cabral, P.D.; Schilling, W.P.; Schmidt, Z.W.; Uddin, A.N.; Gingras, A.; Madhavan, S.M.; Garvin, J.L.; Schelling, J.R. Kidney Proximal Tubule Lipoapoptosis Is Regulated by Fatty Acid Transporter-2 (FATP2). J. Am. Soc. Nephrol. 2018, 29, 81–91. [Google Scholar] [CrossRef]
- Stevens, B.M.; Jones, C.L.; Pollyea, D.A.; Culp-Hill, R.; D’Alessandro, A.; Winters, A.; Krug, A.; Abbott, D.; Goosman, M.; Pei, S.; et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat. Cancer 2020, 1, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.L.; Su, Y.; Qiao, X.; Zhao, J.; Wang, G.; Howard, M.; Edwards, H.; Bao, X.; Li, J.; Hüttemann, M.; et al. Acquired resistance to venetoclax plus azacitidine in acute myeloid leukemia: In vitro models and mechanisms. Biochem. Pharmacol. 2023, 216, 115759. [Google Scholar] [CrossRef] [PubMed]
- Flores-Sierra, J.J.; Muciño-Arellano, M.D.R.; Romo-Morales, G.D.C.; Sánchez-Palafox, J.E.; Correa-Navarro, V.A.; Colín-Castelán, D.; Pérez-Vázquez, V.; Rangel-Salazar, R.; Rivera-Bustamante, R.; de la Rocha, C.; et al. The DNA methyltransferase inhibitor decitabine blunts the response to a high-animal fat and protein diet in mice. J. Lipid Res. 2024, 65, 100586. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Ruiz-Andres, O.; Poveda, J.; Sanchez-Niño, M.D.; Valiño-Rivas, L.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Targeting epigenetic DNA and histone modifications to treat kidney disease. Nephrol. Dial. Transplant. 2018, 33, 1875–1886. [Google Scholar] [CrossRef]
- Xu, X.; Tan, X.; Tampe, B.; Wilhelmi, T.; Hulshoff, M.S.; Saito, S.; Moser, T.; Kalluri, R.; Hasenfuss, G.; Zeisberg, E.M.; et al. High-fidelity CRISPR/Cas9- based gene-specific hydroxymethylation rescues gene expression and attenuates renal fibrosis. Nat. Commun. 2018, 9, 3509. [Google Scholar] [CrossRef]
- Sapozhnikov, D.M.; Szyf, M. Unraveling the functional role of DNA demethylation at specific promoters by targeted steric blockage of DNA methyltransferase with CRISPR/dCas9. Nat. Commun. 2021, 12, 5711. [Google Scholar] [CrossRef]
- Broche, J.; Kungulovski, G.; Bashtrykov, P.; Rathert, P.; Jeltsch, A. Genome-wide investigation of the dynamic changes of epigenome modifications after global DNA methylation editing. Nucleic Acids Res. 2021, 49, 158–176. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, X.; Guo, X.; Chen, Y.; Zhuang, K.; Gong, N.; Fu, Y.; Liang, Y.; Xu, Y.; Wang, S.; Wang, W.; et al. Integration of DNA Methylome and Transcriptome Analysis to Identify Novel Epigenetic Targets in the Acute Kidney Injury–Chronic Kidney Disease Transition. Biomolecules 2025, 15, 498. https://doi.org/10.3390/biom15040498
Zheng X, Guo X, Chen Y, Zhuang K, Gong N, Fu Y, Liang Y, Xu Y, Wang S, Wang W, et al. Integration of DNA Methylome and Transcriptome Analysis to Identify Novel Epigenetic Targets in the Acute Kidney Injury–Chronic Kidney Disease Transition. Biomolecules. 2025; 15(4):498. https://doi.org/10.3390/biom15040498
Chicago/Turabian StyleZheng, Xumin, Xinru Guo, Yuhao Chen, Kaiting Zhuang, Na Gong, Yifei Fu, Yanjun Liang, Yue Xu, Siyang Wang, Wenjuan Wang, and et al. 2025. "Integration of DNA Methylome and Transcriptome Analysis to Identify Novel Epigenetic Targets in the Acute Kidney Injury–Chronic Kidney Disease Transition" Biomolecules 15, no. 4: 498. https://doi.org/10.3390/biom15040498
APA StyleZheng, X., Guo, X., Chen, Y., Zhuang, K., Gong, N., Fu, Y., Liang, Y., Xu, Y., Wang, S., Wang, W., Chen, X., & Cai, G. (2025). Integration of DNA Methylome and Transcriptome Analysis to Identify Novel Epigenetic Targets in the Acute Kidney Injury–Chronic Kidney Disease Transition. Biomolecules, 15(4), 498. https://doi.org/10.3390/biom15040498