
Functional Analysis of Direct In Vitro Effect of Phosphorylated Tau on Mitochondrial Respiration and Hydrogen Peroxide Production


{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Mitochondrial Dysfunction in Alzheimer’s Disease
1.2. Tau Neurotoxicity
1.3. Mitochondrial Toxicity of Tau
1.4. Aim
2. Materials and Methods
2.1. Media and Chemicals
2.2. Mitochondria Isolation
2.3. P-Tau Oligomer Preparation
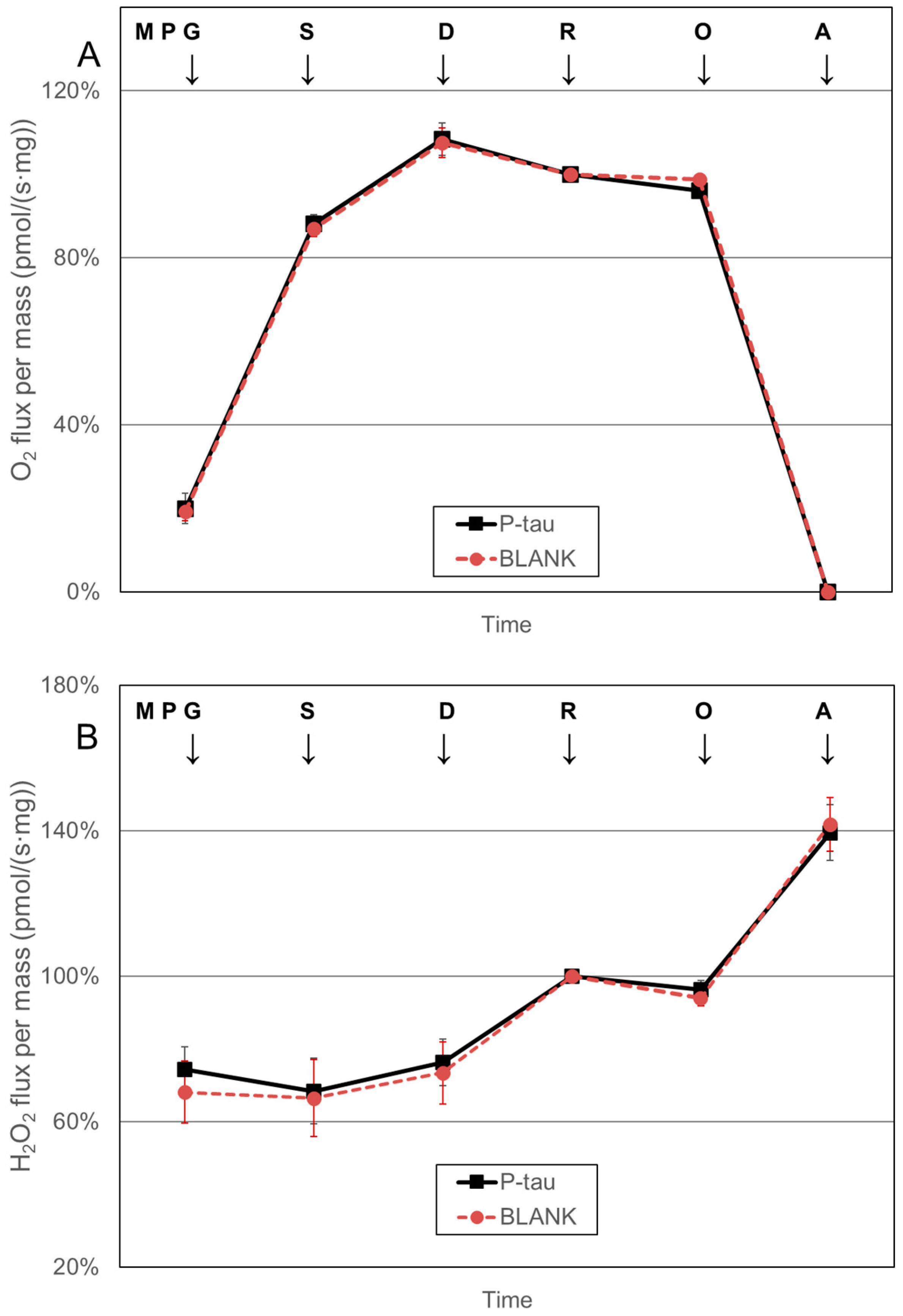
2.4. Mitochondrial Respiration and Hydrogen Peroxide Production
2.5. Data Analysis
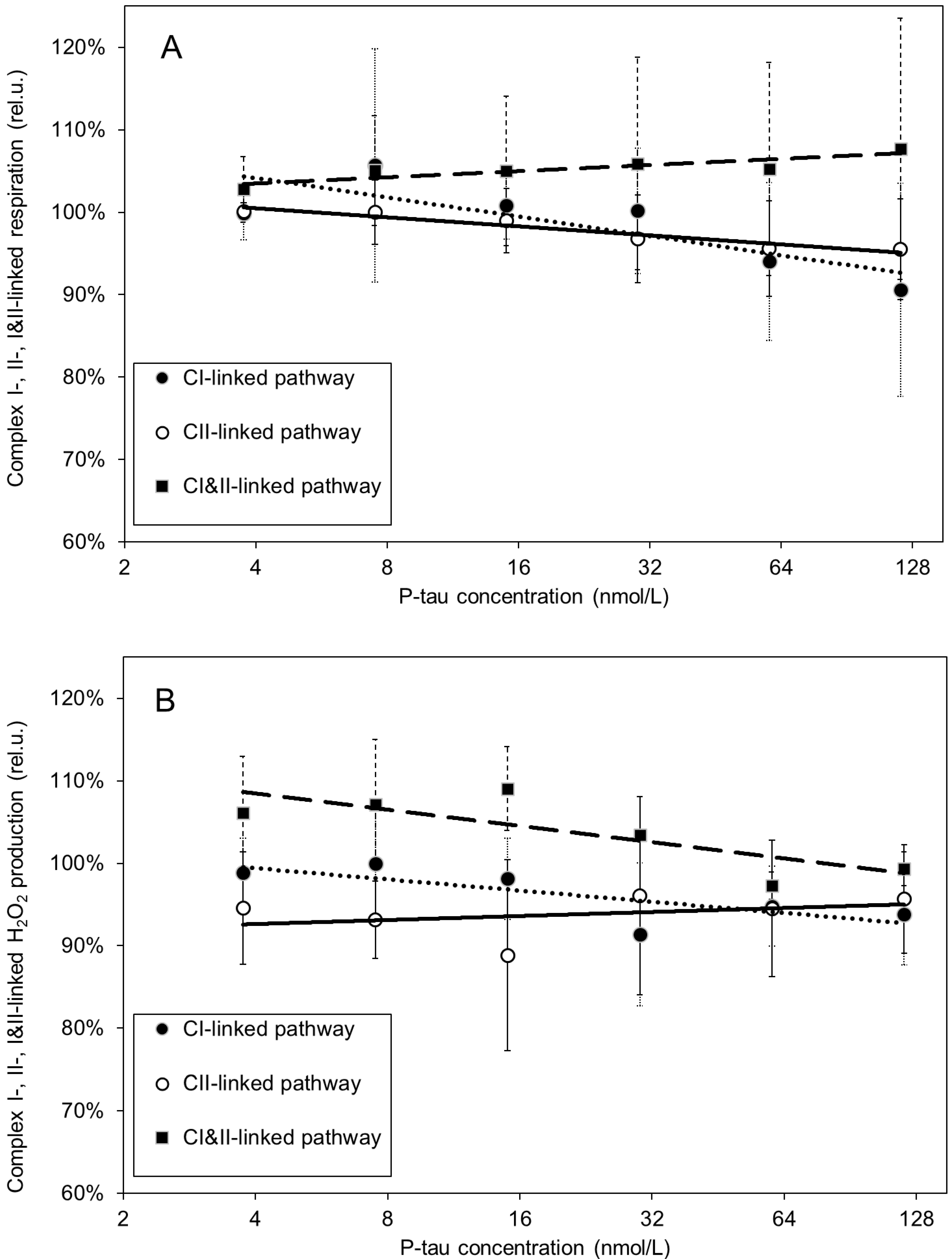
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zia, A.; Pourbagher-Shahri, A.M.; Farkhondeh, T.; Samarghandian, S. Molecular and cellular pathways contributing to brain aging. Behav. Brain Funct. 2021, 17, 6. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: From a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef]
- Fišar, Z.; Hroudová, J.; Zvěřová, M.; Jirák, R.; Raboch, J.; Kitzlerová, E. Age-dependent alterations in platelet mitochondrial respiration. Biomedicines 2023, 11, 1564. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.F.; Santos, A.E.; Moreira, P.I.; Pereira, A.C.; Sousa, F.J.; Cardoso, S.M.; Cruz, M.T. Is Alzheimer’s disease an inflammasomopathy? Ageing Res. Rev. 2019, 56, 100966. [Google Scholar] [CrossRef]
- 2024 Alzheimer’s disease facts and figures. Alzheimers Dement. 2024, 20, 3708–3821. [CrossRef]
- Ma, C.; Hong, F.; Yang, S. Amyloidosis in Alzheimer’s disease: Pathogeny, etiology, and related therapeutic directions. Molecules 2022, 27, 1210. [Google Scholar] [CrossRef]
- Devi, G. The tauopathies. Handb. Clin. Neurol. 2023, 196, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Blömeke, L.; Rehn, F.; Kraemer-Schulien, V.; Kutzsche, J.; Pils, M.; Bujnicki, T.; Lewczuk, P.; Kornhuber, J.; Freiesleben, S.D.; Schneider, L.S.; et al. Aβ oligomers peak in early stages of Alzheimer’s disease preceding tau pathology. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2024, 16, e12589. [Google Scholar] [CrossRef]
- Wang, C.; Zong, S.; Cui, X.; Wang, X.; Wu, S.; Wang, L.; Liu, Y.; Lu, Z. The effects of microglia-associated neuroinflammation on Alzheimer’s disease. Front. Immunol. 2023, 14, 1117172. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative stress in neurodegenerative diseases: From molecular mechanisms to clinical applications. Oxid. Med. Cell Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef]
- Bhatia, V.; Sharma, S. Role of mitochondrial dysfunction, oxidative stress and autophagy in progression of Alzheimer’s disease. J. Neurol. Sci. 2021, 421, 117253. [Google Scholar] [CrossRef]
