
Binding of Pro-Inflammatory Proteins S100A8 or S100A9 to Amyloid-β Peptide Suppresses Its Fibrillation

 , ,
, ,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Bio-Layer Interferometry Studies
2.3. ThT Fluorescence Assay
2.4. Transmission Electron Microscopy
2.5. Structural Modeling of Aβ40-S100 Complexes
2.6. Dynamic Light Scattering Measurements
2.7. Chemical Cross-Linking of Proteins
3. Results
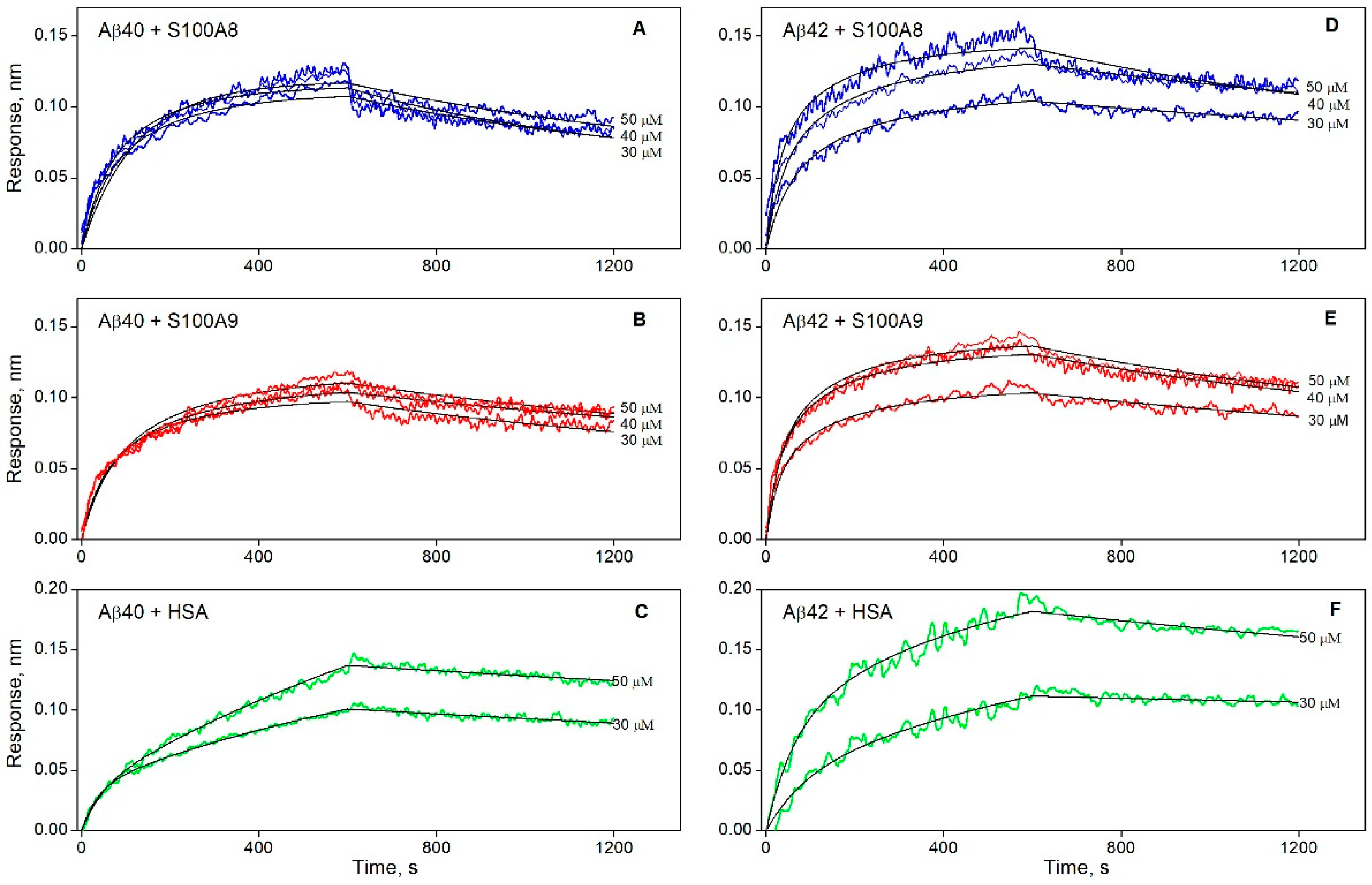
3.1. The Interaction of S100A8/S100A9 Proteins with Aβ Peptides
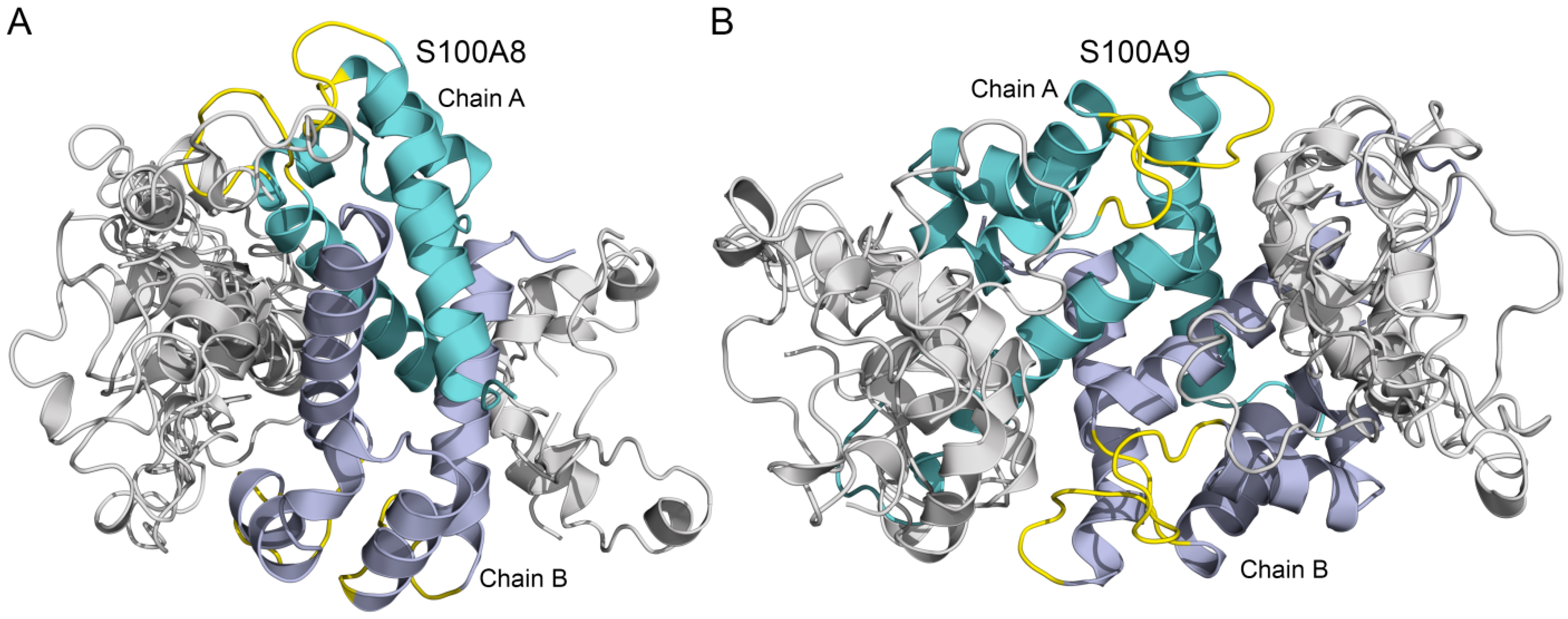
3.2. Modeling of Aβ40-S100A8/S100A9 Complexes
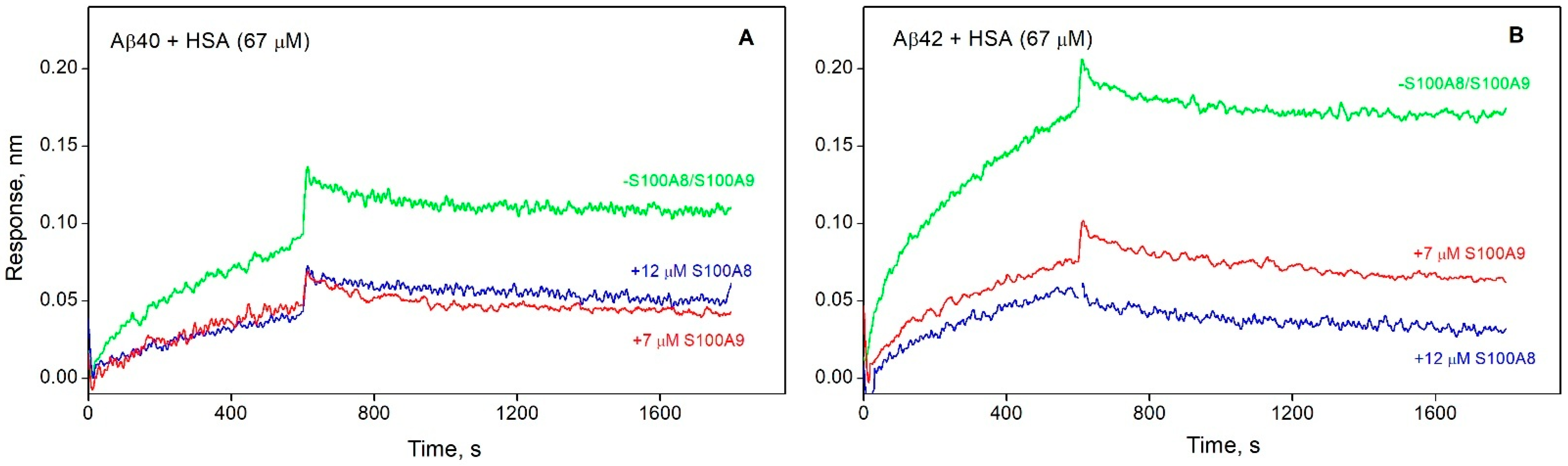
3.3. S100A8/S100A9 Prevent HSA Interaction with Monomeric Aβ
3.4. Effect of S100A8/S100A9 on Aβ Fibrillation In Vitro
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Evaluation of Distribution of Aβ40 and Aβ42 Between S100A8, S100A9, and HSA
Aβ40 + S100A8 Aβ40·S100A8 Aβ40·S100A8 | (Table 1) | (Table 1) | ||
| a | l | x | ||
| Aβ40 + S100A9 Aβ40·S100A9 | (Table 1) | (Table 1) | ||
| a | m | y | ||
| Aβ40 + HSA Aβ40·HSA | (Table 2) | (Table 2) | ||
| a | n | z | ||
| Aβ42 + S100A8 Aβ42·S100A8 | (Table 1) | (Table 1) | ||
| b | l | p | ||
| Aβ42 + S100A9 Aβ42·S100A9 | (Table 1) | (Table 1) | ||
| b | m | q | ||
| Aβ42 + HSA Aβ42·HSA | (Table 2) | (Table 2) | ||
| b | n | r | ||
- The interactions of Aβ40, Aβ42, S100A8, S100A9, and HSA with other molecules are neglected.
