
The Oncoprotein Mucin 1 in Pancreatic Cancer Onset and Progression: Potential Clinical Implications

 , , , , ,
, , , , ,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
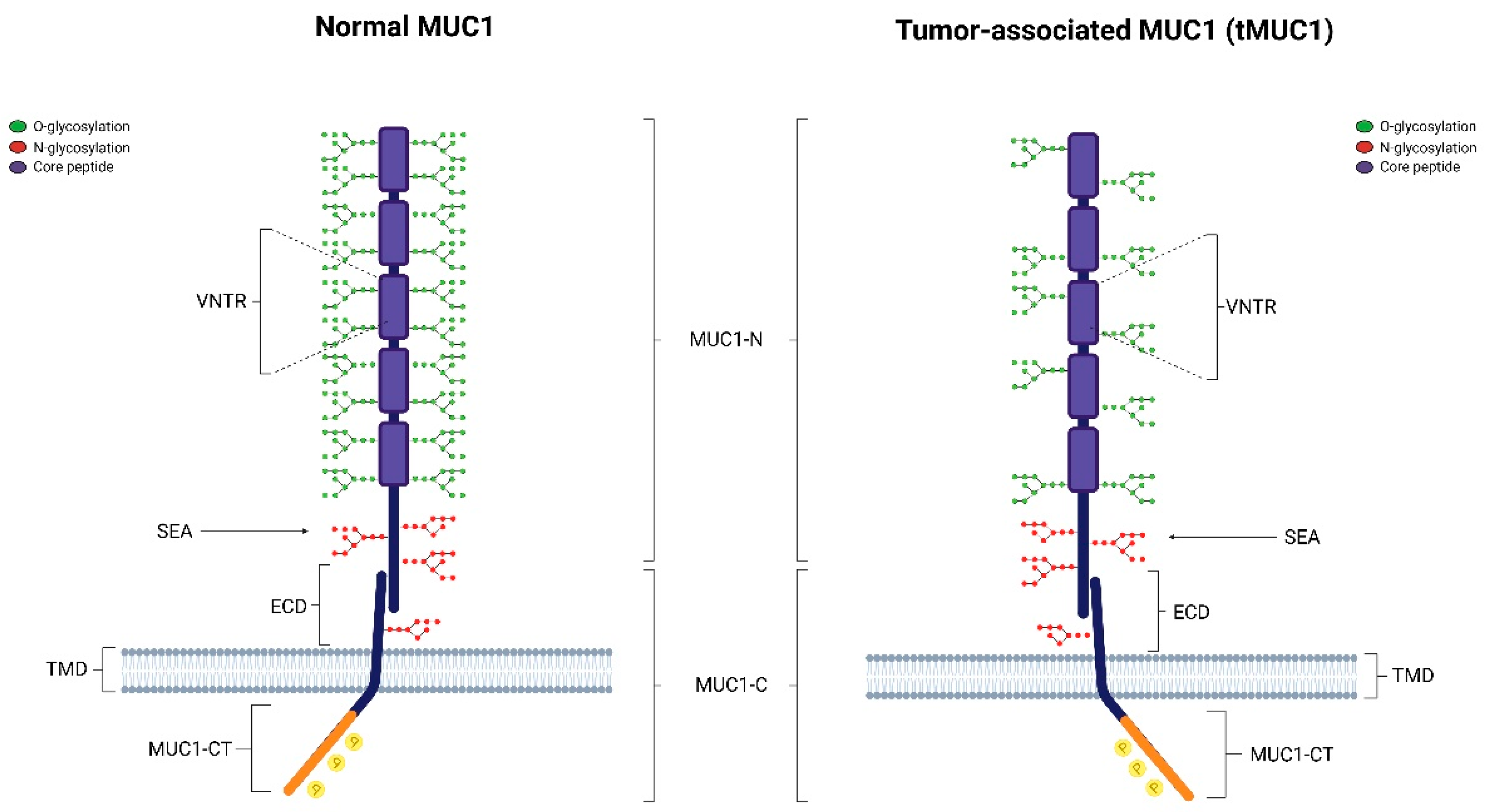
2. Structure of MUC1
3. MUC1 Role in Cancer Tissues
3.1. MUC1 Promotes Tumor Cells Immune Escape
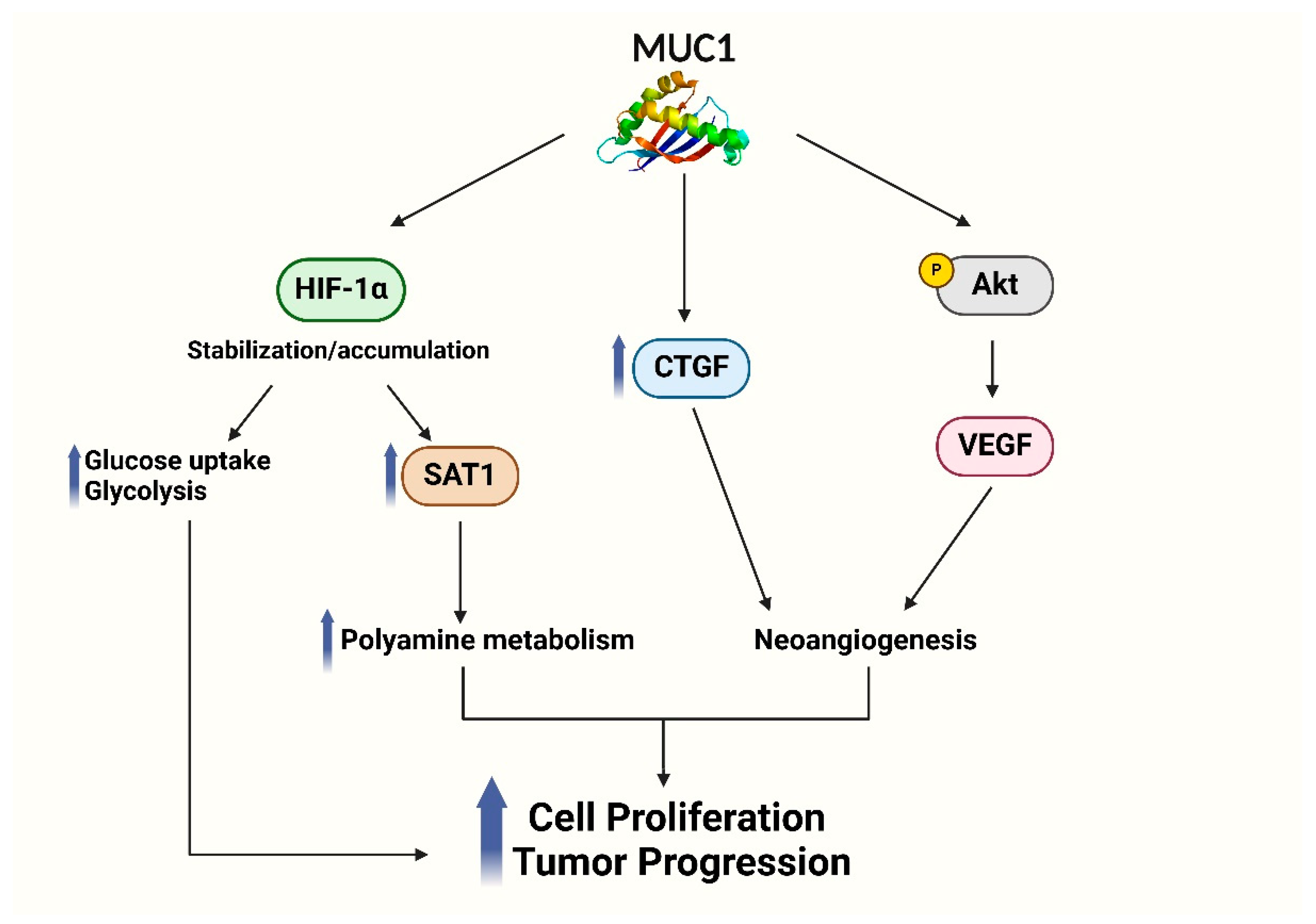
3.2. MUC1 Influences Hypoxic Tumor Microenvironment
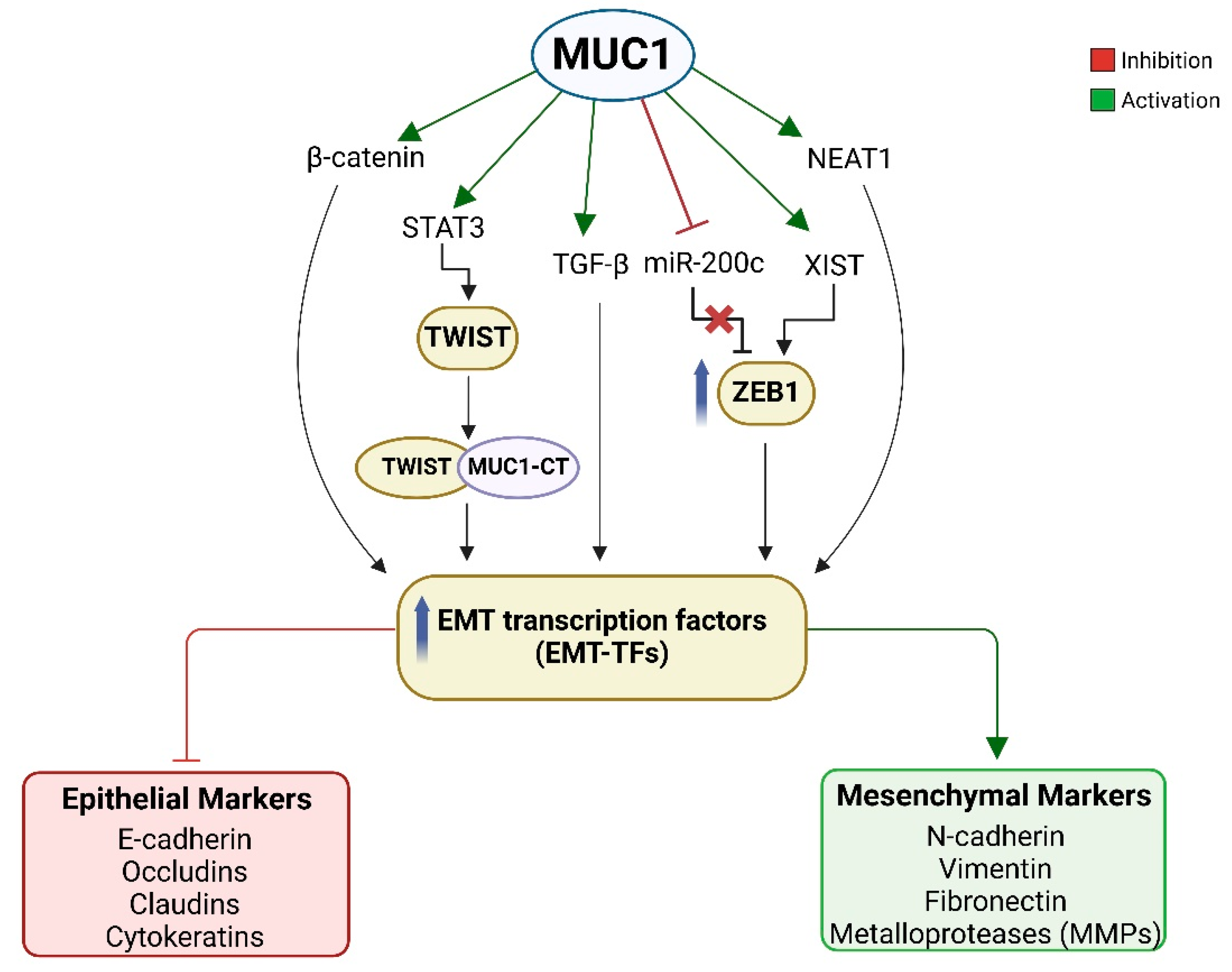
3.3. MUC1 Promotes Epithelial to Mesenchymal Transition
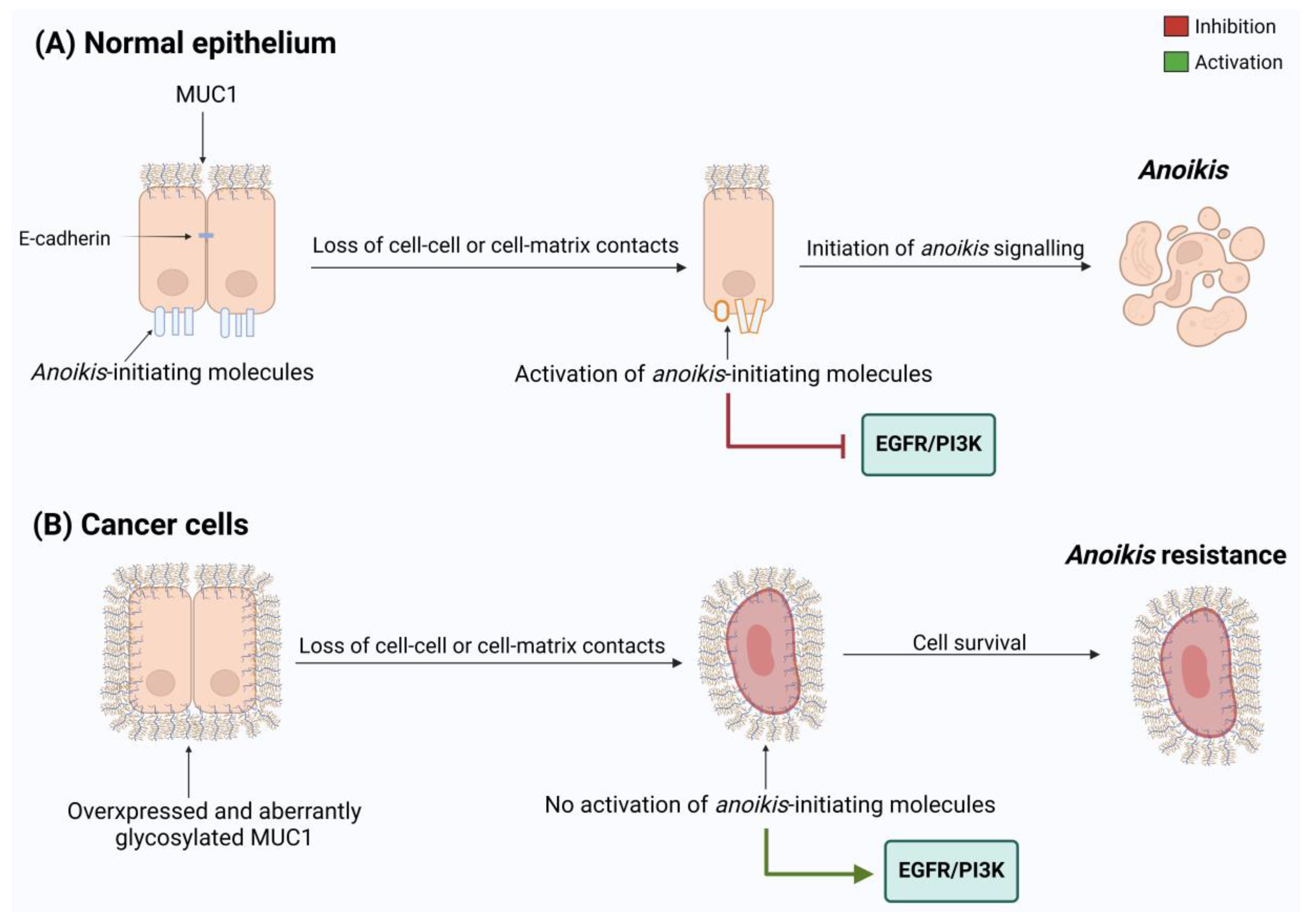
3.4. MUC1 Promotes Anoikis
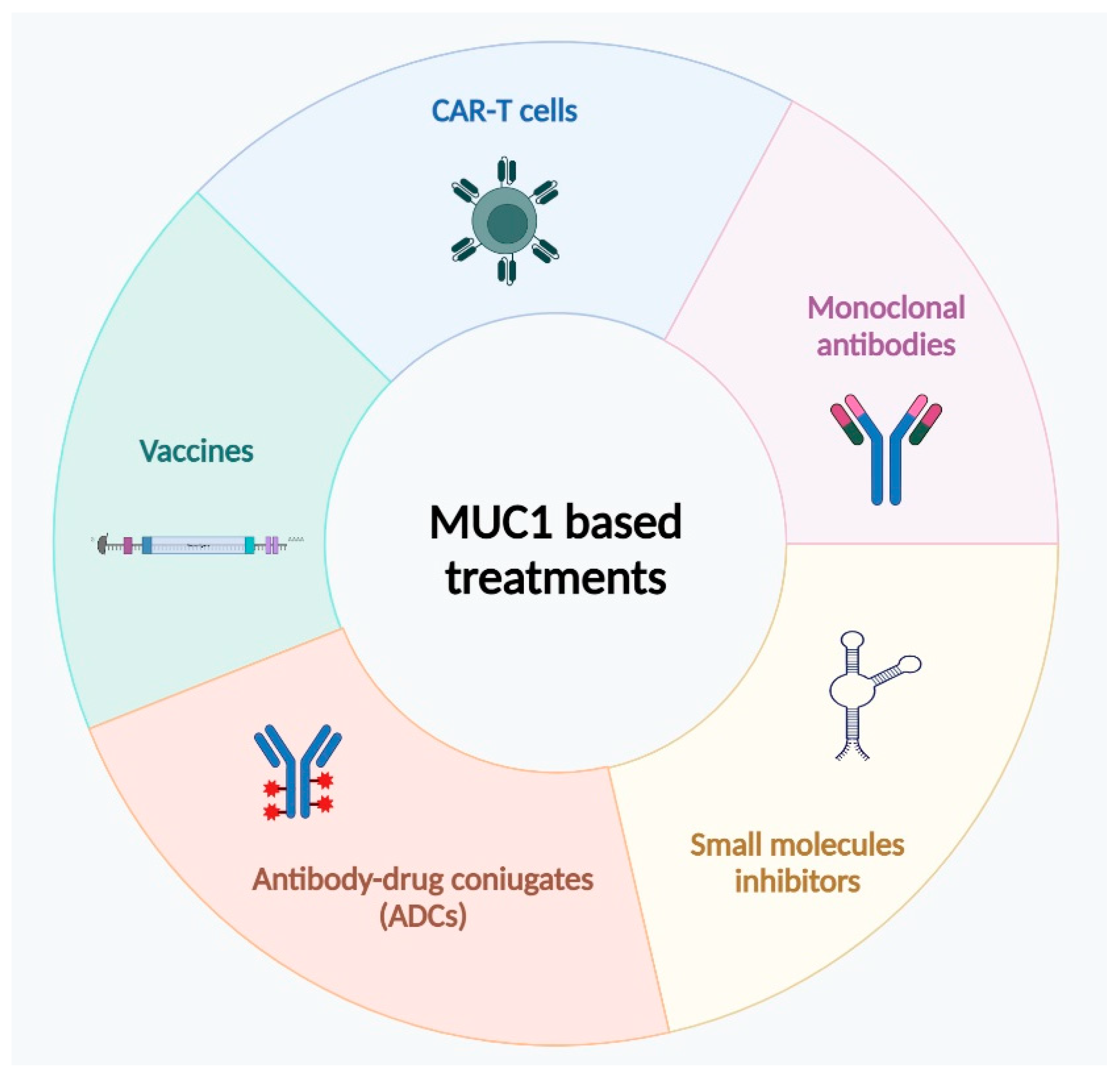
4. MUC1 as Prognostic Marker for Target Therapies
4.1. Immunotherapeutic Approaches
4.2. Antibodies
4.3. MUC1 Inhibitors
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic Cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Partyka, O.; Pajewska, M.; Kwaśniewska, D.; Czerw, A.; Deptała, A.; Budzik, M.; Cipora, E.; Gąska, I.; Gazdowicz, L.; Mielnik, A.; et al. Overview of Pancreatic Cancer Epidemiology in Europe and Recommendations for Screening in High-Risk Populations. Cancers 2023, 15, 3634. [Google Scholar] [CrossRef]
- Michalak, N.; Małecka-Wojciesko, E. Modifiable Pancreatic Ductal Adenocarcinoma (PDAC) Risk Factors. J. Clin. Med. 2023, 12, 4318. [Google Scholar] [CrossRef]
- Schawkat, K.; Manning, M.A.; Glickman, J.N.; Mortele, K.J. Pancreatic Ductal Adenocarcinoma and Its Variants: Pearls and Perils. Radiographics 2020, 40, 1219–1239. [Google Scholar] [CrossRef] [PubMed]
- Delpu, Y.; Hanoun, N.; Lulka, H.; Sicard, F.; Selves, J.; Buscail, L.; Torrisani, J.; Cordelier, P. Genetic and Epigenetic Alterations in Pancreatic Carcinogenesis. Curr. Genom. 2011, 12, 15–24. [Google Scholar] [CrossRef]
- Van Den Broeck, A.; Vankelecom, H.; Van Eijsden, R.; Govaere, O.; Topal, B. Molecular Markers Associated with Outcome and Metastasis in Human Pancreatic Cancer. J. Exp. Clin. Cancer Res. 2012, 31, 68. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.H. Nanomedicine Developments in the Treatment of Metastatic Pancreatic Cancer: Focus on Nanoliposomal Irinotecan. Int. J. Nanomed. 2016, 11, 1225–1235. [Google Scholar] [CrossRef]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Lemire, M.; Zhang, A.; Chan-Seng-Yue, M.; Wilson, G.; Grant, R.C.; Merico, D.; Lungu, I.; et al. Integration of Genomic and Transcriptional Features in Pancreatic Cancer Reveals Increased Cell Cycle Progression in Metastases. Cancer Cell 2019, 35, 267–282.e7. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan Impairs Vascular Function and Drug Delivery in a Mouse Model of Pancreatic Cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Pandol, S.; Edderkaoui, M.; Gukovsky, I.; Lugea, A.; Gukovskaya, A. Desmoplasia of Pancreatic Ductal Adenocarcinoma. Clin. Gastroenterol. Hepatol. 2009, 7, S44–S47. [Google Scholar] [CrossRef] [PubMed]
- Maneshi, P.; Mason, J.; Dongre, M.; Öhlund, D. Targeting Tumor-Stromal Interactions in Pancreatic Cancer: Impact of Collagens and Mechanical Traits. Front. Cell Dev. Biol. 2021, 9, 787485. [Google Scholar] [CrossRef]
