
The Balance of MFN2 and OPA1 in Mitochondrial Dynamics, Cellular Homeostasis, and Disease


Abstract
1. Introduction
2. Structural and Functional Insights into MFN2 and OPA1
2.1. MFN2: Structure and Function
2.2. OPA1: Isoforms and Functional Domains
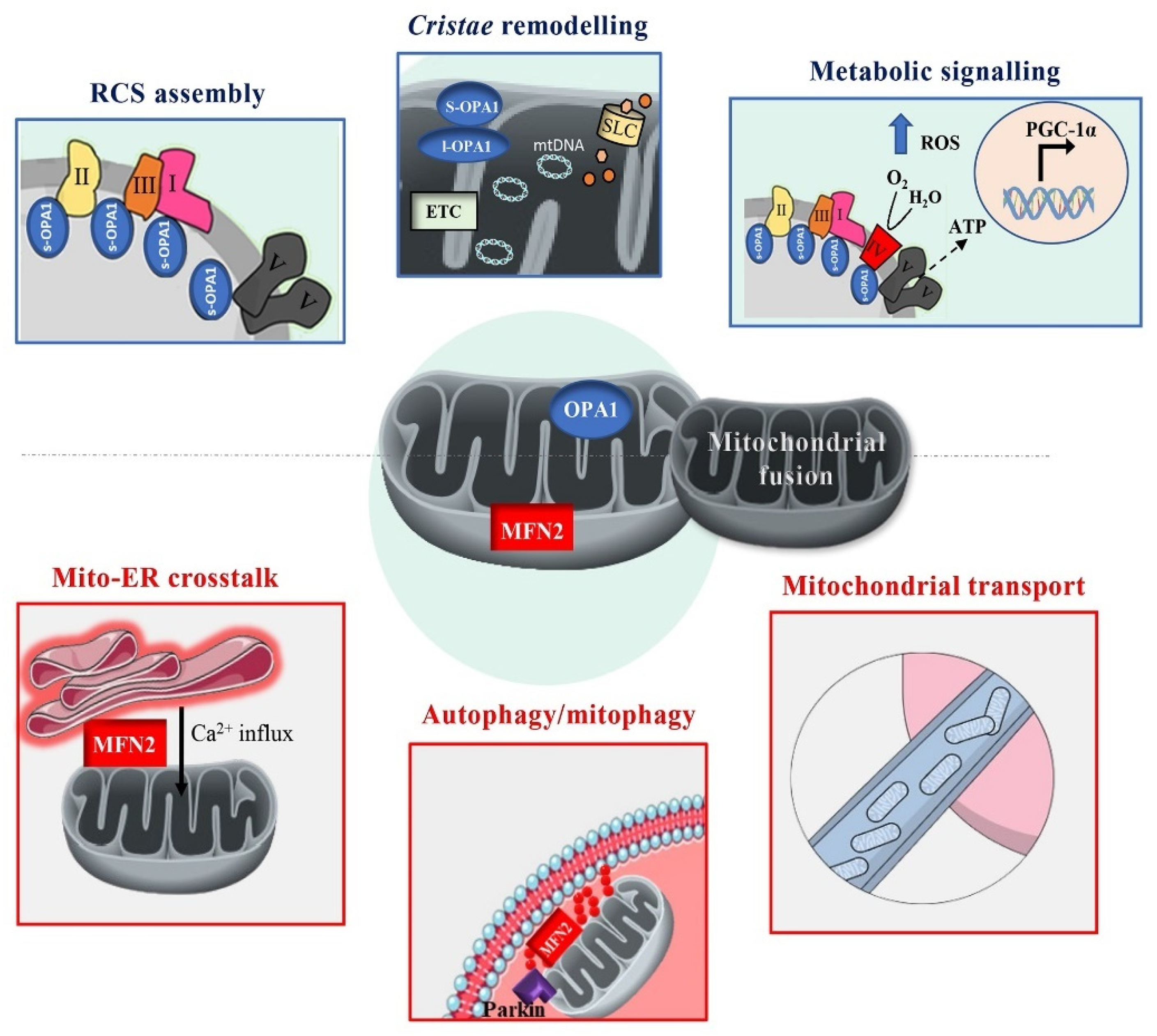
2.3. MFN2 and OPA1 in the Mitochondrial Fusion Process
3. Mitochondrial Fusion Proteins in the Regulation of Oxidative Phosphorylation
3.1. MFN2: A Key Regulator of Fusion, Bioenergetics and Beyond
3.2. OPA1: Master Regulator of Cristae Structure and Supercomplexes Assembly
3.3. Metabolic Cues Driving Mitochondrial Fusion
3.4. Regulation by Metabolic Sensor Kinases and Post-Translational Modifications
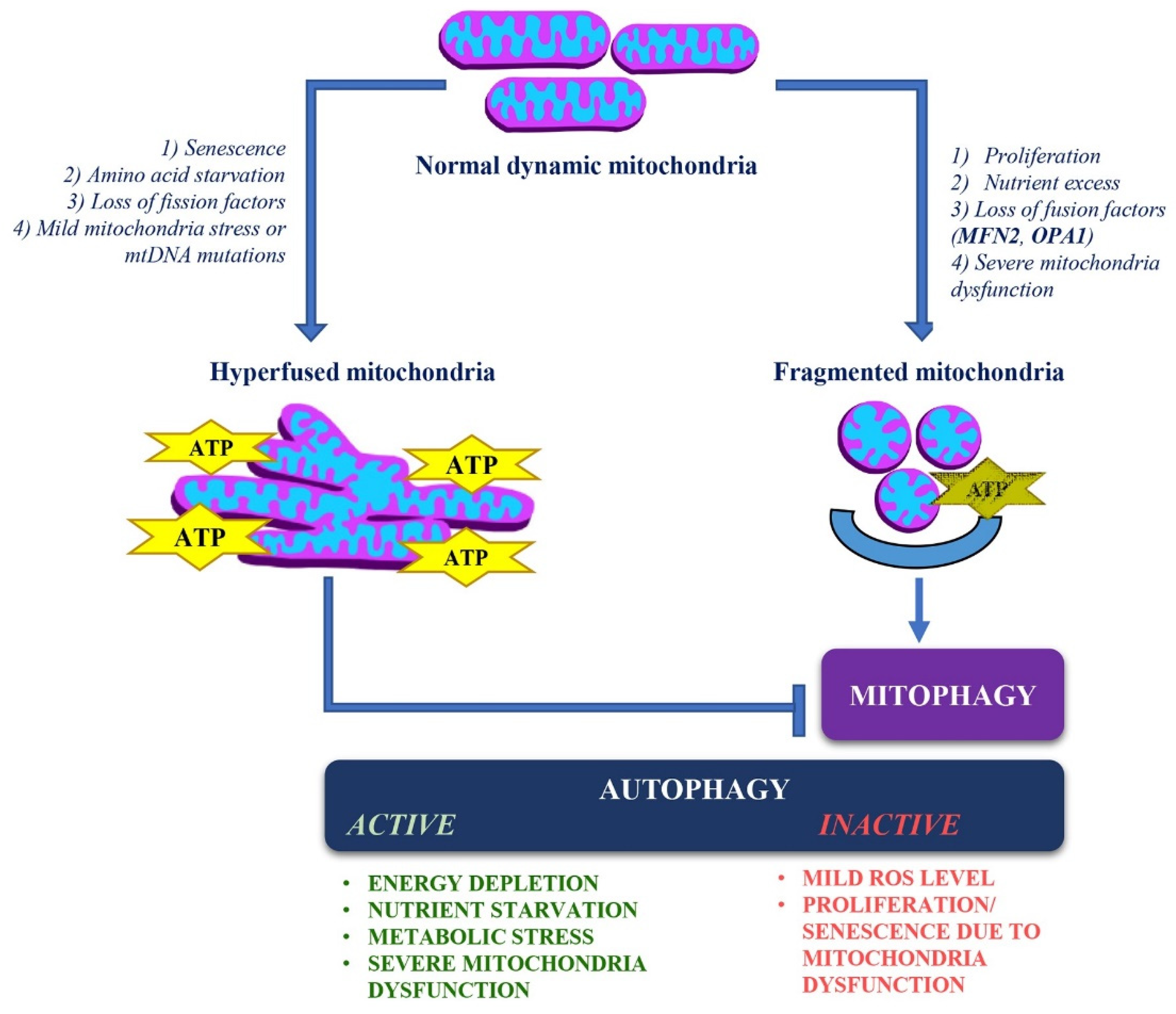
4. Dynamic Changes in Mitochondrial Structure and Quality Control
4.1. Mitochondrial Retrograde Signaling
4.2. Stress-Induced Mitochondrial Hyperfusion (SIMH)
4.3. Integrated Stress Response (ISR) as a Key Regulator of Stress Adaptation
4.4. Proteolytic Systems and Mitochondrial Proteostasis
5. Autophagy/Mitophagy and Mitochondrial Dynamics
6. Mitochondrial Dynamics in Neuronal Differentiation and Bioenergetics
7. Pathological Implications of MFN2 and OPA1 Mutations
7.1. MFN2 Mutations and Charcot-Marie-Tooth Disease Type 2A (CMT2A)
7.2. OPA1 Mutations and Autosomal Dominant Optic Atrophy (ADOA)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | MFN2 Mutation | Remarks | References |
|---|---|---|---|
| Skin fibroblasts from patients with CMT2A | (1) P123L | (1) Mitochondrial fragmentation and abnormal mitochondrial accumulation around the nucleus | [163] |
| (2) L92P | (2) Mitochondrial fragmentation | ||
| Skin fibroblasts from 4 patients with CMT2A2 | M21V | Reduced cellular respiration: uncoupling causing decreased ATP/OReduced ΔѰm | [191] |
| R364Q | |||
| A166T | |||
| Skin fibroblasts from patients with CMT2A | T105M | Mitochondrial morphology, mtDNA integrity and respiratory enzyme activities are unchangedExtensive mitochondrial fusion | [192] |
| I213T | |||
| V273G | |||
| Skin fibroblasts from patients with CMT2A | p.D210V | Respiratory chain defects | [193] |
| Multiple mtDNA deletions | |||
| Defect in mtDNA damage repair system | |||
| Fragmentation of the mitochondrial network | |||
| Skin fibroblasts derived from 4 patients with CMT2A | M376V | Mitochondrial respiratory chain dysfunctionDecreased mtDNA copy numberHigh levels of mtDNA depletion | [194] |
| R707P | |||
| V226_S229del | |||
| Q74R | |||
| Motor neurons derived from iPSCs of patients with CMT2A | R364W | Reduced mitochondrial trafficking with slower anterograde and retrograde velocities along axons | [195] |
| Electrophysiological impairments including increased excitability, higher sodium current density, and reduced inactivation of voltage-dependent sodium and calcium channel | |||
| Motor neurons derived from iPSCs of patients with CMT2A | A383V | Decreased respiratory chain activity: complexes II and III | [173] |
| Reduced mitochondrial mass and mtDNA content | |||
| No mtDNA alterations | |||
| Decrease of mitochondrial trafficking leading to perinuclear aggregation | |||
| No survival and morphometric defects | |||
| Increased resistance to apoptosis | |||
| Increased autophagic and mitophagic flux | |||
| Motor neurons derived from iPSCs of patients with CMT2A | R94Q | Abnormal mitochondrial morphology: shorter mitochondrial length within neurites, presence of abnormal mitochondria with loss of crista | [196] |
| Reduction in the percentage of moving mitochondria | |||
| Decrease in ATP levels in neurites | |||
| Increased toxicity sensitivity to vincristine and paclitaxel | |||
| Motor neurons derived from iPSCs of patients with CMT2A | R94Q | Hyper-connectivity: increase in burst rate | [172] |