- The system is in equilibrium, and the equilibrium constants are independent of the other processes in the system.
- Only the highest equilibrium association constant is taken into consideration for Aβ binding to HSA.
- The following total concentrations of the proteins were used (the Aβ and HSA concentrations equivalent to those in CSF; the total S100A8 and S100A9 contents corresponding to those in the inflammatory focus):
| Protein | Mass Balance | Total Concentration, M | References |
|---|---|---|---|
| Aβ40 | a + x + y + z | 1.7·10−9 | [19,83] |
| Aβ42 | b + p + q + r | 9·10−11 | [19,83] |
| S100A8 | l + x + p | 9.3·10−7 | [86] |
| S100A9 | m + y + q | 3.8·10−6 | [86] |
| HSA | n + z + r | 3.0·10−6 | [89,90] |
| Component | Concentration, M | Concentration, M | Symbol |
| Aβ40 | 1.0·10−10 | 1.02·10−10 | a |
| Aβ40·S100A8 | 1.9·10−11 | 1.88·10−11 | x |
| Aβ40·S100A9 | 1.8·10−10 | 1.83·10−10 | y |
| Aβ40·HSA | 1.4·10−9 | 1.39·10−9 | z |
| Aβ42 | 8.0·10−12 | 8.00·10−12 | b |
| Aβ42·S100A8 | 5.6·10−12 | 5.59·10−12 | p |
| Aβ42·S100A9 | 2.6·10−11 | 2.64·10−11 | q |
| Aβ42·HSA | 5.0·10−11 | 5.00·10−11 | r |
References
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014, 88, 640–651. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Dorszewska, J.; Prendecki, M.; Oczkowska, A.; Dezor, M.; Kozubski, W. Molecular Basis of Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 952–963. [Google Scholar] [CrossRef]
- Hosseininasab, S.S.M.; Ebrahimi, R.; Yaghoobpoor, S.; Kazemi, K.; Khakpour, Y.; Hajibeygi, R.; Mohamadkhani, A.; Fathi, M.; Vakili, K.; Tavasol, A.; et al. Alzheimer’s disease and infectious agents: A comprehensive review of pathogenic mechanisms and microRNA roles. Front. Neurosci. 2024, 18, 1513095. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox. Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Yang, Z.; Zou, Y.; Wang, L. Neurotransmitters in Prevention and Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 3841. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. eBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef]
- Liu, F.; Liang, Z.H.; Gong, C.-X. Hyperphosphorylation of tau and protein phosphatases in Alzheimer disease. Panminerva Medica 2006, 48, 97–108. [Google Scholar] [PubMed]
- Li, X.; Ba, Z.; Huang, J.; Chen, J.; Jiang, J.; Huang, N.; Luo, Y. Comprehensive review on Alzheimer’s disease: From the posttranslational modifications of Tau to corresponding treatments. Ibrain 2024, 10, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.P.; Clark, I.A.; Vissel, B. Questions concerning the role of amyloid-β in the definition, aetiology and diagnosis of Alzheimer’s disease. Acta Neuropathol. 2018, 136, 663–689. [Google Scholar] [CrossRef]
- Paroni, G.; Bisceglia, P.; Seripa, D. Understanding the Amyloid Hypothesis in Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2019, 68, 493–510. [Google Scholar] [CrossRef]
- Karran, E.; De Strooper, B. The amyloid hypothesis in Alzheimer disease: New insights from new therapeutics. Nat. Rev. Drug Discov. 2022, 21, 306–318. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Datta, D.; Del Tredici, K.; Braak, H. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2021, 17, 115–124. [Google Scholar] [CrossRef]
- Qi, X.; Nizamutdinov, D.; Yi, S.S.; Wu, E.; Huang, J.H. Disease Modifying Monoclonal Antibodies and Symptomatic Pharmacological Treatment for Alzheimer’s Disease. Biomedicines 2024, 12, 2636. [Google Scholar] [CrossRef]
- Bibl, M.; Gallus, M.; Welge, V.; Lehmann, S.; Sparbier, K.; Esselmann, H.; Wiltfang, J. Characterization of cerebrospinal fluid aminoterminally truncated and oxidized amyloid-β peptides. PROTEOMICS—Clin. Appl. 2012, 6, 163–169. [Google Scholar] [CrossRef]
- van Steenoven, I.; van der Flier, W.M.; Scheltens, P.; Teunissen, C.E.; Lemstra, A.W. Amyloid-β peptides in cerebrospinal fluid of patients with dementia with Lewy bodies. Alzheimer’s Res. Ther. 2019, 11, 83. [Google Scholar] [CrossRef]