- Diaz, B.; Yuen, A. The Impact of Hypoxia in Pancreatic Cancer Invasion and Metastasis. Hypoxia 2014, 2, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Sahin, I.H.; Elias, H.; Chou, J.F.; Capanu, M.; O’Reilly, E.M. Pancreatic Adenocarcinoma: Insights into Patterns of Recurrence and Disease Behavior. BMC Cancer 2018, 18, 769. [Google Scholar] [CrossRef] [PubMed]
- Daamen, L.A.; Groot, V.P.; Intven, M.P.W.; Besselink, M.G.; Busch, O.R.; Koerkamp, B.G.; Mohammad, N.H.; Hermans, J.J.; van Laarhoven, H.W.M.; Nuyttens, J.J.; et al. Postoperative Surveillance of Pancreatic Cancer Patients. Eur. J. Surg. Oncol. 2019, 45, 1770–1777. [Google Scholar] [CrossRef]
- Brunner, M.; Wu, Z.; Krautz, C.; Pilarsky, C.; Grützmann, R.; Weber, G.F. Current clinical strategies of pancreatic cancer treatment and open molecular questions. Int. J. Mol. Sci. 2019, 20, 4543. [Google Scholar] [CrossRef]
- O’Kane, G.M.; Ladak, F.; Gallinger, S. Advances in the Management of Pancreatic Ductal Adenocarcinoma. CMAJ 2021, 193, E844–E851. [Google Scholar] [CrossRef]
- Lee, D.H.; Choi, S.; Park, Y.; Jin, H.S. Mucin1 and Mucin16: Therapeutic Targets for Cancer Therapy. Pharmaceuticals 2021, 14, 1053. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Mukherjee, P. MUC1: A Multifaceted Oncoprotein with a Key Role in Cancer Progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.H.; Triyana, S.; Wang, R.; Das, I.; Gerloff, K.; Florin, T.H.; Sutton, P.; Mcguckin, M.A. MUC1 and MUC13 Differentially Regulate Epithelial Inflammation in Response to Inflammatory and Infectious Stimuli. Soc. Mucosal Immunol. 2013, 6, 557–568. [Google Scholar] [CrossRef]
- Altschuler, Y.; Kinlough, C.L.; Poland, P.A.; Bruns, J.B.; Apodaca, G.; Weisz, O.A.; Hughey, R.P. Clathrin-Mediated Endocytosis of MUC1 Is Modulated by Its Glycosylation State. Mol. Biol. Cell 2000, 11, 819–831. [Google Scholar] [CrossRef]
- Nielsen, M.F.B.; Mortensen, M.B.; Detlefsen, S. Key Players in Pancreatic Cancer-Stroma Interaction: Cancer-Associated Fibroblasts, Endothelial and Inflammatory Cells. World J. Gastroenterol. 2016, 22, 2678–2700. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; Lobello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; Von Hoff, D.D.; Han, H. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin. Cancer Res. 2015, 21, 3561–3568. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Vega, A.; Maldonado-Lagunas, V.; Mitre-Aguilar, I.B.; Melendez-Zajgla, J. Tumor Microenvironment Role in Pancreatic Cancer Stem Cells. Cells 2023, 12, 1560. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Bosch, N.; Vinaixa, J.; Navarro, P. Immune Evasion in Pancreatic Cancer: From Mechanisms to Therapy. Cancers 2018, 10, 6. [Google Scholar] [CrossRef]
- Johnson, B.A.; Yarchoan, M.; Lee, V.; Laheru, D.A.; Jaffee, E.M. Strategies for Increasing Pancreatic Tumor Immunogenicity. Clin. Cancer Res. 2017, 23, 1656–1669. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Radhakrishnan, P. Tumor-Stromal Crosstalk in Pancreatic Cancer and Tissue Fibrosis. Mol. Cancer 2019, 18, 14. [Google Scholar] [CrossRef]