| Alterations in mitochondrial morphology: reduced mitochondrial elongation and increase in circularity | |||
| Impairment in axonal transport: decrease in the speed of mitochondria and lysosomes and the proportion of active mitochondria and lysosomes moving within the cells | |||
| Defects in OxPhos: decrease in mitochondrial basal respiration | |||
| Transcriptomic analysis: enrichment in PI3K-AKT signaling and respiratory chain pathway | |||
| Skin fibroblasts derived from CMT2A patients | R364W | Mitochondrial mass and mtDNA levels are unchanged | [168] |
| M376V | Moderate disturbances in Ca2+ homeostasis | ||
| W740S | Reduced ER-mitochondria contacts | ||
| Skin fibroblasts from CMT2A patient | C217F | Mitochondrial mass and mtDNA levels and integrity unchanged | [197] |
| Mitochondrial fragmentation | |||
| Reduced ΔѰm | |||
| Reduction of respiratory chain complexes activity | |||
| Transcriptomic analysis: enrichment in PI3K-AKT signaling | |||
| Reduced autophagy and increased cellular proliferation (mTORC2/AKT activation) |
| Cell Type | OPA1 Mutation | Remarks | References |
|---|---|---|---|
| Fibroblasts from 3 patients with ADOA plus | V903Gfs3,E221K QT86Sfs15, H957Y QT86Sfs*15, H957Y | Increased mitochondrial fragmentation | [178] |
| Depletion of mtDNA | |||
| Altered mitochondrial localization | |||
| Increased mitophagy flux | |||
| Fibroblasts from 7 ADOA patients |
| (1), (2) Fragmented and punctiform mitochondria (3) Mild mitochondrial fragmentation (1), (2), (3) Loss of mitochondrial volume (1), (2) Mild uncoupling of oxidative phosphorylation (3), (4) Severe defects in oxidative phosphorylation Altered autophagy (4), (5) Mild mtDNA depletion | [175] |
| Lymphoblastoid cells derived from ADOA patients | P400A | Decreased mtDNA copy number | [198] |
| Reduced levels of 4 mtDNA-encoded polypeptides | |||
| Respiratory capacity defects | |||
| ATP synthesis defects | |||
| Altered mitochondrial membrane potential | |||
| Increased ROS production | |||
| Increased apoptosis | |||
| Mitochondrial morphological defects (fragmentation and swelling) | |||
| Lymphoblastoid cell lines from ADOA patient | c.1444–2A>C (splicing variants, deletion of the 15th exon in mRNA transcript, low protein levels) | Respiratory chain activity defects | [177] |
| More punctate mitochondria clustered in the perinuclear region | |||
| No marked depletion of mtDNA or mitochondrial mass | |||
| Reduced ATP synthesis | |||
| Reduced ΔѰm | |||
| Increased ROS production | |||
| Increased mitophagy | |||
| Skin fibroblasts from ADOA plus patient | H42Y | Mitochondrial mass unchanged | [199] |
| mtDNA levels slightly increased | |||
| mtDNA integrity unchanged | |||
| Mitochondrial fragmentation | |||
| Reduced ΔѰm | |||
| Reduction of respiratory chain complexes activity | |||
| Increased ROS production | |||
| Transcriptomic analysis: enrichment in p21WAF1/CIP1 and p53 pathways along with downregulation of mitotic cell cycle genes | |||
| Reduced autophagy and increased expression of senescence markers (SA-β galactosidase; p53 and p21) associated with lower mTORC2 activity |
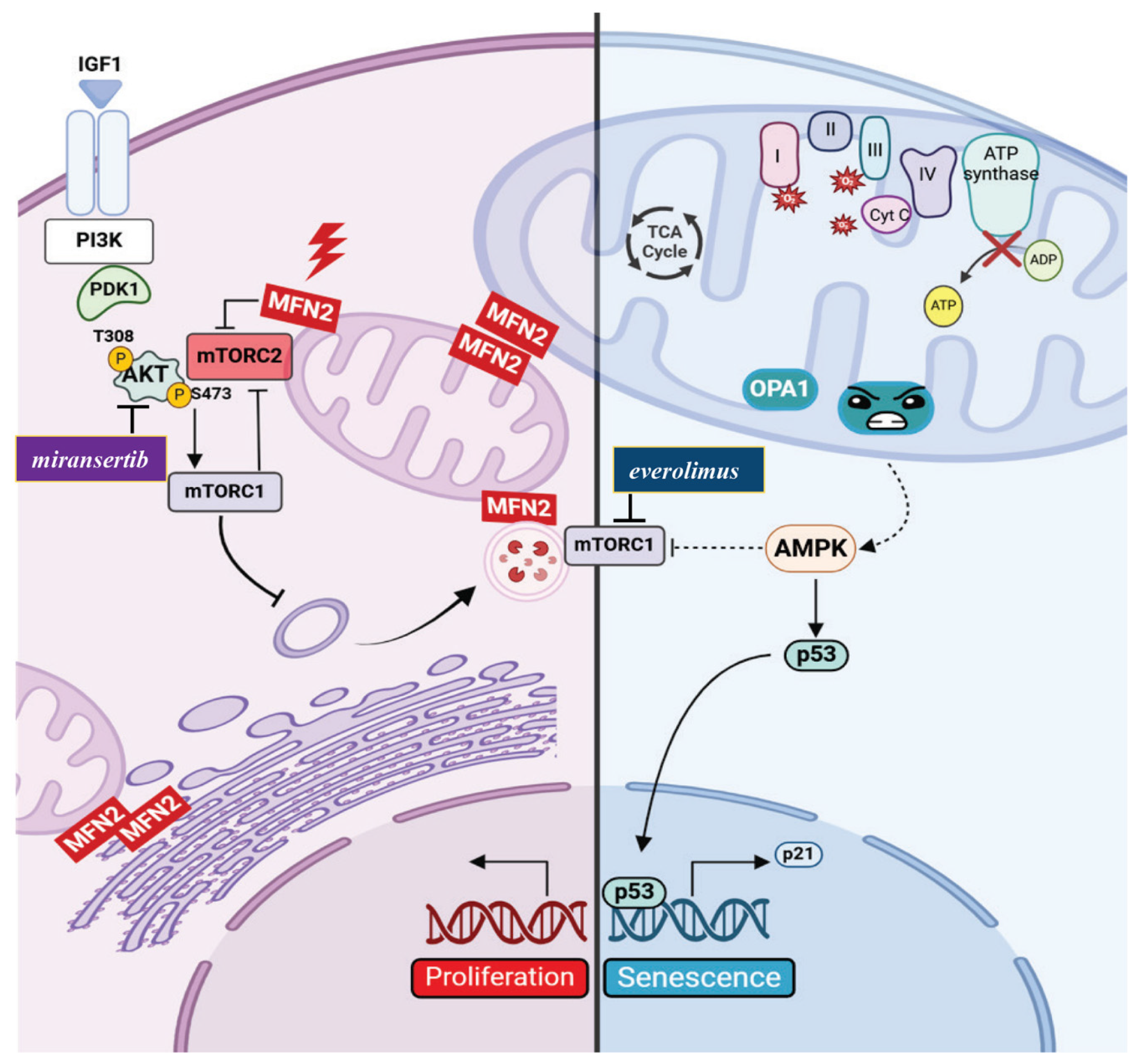
8. Distinct Cellular Outcomes of MFN2 and OPA1 Mutations
Therapeutic Implications
9. Discussion
10. Conclusions
11. Future Directions
- •
- Could the proliferative changes observed in CMT2A2 fibroblasts and cancer models of MFN2 dysfunction also occur in neuronal stem cells affected by CMT2A?
- •
- Does senescence caused by OPA1 deficiency directly contribute to neurodegeneration in ADOA, and is this related to impaired autophagy?
- •
- How does metabolic reprogramming influence the progression of mitochondrial diseases, and can interventions be implemented to restore the balance of autophagy and mitophagy?