- Liu, Z.; Li, T.; Li, P.; Wei, N.; Zhao, Z.; Liang, H.; Ji, X.; Chen, W.; Xue, M.; Wei, J. The ambiguous relationship of oxidative stress, tau hyperphosphorylation, and autophagy dysfunction in Alzheimer’s disease. Oxid. Med. Cell Longev. 2015, 2015, 352723. [Google Scholar] [CrossRef] [PubMed]
- Leuzy, A.; Chiotis, K.; Lemoine, L.; Gillberg, P.G.; Almkvist, O.; Rodriguez-Vieitez, E.; Nordberg, A. Tau PET imaging in neurodegenerative tauopathies-still a challenge. Mol. Psychiatry 2019, 24, 1112–1134. [Google Scholar] [CrossRef]
- Medina, M.; Hernandez, F.; Avila, J. New features about tau function and dysfunction. Biomolecules 2016, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.K.; Jara, C.; Park-Kang, H.S.; Polanco, C.M.; Tapia, D.; Alarcon, F.; de la Pena, A.; Llanquinao, J.; Vargas-Mardones, G.; Indo, J.A.; et al. Synaptic mitochondria: An early target of amyloid-beta and tau in Alzheimer’s disease. J. Alzheimers Dis. 2021, 84, 1391–1414. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Bai, F. The association of tau with mitochondrial dysfunction in Alzheimer’s disease. Front. Neurosci. 2018, 12, 163. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef]
- Supnet, C.; Bezprozvanny, I. The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium 2010, 47, 183–189. [Google Scholar] [CrossRef]
- Musiek, E.S.; Holtzman, D.M. Three dimensions of the amyloid hypothesis: Time, space and ‘wingmen’. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef]
- Swerdlow, R.H. The mitochondrial hypothesis: Dysfunction, bioenergetic defects, and the metabolic link to Alzheimer’s disease. Int. Rev. Neurobiol. 2020, 154, 207–233. [Google Scholar] [CrossRef]
- Karran, E.; De Strooper, B. The amyloid cascade hypothesis: Are we poised for success or failure? J. Neurochem. 2016, 139 (Suppl. S2), 237–252. [Google Scholar] [CrossRef]
- Liu, P.P.; Xie, Y.; Meng, X.Y.; Kang, J.S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and mitochondrial cascades in Alzheimer’s disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef]
- Fišar, Z. Linking the amyloid, tau, and mitochondrial hypotheses of Alzheimer’s disease and identifying promising drug targets. Biomolecules 2022, 12, 1676. [Google Scholar] [CrossRef] [PubMed]
- Bonda, D.J.; Wang, X.; Lee, H.G.; Smith, M.A.; Perry, G.; Zhu, X. Neuronal failure in Alzheimer’s disease: A view through the oxidative stress looking-glass. Neurosci. Bull. 2014, 30, 243–252. [Google Scholar] [CrossRef]