- Hoshi, M.; Sato, M.; Matsumoto, S.; Noguchi, A.; Yasutake, K.; Yoshida, N.; Sato, K. Spherical aggregates of beta-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3beta. Proc. Natl. Acad. Sci. USA 2003, 100, 6370–6375. [Google Scholar] [CrossRef]
- Bitan, G.; Kirkitadze, M.D.; Lomakin, A.; Vollers, S.S.; Benedek, G.B.; Teplow, D.B. Amyloid beta -protein (Abeta) assembly: Abeta 40 and Abeta 42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA 2003, 100, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Iwatsubo, T.; Odaka, A.; Suzuki, N.; Mizusawa, H.; Nukina, N.; Ihara, Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: Evidence that an initially deposited species is A beta 42(43). Neuron 1994, 13, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Klafki, H.-W.; Morgado, B.; Wirths, O.; Jahn, O.; Bauer, C.; Esselmann, H.; Schuchhardt, J.; Wiltfang, J. Is plasma amyloid-β 1–42/1–40 a better biomarker for Alzheimer’s disease than AβX–42/X–40? Fluids Barriers CNS 2022, 19, 96. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Lendel, C. Extracellular protein components of amyloid plaques and their roles in Alzheimer’s disease pathology. Mol. Neurodegener. 2021, 16, 59. [Google Scholar] [CrossRef]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Hagmeyer, S.; Romao, M.A.; Cristovao, J.S.; Vilella, A.; Zoli, M.; Gomes, C.M.; Grabrucker, A.M. Distribution and Relative Abundance of S100 Proteins in the Brain of the APP23 Alzheimer’s Disease Model Mice. Front. Neurosci. 2019, 13, 640. [Google Scholar] [CrossRef]
- Shepherd, C.E.; Goyette, J.; Utter, V.; Rahimi, F.; Yang, Z.; Geczy, C.L.; Halliday, G.M. Inflammatory S100A9 and S100A12 proteins in Alzheimer’s disease. Neurobiol. Aging 2006, 27, 1554–1563. [Google Scholar] [CrossRef]
- Biere, A.L.; Ostaszewski, B.; Stimson, E.R.; Hyman, B.T.; Maggio, J.E.; Selkoe, D.J. Amyloid β-Peptide Is Transported on Lipoproteins and Albumin in Human Plasma. J. Biol. Chem. 1996, 271, 32916–32922. [Google Scholar] [CrossRef]
- Stanyon, H.F.; Viles, J.H. Human serum albumin can regulate amyloid-β peptide fiber growth in the brain interstitium: Implications for Alzheimer disease. J. Biol. Chem. 2012, 287, 28163–28168. [Google Scholar] [CrossRef]
- Litus, E.A.; Kazakov, A.S.; Sokolov, A.S.; Nemashkalova, E.L.; Galushko, E.I.; Dzhus, U.F.; Marchenkov, V.V.; Galzitskaya, O.V.; Permyakov, E.A.; Permyakov, S.E. The binding of monomeric amyloid β peptide to serum albumin is affected by major plasma unsaturated fatty acids. Biochem. Biophys. Res. Commun. 2019, 510, 248–253. [Google Scholar] [CrossRef]
- Milojevic, J.; Melacini, G. Stoichiometry and affinity of the human serum albumin-Alzheimer’s Aβ peptide interactions. Biophys. J. 2011, 100, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Cheng, F.; Xu, L.; Jia, L. HSA targets multiple Aβ42 species and inhibits the seeding-mediated aggregation and cytotoxicity of Aβ42 aggregates. RSC Adv. 2016, 6, 71165–71175. [Google Scholar] [CrossRef]
- Ezra, A.; Rabinovich-Nikitin, I.; Rabinovich-Toidman, P.; Solomon, B. Multifunctional Effect of Human Serum Albumin Reduces Alzheimer’s Disease Related Pathologies in the 3xTg Mouse Model. J. Alzheimer’s Dis. JAD 2016, 50, 175–188. [Google Scholar] [CrossRef]
- Boada, M.; Ortiz, P.; Anaya, F.; Hernandez, I.; Munoz, J.; Nunez, L.; Olazaran, J.; Roca, I.; Cuberas, G.; Tarraga, L.; et al. Amyloid-targeted therapeutics in Alzheimer’s disease: Use of human albumin in plasma exchange as a novel approach for Abeta mobilization. Drug News Perspect. 2009, 22, 1395256. [Google Scholar] [CrossRef]
- Boada, M.; López, O.L.; Olazarán, J.; Núñez, L.; Pfeffer, M.; Paricio, M.; Lorites, J.; Piñol-Ripoll, G.; Gámez, J.E.; Anaya, F.; et al. A randomized, controlled clinical trial of plasma exchange with albumin replacement for Alzheimer’s disease: Primary results of the AMBAR Study. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2020, 16, 1412–1425. [Google Scholar] [CrossRef]