- Zhao, Q.; Guo, X.; Nash, G.B.; Stone, P.C.; Hilkens, J.; Rhodes, J.M.; Yu, L.G. Circulating Galectin-3 Promotes Metastasis by Modifying MUC1 Localization on Cancer Cell Surface. Cancer Res. 2009, 69, 6799–6806. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Kumar, S.; Momi, N.; Sasson, A.R.; Batra, S.K. Mucins in Pancreatic Cancer and Its Microenvironment. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Sindrewicz, P.; Lian, L.Y.; Yu, L.G. Interaction of the Oncofetal Thomsen-Friedenreich Antigen with Galectins in Cancer Progression and Metastasis. Front. Oncol. 2016, 6, 79. [Google Scholar] [CrossRef] [PubMed]
- Radziejewska, I. Galectin-3 and Epithelial MUC1 Mucin—Interactions Supporting Cancer Development. Cancers 2023, 15, 2680. [Google Scholar] [CrossRef] [PubMed]
- Chou, F.C.; Chen, H.Y.; Kuo, C.C.; Sytwu, H.K. Role of Galectins in Tumors and in Clinical Immunotherapy. Int. J. Mol. Sci. 2018, 19, 430. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Barclay, M.; Hilkens, J.; Guo, X.; Barrow, H.; Rhodes, J.M.; Yu, L.G. Interaction Between Circulating Galectin-3 and Cancer-Associated MUC1 Enhances Tumour Cell Homotypic Aggregation and Prevents Anoikis. Mol. Cancer 2010, 9, 154. [Google Scholar] [CrossRef]
- Duckworth, C.A.; Guimond, S.E.; Sindrewicz, P.; Ashley, J.; French, N.S.; Lian, L.Y.; Yates, E.A.; Pritchard, D.M.; Rhodes, J.M.; Turnbull, J.E.; et al. Chemically Modified, Non-Anticoagulant Heparin Derivatives Are Potent Galectin-3 Binding Inhibitors and Inhibit Circulating Galectin-3-Promoted Metastasis Chemically Modified, Non-Anticoagulant Heparin Derivatives Are Potent Galectin-3 Binding Inhibitor and Inhibit Circulating Galectin-3-Promoted Metastasis. Oncotarget 2015, 6, 23671. [Google Scholar] [CrossRef]
- Zhuang, Y.; Liu, K.; He, Q.; Gu, X.; Jiang, C.; Wu, J. Hypoxia signaling in cancer: Implications for therapeutic interventions. MedComm 2023, 4, e203. [Google Scholar] [CrossRef] [PubMed]
- Zoa, A.; Yang, Y.; Huang, W.; Yang, J.; Wang, J.; Wang, H.; Dong, M.; Tian, Y. High Expression of Hypoxia-Inducible Factor 1-Alpha Predicts Poor Prognosis in Pancreatic Ductal Adenocarcinoma: A Meta-Analysis and Database Validation Protocol. Transl. Cancer Res. 2022, 11, 3080–3091. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Chen, J.; Yang, Y.; Hu, W. Hypoxia Correlates With Poor Survival and M2 Macrophage Infiltration in Colorectal Cancer. Front. Oncol. 2020, 10, 566430. [Google Scholar] [CrossRef]
- Brown, J.M.; Wilson, W.R. Exploiting Tumour Hypoxia in Cancer Treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef]
- Utispan, K.; Koontongkaew, S. Mucin 1 Regulates the Hypoxia Response in Head and Neck Cancer Cells. J. Pharmacol. Sci. 2021, 147, 331–339. [Google Scholar] [CrossRef]
- Kitamoto, S.; Yokoyama, S.; Higashi, M.; Yamada, N.; Takao, S.; Yonezawa, S. MUC1 Enhances Hypoxia-Driven Angiogenesis through the Regulation of Multiple Proangiogenic Factors. Oncogene 2013, 32, 4614–4621. [Google Scholar] [CrossRef] [PubMed]
- Behrens, M.E.; Grandgenett, P.M.; Bailey, J.M.; Singh, P.K.; Yi, C.H.; Yu, F.; Hollingsworth, M.A. The Reactive Tumor Microenvironment: MUC1 Signaling Directly Reprograms Transcription of CTGF. Oncogene 2010, 29, 5667–5677. [Google Scholar] [CrossRef] [PubMed]