- Tapia-Rojas, C.; Cabezas-Opazo, F.; Deaton, C.A.; Vergara, E.H.; Johnson, G.V.W.; Quintanilla, R.A. It’s all about tau. Prog. Neurobiol. 2019, 175, 54–76. [Google Scholar] [CrossRef]
- Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur. J. Neurosci. 2005, 22, 1942–1950. [Google Scholar] [CrossRef]
- Leroy, K.; Yilmaz, Z.; Brion, J.P. Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 2007, 33, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef]
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Datta, D.; Del Tredici, K.; Braak, H. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimers Dement. 2021, 17, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Kametani, F.; Hasegawa, M. Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Terro, F. Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem. Int. 2011, 58, 458–471. [Google Scholar] [CrossRef]
- Takashima, A. Tauopathies and tau oligomers. J. Alzheimers Dis. 2013, 37, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.M.; Himmelstein, D.S.; Lancia, J.K.; Binder, L.I. Tau oligomers and tau toxicity in neurodegenerative disease. Biochem. Soc. Trans. 2012, 40, 667–671. [Google Scholar] [CrossRef]
- Cardenas-Aguayo Mdel, C.; Gomez-Virgilio, L.; DeRosa, S.; Meraz-Rios, M.A. The role of tau oligomers in the onset of Alzheimer’s disease neuropathology. ACS Chem. Neurosci. 2014, 5, 1178–1191. [Google Scholar] [CrossRef]
- Cowan, C.M.; Mudher, A. Are tau aggregates toxic or protective in tauopathies? Front. Neurol. 2013, 4, 114. [Google Scholar] [CrossRef]
- Szabo, L.; Eckert, A.; Grimm, A. Insights into disease-associated tau impact on mitochondria. Int. J. Mol. Sci. 2020, 21, 6344. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Sengupta, U.; Castillo-Carranza, D.; Gerson, J.E.; Guerrero-Munoz, M.; Troncoso, J.C.; Jackson, G.R.; Kayed, R. The formation of tau pore-like structures is prevalent and cell specific: Possible implications for the disease phenotypes. Acta Neuropathol. Commun. 2014, 2, 56. [Google Scholar] [CrossRef]
- Clavaguera, F.; Akatsu, H.; Fraser, G.; Crowther, R.A.; Frank, S.; Hench, J.; Probst, A.; Winkler, D.T.; Reichwald, J.; Staufenbiel, M.; et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. USA 2013, 110, 9535–9540. [Google Scholar] [CrossRef]
- Lewis, J.; Dickson, D.W. Propagation of tau pathology: Hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2016, 131, 27–48. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Karikari, T.K.; Nagel, D.A.; Grainger, A.; Clarke-Bland, C.; Hill, E.J.; Moffat, K.G. Preparation of stable tau oligomers for cellular and biochemical studies. Anal. Biochem. 2019, 566, 67–74. [Google Scholar] [CrossRef]