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liesa, M.; Shirihai, O.S. Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef] [PubMed]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef]
- Santel, A.; Fuller, M.T. Control of Mitochondrial Morphology by a Human Mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar] [CrossRef]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial Fragmentation in Neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505–518. [Google Scholar] [CrossRef]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The Cell Biology of Mitochondrial Membrane Dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial Fusion and Fission in Mammals. Annu. Rev. Cell Dev. Biol. 2006, 22, 79–99. [Google Scholar] [CrossRef]
- Gomes, L.C.; Scorrano, L. Mitochondrial Morphology in Mitophagy and Macroautophagy. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2013, 1833, 205–212. [Google Scholar] [CrossRef]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to Metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef]
- Filadi, R.; Pendin, D.; Pizzo, P. Mitofusin 2: From Functions to Disease. Cell Death Dis. 2018, 9, 330. [Google Scholar] [CrossRef]
- Gilkerson, R.; De La Torre, P.; St. Vallier, S. Mitochondrial OMA1 and OPA1 as Gatekeepers of Organellar Structure/Function and Cellular Stress Response. Front. Cell Dev. Biol. 2021, 9, 626117. [Google Scholar] [CrossRef] [PubMed]
- Rojo, M.; Legros, F.; Chateau, D.; Lombès, A. Membrane Topology and Mitochondrial Targeting of Mitofusins, Ubiquitous Mammalian Homologs of the Transmembrane GTPase Fzo. J. Cell Sci. 2002, 115, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Chandhok, G.; Lazarou, M.; Neumann, B. Structure, Function, and Regulation of Mitofusin-2 in Health and Disease. Biol. Rev. 2018, 93, 933–949. [Google Scholar] [CrossRef] [PubMed]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase Superfamily: A Conserved Switch for Diverse Cell Functions. Nature 1990, 348, 125–132. [Google Scholar] [CrossRef]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase Superfamily: Conserved Structure and Molecular Mechanism. Nature 1991, 349, 117–127. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 Coordinately Regulate Mitochondrial Fusion and Are Essential for Embryonic Development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural Basis of Mitochondrial Tethering by Mitofusin Complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef]
- Chen, K.-H.; Guo, X.; Ma, D.; Guo, Y.; Li, Q.; Yang, D.; Li, P.; Qiu, X.; Wen, S.; Xiao, R.-P.; et al. Dysregulation of HSG Triggers Vascular Proliferative Disorders. Nat. Cell Biol. 2004, 6, 872–883. [Google Scholar] [CrossRef]
- Eura, Y. Two Mitofusin Proteins, Mammalian Homologues of FZO, with Distinct Functions Are Both Required for Mitochondrial Fusion. J. Biochem. 2003, 134, 333–344. [Google Scholar] [CrossRef]
- Qi, Y.; Yan, L.; Yu, C.; Guo, X.; Zhou, X.; Hu, X.; Huang, X.; Rao, Z.; Lou, Z.; Hu, J. Structures of Human Mitofusin 1 Provide Insight into Mitochondrial Tethering. J. Cell Biol. 2016, 215, 621–629. [Google Scholar] [CrossRef]
- Cao, Y.-L.; Meng, S.; Chen, Y.; Feng, J.-X.; Gu, D.-D.; Yu, B.; Li, Y.-J.; Yang, J.-Y.; Liao, S.; Chan, D.C.; et al. MFN1 Structures Reveal Nucleotide-Triggered Dimerization Critical for Mitochondrial Fusion. Nature 2017, 542, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-J.; Cao, Y.-L.; Feng, J.-X.; Qi, Y.; Meng, S.; Yang, J.-F.; Zhong, Y.-T.; Kang, S.; Chen, X.; Lan, L.; et al. Structural Insights of Human Mitofusin-2 into Mitochondrial Fusion and CMT2A Onset. Nat. Commun. 2019, 10, 4914. [Google Scholar] [CrossRef] [PubMed]
- Von Der Malsburg, A.; Sapp, G.M.; Zuccaro, K.E.; Von Appen, A.; Moss, F.R.; Kalia, R.; Bennett, J.A.; Abriata, L.A.; Dal Peraro, M.; Van Der Laan, M.; et al. Structural Mechanism of Mitochondrial Membrane Remodelling by Human OPA1. Nature 2023, 620, 1101–1108. [Google Scholar] [CrossRef]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K. Regulation of Mitochondrial Morphology through Proteolytic Cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [Google Scholar] [CrossRef]
- Amati-Bonneau, P.; Milea, D.; Bonneau, D.; Chevrollier, A.; Ferré, M.; Guillet, V.; Gueguen, N.; Loiseau, D.; Crescenzo, M.-A.P.D.; Verny, C.; et al. OPA1-Associated Disorders: Phenotypes and Pathophysiology. Int. J. Biochem. Cell Biol. 2009, 41, 1855–1865. [Google Scholar] [CrossRef]
- Baker, M.J.; Lampe, P.A.; Stojanovski, D.; Korwitz, A.; Anand, R.; Tatsuta, T.; Langer, T. Stress-Induced OMA1 Activation and Autocatalytic Turnover Regulate OPA1-Dependent Mitochondrial Dynamics. EMBO J. 2014, 33, 578–593. [Google Scholar] [CrossRef]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA Protease YME1L and OMA1 Cleave OPA1 to Balance Mitochondrial Fusion and Fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef]
- Van Der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of Mitochondrial Fission and Fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef]
- Yan, L.; Qi, Y.; Huang, X.; Yu, C.; Lan, L.; Guo, X.; Rao, Z.; Hu, J.; Lou, Z. Structural Basis for GTP Hydrolysis and Conformational Change of MFN1 in Mediating Membrane Fusion. Nat. Struct. Mol. Biol. 2018, 25, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Mattie, S.; Riemer, J.; Wideman, J.G.; McBride, H.M. A New Mitofusin Topology Places the Redox-Regulated C Terminus in the Mitochondrial Intermembrane Space. J. Cell Biol. 2018, 217, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Duvezin-Caubet, S.; Jagasia, R.; Wagener, J.; Hofmann, S.; Trifunovic, A.; Hansson, A.; Chomyn, A.; Bauer, M.F.; Attardi, G.; Larsson, N.-G.; et al. Proteolytic Processing of OPA1 Links Mitochondrial Dysfunction to Alterations in Mitochondrial Morphology. J. Biol. Chem. 2006, 281, 37972–37979. [Google Scholar] [CrossRef] [PubMed]
- Brandt, T.; Cavellini, L.; Kühlbrandt, W.; Cohen, M.M. A Mitofusin-Dependent Docking Ring Complex Triggers Mitochondrial Fusion in Vitro. eLife 2016, 5, e14618. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and Selective Fusion Govern Mitochondrial Segregation and Elimination by Autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial Fusion Is Required for mtDNA Stability in Skeletal Muscle and Tolerance of mtDNA Mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Physiological Functions of Mitochondrial Fusion. Ann. N. Y. Acad. Sci. 2010, 1201, 21–25. [Google Scholar] [CrossRef]
- Rodríguez-Nuevo, A.; Díaz-Ramos, A.; Noguera, E.; Díaz-Sáez, F.; Duran, X.; Muñoz, J.P.; Romero, M.; Plana, N.; Sebastián, D.; Tezze, C.; et al. Mitochondrial DNA and TLR9 Drive Muscle Inflammation upon Opa1 Deficiency. EMBO J. 2018, 37, e96553. [Google Scholar] [CrossRef]
- Yao, C.-H.; Wang, R.; Wang, Y.; Kung, C.-P.; Weber, J.D.; Patti, G.J. Mitochondrial Fusion Supports Increased Oxidative Phosphorylation during Cell Proliferation. eLife 2019, 8, e41351. [Google Scholar] [CrossRef]
- Gomes, L.C.; Benedetto, G.D.; Scorrano, L. During Autophagy Mitochondria Elongate, Are Spared from Degradation and Sustain Cell Viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef]
- Molina, A.J.A.; Wikstrom, J.D.; Stiles, L.; Las, G.; Mohamed, H.; Elorza, A.; Walzer, G.; Twig, G.; Katz, S.; Corkey, B.E.; et al. Mitochondrial Networking Protects β-Cells From Nutrient-Induced Apoptosis. Diabetes 2009, 58, 2303–2315. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhao, H.; Li, Y. Mitochondrial Dynamics in Health and Disease: Mechanisms and Potential Targets. Signal Transduct. Target. Ther. 2023, 8, 333. [Google Scholar] [CrossRef] [PubMed]
- Jheng, H.-F.; Tsai, P.-J.; Guo, S.-M.; Kuo, L.-H.; Chang, C.-S.; Su, I.-J.; Chang, C.-R.; Tsai, Y.-S. Mitochondrial Fission Contributes to Mitochondrial Dysfunction and Insulin Resistance in Skeletal Muscle. Mol. Cell. Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Ryu, K.W.; Fung, T.S.; Baker, D.C.; Saoi, M.; Park, J.; Febres-Aldana, C.A.; Aly, R.G.; Cui, R.; Sharma, A.; Fu, Y.; et al. Cellular ATP Demand Creates Metabolically Distinct Subpopulations of Mitochondria. Nature 2024, 635, 746–754. [Google Scholar] [CrossRef]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 Tethers Endoplasmic Reticulum to Mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Hernández-Alvarez, M.I.; Sebastián, D.; Vives, S.; Ivanova, S.; Bartoccioni, P.; Kakimoto, P.; Plana, N.; Veiga, S.R.; Hernández, V.; Vasconcelos, N.; et al. Deficient Endoplasmic Reticulum-Mitochondrial Phosphatidylserine Transfer Causes Liver Disease. Cell 2019, 177, 881–895.e17. [Google Scholar] [CrossRef]