- Cristovao, J.S.; Gomes, C.M. S100 Proteins in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 463. [Google Scholar] [CrossRef]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef]
- Singh, P.; Ali, S.A. Multifunctional Role of S100 Protein Family in the Immune System: An Update. Cells 2022, 11, 2274. [Google Scholar] [CrossRef]
- Gonzalez, L.L.; Garrie, K.; Turner, M.D. Role of S100 proteins in health and disease. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2020, 1867, 118677. [Google Scholar] [CrossRef]
- Sreejit, G.; Flynn, M.C.; Patil, M.; Krishnamurthy, P.; Murphy, A.J.; Nagareddy, P.R. S100 family proteins in inflammation and beyond. Adv. Clin. Chem. 2020, 98, 173–231. [Google Scholar] [CrossRef]
- Persechini, A.; Moncrief, N.D.; Kretsinger, R.H. The EF-hand family of calcium-modulated proteins. Trends Neurosci. 1989, 12, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Gifford, J.L.; Walsh, M.P.; Vogel, H.J. Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem. J. 2007, 405, 199–221. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Klechikov, A.G.; Gharibyan, A.L.; Warmlander, S.K.; Jarvet, J.; Zhao, L.; Jia, X.; Narayana, V.K.; Shankar, S.K.; Olofsson, A.; et al. The role of pro-inflammatory S100A9 in Alzheimer’s disease amyloid-neuroinflammatory cascade. Acta Neuropathol. 2014, 127, 507–522. [Google Scholar] [CrossRef]
- Boom, A.; Pochet, R.; Authelet, M.; Pradier, L.; Borghgraef, P.; Van Leuven, F.; Heizmann, C.W.; Brion, J.P. Astrocytic calcium/zinc binding protein S100A6 over expression in Alzheimer’s disease and in PS1/APP transgenic mice models. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2004, 1742, 161–168. [Google Scholar] [CrossRef]
- Tian, Z.Y.; Wang, C.Y.; Wang, T.; Li, Y.C.; Wang, Z.Y. Glial S100A6 Degrades beta-amyloid Aggregation through Targeting Competition with Zinc Ions. Aging Dis. 2019, 10, 756–769. [Google Scholar] [CrossRef]
- Ma, L.; Sun, P.; Zhang, J.C.; Zhang, Q.; Yao, S.L. Proinflammatory effects of S100A8/A9 via TLR4 and RAGE signaling pathways in BV-2 microglial cells. Int. J. Mol. Med. 2017, 40, 31–38. [Google Scholar] [CrossRef]
- Venegas, C.; Heneka, M.T. Danger-associated molecular patterns in Alzheimer’s disease. J. Leukoc. Biol. 2017, 101, 87–98. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Kummer, M.P.; Vogl, T.; Axt, D.; Griep, A.; Vieira-Saecker, A.; Jessen, F.; Gelpi, E.; Roth, J.; Heneka, M.T. Mrp14 deficiency ameliorates amyloid beta burden by increasing microglial phagocytosis and modulation of amyloid precursor protein processing. J. Neurosci. 2012, 32, 17824–17829. [Google Scholar] [CrossRef]
- Ha, T.Y.; Chang, K.A.; Kim, J.; Kim, H.S.; Kim, S.; Chong, Y.H.; Suh, Y.H. S100a9 knockdown decreases the memory impairment and the neuropathology in Tg2576 mice, AD animal model. PLoS ONE 2010, 5, e8840. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, D.; Zhou, X.; Zhang, C.; Yin, Q.; Chen, L.; Tang, Y.; Liu, Y.; Morozova-Roche, L.A. Proinflammatory S100A9 stimulates TLR4/NF-kappaB signaling pathways causing enhanced phagocytic capacity of microglial cells. Immunol. Lett. 2023, 255, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, Y.; Gilthorpe, J.; van der Maarel, J.R. MRP14 (S100A9) protein interacts with Alzheimer beta-amyloid peptide and induces its fibrillization. PLoS ONE 2012, 7, e32953. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Iashchishyn, I.A.; Pansieri, J.; Nystrom, S.; Klementieva, O.; Kara, J.; Horvath, I.; Moskalenko, R.; Rofougaran, R.; Gouras, G.; et al. S100A9-Driven Amyloid-Neuroinflammatory Cascade in Traumatic Brain Injury as a Precursor State for Alzheimer’s Disease. Sci. Rep. 2018, 8, 12836. [Google Scholar] [CrossRef] [PubMed]
- Mielke, M.M.; Ransom, J.E.; Mandrekar, J.; Turcano, P.; Savica, R.; Brown, A.W. Traumatic Brain Injury and Risk of Alzheimer’s Disease and Related Dementias in the Population. J. Alzheimer’s Dis. JAD 2022, 88, 1049–1059. [Google Scholar] [CrossRef]