- Khodabakhsh, F.; Merikhian, P.; Eisavand, M.R.; Farahmand, L. Crosstalk between MUC1 and VEGF in Angiogenesis and Metastasis: A Review Highlighting Roles of the MUC1 with an Emphasis on Metastatic and Angiogenic Signaling. Cancer Cell Int. 2021, 21, 200. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.K.; Purohit, V.; Mehla, K.; Gunda, V.; Nina, V.; Vernucci, E.; King, R.J.; Abrego, J.; Goode, G.D.; Illies, A.L.; et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 2017, 32, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E. Functions of Polyamines in Mammals. J. Biol. Chem. 2016, 291, 14904–14912. [Google Scholar] [CrossRef]
- Sagar, N.A.; Tarafdar, S.; Agarwal, S.; Tarafdar, A.; Sharma, S. Polyamines: Functions, Metabolism, and Role in Human Disease Management. Med. Sci. 2021, 9, 44. [Google Scholar] [CrossRef]
- Murthy, D.; Attri, K.S.; Suresh, V.; Rajacharya, G.H.; Valenzuela, C.A.; Thakur, R.; Zhao, J.; Shukla, S.K.; Chaika, N.V.; LaBreck, D.; et al. The MUC1–HIF-1α Signaling Axis Regulates Pancreatic Cancer Pathogenesis through Polyamine Metabolism Remodeling. Proc. Natl. Acad. Sci. USA 2024, 121, e2315509121. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. EMT: When Epithelial Cells Decide to Become Mesenchymal-like Cells. J. Clin. Investig. 2009, 119, 1417–1419. [Google Scholar] [CrossRef] [PubMed]
- Babaei, G.; Aziz, S.G.G.; Jaghi, N.Z.Z. EMT, Cancer Stem Cells and Autophagy; The Three Main Axes of Metastasis. Biomed. Pharmacother. 2021, 133, 110909. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in Cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Bulle, A.; Dekervel, J.; Libbrecht, L.; Nittner, D.; Deschuttere, L.; Lambrecht, D. Gemcitabine Induces Epithelial-to-Mesenchymal Transition in Patient-Derived Pancreatic Ductal Adenocarcinoma Xenografts. Am. J. Transl. Res. 2019, 11, 765–779. [Google Scholar] [PubMed]
- Dardare, J.; Witz, A.; Merlin, J.L.; Bochnakian, A.; Toussaint, P.; Gilson, P.; Harlé, A. Epithelial to Mesenchymal Transition in Patients with Pancreatic Ductal Adenocarcinoma: State-of-the-Art and Therapeutic Opportunities. Pharmaceuticals 2021, 14, 740. [Google Scholar] [CrossRef]
- Yang, R.M.; Zhan, M.; Xu, S.W.; Long, M.M.; Yang, L.H.; Chen, W.; Huang, S.; Liu, Q.; Zhou, J.; Zhu, J.; et al. Mir-3656 Expression Enhances the Chemosensitivity of Pancreatic Cancer to Gemcitabine through Modulation of the Rhof/Emt Axis. Cell Death Dis. 2017, 8, e3129. [Google Scholar] [CrossRef]
- Roy, L.D.; Sahraei, M.; Subramani, D.B.; Besmer, D.; Nath, S.; Tinder, T.L.; Bajaj, E.; Shanmugam, K.; Lee, Y.Y.; Hwang, S.I.L.; et al. MUC1 Enhances Invasiveness of Pancreatic Cancer Cells by Inducing Epithelial to Mesenchymal Transition. Oncogene 2011, 30, 1449–1459. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Wen, Y.; Swanson, B.J.; Shanmugam, K.; Kazlauskas, A.; Cerny, R.L.; Gendler, S.J.; Hollingsworth, M.A. Platelet-Derived Growth Factor Receptor β-Mediated Phosphorylation of MUC1 Enhances Invasiveness in Pancreatic Adenocarcinoma Cells. Cancer Res. 2007, 67, 5201–5210. [Google Scholar] [CrossRef] [PubMed]
- Kufe, D.W. Functional Targeting of the MUC1 Oncogen in Human Cancers. Cancer Biol. Ther. 2009, 8, 1197–1203. [Google Scholar] [CrossRef] [PubMed]