- Cieri, D.; Vicario, M.; Vallese, F.; D’Orsi, B.; Berto, P.; Grinzato, A.; Catoni, C.; De Stefani, D.; Rizzuto, R.; Brini, M.; et al. Tau localises within mitochondrial sub-compartments and its caspase cleavage affects ER-mitochondria interactions and cellular Ca(2+) handling. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3247–3256. [Google Scholar] [CrossRef]
- Shafiei, S.S.; Guerrero-Munoz, M.J.; Castillo-Carranza, D.L. Tau oligomers: Cytotoxicity, propagation, and mitochondrial damage. Front. Aging Neurosci. 2017, 9, 83. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta 2014, 1837, 408–417. [Google Scholar] [CrossRef]
- Camilleri, A.; Ghio, S.; Caruana, M.; Weckbecker, D.; Schmidt, F.; Kamp, F.; Leonov, A.; Ryazanov, S.; Griesinger, C.; Giese, A.; et al. Tau-induced mitochondrial membrane perturbation is dependent upon cardiolipin. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183064. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Clos, A.L.; Jackson, G.R.; Kayed, R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 2011, 6, 39. [Google Scholar] [CrossRef]
- Britti, E.; Ros, J.; Esteras, N.; Abramov, A.Y. Tau inhibits mitochondrial calcium efflux and makes neurons vulnerable to calcium-induced cell death. Cell Calcium 2020, 86, 102150. [Google Scholar] [CrossRef]
- Eckert, A.; Nisbet, R.; Grimm, A.; Gotz, J. March separate, strike together—Role of phosphorylated TAU in mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1258–1266. [Google Scholar] [CrossRef]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Drose, S.; Brandt, U.; et al. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef]
- David, D.C.; Hauptmann, S.; Scherping, I.; Schuessel, K.; Keil, U.; Rizzu, P.; Ravid, R.; Drose, S.; Brandt, U.; Muller, W.E.; et al. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J. Biol. Chem. 2005, 280, 23802–23814. [Google Scholar] [CrossRef]
- Schulz, K.L.; Eckert, A.; Rhein, V.; Mai, S.; Haase, W.; Reichert, A.S.; Jendrach, M.; Muller, W.E.; Leuner, K. A new link to mitochondrial impairment in tauopathies. Mol. Neurobiol. 2012, 46, 205–216. [Google Scholar] [CrossRef]
- Grimm, A.; Biliouris, E.E.; Lang, U.E.; Gotz, J.; Mensah-Nyagan, A.G.; Eckert, A. Sex hormone-related neurosteroids differentially rescue bioenergetic deficits induced by amyloid-beta or hyperphosphorylated tau protein. Cell Mol. Life Sci. 2016, 73, 201–215. [Google Scholar] [CrossRef]
- Grimm, A.; Lejri, I.; Halle, F.; Schmitt, M.; Gotz, J.; Bihel, F.; Eckert, A. Mitochondria modulatory effects of new TSPO ligands in a cellular model of tauopathies. J. Neuroendocr. 2020, 32, e12796. [Google Scholar] [CrossRef]