- Lodeiro, M.; Puerta, E.; Ismail, M.A.; Rodriguez-Rodriguez, P.; Ronnback, A.; Codita, A.; Parrado-Fernandez, C.; Maioli, S.; Gil-Bea, F.; Merino-Serrais, P.; et al. Aggregation of the Inflammatory S100A8 Precedes Abeta Plaque Formation in Transgenic APP Mice: Positive Feedback for S100A8 and Abeta Productions. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 72, 319–328. [Google Scholar] [CrossRef]
- Litus, E.; Shevelyova, M.; Vologzhannikova, A.; Deryusheva, E.; Nemashkalova, E.; Machulin, A.; Nazipova, A.; Permyakova, M.; Permyakov, S. S100A8 Interaction with Amyloid-β Peptide Suppresses Its Fibrillation. Proceedings 2024, 103, 1. [Google Scholar] [CrossRef]
- Horvath, I.; Jia, X.; Johansson, P.; Wang, C.; Moskalenko, R.; Steinau, A.; Forsgren, L.; Wagberg, T.; Svensson, J.; Zetterberg, H.; et al. Pro-inflammatory S100A9 Protein as a Robust Biomarker Differentiating Early Stages of Cognitive Impairment in Alzheimer’s Disease. ACS Chem. Neurosci. 2016, 7, 34–39. [Google Scholar] [CrossRef]
- Sedov, I.; Khaibrakhmanova, D. Molecular Mechanisms of Inhibition of Protein Amyloid Fibril Formation: Evidence and Perspectives Based on Kinetic Models. Int. J. Mol. Sci. 2022, 23, 13428. [Google Scholar] [CrossRef]
- Deryusheva, E.I.; Shevelyova, M.P.; Rastrygina, V.A.; Nemashkalova, E.L.; Vologzhannikova, A.A.; Machulin, A.V.; Nazipova, A.A.; Permyakova, M.E.; Permyakov, S.E.; Litus, E.A. In Search for Low-Molecular-Weight Ligands of Human Serum Albumin That Affect Its Affinity for Monomeric Amyloid beta Peptide. Int. J. Mol. Sci. 2024, 25, 4975. [Google Scholar] [CrossRef]
- Travis, J.; Pannell, R. Selective removal of albumin from plasma by affinity chromatography. Clin. Chim. Acta 1973, 49, 49–52. [Google Scholar] [CrossRef]
- Litus, E.A.; Kazakov, A.S.; Deryusheva, E.I.; Nemashkalova, E.L.; Shevelyova, M.P.; Machulin, A.V.; Nazipova, A.A.; Permyakova, M.E.; Uversky, V.N.; Permyakov, S.E. Ibuprofen Favors Binding of Amyloid-beta Peptide to Its Depot, Serum Albumin. Int. J. Mol. Sci. 2022, 23, 6168. [Google Scholar] [CrossRef] [PubMed]
- Kazakov, A.S.; Sofin, A.D.; Avkhacheva, N.V.; Denesyuk, A.I.; Deryusheva, E.I.; Rastrygina, V.A.; Sokolov, A.S.; Permyakova, M.E.; Litus, E.A.; Uversky, V.N.; et al. Interferon Beta Activity Is Modulated via Binding of Specific S100 Proteins. Int. J. Mol. Sci. 2020, 21, 9473. [Google Scholar] [CrossRef] [PubMed]
- Catanzariti, A.M.; Soboleva, T.A.; Jans, D.A.; Board, P.G.; Baker, R.T. An efficient system for high-level expression and easy purification of authentic recombinant proteins. Protein Sci. 2004, 13, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Vajdos, F.; Fee, L.; Grimsley, G.; Gray, T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995, 4, 2411–2423. [Google Scholar] [CrossRef]
- De Ferrari, G.V.; Mallender, W.D.; Inestrosa, N.C.; Rosenberry, T.L. Thioflavin T is a fluorescent probe of the acetylcholinesterase peripheral site that reveals conformational interactions between the peripheral and acylation sites. J. Biol. Chem. 2001, 276, 23282–23287. [Google Scholar] [CrossRef]
- Litus, E.A.; Kazakov, A.S.; Deryusheva, E.I.; Nemashkalova, E.L.; Shevelyova, M.P.; Nazipova, A.A.; Permyakova, M.E.; Raznikova, E.V.; Uversky, V.N.; Permyakov, S.E. Serotonin Promotes Serum Albumin Interaction with the Monomeric Amyloid beta Peptide. Int. J. Mol. Sci. 2021, 22, 5896. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Desta, I.T.; Porter, K.A.; Xia, B.; Kozakov, D.; Vajda, S. Performance and Its Limits in Rigid Body Protein-Protein Docking. Structure 2020, 28, 1071–1081.e3. [Google Scholar] [CrossRef]
- Kazakov, A.S.; Deryusheva, E.I.; Sokolov, A.S.; Permyakova, M.E.; Litus, E.A.; Rastrygina, V.A.; Uversky, V.N.; Permyakov, E.A.; Permyakov, S.E. Erythropoietin Interacts with Specific S100 Proteins. Biomolecules 2022, 12, 120. [Google Scholar] [CrossRef]
- Schrodinger, L.L.C. The PyMOL Molecular Graphics System. Version 2015, 1, 8. [Google Scholar]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef] [PubMed]