- Tolomeo, M.; Cascio, A. The Multifaced Role of Stat3 in Cancer and Its Implication for Anticancer Therapy. Int. J. Mol. Sci. 2021, 22, 603. [Google Scholar] [CrossRef]
- Huang, B.; Lang, X.; Li, X. The Role of IL-6/JAK2/STAT3 Signaling Pathway in Cancers. Front. Oncol. 2022, 12, 1023177. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Contino, G.; Deshpande, V.; Tzatsos, A.; Conrad, C.; Benes, C.H.; Levy, D.E.; Settleman, J.; Engelman, J.A.; Bardeesy, N. STAT3 Plays a Critical Role in KRAS-Induced Pancreatic Tumorigenesis. Cancer Res. 2011, 71, 5020–5029. [Google Scholar] [CrossRef]
- Li, B.; Huang, C. Regulation of EMT by STAT3 in Gastrointestinal Cancer (Review). Int. J. Oncol. 2017, 50, 753–767. [Google Scholar] [CrossRef]
- Guo, H.; Hu, Z.; Yang, X.; Yuan, Z.; Gao, Y.; Chen, J.; Xie, L.; Chen, C.; Guo, Y.; Bai, Y. STAT3 Inhibition Enhances Gemcitabine Sensitivity in Pancreatic Cancer by Suppressing EMT, Immune Escape and Inducing Oxidative Stress Damage. Int. Immunopharmacol. 2023, 123, 110709. [Google Scholar] [CrossRef]
- Hamel, Z.; Sanchez, S.; Standing, D.; Anant, S. Role of STAT3 in Pancreatic Cancer. Explor. Target. Anti-Tumor Ther. 2024, 5, 20–33. [Google Scholar] [CrossRef]
- Bose, M.; Sanders, A.; Handa, A.; Vora, A.; Cardona, M.R.; Brouwer, C.; Mukherjee, P. Molecular Crosstalk between MUC1 and STAT3 Influences the Anti-Proliferative Effect of Napabucasin in Epithelial Cancers. Sci. Rep. 2024, 14, 3178. [Google Scholar] [CrossRef] [PubMed]
- Hata, T.; Rajabi, H.; Yamamoto, M.; Jin, C.; Ahmad, R.; Zhang, Y.; Kui, L.; Li, W.; Yasumizu, Y.; Hong, D.; et al. Targeting MUC1-C Inhibits TWIst1 Signaling in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2019, 18, 1744–1754. [Google Scholar] [CrossRef] [PubMed]
- Cano, C.; Motoo, Y.; Iovanna, J.L. Epithelial-to-Mesenchymal Transition in Pancreatic Adenocarcinoma. Sci. World J. 2010, 10, 1047–1057. [Google Scholar] [CrossRef]
- Wang, H.; Wu, J.; Zhang, Y.; Xue, X.; Tang, D.; Yuan, Z.; Chen, M.; Wei, J.; Zhang, J.; Miao, Y. Transforming Growth Factor β-Induced Epithelial-Mesenchymal Transition Increases Cancer Stem-like Cells in the PANC-1 Cell Line. Oncol. Lett. 2012, 3, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Thürling, I.; Färber, B.; Kowalke, T.; Fischer, T.; De Assis, L.V.M.; Braun, R.; Castven, D.; Oster, H.; Konukiewitz, B.; et al. The Quasimesenchymal Pancreatic Ductal Epithelial Cell Line PANC-1—A Useful Model to Study Clonal Heterogeneity and EMT Subtype Shifting. Cancers 2022, 14, 2057. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Li, Y.; Tan, D.; Dong, X.; Chang, P.; Kar, S.; Li, D. Biomarkers of TGF-β Signaling Pathway and Prognosis of Pancreatic Cancer. PLoS ONE 2014, 9, e85942. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liang, Y.; Yin, Q.; Liu, S.; Wang, Q.; Tang, Y.; Cao, C. Clinical and Prognostic Significance of Serum Transforming Growth Factor-Beta1 Levels in Patients with Pancreatic Ductal Adenocarcinoma. J. Med. Biol. Res. 2016, 49, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Bradshaw, A.D.; Gera, S.; Zahidunnabi Dewan, M.; Xu, R. The TGF-β/Smad4 Signaling Pathway in Pancreatic Carcinogenesis and Its Clinical Significance. J. Clin. Med. 2017, 6, 5. [Google Scholar] [CrossRef]