- Zorzano, A.; Hernández-Alvarez, M.I.; Sebastián, D.; Muñoz, J.P. Mitofusin 2 as a Driver That Controls Energy Metabolism and Insulin Signaling. Antioxid. Redox Signal. 2015, 22, 1020–1031. [Google Scholar] [CrossRef]
- Kaufman, R.J.; Malhotra, J.D. Calcium Trafficking Integrates Endoplasmic Reticulum Function with Mitochondrial Bioenergetics. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2014, 1843, 2233–2239. [Google Scholar] [CrossRef]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.; Witkiewicz, A.K.; Howell, A.; Pavlides, S.; Tsirigos, A.; Ertel, A.; Pestell, R.G.; et al. Hyperactivation of Oxidative Mitochondrial Metabolism in Epithelial Cancer Cells in Situ: Visualizing the Therapeutic Effects of Metformin in Tumor Tissue. Cell Cycle 2011, 10, 4047–4064. [Google Scholar] [CrossRef]
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernández-Alvarez, M.I.; et al. Critical Reappraisal Confirms That Mitofusin 2 Is an Endoplasmic Reticulum–Mitochondria Tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Mitofusin 2 Ablation Increases Endoplasmic Reticulum–Mitochondria Coupling. Proc. Natl. Acad. Sci. USA 2015, 112, E2174–E2181. [Google Scholar] [CrossRef] [PubMed]
- Naon, D.; Scorrano, L. At the Right Distance: ER-Mitochondria Juxtaposition in Cell Life and Death. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2014, 1843, 2184–2194. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Theurey, P.; Pizzo, P. The Endoplasmic Reticulum-Mitochondria Coupling in Health and Disease: Molecules, Functions and Significance. Cell Calcium 2017, 62, 1–15. [Google Scholar] [CrossRef]
- Lin, M.-Y.; Sheng, Z.-H. Regulation of Mitochondrial Transport in Neurons. Exp. Cell Res. 2015, 334, 35–44. [Google Scholar] [CrossRef]
- Misko, A.; Jiang, S.; Wegorzewska, I.; Milbrandt, J.; Baloh, R.H. Mitofusin 2 Is Necessary for Transport of Axonal Mitochondria and Interacts with the Miro/Milton Complex. J. Neurosci. 2010, 30, 4232–4240. [Google Scholar] [CrossRef]
- Mou, Y.; Dein, J.; Chen, Z.; Jagdale, M.; Li, X.-J. MFN2 Deficiency Impairs Mitochondrial Transport and Downregulates Motor Protein Expression in Human Spinal Motor Neurons. Front. Mol. Neurosci. 2021, 14, 727552. [Google Scholar] [CrossRef] [PubMed]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acín-Pérez, R.; Latorre-Pellicer, A.; Colás, C.; Balsa, E.; Perales-Clemente, E.; Quirós, P.M.; Calvo, E.; Rodríguez-Hernández, M.A.; et al. Supercomplex Assembly Determines Electron Flux in the Mitochondrial Electron Transport Chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial Cristae Shape Determines Respiratory Chain Supercomplexes Assembly and Respiratory Efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef]
- Patten, D.A.; Wong, J.; Khacho, M.; Soubannier, V.; Mailloux, R.J.; Pilon-Larose, K.; MacLaurin, J.G.; Park, D.S.; McBride, H.M.; Trinkle-Mulcahy, L.; et al. OPA1-dependent Cristae Modulation Is Essential for Cellular Adaptation to Metabolic Demand. EMBO J. 2014, 33, 2676–2691. [Google Scholar] [CrossRef]
- Sauvanet, C.; Duvezin-Caubet, S.; Di Rago, J.-P.; Rojo, M. Energetic Requirements and Bioenergetic Modulation of Mitochondrial Morphology and Dynamics. Semin. Cell Dev. Biol. 2010, 21, 558–565. [Google Scholar] [CrossRef]
- Sood, A.; Jeyaraju, D.V.; Prudent, J.; Caron, A.; Lemieux, P.; McBride, H.M.; Laplante, M.; Tóth, K.; Pellegrini, L. A Mitofusin-2–Dependent Inactivating Cleavage of Opa1 Links Changes in Mitochondria Cristae and ER Contacts in the Postprandial Liver. Proc. Natl. Acad. Sci. USA 2014, 111, 16017–16022. [Google Scholar] [CrossRef]
- Ehses, S.; Raschke, I.; Mancuso, G.; Bernacchia, A.; Geimer, S.; Tondera, D.; Martinou, J.-C.; Westermann, B.; Rugarli, E.I.; Langer, T. Regulation of OPA1 Processing and Mitochondrial Fusion by m -AAA Protease Isoenzymes and OMA1. J. Cell Biol. 2009, 187, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Morita, M.; Prudent, J.; Basu, K.; Goyon, V.; Katsumura, S.; Hulea, L.; Pearl, D.; Siddiqui, N.; Strack, S.; McGuirk, S.; et al. mTOR Controls Mitochondrial Dynamics and Cell Survival via MTFP1. Mol. Cell 2017, 67, 922–935.e5. [Google Scholar] [CrossRef] [PubMed]
- Parra, V.; Verdejo, H.E.; Iglewski, M.; Del Campo, A.; Troncoso, R.; Jones, D.; Zhu, Y.; Kuzmicic, J.; Pennanen, C.; Lopez-Crisosto, C.; et al. Insulin Stimulates Mitochondrial Fusion and Function in Cardiomyocytes via the Akt-mTOR-NFκB-Opa-1 Signaling Pathway. Diabetes 2014, 63, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.J.; Chen, Y.; Shi, L.X.; Cheng, H.R.; Banda, I.; Ji, Y.H.; Wang, Y.T.; Li, X.M.; Mao, Y.X.; Zhang, D.F.; et al. AKT-GSK3 β Signaling Pathway Regulates Mitochondrial Dysfunction-Associated OPA1 Cleavage Contributing to Osteoblast Apoptosis: Preventative Effects of Hydroxytyrosol. Oxidative Med. Cell. Longev. 2019, 2019, 4101738. [Google Scholar] [CrossRef]
- Cribbs, J.T.; Strack, S. Reversible Phosphorylation of Drp1 by Cyclic AMP-dependent Protein Kinase and Calcineurin Regulates Mitochondrial Fission and Cell Death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef]
- Escobar-Henriques, M.; Joaquim, M. Mitofusins: Disease Gatekeepers and Hubs in Mitochondrial Quality Control by E3 Ligases. Front. Physiol. 2019, 10, 517. [Google Scholar] [CrossRef]
- Eisner, V.; Picard, M.; Hajnóczky, G. Mitochondrial Dynamics in Adaptive and Maladaptive Cellular Stress Responses. Nat. Cell Biol. 2018, 20, 755–765. [Google Scholar] [CrossRef]
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial Fusion and Fission: The Fine-tune Balance for Cellular Homeostasis. FASEB J. 2021, 35, e21620. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial Fusion and Fission in Cell Life and Death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Mottis, A.; Jovaisaite, V.; Auwerx, J. The Mitochondrial Unfolded Protein Response in Mammalian Physiology. Mamm. Genome 2014, 25, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Evans, R. PPARs and ERRs: Molecular Mediators of Mitochondrial Metabolism. Curr. Opin. Cell Biol. 2015, 33, 49–54. [Google Scholar] [CrossRef]
- Cardamone, M.D.; Tanasa, B.; Cederquist, C.T.; Huang, J.; Mahdaviani, K.; Li, W.; Rosenfeld, M.G.; Liesa, M.; Perissi, V. Mitochondrial Retrograde Signaling in Mammals Is Mediated by the Transcriptional Cofactor GPS2 via Direct Mitochondria-to-Nucleus Translocation. Mol. Cell 2018, 69, 757–772.e7. [Google Scholar] [CrossRef]
- Hock, M.B.; Kralli, A. Transcriptional Control of Mitochondrial Biogenesis and Function. Annu. Rev. Physiol. 2009, 71, 177–203. [Google Scholar] [CrossRef]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional Integration of Mitochondrial Biogenesis. Trends Endocrinol. Metab. 2012, 23, 459–466. [Google Scholar] [CrossRef]
- Finley, L.W.S.; Haigis, M.C. The Coordination of Nuclear and Mitochondrial Communication during Aging and Calorie Restriction. Ageing Res. Rev. 2009, 8, 173–188. [Google Scholar] [CrossRef]
- Guha, M.; Avadhani, N.G. Mitochondrial Retrograde Signaling at the Crossroads of Tumor Bioenergetics, Genetics and Epigenetics. Mitochondrion 2013, 13, 577–591. [Google Scholar] [CrossRef]
- Bohovych, I.; Khalimonchuk, O. Sending Out an SOS: Mitochondria as a Signaling Hub. Front. Cell Dev. Biol. 2016, 4, 109. [Google Scholar] [CrossRef]
- Chandel, N.S. Evolution of Mitochondria as Signaling Organelles. Cell Metab. 2015, 22, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Butow, R.A.; Avadhani, N.G. Mitochondrial Signaling. Mol. Cell 2004, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Butow, R.A. Mitochondrial Retrograde Signaling. Annu. Rev. Genet. 2006, 40, 159–185. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. Essential Amino Acids and Glutamine Regulate Induction of Mitochondrial Elongation during Autophagy. Cell Cycle 2011, 10, 2635–2639. [Google Scholar] [CrossRef]
- Tondera, D.; Grandemange, S.; Jourdain, A.; Karbowski, M.; Mattenberger, Y.; Herzig, S.; Da Cruz, S.; Clerc, P.; Raschke, I.; Merkwirth, C.; et al. SLP-2 Is Required for Stress-Induced Mitochondrial Hyperfusion. EMBO J. 2009, 28, 1589–1600. [Google Scholar] [CrossRef]
- Tezze, C.; Romanello, V.; Desbats, M.A.; Fadini, G.P.; Albiero, M.; Favaro, G.; Ciciliot, S.; Soriano, M.E.; Morbidoni, V.; Cerqua, C.; et al. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab. 2017, 25, 1374–1389.e6. [Google Scholar] [CrossRef]
- Yoon, Y.; Galloway, C.A.; Jhun, B.S.; Yu, T. Mitochondrial Dynamics in Diabetes. Antioxid. Redox Signal. 2011, 14, 439–457. [Google Scholar] [CrossRef]
- Yoon, Y. Morphological Control of Mitochondrial Bioenergetics. Front. Biosci. 2015, 20, 229–246. [Google Scholar] [CrossRef]