- Eckert, A.; Schmitt, K.; Gotz, J. Mitochondrial dysfunction—The beginning of the end in Alzheimer’s disease? Separate and synergistic modes of tau and amyloid-beta toxicity. Alzheimers Res. Ther. 2011, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.J.; Jara, C.; Quintanilla, R.A. Contribution of tau pathology to mitochondrial impairment in neurodegeneration. Front. Neurosci. 2018, 12, 441. [Google Scholar] [CrossRef]
- Plascencia-Villa, G.; Perry, G. Roles of oxidative stress in synaptic dysfunction and neuronal cell death in Alzheimer’s disease. Antioxidants 2023, 12, 1628. [Google Scholar] [CrossRef]
- Li, X.C.; Hu, Y.; Wang, Z.H.; Luo, Y.; Zhang, Y.; Liu, X.P.; Feng, Q.; Wang, Q.; Ye, K.; Liu, G.P.; et al. Human wild-type full-length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci. Rep. 2016, 6, 24756. [Google Scholar] [CrossRef]
- Atlante, A.; Amadoro, G.; Bobba, A.; de Bari, L.; Corsetti, V.; Pappalardo, G.; Marra, E.; Calissano, P.; Passarella, S. A peptide containing residues 26-44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochim. Biophys. Acta 2008, 1777, 1289–1300. [Google Scholar] [CrossRef]
- Pesta, D.; Gnaiger, E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol. Biol. 2012, 810, 25–58. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Brandacher, G.; Steurer, W.; Margreiter, R.; Gnaiger, E. Isolated rat heart mitochondria and whole rat heart as models for mitochondrial cold ischemia-reperfusion injury. Transpl. Proc. 2000, 32, 45. [Google Scholar] [CrossRef] [PubMed]
- Hroudová, J.; Fišar, Z. Assessment of the effects of drugs on mitochondrial respiration. Methods Mol. Biol. 2021, 2277, 133–142. [Google Scholar] [CrossRef]
- Fišar, Z.; Hroudová, J. Pig brain mitochondria as a biological model for study of mitochondrial respiration. Folia Biol. 2016, 62, 15–25. [Google Scholar] [CrossRef]
- Krumschnabel, G.; Fontana-Ayoub, M.; Sumbalova, Z.; Heidler, J.; Gauper, K.; Fasching, M.; Gnaiger, E. Simultaneous high-resolution measurement of mitochondrial respiration and hydrogen peroxide production. Methods Mol. Biol. 2015, 1264, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Fišar, Z.; Hroudová, J.; Singh, N.; Kopřivová, A.; Macečková, D. Effect of simvastatin, coenzyme Q10, resveratrol, acetylcysteine and acetylcarnitine on mitochondrial respiration. Folia Biol. 2016, 62, 53–66. [Google Scholar] [CrossRef]