- Nemashkalova, E.L.; Permyakov, E.A.; Permyakov, S.E.; Litus, E.A. Modulation of linoleic acid-binding properties of human serum albumin by divalent metal cations. Biometals 2017, 30, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Streicher, W.W.; Lopez, M.M.; Makhatadze, G.I. Modulation of quaternary structure of S100 proteins by calcium ions. Biophys. Chem. 2010, 151, 181–186. [Google Scholar] [CrossRef]
- Chang, C.C.; Khan, I.; Tsai, K.L.; Li, H.; Yang, L.W.; Chou, R.H.; Yu, C. Blocking the interaction between S100A9 and RAGE V domain using CHAPS molecule: A novel route to drug development against cell proliferation. Biochim. Biophys. Acta 2016, 1864, 1558–1569. [Google Scholar] [CrossRef]
- Katte, R.; Yu, C. Blocking the interaction between S100A9 protein and RAGE V domain using S100A12 protein. PLoS ONE 2018, 13, e0198767. [Google Scholar] [CrossRef]
- Paramasivam, S.; Murugesan, J.; Vedagiri, H.; Perumal, S.S.; Ekambaram, S.P. Virtual Probing on the Influence of Ca(2+) and Zn(2+) Bound S100A8 and S100A9 Proteins Towards their Interaction Against Pattern Recognition Receptors Aggravating Rheumatoid Arthritis. Cell Biochem. Biophys. 2024, 1–23. [Google Scholar] [CrossRef]
- Kazakov, A.S.; Deryusheva, E.I.; Rastrygina, V.A.; Sokolov, A.S.; Permyakova, M.E.; Litus, E.A.; Uversky, V.N.; Permyakov, E.A.; Permyakov, S.E. Interaction of S100A6 Protein with the Four-Helical Cytokines. Biomolecules 2023, 13, 1345. [Google Scholar] [CrossRef]
- Kazakov, A.S.; Deryusheva, E.I.; Permyakova, M.E.; Sokolov, A.S.; Rastrygina, V.A.; Uversky, V.N.; Permyakov, E.A.; Permyakov, S.E. Calcium-Bound S100P Protein Is a Promiscuous Binding Partner of the Four-Helical Cytokines. Int. J. Mol. Sci. 2022, 23, 12000. [Google Scholar] [CrossRef]
- Permyakov, S.E.; Ismailov, R.G.; Xue, B.; Denesyuk, A.I.; Uversky, V.N.; Permyakov, E.A. Intrinsic disorder in S100 proteins. Mol. Biosyst. 2011, 7, 2164–2180. [Google Scholar] [CrossRef]
- Wisniewski, H.M.; Kozlowski, P.B. Evidence for blood-brain barrier changes in senile dementia of the Alzheimer type (SDAT). Ann. N. Y. Acad. Sci. 1982, 396, 119–129. [Google Scholar] [CrossRef]
- Kim, H.J.; Park, K.W.; Kim, T.E.; Im, J.Y.; Shin, H.S.; Kim, S.; Lee, D.H.; Ye, B.S.; Kim, J.H.; Kim, E.J.; et al. Elevation of the Plasma Abeta40/Abeta42 Ratio as a Diagnostic Marker of Sporadic Early-Onset Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2015, 48, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Mayeux, R.; Honig, L.S.; Tang, M.X.; Manly, J.; Stern, Y.; Schupf, N.; Mehta, P.D. Plasma A[beta]40 and A[beta]42 and Alzheimer’s disease: Relation to age, mortality, and risk. Neurology 2003, 61, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.D.; Pirttila, T.; Mehta, S.P.; Sersen, E.A.; Aisen, P.S.; Wisniewski, H.M. Plasma and cerebrospinal fluid levels of amyloid beta proteins 1-40 and 1-42 in Alzheimer disease. Arch. Neurol. 2000, 57, 100–105. [Google Scholar] [CrossRef]
- Seeliger, T.; Gingele, S.; Guzeloglu, Y.E.; Heitmann, L.; Luling, B.; Kohle, F.; Pressler, H.; Stascheit, F.; Motte, J.; Fisse, A.L.; et al. Comparative analysis of albumin quotient and total CSF protein in immune-mediated neuropathies: A multicenter study on diagnostic implications. Front. Neurol. 2023, 14, 1330484. [Google Scholar] [CrossRef]
- Srsen, S.; Held, M.; Sestan, M.; Kifer, N.; Kozmar, A.; Supe Domic, D.; Benzon, B.; Gagro, A.; Frkovic, M.; Jelusic, M. Serum Levels of S100A8/A9 as a Biomarker of Disease Activity in Patients with IgA Vasculitis. Biomedicines 2024, 12, 750. [Google Scholar] [CrossRef]
- Frosch, M.; Strey, A.; Vogl, T.; Wulffraat, N.M.; Kuis, W.; Sunderkotter, C.; Harms, E.; Sorg, C.; Roth, J. Myeloid-related proteins 8 and 14 are specifically secreted during interaction of phagocytes and activated endothelium and are useful markers for monitoring disease activity in pauciarticular-onset juvenile rheumatoid arthritis. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2000, 43, 628–637. [Google Scholar] [CrossRef]