- Lee, H.; Smith, S.B.; Yoon, Y. The Short Variant of the Mitochondrial Dynamin OPA1 Maintains Mitochondrial Energetics and Cristae Structure. J. Biol. Chem. 2017, 292, 7115–7130. [Google Scholar] [CrossRef]
- Sessions, D.T.; Kim, K.-B.; Kashatus, J.A.; Churchill, N.; Park, K.-S.; Mayo, M.W.; Sesaki, H.; Kashatus, D.F. Opa1 and Drp1 Reciprocally Regulate Cristae Morphology, ETC Function, and NAD+ Regeneration in KRas-Mutant Lung Adenocarcinoma. Cell Rep. 2022, 41, 111818. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; Martins De Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 Controls Apoptotic Cristae Remodeling Independently from Mitochondrial Fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, Y.; Jiang, X.; Zhang, H.; Gao, Z.; Li, Y.; Fu, R.; Li, L.; Li, J.; Cui, H.; et al. ROS-Mediated Activation and Mitochondrial Translocation of CaMKII Contributes to Drp1-Dependent Mitochondrial Fission and Apoptosis in Triple-Negative Breast Cancer Cells by Isorhamnetin and Chloroquine. J. Exp. Clin. Cancer Res. 2019, 38, 225. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Hardie, D.G. New Insights into Activation and Function of the AMPK. Nat. Rev. Mol. Cell Biol. 2023, 24, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Koju, N.; Sheng, R. Mammalian Integrated Stress Responses in Stressed Organelles and Their Functions. Acta Pharmacol. Sin. 2024, 45, 1095–1114. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Shaw, R.J. The LKB1–AMPK Pathway: Metabolism and Growth Control in Tumour Suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef]
- Zhao, B.; Qiang, L.; Joseph, J.; Kalyanaraman, B.; Viollet, B.; He, Y.-Y. Mitochondrial Dysfunction Activates the AMPK Signaling and Autophagy to Promote Cell Survival. Genes Dis. 2016, 3, 82–87. [Google Scholar] [CrossRef]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular Network Formation Protects Mitochondria from Autophagosomal Degradation during Nutrient Starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef]
- Kang, S.W.S.; Haydar, G.; Taniane, C.; Farrell, G.; Arias, I.M.; Lippincott-Schwartz, J.; Fu, D. AMPK Activation Prevents and Reverses Drug-Induced Mitochondrial and Hepatocyte Injury by Promoting Mitochondrial Fusion and Function. PLoS ONE 2016, 11, e0165638. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; Van Haastert, E.S.; Eikelenboom, P.; De Vos, R.A.I.; Rozemuller, J.M.; Scheper, W. Activation of the Unfolded Protein Response in Parkinson’s Disease. Biochem. Biophys. Res. Commun. 2007, 354, 707–711. [Google Scholar] [CrossRef]
- Ma, T.; Trinh, M.A.; Wexler, A.J.; Bourbon, C.; Gatti, E.; Pierre, P.; Cavener, D.R.; Klann, E. Suppression of eIF2α Kinases Alleviates Alzheimer’s Disease–Related Plasticity and Memory Deficits. Nat. Neurosci. 2013, 16, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Millot, P.; Pujol, C.; Paquet, C.; Mouton-Liger, F. Impaired Mitochondrial Dynamics in the Blood of Patients with Alzheimer’s Disease and Lewy Body Dementia. Alzheimer’s Dement. 2023, 19, e075795. [Google Scholar] [CrossRef]
- Bilen, M.; Benhammouda, S.; Slack, R.S.; Germain, M. The Integrated Stress Response as a Key Pathway Downstream of Mitochondrial Dysfunction. Curr. Opin. Physiol. 2022, 27, 100555. [Google Scholar] [CrossRef]
- Oliveira, M.M.; Lourenco, M.V. Integrated Stress Response: Connecting ApoE4 to Memory Impairment in Alzheimer’s Disease. J. Neurosci. 2016, 36, 1053–1055. [Google Scholar] [CrossRef]
- Shpilka, T.; Haynes, C.M. The Mitochondrial UPR: Mechanisms, Physiological Functions and Implications in Ageing. Nat. Rev. Mol. Cell Biol. 2018, 19, 109–120. [Google Scholar] [CrossRef]
- Song, J.; Herrmann, J.M.; Becker, T. Quality Control of the Mitochondrial Proteome. Nat. Rev. Mol. Cell Biol. 2021, 22, 54–70. [Google Scholar] [CrossRef]
- Vannuvel, K.; Renard, P.; Raes, M.; Arnould, T. Functional and Morphological Impact of ER Stress on Mitochondria. J. Cell. Physiol. 2013, 228, 1802–1818. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, Autophagy, and Mitochondria Crosstalk Underlies the ER Stress Response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef]
- Arnould, T.; Michel, S.; Renard, P. Mitochondria Retrograde Signaling and the UPRmt: Where Are We in Mammals? Int. J. Mol. Sci. 2015, 16, 18224–18251. [Google Scholar] [CrossRef]
- Nandi, D.; Tahiliani, P.; Kumar, A.; Chandu, D. The Ubiquitin-Proteasome System. J. Biosci. 2006, 31, 137–155. [Google Scholar] [CrossRef]
- Grice, G.L.; Nathan, J.A. The Recognition of Ubiquitinated Proteins by the Proteasome. Cell. Mol. Life Sci. 2016, 73, 3497–3506. [Google Scholar] [CrossRef]
- Hsu, M.-C.; Kinefuchi, H.; Lei, L.; Kikuchi, R.; Yamano, K.; Youle, R.J. Mitochondrial YME1L1 Governs Unoccupied Protein Translocase Channels. Nat. Cell Biol. 2025, 27, 309–321. [Google Scholar] [CrossRef]
- Jin, S.M.; Youle, R.J. The Accumulation of Misfolded Proteins in the Mitochondrial Matrix Is Sensed by PINK1 to Induce PARK2/Parkin-Mediated Mitophagy of Polarized Mitochondria. Autophagy 2013, 9, 1750–1757. [Google Scholar] [CrossRef] [PubMed]
- Mohan, J.; Wollert, T. Human Ubiquitin-like Proteins as Central Coordinators in Autophagy. Interface Focus 2018, 8, 20180025. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Jensen, L.E.; Hurley, J.H. Autophagosome Biogenesis Comes out of the Black Box. Nat. Cell Biol. 2021, 23, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Simonsen, A. Diversity of Mitophagy Pathways at a Glance. J. Cell Sci. 2022, 135, jcs259748. [Google Scholar] [CrossRef]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome Formation from Membrane Compartments Enriched in Phosphatidylinositol 3-Phosphate and Dynamically Connected to the Endoplasmic Reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef]
- Simonsen, A.; Tooze, S.A. Coordination of Membrane Events during Autophagy by Multiple Class III PI3-Kinase Complexes. J. Cell Biol. 2009, 186, 773–782. [Google Scholar] [CrossRef]
- Markaki, M.; Tsagkari, D.; Tavernarakis, N. Mitophagy Mechanisms in Neuronal Physiology and Pathology during Ageing. Biophys. Rev. 2021, 13, 955–965. [Google Scholar] [CrossRef]
- Wang, S.; Long, H.; Hou, L.; Feng, B.; Ma, Z.; Wu, Y.; Zeng, Y.; Cai, J.; Zhang, D.; Zhao, G. The Mitophagy Pathway and Its Implications in Human Diseases. Signal Transduct. Target. Ther. 2023, 8, 304. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial Membrane Potential Regulates PINK1 Import and Proteolytic Destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.-F.; Karbowski, M.; Youle, R.J. Proteasome and P97 Mediate Mitophagy and Degradation of Mitofusins Induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Cooper, J.M.; Chau, K.-Y.; Rojo, M.; Schapira, A.H.V.; Taanman, J.-W. Mitofusin 1 and Mitofusin 2 Are Ubiquitinated in a PINK1/Parkin-Dependent Manner upon Induction of Mitophagy. Hum. Mol. Genet. 2010, 19, 4861–4870. [Google Scholar] [CrossRef] [PubMed]
- Ziviani, E.; Tao, R.N.; Whitworth, A.J. Drosophila Parkin Requires PINK1 for Mitochondrial Translocation and Ubiquitinates Mitofusin. Proc. Natl. Acad. Sci. USA 2010, 107, 5018–5023. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin Is Recruited Selectively to Impaired Mitochondria and Promotes Their Autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 Stabilized by Mitochondrial Depolarization Recruits Parkin to Damaged Mitochondria and Activates Latent Parkin for Mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Cohen, M.M.J.; Leboucher, G.P.; Livnat-Levanon, N.; Glickman, M.H.; Weissman, A.M. Ubiquitin–Proteasome-Dependent Degradation of a Mitofusin, a Critical Regulator of Mitochondrial Fusion. MBoC 2008, 19, 2457–2464. [Google Scholar] [CrossRef]
- Sugiura, A.; Nagashima, S.; Tokuyama, T.; Amo, T.; Matsuki, Y.; Ishido, S.; Kudo, Y.; McBride, H.M.; Fukuda, T.; Matsushita, N.; et al. MITOL Regulates Endoplasmic Reticulum-Mitochondria Contacts via Mitofusin2. Mol. Cell 2013, 51, 20–34. [Google Scholar] [CrossRef]
- Kissová, I.; Deffieu, M.; Manon, S.; Camougrand, N. Uth1p Is Involved in the Autophagic Degradation of Mitochondria. J. Biol. Chem. 2004, 279, 39068–39074. [Google Scholar] [CrossRef]
- Princely Abudu, Y.; Pankiv, S.; Mathai, B.J.; Håkon Lystad, A.; Bindesbøll, C.; Brenne, H.B.; Yoke Wui Ng, M.; Thiede, B.; Yamamoto, A.; Mutugi Nthiga, T.; et al. NIPSNAP1 and NIPSNAP2 Act as “Eat Me” Signals for Mitophagy. Dev. Cell 2019, 49, 509–525.e12. [Google Scholar] [CrossRef]
- Li, A.; Gao, M.; Liu, B.; Qin, Y.; Chen, L.; Liu, H.; Wu, H.; Gong, G. Mitochondrial Autophagy: Molecular Mechanisms and Implications for Cardiovascular Disease. Cell Death Dis. 2022, 13, 444. [Google Scholar] [CrossRef]