- Fišar, Z.; Singh, N.; Hroudová, J. Cannabinoid-induced changes in respiration of brain mitochondria. Toxicol. Lett. 2014, 231, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Hroudová, J.; Fišar, Z. In vitro inhibition of mitochondrial respiratory rate by antidepressants. Toxicol. Lett. 2012, 213, 345–352. [Google Scholar] [CrossRef]
- Singh, N.; Hroudová, J.; Fišar, Z. In vitro effects of cognitives and nootropics on mitochondrial respiration and monoamine oxidase activity. Mol. Neurobiol. 2017, 54, 5894–5904. [Google Scholar] [CrossRef]
- Rawat, P.; Sehar, U.; Bisht, J.; Selman, A.; Culberson, J.; Reddy, P.H. Phosphorylated tau in Alzheimer’s disease and other tauopathies. Int. J. Mol. Sci. 2022, 23, 12841. [Google Scholar] [CrossRef]
- Pickett, E.K.; Rose, J.; McCrory, C.; McKenzie, C.A.; King, D.; Smith, C.; Gillingwater, T.H.; Henstridge, C.M.; Spires-Jones, T.L. Region-specific depletion of synaptic mitochondria in the brains of patients with Alzheimer’s disease. Acta Neuropathol. 2018, 136, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Reddy, P.H. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef]
- Di, J.; Cohen, L.S.; Corbo, C.P.; Phillips, G.R.; El Idrissi, A.; Alonso, A.D. Abnormal tau induces cognitive impairment through two different mechanisms: Synaptic dysfunction and neuronal loss. Sci. Rep. 2016, 6, 20833. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Cummings, J.; Zhou, Y.; Lee, G.; Zhong, K.; Fonseca, J.; Cheng, F. Alzheimer’s disease drug development pipeline: 2023. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2023, 9, e12385. [Google Scholar] [CrossRef]
- Reddy, A.P.; Reddy, P.H. Mitochondria-targeted molecules as potential drugs to treat patients with Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 2017, 146, 173–201. [Google Scholar] [CrossRef]
- Mashal, Y.; Abdelhady, H.; Iyer, A.K. Comparison of tau and amyloid-β targeted immunotherapy nanoparticles for Alzheimer’s disease. Biomolecules 2022, 12, 1001. [Google Scholar] [CrossRef]
- Tolar, M.; Hey, J.; Power, A.; Abushakra, S. Neurotoxic soluble amyloid oligomers drive Alzheimer’s pathogenesis and represent a clinically validated target for slowing disease progression. Int. J. Mol. Sci. 2021, 22, 6355. [Google Scholar] [CrossRef]
- Aillaud, I.; Funke, S.A. Tau aggregation inhibiting peptides as potential therapeutics for Alzheimer disease. Cell Mol. Neurobiol. 2023, 43, 951–961. [Google Scholar] [CrossRef]
- Fišar, Z.; Hroudová, J. CoQ10 and mitochondrial dysfunction in Alzheimer’s disease. Antioxidants 2024, 13, 191. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Bharti; Kumar, R.; Pavlov, P.F.; Winblad, B. Small molecule therapeutics for tauopathy in Alzheimer’s disease: Walking on the path of most resistance. Eur. J. Med. Chem. 2021, 209, 112915. [Google Scholar] [CrossRef]