- Grover, P.; Nath, S.; Nye, M.D.; Zhou, R.; Ahmad, M.; Mukherjee, P. SMAD4-Independent Activation of TGF-β Signaling by MUC1 in a Human Pancreatic Cancer Cell Line. Oncotarget 2018, 9, 6897–6910. [Google Scholar] [CrossRef] [PubMed]
- Bose, M.; Grover, P.; Sanders, A.J.; Zhou, R.; Ahmad, M.; Shwartz, S.; Lala, P.; Nath, S.; Yazdanifar, M.; Brouwer, C.; et al. Overexpression of MUC1 Induces Non-Canonical TGF-β Signaling in Pancreatic Ductal Adenocarcinoma. Front. Cell Dev. Biol. 2022, 10, 821875. [Google Scholar] [CrossRef]
- Giulietti, M.; Righetti, A.; Principato, G.; Piva, F. LncRNA Co-Expression Network Analysis Reveals Novel Biomarkers for Pancreatic Cancer. Carcinogenesis 2018, 39, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Sempere, L.F.; Powell, K.; Rana, J.; Brock, A.A.; Schmittgen, T.D. Role of Non-Coding RNAs in Tumor Progression and Metastasis in Pancreatic Cancer. Cancer Metastasis Rev. 2021, 40, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Koyama, K.; Ota, Y.; Iwamoto, H.; Yamakita, K.; Fujii, S.; Kitano, Y. The Interaction Between Long Non-Coding RNA HULC and MicroRNA-622 via Transfer by Extracellular Vesicles Regulates Cell Invasion and Migration in Human Pancreatic Cancer. Front. Oncol. 2020, 10, 1013. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Molecular Mechanisms of Long Noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Klicka, K.; Grzywa, T.M.; Mielniczuk, A.; Klinke, A.; Włodarski, P.K. The Role of MiR-200 Family in the Regulation of Hallmarks of Cancer. Front. Oncol. 2022, 12, 965231. [Google Scholar] [CrossRef] [PubMed]
- Cavallari, I.; Ciccarese, F.; Sharova, E.; Urso, L.; Raimondi, V.; Silic-Benussi, M.; D’Agostino, D.M.; Ciminale, V. The MiR-200 Family of MicroRNAs: Fine Tuners of Epithelial-Mesenchymal Transition and Circulating Cancer Biomarkers. Cancers 2021, 13, 5874. [Google Scholar] [CrossRef] [PubMed]
- Shichiri, K.; Oshi, M.; Ziazadeh, D.; Endo, I.; Takabe, K. MiR-200c in Gastric Cancer Patients. Am. J. Cancer Res. 2023, 13, 3027–3040. [Google Scholar]
- Diaz-Riascos, Z.V.; Ginesta, M.M.; Fabregat, J.; Serrano, T.; Busquets, J.; Buscail, L.; Cordelier, P.; Capellá, G. Expression and Role of MicroRNAs from the MiR-200 Family in the Tumor Formation and Metastatic Propensity of Pancreatic Cancer. Mol. Ther. Nucleic Acids 2019, 17, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Katoch, A.; Jamwal, V.L.; Faheem, M.M.; Kumar, S.; Senapati, S.; Yadav, G.; Gandhi, S.G.; Goswami, A. Overlapping Targets Exist between the Par-4 and MiR-200c Axis Which Regulate EMT and Proliferation of Pancreatic Cancer Cells. Transl. Oncol. 2021, 14, 100879. [Google Scholar] [CrossRef]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A Reciprocal Repression between ZEB1 and Members of the MiR-200 Family Promotes EMT and Invasion in Cancer Cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef]
- Lu, Y.; Lu, J.; Li, X.; Zhu, H.; Fan, X.; Zhu, S.; Wang, Y.; Guo, Q.; Wang, L.; Huang, Y.; et al. MiR-200a Inhibits Epithelial-Mesenchymal Transition of Pancreatic Cancer Stem Cell. BMC Cancer 2014, 14, 85. [Google Scholar] [CrossRef]