- Chen, M.; Chen, Z.; Wang, Y.; Tan, Z.; Zhu, C.; Li, Y.; Han, Z.; Chen, L.; Gao, R.; Liu, L.; et al. Mitophagy Receptor FUNDC1 Regulates Mitochondrial Dynamics and Mitophagy. Autophagy 2016, 12, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Landes, T.; Emorine, L.J.; Courilleau, D.; Rojo, M.; Belenguer, P.; Arnauné-Pelloquin, L. The BH3-only Bnip3 Binds to the Dynamin Opa1 to Promote Mitochondrial Fragmentation and Apoptosis by Distinct Mechanisms. EMBO Rep. 2010, 11, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, A.; McLelland, G.; Fon, E.A.; McBride, H.M. A New Pathway for Mitochondrial Quality Control: Mitochondrial-derived Vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef] [PubMed]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria Supply Membranes for Autophagosome Biogenesis during Starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes Form at ER–Mitochondria Contact Sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Zhao, T.; Huang, X.; Han, L.; Wang, X.; Cheng, H.; Zhao, Y.; Chen, Q.; Chen, J.; Cheng, H.; Xiao, R.; et al. Central Role of Mitofusin 2 in Autophagosome-Lysosome Fusion in Cardiomyocytes. J. Biol. Chem. 2012, 287, 23615–23625. [Google Scholar] [CrossRef]
- Ding, Y.; Gao, H.; Zhao, L.; Wang, X.; Zheng, M. Mitofusin 2-Deficiency Suppresses Cell Proliferation through Disturbance of Autophagy. PLoS ONE 2015, 10, e0121328. [Google Scholar] [CrossRef]
- Pich, S.; Bach, D.; Briones, P.; Liesa, M.; Camps, M.; Testar, X.; Palacín, M.; Zorzano, A. The Charcot–Marie–Tooth Type 2A Gene Product, Mfn2, up-Regulates Fuel Oxidation through Expression of OXPHOS System. Hum. Mol. Genet. 2005, 14, 1405–1415. [Google Scholar] [CrossRef]
- Gordaliza-Alaguero, I.; Sànchez-Fernàndez-de-Landa, P.; Radivojevikj, D.; Villarreal, L.; Arauz-Garofalo, G.; Gay, M.; Martinez-Vicente, M.; Seco, J.; Martín-Malpartida, P.; Vilaseca, M.; et al. Endogenous Interactomes of MFN1 and MFN2 Provide Novel Insights into Interorganelle Communication and Autophagy. Autophagy 2024, 1–22. [Google Scholar] [CrossRef]
- Khacho, M.; Clark, A.; Svoboda, D.S.; Azzi, J.; MacLaurin, J.G.; Meghaizel, C.; Sesaki, H.; Lagace, D.C.; Germain, M.; Harper, M.-E.; et al. Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell 2016, 19, 232–247. [Google Scholar]
- Inak, G.; Rybak-Wolf, A.; Lisowski, P.; Pentimalli, T.M.; Jüttner, R.; Glažar, P.; Uppal, K.; Bottani, E.; Brunetti, D.; Secker, C.; et al. Defective metabolic programming impairs early neuronal morphogenesis in neural cultures and an organoid model of Leigh syndrome. Nat. Commun. 2021, 12, 1929. [Google Scholar]
- Kawano, I.; Bazila, B.; Ježek, P.; Dlasková, A. Mitochondrial Dynamics and Cristae Shape Changes During Metabolic Reprogramming. Antioxid. Redox Signal. 2023, 39, 684–707. [Google Scholar]
- Choi, H.W.; Kim, J.H.; Chung, M.K.; Hong, Y.J.; Jang, H.S.; Seo, B.J.; Jung, T.H.; Kim, J.S.; Chung, H.M.; Byun, S.J.; et al. Mitochondrial and Metabolic Remodeling During Reprogramming and Differentiation of the Reprogrammed Cells. Stem Cells Dev. 2015, 24, 1366–1373. [Google Scholar] [CrossRef]
- Prigione, A.; Fauler, B.; Lurz, R.; Lehrach, H.; Adjaye, J. The Senescence-Related Mitochondrial/Oxidative Stress Pathway Is Repressed in Human Induced Pluripotent Stem Cells. Stem Cells 2010, 28, 721–733. [Google Scholar] [CrossRef]
- Kasahara, A.; Cipolat, S.; Chen, Y.; Dorn, G.W.; Scorrano, L. Mitochondrial Fusion Directs Cardiomyocyte Differentiation via Calcineurin and Notch Signaling. Science 2013, 342, 734–737. [Google Scholar] [CrossRef]
- Yi, S.; Cui, C.; Huang, X.; Yin, X.; Li, Y.; Wen, J.; Luan, Q. MFN2 Silencing Promotes Neural Differentiation of Embryonic Stem Cells via the Akt Signaling Pathway. J. Cell. Physiol. 2020, 235, 1051–1064. [Google Scholar] [CrossRef]
- Fang, D.; Yan, S.; Yu, Q.; Chen, D.; Yan, S.S. Mfn2 Is Required for Mitochondrial Development and Synapse Formation in Human Induced Pluripotent Stem Cells/hiPSC Derived Cortical Neurons. Sci. Rep. 2016, 6, 31462. [Google Scholar] [CrossRef]
- Chung, S.; Dzeja, P.P.; Faustino, R.S.; Perez-Terzic, C.; Behfar, A.; Terzic, A. Mitochondrial Oxidative Metabolism Is Required for the Cardiac Differentiation of Stem Cells. Nat. Rev. Cardiol. 2007, 4, S60–S67. [Google Scholar] [CrossRef]
- Özbudak, E.M.; Tassy, O.; Pourquié, O. Spatiotemporal Compartmentalization of Key Physiological Processes during Muscle Precursor Differentiation. Proc. Natl. Acad. Sci. USA 2010, 107, 4224–4229. [Google Scholar] [CrossRef]
- Forni, M.F.; Peloggia, J.; Trudeau, K.; Shirihai, O.; Kowaltowski, A.J. Murine Mesenchymal Stem Cell Commitment to Differentiation Is Regulated by Mitochondrial Dynamics. Stem Cells 2016, 34, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.M.; Kwon, S.; Pak, Y.K.; Seol, H.W.; Choi, Y.M.; Park, D.J.; Park, K.S.; Lee, H.K. Dynamic Changes in Mitochondrial Biogenesis and Antioxidant Enzymes during the Spontaneous Differentiation of Human Embryonic Stem Cells. Biochem. Biophys. Res. Commun. 2006, 348, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, Z.; Chang, C.; Yang, Z.; Wang, P.; Fu, H.; Wei, X.; Chen, E.; Tan, S.; Huang, W.; et al. A Bioenergetic Shift Is Required for Spermatogonial Differentiation. Cell Discov. 2020, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Sun, Y.; Sun, Q.; Zhang, J.; Jiang, M.; Chang, C.; Huang, X.; Wang, C.; Wang, P.; Zhang, Z.; et al. MFN2 Plays a Distinct Role from MFN1 in Regulating Spermatogonial Differentiation. Stem Cell Rep. 2020, 14, 803–817. [Google Scholar] [CrossRef]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef]
- Youle, R.J.; Van Der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Kochan, S.M.V.; Malo, M.C.; Jevtic, M.; Jahn-Kelleter, H.M.; Wani, G.A.; Ndoci, K.; Pérez-Revuelta, L.; Gaedke, F.; Schäffner, I.; Lie, D.C.; et al. Enhanced Mitochondrial Fusion during a Critical Period of Synaptic Plasticity in Adult-Born Neurons. Neuron 2024, 112, 1997–2014.e6. [Google Scholar] [CrossRef]
- Virga, D.M.; Hamilton, S.; Osei, B.; Morgan, A.; Kneis, P.; Zamponi, E.; Park, N.J.; Hewitt, V.L.; Zhang, D.; Gonzalez, K.C.; et al. Activity-Dependent Compartmentalization of Dendritic Mitochondria Morphology through Local Regulation of Fusion-Fission Balance in Neurons in Vivo. Nat. Commun. 2024, 15, 2142. [Google Scholar] [CrossRef]
- Zaninello, M.; Baptista, P.; Duarte, F.V. Mitochondrial Dynamics and mRNA Translation: A Local Synaptic Tale. Biology 2024, 13, 746. [Google Scholar] [CrossRef]
- Cioni, J.-M.; Lin, J.Q.; Holtermann, A.V.; Koppers, M.; Jakobs, M.A.H.; Azizi, A.; Turner-Bridger, B.; Shigeoka, T.; Franze, K.; Harris, W.A.; et al. Late Endosomes Act as mRNA Translation Platforms and Sustain Mitochondria in Axons. Cell 2019, 176, 56–72.e15. [Google Scholar] [CrossRef] [PubMed]
- Loedige, I.; Baranovskii, A.; Mendonsa, S.; Dantsuji, S.; Popitsch, N.; Breimann, L.; Zerna, N.; Cherepanov, V.; Milek, M.; Ameres, S.; et al. mRNA Stability and m6A Are Major Determinants of Subcellular mRNA Localization in Neurons. Mol. Cell 2023, 83, 2709–2725.e10. [Google Scholar] [CrossRef] [PubMed]
- Niu, M.; Cao, W.; Wang, Y.; Zhu, Q.; Luo, J.; Wang, B.; Zheng, H.; Weitz, D.A.; Zong, C. Droplet-Based Transcriptome Profiling of Individual Synapses. Nat. Biotechnol. 2023, 41, 1332–1344. [Google Scholar] [CrossRef] [PubMed]
- Züchner, S.; De Jonghe, P.; Jordanova, A.; Claeys, K.G.; Guergueltcheva, V.; Cherninkova, S.; Hamilton, S.R.; Van Stavern, G.; Krajewski, K.M.; Stajich, J.; et al. Axonal Neuropathy with Optic Atrophy Is Caused by Mutations in Mitofusin 2. Ann. Neurol. 2006, 59, 276–281. [Google Scholar] [CrossRef]
- Vallat, J.-M.; Ouvrier, R.A.; Pollard, J.D.; Magdelaine, C.; Zhu, D.; Nicholson, G.A.; Grew, S.; Ryan, M.M.; Funalot, B. Histopathological Findings in Hereditary Motor and Sensory Neuropathy of Axonal Type With Onset in Early Childhood Associated With Mitofusin 2 Mutations. J. Neuropathol. Exp. Neurol. 2008, 67, 1097–1102. [Google Scholar] [CrossRef]
- Verhoeven, K. MFN2 Mutation Distribution and Genotype/Phenotype Correlation in Charcot-Marie-Tooth Type 2. Brain 2006, 129, 2093–2102. [Google Scholar] [CrossRef]
- Baloh, R.H.; Schmidt, R.E.; Pestronk, A.; Milbrandt, J. Altered Axonal Mitochondrial Transport in the Pathogenesis of Charcot-Marie-Tooth Disease from Mitofusin 2 Mutations. J. Neurosci. 2007, 27, 422–430. [Google Scholar] [CrossRef]