- Ahn, S.M.; Byun, K.; Cho, K.; Kim, J.Y.; Yoo, J.S.; Kim, D.; Paek, S.H.; Kim, S.U.; Simpson, R.J.; Lee, B. Human microglial cells synthesize albumin in brain. PLoS ONE 2008, 3, e2829. [Google Scholar] [CrossRef]
- Raskatov, J.A. What Is the “Relevant” Amyloid β42 Concentration? Chembiochem A Eur. J. Chem. Biol. 2019, 20, 1725–1726. [Google Scholar] [CrossRef]
- Schilde, L.M.; Kösters, S.; Steinbach, S.; Schork, K.; Eisenacher, M.; Galozzi, S.; Turewicz, M.; Barkovits, K.; Mollenhauer, B.; Marcus, K.; et al. Protein variability in cerebrospinal fluid and its possible implications for neurological protein biomarker research. PLoS ONE 2018, 13, e0206478. [Google Scholar] [CrossRef]
- Bohrmann, B.; Tjernberg, L.; Kuner, P.; Poli, S.; Levet-Trafit, B.; Näslund, J.; Richards, G.; Huber, W.; Döbeli, H.; Nordstedt, C. Endogenous proteins controlling amyloid beta-peptide polymerization. Possible implications for beta-amyloid formation in the central nervous system and in peripheral tissues. J. Biol. Chem. 1999, 274, 15990–15995. [Google Scholar] [CrossRef]
- Baronaite, I.; Sulskis, D.; Kopu Stas, A.; Tutkus, M.; Smirnovas, V. Formation of Calprotectin Inhibits Amyloid Aggregation of S100A8 and S100A9 Proteins. ACS Chem. Neurosci. 2024, 15, 1915–1925. [Google Scholar] [CrossRef] [PubMed]
- Pansieri, J.; Iashchishyn, I.A.; Fakhouri, H.; Ostojic, L.; Malisauskas, M.; Musteikyte, G.; Smirnovas, V.; Schneider, M.M.; Scheidt, T.; Xu, C.K.; et al. Templating S100A9 amyloids on Abeta fibrillar surfaces revealed by charge detection mass spectrometry, microscopy, kinetic and microfluidic analyses. Chem. Sci. 2020, 11, 7031–7039. [Google Scholar] [CrossRef] [PubMed]
- Bode, D.C.; Stanyon, H.F.; Hirani, T.; Baker, M.D.; Nield, J.; Viles, J.H. Serum Albumin’s Protective Inhibition of Amyloid-β Fiber Formation Is Suppressed by Cholesterol, Fatty Acids and Warfarin. J. Mol. Biol. 2018, 430, 919–934. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KD × 106, M | ka × 10−2, M−1s−1 | kd × 104, s−1 | |
| Aβ40 | |||
| S100A8 | 5.1 ± 2.3 | 2.2 ± 0.7 | 10.9 ± 1.5 |
| S100A9 | 2.1 ± 1.3 | 2.6 ± 1.0 | 5.4 ± 1.0 |
| Aβ42 | |||
| S100A8 | 1.3 ± 0.9 | 3.4 ± 0.9 | 4.6 ± 1.9 |
| S100A9 | 1.2 ± 0.6 | 4.5 ± 1.3 | 5.1 ± 1.1 |
| KD1 × 106, M | ka1 × 10−2, M−1s−1 | kd1 × 104, s−1 | KD2 × 106, M | ka2 × 10−2, M−1s−1 | kd2 × 104, s−1 |
| Aβ40 | |||||
| 0.22 ± 0.12 | 7.9 ± 4.4 | 1.72 ± 0.04 | 6.7 ± 4.5 | 0.36 ± 0.24 | 2.38 ± 0.06 |
| Aβ42 | |||||
| 0.48 ± 0.09 | 3.5 ± 0.5 | 1.66 ± 0.10 | 5.6 ± 4.6 | 0.28 ± 0.18 | 1.58 ± 0.26 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Litus, E.A.; Shevelyova, M.P.; Vologzhannikova, A.A.; Deryusheva, E.I.; Machulin, A.V.; Nemashkalova, E.L.; Permyakova, M.E.; Sokolov, A.S.; Alikova, V.D.; Uversky, V.N.; et al. Binding of Pro-Inflammatory Proteins S100A8 or S100A9 to Amyloid-β Peptide Suppresses Its Fibrillation. Biomolecules 2025, 15, 431. https://doi.org/10.3390/biom15030431
Litus EA, Shevelyova MP, Vologzhannikova AA, Deryusheva EI, Machulin AV, Nemashkalova EL, Permyakova ME, Sokolov AS, Alikova VD, Uversky VN, et al. Binding of Pro-Inflammatory Proteins S100A8 or S100A9 to Amyloid-β Peptide Suppresses Its Fibrillation. Biomolecules. 2025; 15(3):431. https://doi.org/10.3390/biom15030431
Chicago/Turabian StyleLitus, Ekaterina A., Marina P. Shevelyova, Alisa A. Vologzhannikova, Evgenia I. Deryusheva, Andrey V. Machulin, Ekaterina L. Nemashkalova, Maria E. Permyakova, Andrey S. Sokolov, Valeria D. Alikova, Vladimir N. Uversky, and et al. 2025. "Binding of Pro-Inflammatory Proteins S100A8 or S100A9 to Amyloid-β Peptide Suppresses Its Fibrillation" Biomolecules 15, no. 3: 431. https://doi.org/10.3390/biom15030431
APA StyleLitus, E. A., Shevelyova, M. P., Vologzhannikova, A. A., Deryusheva, E. I., Machulin, A. V., Nemashkalova, E. L., Permyakova, M. E., Sokolov, A. S., Alikova, V. D., Uversky, V. N., & Permyakov, S. E. (2025). Binding of Pro-Inflammatory Proteins S100A8 or S100A9 to Amyloid-β Peptide Suppresses Its Fibrillation. Biomolecules, 15(3), 431. https://doi.org/10.3390/biom15030431