- Ghazanfari, D.; Noori, M.S.; Bergmeier, S.C.; Hines, J.V.; McCall, K.D.; Goetz, D.J. A novel GSK-3 inhibitor binds to GSK-3β via a reversible, time and Cys-199-dependent mechanism. Bioorg Med. Chem. 2021, 40, 116179. [Google Scholar] [CrossRef]
- Medina, M. An Overview on the clinical development of tau-based therapeutics. Int. J. Mol. Sci. 2018, 19, 1160. [Google Scholar] [CrossRef] [PubMed]
- Soeda, Y.; Takashima, A. New insights into drug discovery targeting tau protein. Front. Mol. Neurosci. 2020, 13, 590896. [Google Scholar] [CrossRef]
- Tolar, M.; Abushakra, S.; Sabbagh, M. The path forward in Alzheimer’s disease therapeutics: Reevaluating the amyloid cascade hypothesis. Alzheimers Dement. 2020, 16, 1553–1560. [Google Scholar] [CrossRef]
- Mamsa, S.S.A.; Meloni, B.P. Arginine and arginine-rich peptides as modulators of protein aggregation and cytotoxicity associated with Alzheimer’s disease. Front. Mol. Neurosci. 2021, 14, 759729. [Google Scholar] [CrossRef]
- Andreux, P.A.; Houtkooper, R.H.; Auwerx, J. Pharmacological approaches to restore mitochondrial function. Nat. Rev. Drug Discov. 2013, 12, 465–483. [Google Scholar] [CrossRef]
- Hroudová, J.; Singh, N.; Fišar, Z.; Ghosh, K.K. Progress in drug development for Alzheimer’s disease: An overview in relation to mitochondrial energy metabolism. Eur. J. Med. Chem. 2016, 121, 774–784. [Google Scholar] [CrossRef]
- Weissig, V. Drug development for the therapy of mitochondrial diseases. Trends Mol. Med. 2020, 26, 40–57. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fišar, Z.; Hroudová, J. Functional Analysis of Direct In Vitro Effect of Phosphorylated Tau on Mitochondrial Respiration and Hydrogen Peroxide Production. Biomolecules 2025, 15, 495. https://doi.org/10.3390/biom15040495
Fišar Z, Hroudová J. Functional Analysis of Direct In Vitro Effect of Phosphorylated Tau on Mitochondrial Respiration and Hydrogen Peroxide Production. Biomolecules. 2025; 15(4):495. https://doi.org/10.3390/biom15040495
Chicago/Turabian StyleFišar, Zdeněk, and Jana Hroudová. 2025. "Functional Analysis of Direct In Vitro Effect of Phosphorylated Tau on Mitochondrial Respiration and Hydrogen Peroxide Production" Biomolecules 15, no. 4: 495. https://doi.org/10.3390/biom15040495
APA StyleFišar, Z., & Hroudová, J. (2025). Functional Analysis of Direct In Vitro Effect of Phosphorylated Tau on Mitochondrial Respiration and Hydrogen Peroxide Production. Biomolecules, 15(4), 495. https://doi.org/10.3390/biom15040495