- Detmer, S.A.; Chan, D.C. Complementation between Mouse Mfn1 and Mfn2 Protects Mitochondrial Fusion Defects Caused by CMT2A Disease Mutations. J. Cell Biol. 2007, 176, 405–414. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Critical Dependence of Neurons on Mitochondrial Dynamics. Curr. Opin. Cell Biol. 2006, 18, 453–459. [Google Scholar] [CrossRef]
- Feely, S.M.E.; Laura, M.; Siskind, C.E.; Sottile, S.; Davis, M.; Gibbons, V.S.; Reilly, M.M.; Shy, M.E. MFN2 Mutations Cause Severe Phenotypes in Most Patients with CMT2A. Neurology 2011, 76, 1690–1696. [Google Scholar] [CrossRef]
- Larrea, D.; Pera, M.; Gonnelli, A.; Quintana–Cabrera, R.; Akman, H.O.; Guardia-Laguarta, C.; Velasco, K.R.; Area-Gomez, E.; Dal Bello, F.; De Stefani, D.; et al. MFN2 Mutations in Charcot–Marie–Tooth Disease Alter Mitochondria-Associated ER Membrane Function but Do Not Impair Bioenergetics. Hum. Mol. Genet. 2019, 28, 1782–1800. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W. Mitofusin 2 Dysfunction and Disease in Mice and Men. Front. Physiol. 2020, 11, 782. [Google Scholar] [CrossRef]
- Rizzo, F.; Ronchi, D.; Salani, S.; Nizzardo, M.; Fortunato, F.; Bordoni, A.; Stuppia, G.; Del Bo, R.; Piga, D.; Fato, R.; et al. Selective Mitochondrial Depletion, Apoptosis Resistance, and Increased Mitophagy in Human Charcot-Marie-Tooth 2A Motor Neurons. Hum. Mol. Genet. 2016, 25, 4266–4281. [Google Scholar] [CrossRef]
- Hoshino, A.; Ariyoshi, M.; Okawa, Y.; Kaimoto, S.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Ikeda, K.; Ueyama, T.; Ogata, T.; et al. Inhibition of P53 Preserves Parkin-Mediated Mitophagy and Pancreatic β-Cell Function in Diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 3116–3121. [Google Scholar] [CrossRef] [PubMed]
- Van Lent, J.; Verstraelen, P.; Asselbergh, B.; Adriaenssens, E.; Mateiu, L.; Verbist, C.; De Winter, V.; Eggermont, K.; Van Den Bosch, L.; De Vos, W.H.; et al. Induced Pluripotent Stem Cell-Derived Motor Neurons of CMT Type 2 Patients Reveal Progressive Mitochondrial Dysfunction. Brain 2021, 144, 2471–2485. [Google Scholar] [CrossRef]
- Yang, T.-H.; Kang, E.Y.-C.; Lin, P.-H.; Yu, B.B.-C.; Wang, J.H.-H.; Chen, V.; Wang, N.-K. Mitochondria in Retinal Ganglion Cells: Unraveling the Metabolic Nexus and Oxidative Stress. IJMS 2024, 25, 8626. [Google Scholar] [CrossRef]
- Carelli, V.; Musumeci, O.; Caporali, L.; Zanna, C.; La Morgia, C.; Del Dotto, V.; Porcelli, A.M.; Rugolo, M.; Valentino, M.L.; Iommarini, L.; et al. Syndromic Parkinsonism and Dementia Associated with OPA 1 Missense Mutations. Ann. Neurol. 2015, 78, 21–38. [Google Scholar] [CrossRef]
- Kane, M.S.; Alban, J.; Desquiret-Dumas, V.; Gueguen, N.; Ishak, L.; Ferre, M.; Amati-Bonneau, P.; Procaccio, V.; Bonneau, D.; Lenaers, G.; et al. Autophagy Controls the Pathogenicity of OPA 1 Mutations in Dominant Optic Atrophy. J. Cell. Mol. Med. 2017, 21, 2284–2297. [Google Scholar] [CrossRef]
- Sun, S.; Erchova, I.; Sengpiel, F.; Votruba, M. Opa1 Deficiency Leads to Diminished Mitochondrial Bioenergetics With Compensatory Increased Mitochondrial Motility. Investig. Ophthalmol. Vis. Sci. 2020, 61, 42. [Google Scholar] [CrossRef]
- Sun, C.; Wu, X.; Bai, H.-X.; Wang, C.; Liu, Z.; Yang, C.; Lu, Y.; Jiang, P. OPA1 Haploinsufficiency Due to a Novel Splicing Variant Resulting in Mitochondrial Dysfunction without Mitochondrial DNA Depletion. Ophthalmic Genet. 2021, 42, 45–52. [Google Scholar] [CrossRef]
- Liao, C.; Ashley, N.; Diot, A.; Morten, K.; Phadwal, K.; Williams, A.; Fearnley, I.; Rosser, L.; Lowndes, J.; Fratter, C.; et al. Dysregulated Mitophagy and Mitochondrial Organization in Optic Atrophy Due to OPA1 Mutations. Neurology 2017, 88, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Sarzi, E.; Angebault, C.; Seveno, M.; Gueguen, N.; Chaix, B.; Bielicki, G.; Boddaert, N.; Mausset-Bonnefont, A.-L.; Cazevieille, C.; Rigau, V.; et al. The Human OPA1delTTAG Mutation Induces Premature Age-Related Systemic Neurodegeneration in Mouse. Brain 2012, 135, 3599–3613. [Google Scholar] [CrossRef] [PubMed]
- Diot, A.; Agnew, T.; Sanderson, J.; Liao, C.; Carver, J.; Neves, R.P.D.; Gupta, R.; Guo, Y.; Waters, C.; Seto, S.; et al. Validating the RedMIT/GFP-LC3 Mouse Model by Studying Mitophagy in Autosomal Dominant Optic Atrophy Due to the OPA1Q285STOP Mutation. Front. Cell Dev. Biol. 2018, 6, 103. [Google Scholar] [CrossRef] [PubMed]
- Zaninello, M.; Palikaras, K.; Naon, D.; Iwata, K.; Herkenne, S.; Quintana-Cabrera, R.; Semenzato, M.; Grespi, F.; Ross-Cisneros, F.N.; Carelli, V.; et al. Inhibition of Autophagy Curtails Visual Loss in a Model of Autosomal Dominant Optic Atrophy. Nat. Commun. 2020, 11, 4029. [Google Scholar] [CrossRef]
- Zaninello, M.; Palikaras, K.; Sotiriou, A.; Tavernarakis, N.; Scorrano, L. Sustained Intracellular Calcium Rise Mediates Neuronal Mitophagy in Models of Autosomal Dominant Optic Atrophy. Cell Death Differ. 2022, 29, 167–177. [Google Scholar] [CrossRef]
- Moulis, M.F.; Millet, A.M.; Daloyau, M.; Miquel, M.; Ronsin, B.; Wissinger, B.; Arnauné-Pelloquin, L.; Belenguer, P. OPA1 Haploinsufficiency Induces a BNIP3-dependent Decrease in Mitophagy in Neurons: Relevance to Dominant Optic Atrophy. J. Neurochem. 2017, 140, 485–494. [Google Scholar] [CrossRef]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, Å.B. Microtubule-Associated Protein 1 Light Chain 3 (LC3) Interacts with Bnip3 Protein to Selectively Remove Endoplasmic Reticulum and Mitochondria via Autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef]
- Wang, D.; Wang, J.; Bonamy, G.M.C.; Meeusen, S.; Brusch, R.G.; Turk, C.; Yang, P.; Schultz, P.G. A Small Molecule Promotes Mitochondrial Fusion in Mammalian Cells. Angew. Chem. Int. Ed. 2012, 51, 9302–9305. [Google Scholar] [CrossRef]
- Lin, Y.; Wang, D.; Li, B.; Wang, J.; Xu, L.; Sun, X.; Ji, K.; Yan, C.; Liu, F.; Zhao, Y. Targeting DRP1 with Mdivi-1 to Correct Mitochondrial Abnormalities in ADOA+ Syndrome. JCI Insight 2024, 9, e180582. [Google Scholar] [CrossRef]
- Szabo, A.; Sumegi, K.; Fekete, K.; Hocsak, E.; Debreceni, B.; Setalo, G.; Kovacs, K.; Deres, L.; Kengyel, A.; Kovacs, D.; et al. Activation of Mitochondrial Fusion Provides a New Treatment for Mitochondria-Related Diseases. Biochem. Pharmacol. 2018, 150, 86–96. [Google Scholar] [CrossRef]
- Alberti, C.; Rizzo, F.; Anastasia, A.; Comi, G.; Corti, S.; Abati, E. Charcot-Marie-Tooth Disease Type 2A: An Update on Pathogenesis and Therapeutic Perspectives. Neurobiol. Dis. 2024, 193, 106467. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.S.V.; Trigano, M.; Freeman, T.; Varrichio, C.; Kandaswamy, D.K.; Newland, B.; Brancale, A.; Rozanowska, M.; Votruba, M. New Avenues for Therapy in Mitochondrial Optic Neuropathies. Ther. Adv. Rare Dis. 2021, 2, 26330040211029037. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kodam, P.; Maity, S. Targeting Protein Interaction Networks in Mitochondrial Dynamics for Neurodegenerative Diseases. J. Proteins Proteom. 2024, 15, 309–328. [Google Scholar] [CrossRef]
- Loiseau, D.; Chevrollier, A.; Verny, C.; Guillet, V.; Gueguen, N.; Pou De Crescenzo, M.; Ferré, M.; Malinge, M.; Guichet, A.; Nicolas, G.; et al. Mitochondrial Coupling Defect in Charcot–Marie–Tooth Type 2A Disease. Ann. Neurol. 2007, 61, 315–323. [Google Scholar] [CrossRef]
- Amiott, E.A.; Lott, P.; Soto, J.; Kang, P.B.; McCaffery, J.M.; DiMauro, S.; Abel, E.D.; Flanigan, K.M.; Lawson, V.H.; Shaw, J.M. Mitochondrial Fusion and Function in Charcot–Marie–Tooth Type 2A Patient Fibroblasts with Mitofusin 2 Mutations. Exp. Neurol. 2008, 211, 115–127. [Google Scholar] [CrossRef]
- Rouzier, C.; Bannwarth, S.; Chaussenot, A.; Chevrollier, A.; Verschueren, A.; Bonello-Palot, N.; Fragaki, K.; Cano, A.; Pouget, J.; Pellissier, J.-F.; et al. Reply: MFN2 Mutations Cause Compensatory Mitochondrial DNA Proliferation. Brain 2012, 135, e220. [Google Scholar] [CrossRef]
- Vielhaber, S.; Debska-Vielhaber, G.; Peeva, V.; Schoeler, S.; Kudin, A.P.; Minin, I.; Schreiber, S.; Dengler, R.; Kollewe, K.; Zuschratter, W.; et al. Mitofusin 2 Mutations Affect Mitochondrial Function by Mitochondrial DNA Depletion. Acta Neuropathol 2013, 125, 245–256. [Google Scholar] [CrossRef]
- Saporta, M.A.; Dang, V.; Volfson, D.; Zou, B.; Xie, X.; Adebola, A.; Liem, R.K.; Shy, M.; Dimos, J.T. Axonal Charcot–Marie–Tooth Disease Patient-Derived Motor Neurons Demonstrate Disease-Specific Phenotypes Including Abnormal Electrophysiological Properties. Exp. Neurol. 2015, 263, 190–199. [Google Scholar] [CrossRef]
- Ohara, R.; Imamura, K.; Morii, F.; Egawa, N.; Tsukita, K.; Enami, T.; Shibukawa, R.; Mizuno, T.; Nakagawa, M.; Inoue, H. Modeling Drug-Induced Neuropathy Using Human iPSCs for Predictive Toxicology. Clin. Pharmacol. Ther. 2017, 101, 754–762. [Google Scholar] [CrossRef]
- Zanfardino, P.; Longo, G.; Amati, A.; Morani, F.; Picardi, E.; Girolamo, F.; Pafundi, M.; Cox, S.N.; Manzari, C.; Tullo, A.; et al. Mitofusin 2 Mutation Drives Cell Proliferation in Charcot-Marie-Tooth 2A Fibroblasts. Hum. Mol. Genet. 2023, 32, 333–350. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.; Liang, X.; Lu, Y.; Zhu, L.; Fu, R.; Ji, Y.; Fan, W.; Chen, J.; Lin, B.; et al. A Novel ADOA-Associated OPA1 Mutation Alters the Mitochondrial Function, Membrane Potential, ROS Production and Apoptosis. Sci. Rep. 2017, 7, 5704. [Google Scholar] [CrossRef] [PubMed]
- Zanfardino, P.; Amati, A.; Doccini, S.; Cox, S.N.; Tullo, A.; Longo, G.; D’Erchia, A.; Picardi, E.; Nesti, C.; Santorelli, F.M.; et al. OPA 1 Mutation Affects Autophagy and Triggers Senescence in Autosomal Dominant Optic Atrophy plus Fibroblasts. Hum. Mol. Genet. 2024, 33, 768–786. [Google Scholar] [CrossRef] [PubMed]
- Zanfardino, P.; Amati, A.; Petracca, E.A.; Santorelli, F.M.; Petruzzella, V. Torin1 restores proliferation rate in Charcot-Marie-Tooth disease type 2A cells harbouring MFN2 (mitofusin 2) mutation. Acta Myol. 2022, 41, 201–206. [Google Scholar]
- Zanfardino, P.; Petruzzella, V. Autophagy and Proliferation Are Dysregulated in Charcot-Marie-Tooth Disease Type 2A Cells Harboring MFN2 (Mitofusin 2) Mutation. Autophagy Rep. 2022, 1, 537–541. [Google Scholar] [CrossRef]
- Xu, K.; Chen, G.; Li, X.; Wu, X.; Chang, Z.; Xu, J.; Zhu, Y.; Yin, P.; Liang, X.; Dong, L. MFN2 Suppresses Cancer Progression through Inhibition of mTORC2/Akt Signaling. Sci. Rep. 2017, 7, 41718. [Google Scholar] [CrossRef]
- Xue, R.; Meng, Q.; Lu, D.; Liu, X.; Wang, Y.; Hao, J. Mitofusin2 Induces Cell Autophagy of Pancreatic Cancer through Inhibiting the PI3K/Akt/mTOR Signaling Pathway. Oxidative Med. Cell. Longev. 2018, 2018, 2798070. [Google Scholar] [CrossRef]
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK Activation Protects Cells from Oxidative Stress-induced Senescence via Autophagic Flux Restoration and Intracellular NAD+ Elevation. Aging Cell 2016, 15, 416–427. [Google Scholar] [CrossRef]
- Tai, H.; Wang, Z.; Gong, H.; Han, X.; Zhou, J.; Wang, X.; Wei, X.; Ding, Y.; Huang, N.; Qin, J.; et al. Autophagy Impairment with Lysosomal and Mitochondrial Dysfunction Is an Important Characteristic of Oxidative Stress-Induced Senescence. Autophagy 2017, 13, 99–113. [Google Scholar] [CrossRef]
- Kazyken, D.; Magnuson, B.; Bodur, C.; Acosta-Jaquez, H.A.; Zhang, D.; Tong, X.; Barnes, T.M.; Steinl, G.K.; Patterson, N.E.; Altheim, C.H.; et al. AMPK Directly Activates mTORC2 to Promote Cell Survival during Acute Energetic Stress. Sci. Signal. 2019, 12, eaav3249. [Google Scholar] [CrossRef] [PubMed]
- Romanello, V.; Scalabrin, M.; Albiero, M.; Blaauw, B.; Scorrano, L.; Sandri, M. Inhibition of the Fission Machinery Mitigates OPA1 Impairment in Adult Skeletal Muscles. Cells 2019, 8, 597. [Google Scholar] [CrossRef]
- Lee, S.; Jeong, S.-Y.; Lim, W.-C.; Kim, S.; Park, Y.-Y.; Sun, X.; Youle, R.J.; Cho, H. Mitochondrial Fission and Fusion Mediators, hFis1 and OPA1, Modulate Cellular Senescence. J. Biol. Chem. 2007, 282, 22977–22983. [Google Scholar] [CrossRef]
- Stab, B.R.; Martinez, L.; Grismaldo, A.; Lerma, A.; Gutiérrez, M.L.; Barrera, L.A.; Sutachan, J.J.; Albarracín, S.L. Mitochondrial Functional Changes Characterization in Young and Senescent Human Adipose Derived MSCs. Front. Aging Neurosci. 2016, 8, 299. [Google Scholar] [CrossRef]
- Navratil, M.; Terman, A.; Arriaga, E.A. Giant Mitochondria Do Not Fuse and Exchange Their Contents with Normal Mitochondria. Exp. Cell Res. 2008, 314, 164–172. [Google Scholar] [CrossRef]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef]
- Lamming, D.W.; Mihaylova, M.M.; Katajisto, P.; Baar, E.L.; Yilmaz, O.H.; Hutchins, A.; Gultekin, Y.; Gaither, R.; Sabatini, D.M. Depletion of Rictor, an Essential Protein Component of mTORC2, Decreases Male Lifespan. Aging Cell 2014, 13, 911–917. [Google Scholar] [CrossRef]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-Induced Insulin Resistance Is Mediated by mTORC2 Loss and Uncoupled from Longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Rapamycin, Proliferation and Geroconversion to Senescence. Cell Cycle 2018, 17, 2655–2665. [Google Scholar] [CrossRef]
- LaBaer, J.; Garrett, M.D.; Stevenson, L.F.; Slingerland, J.M.; Sandhu, C.; Chou, H.S.; Fattaey, A.; Harlow, E. New Functional Activities for the P21 Family of CDK Inhibitors. Genes Dev. 1997, 11, 847–862. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda Di Fagagna, F. Cellular Senescence: When Bad Things Happen to Good Cells. Nat Rev Mol Cell Biol 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Chen, G.; Ding, X.-F.; Bouamar, H.; Pressley, K.; Sun, L.-Z. Everolimus Induces G1 Cell Cycle Arrest through Autophagy-Mediated Protein Degradation of Cyclin D1 in Breast Cancer Cells. Am. J. Physiol.-Cell Physiol. 2019, 317, C244–C252. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Bhatia-Kiššová, I.; Camougrand, N. Mitophagy Is Not Induced by Mitochondrial Damage but Plays a Role in the Regulation of Cellular Autophagic Activity. Autophagy 2013, 9, 1897–1899. [Google Scholar] [CrossRef]
- Eskelinen, E.-L. Maturation of Autophagic Vacuoles in Mammalian Cells. Autophagy 2005, 1, 1–10. [Google Scholar] [CrossRef]
- Morán, M.; Delmiro, A.; Blázquez, A.; Ugalde, C.; Arenas, J.; Martín, M.A. Bulk Autophagy, but Not Mitophagy, Is Increased in Cellular Model of Mitochondrial Disease. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2014, 1842, 1059–1070. [Google Scholar] [CrossRef]
- Deffieu, M.; Bhatia-Kiššová, I.; Salin, B.; Klionsky, D.J.; Pinson, B.; Manon, S.; Camougrand, N. Increased Levels of Reduced Cytochrome b and Mitophagy Components Are Required to Trigger Nonspecific Autophagy Following Induced Mitochondrial Dysfunction. J. Cell Sci. 2013, 126, 415–426. [Google Scholar] [CrossRef]
- Frank, M.; Duvezin-Caubet, S.; Koob, S.; Occhipinti, A.; Jagasia, R.; Petcherski, A.; Ruonala, M.O.; Priault, M.; Salin, B.; Reichert, A.S. Mitophagy Is Triggered by Mild Oxidative Stress in a Mitochondrial Fission Dependent Manner. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2012, 1823, 2297–2310. [Google Scholar] [CrossRef]
- Hurko, O. Drug Development for Rare Mitochondrial Disorders. Neurotherapeutics 2013, 10, 286–306. [Google Scholar] [CrossRef]
- Thangaraj, K.; Khan, N.; Govindaraj, P.; Meena, A. Mitochondrial Disorders: Challenges in Diagnosis & Treatment. Indian J. Med. Res. 2015, 141, 13. [Google Scholar]
- Civiletto, G.; Dogan, S.A.; Cerutti, R.; Fagiolari, G.; Moggio, M.; Lamperti, C.; Benincá, C.; Viscomi, C.; Zeviani, M. Rapamycin Rescues Mitochondrial Myopathy via Coordinated Activation of Autophagy and Lysosomal Biogenesis. EMBO Mol. Med. 2018, 10, e8799. [Google Scholar] [CrossRef] [PubMed]
- Mannick, J.B.; Del Giudice, G.; Lattanzi, M.; Valiante, N.M.; Praestgaard, J.; Huang, B.; Lonetto, M.A.; Maecker, H.T.; Kovarik, J.; Carson, S.; et al. mTOR Inhibition Improves Immune Function in the Elderly. Sci. Transl. Med. 2014, 6, 268ra179. [Google Scholar] [CrossRef] [PubMed]
- Barriocanal-Casado, E.; Hidalgo-Gutiérrez, A.; Raimundo, N.; González-García, P.; Acuña-Castroviejo, D.; Escames, G.; López, L.C. Rapamycin Administration Is Not a Valid Therapeutic Strategy for Every Case of Mitochondrial Disease. EBioMedicine 2019, 42, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.-X.; Wang, S.-F.; Tan, Y.; Song, J.-X.; Zhu, Z.; Wang, Z.-Y.; Wu, M.-Y.; Cai, C.-Z.; Huang, Z.-J.; Tan, J.-Q.; et al. Pharmacological Enhancement of TFEB-Mediated Autophagy Alleviated Neuronal Death in Oxidative Stress-Induced Parkinson’s Disease Models. Cell Death Dis. 2020, 11, 128. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanfardino, P.; Amati, A.; Perrone, M.; Petruzzella, V. The Balance of MFN2 and OPA1 in Mitochondrial Dynamics, Cellular Homeostasis, and Disease. Biomolecules 2025, 15, 433. https://doi.org/10.3390/biom15030433
Zanfardino P, Amati A, Perrone M, Petruzzella V. The Balance of MFN2 and OPA1 in Mitochondrial Dynamics, Cellular Homeostasis, and Disease. Biomolecules. 2025; 15(3):433. https://doi.org/10.3390/biom15030433
Chicago/Turabian StyleZanfardino, Paola, Alessandro Amati, Mirko Perrone, and Vittoria Petruzzella. 2025. "The Balance of MFN2 and OPA1 in Mitochondrial Dynamics, Cellular Homeostasis, and Disease" Biomolecules 15, no. 3: 433. https://doi.org/10.3390/biom15030433
APA StyleZanfardino, P., Amati, A., Perrone, M., & Petruzzella, V. (2025). The Balance of MFN2 and OPA1 in Mitochondrial Dynamics, Cellular Homeostasis, and Disease. Biomolecules, 15(3), 433. https://doi.org/10.3390/biom15030433