- Xie, H.; Chen, J.; Ma, Z.; Gao, Y.; Zeng, J.; Chen, Y.; Yang, Z.; Xu, S. PrLZ Regulates EMT and Invasion in Prostate Cancer via the TGF-Β1/p-Smad2/MiR-200 Family/ZEB1 Axis. Prostate 2024, 84, 317–328. [Google Scholar] [CrossRef]
- Mohr, A.M.; Bailey, J.M.; Lewallen, M.E.; Liu, X.; Radhakrishnan, P.; Yu, F.; Tapprich, W.; Hollingsworth, M.A. MUC1 Regulates Expression of Multiple MicroRNAs Involved in Pancreatic Tumor Progression, Including the MiR-200c/141 Cluster. PLoS ONE 2013, 8, e73306. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Lafreniere, R.G.; Powers, V.E.; Sebastio, G.; Ballabio, A.; Pettigrew, A.L.; Ledbetter, D.H.; Levy, E.; Craig, I.W.; Willard, H.F. Localization of the X Inactivation Centre on the Human X Chromosome in Xq13. Nature 1991, 349, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Liu, Y.; Lu, Y.; Yang, B.; Tang, L. LncRNA XIST Promotes Pancreatic Cancer Proliferation Through MiR-133a/EGFR. J. Cell. Biochem. 2017, 3358, 3349–3358. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, Y. LncRNA XIST Enhanced TGF-Β2 Expression by Targeting MiR-141-3p to Promote Pancreatic Cancer Cells Invasion. Biosci. Rep. 2019, 39, BSR20190332. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Hong, L.; Yu, D.; Cao, T.; Zhou, Z.; He, S. International Journal of Biochemistry and Cell Biology LncRNA XIST Promotes Pancreatic Cancer Migration, Invasion and EMT by Sponging MiR-429 to Modulate ZEB1 Expression. Int. J. Biochem. Cell Biol. 2019, 113, 17–26. [Google Scholar] [CrossRef]
- Wang, K.; Bhattacharya, A.; Haratake, N.; Daimon, T.; Nakashoji, A.; Ozawa, H.; Peng, B.; Li, W.; Kufe, D. XIST and MUC1-C Form an Auto-Regulatory Pathway in Driving Cancer Progression. Cell Death Dis. 2024, 15, 330. [Google Scholar] [CrossRef]
- Huang, B.; Liu, C.; Wu, Q.; Zhang, J.; Min, Q.; Sheng, T.; Wang, X.; Zou, Y. Long Non-Coding RNA NEAT1 Facilitates Pancreatic Cancer Progression through Negative Modulation of MiR-506-3p. Biochem. Biophys. Res. Commun. 2017, 482, 828–834. [Google Scholar] [CrossRef]
- Clemson, C.M.; Hutchinson, J.N.; Sara, S.A.; Ensminger, A.W.; Fox, A.H.; Chess, A.; Lawrence, J.B. Article An Architectural Role for a Nuclear Noncoding RNA: NEAT1 RNA Is Essential for the Structure of Paraspeckles. Mol. Cell 2009, 33, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Deng, X.; Xiao, W.; Huang, B.; Yi, X.; Zou, Y. Downregulation of NEAT1 Sensitizes Gemcitabine-Resistant Pancreatic Cancer Cells to Gemcitabine through Modulation of the MiR-506-3p/ZEB2/EMT Axis. Am. J. Cancer Res. 2021, 11, 3841–3856. [Google Scholar]
- Bhattacharya, A.; Wang, K.; Penailillo, J.; Chan, C.N.; Fushimi, A.; Yamashita, N.; Daimon, T. MUC1-C Regulates NEAT1 LncRNA Expression and Paraspeckle Formation in Cancer Progression. Oncogene 2024, 43, 2199–2214. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.; Jiang, X.Y.; Tian, H.X.; Rong, D.C.; Song, J.N.; Wang, L.; Chen, Y.S.; Wong, R.C.B.; Guo, C.X.; Wang, L.S.; et al. Anoikis in Cell Fate, Physiopathology, and Therapeutic Interventions. MedComm 2024, 5, 718. [Google Scholar] [CrossRef] [PubMed]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis Molecular Pathways and Its Role in Cancer Progression. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.N.; Koo, K.H.; Sung, J.Y.; Yun, U.J.; Kim, H. Anoikis Resistance: An Essential Prerequisite for Tumor Metastasis. Int. J. Cell Biol. 2012, 2012, 306879. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.J.; Wong, M.K.; Tan, T.Z.; Kuay, K.T.; C Ng, A.H.; Chung, V.Y.; Chu, Y.S.; Matsumura, N.; Lai, H.C.; Lee, Y.F.; et al. An EMT Spectrum Defines an Anoikis-Resistant and Spheroidogenic Intermediate Mesenchymal State That Is Sensitive to e-Cadherin Restoration by a Src-Kinase Inhibitor, Saracatinib (AZD0530). Cell Death Dis. 2013, 4, 915. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.N.; Zhang, Z.T.; Wang, L.; Wei, H.X.; Zhang, T.; Zhang, L.M.; Lin, H.; Zhang, H.; Wang, S.Q. TGF-Β1/SH2B3 Axis Regulates Anoikis Resistance and EMT of Lung Cancer Cells by Modulating JAK2/STAT3 and SHP2/Grb2 Signaling Pathways. Cell Death Dis. 2022, 13, 472. [Google Scholar] [CrossRef]
- Fofaria, N.M.; Srivastava, S.K. STAT3 Induces Anoikis Resistance, Promotes Cell Invasion and Metastatic Potential in Pancreatic Cancer Cells. Carcinogenesis 2015, 36, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cheng, S.; Fleishman, J.S.; Chen, J.; Tang, H.; Chen, Z.; Chen, W.; Ding, M. Targeting Anoikis Resistance as a Strategy for Cancer Therapy. Drug Resist. Updat. 2024, 75, 101099. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Hu, H.; Yuan, Z.; Yao, H. A Prognostic Model for Anoikis-Related Genes in Pancreatic Cancer. Sci. Rep. 2024, 14, 15200. [Google Scholar] [CrossRef] [PubMed]
- Piyush, T.; Rhodes, J.M.; Yu, L. MUC1 O-Glycosylation Contributes to Anoikis Resistance in Epithelial Cancer Cells. Cell Death Discov. 2017, 3, 17044. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Piyush, T.; Chen, C.; Hollingsworth, M.A.; Hilkens, J.; Rhodes, J.M.; Yu, L. MUC1 Extracellular Domain Confers Resistance of Epithelial Cancer Cells to Anoikis. Cell Death Dis. 2014, 5, e1438. [Google Scholar] [CrossRef] [PubMed]
- Yu, L. Cancer Cell Resistance to Anoikis: MUC1 Glycosylation Comes to Play. Cell Death Dis. 2017, 8, e2962. [Google Scholar] [CrossRef] [PubMed]
- Bose, M.; Sanders, A.; De, C.; Zhou, R.; Lala, P.; Shwartz, S.; Mitra, B.; Brouwer, C.; Mukherjee, P. Targeting Tumor-Associated MUC1 Overcomes Anoikis-Resistance in Pancreatic Cancer. Transl. Res. 2023, 253, 41–56. [Google Scholar] [CrossRef]
- Torres, M.P.; Chakraborty, S.; Souchek, J.; Batra, S.K. Mucin-Based Targeted Pancreatic Cancer Therapy. Curr. Pharm. Des. 2012, 18, 2472–2481. [Google Scholar] [CrossRef]
- Hepatology, T.L. The Lancet Gastroenterology & Hepatology Pancreatic Cancer: A State of Emergency? Lancet Gastroenterol. Hepatol. 2021, 6, 81. [Google Scholar] [CrossRef]
- Hawkes, N. Cancer Survival Data Emphasise Importance of Early Diagnosis. BMJ 2019, 364, l408. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; You, L.; Dai, M.; Zhao, Y. Mucins in Pancreatic Cancer: A Well-Established but Promising Family for Diagnosis, Prognosis and Therapy. J. Cell. Mol. Med. 2020, 24, 10279–10289. [Google Scholar] [CrossRef]
- Rachagani, S.; Torres, M.P.; Kumar, S.; Haridas, D.; Baine, M.; MacHa, M.A.; Kaur, S.; Ponnusamy, M.P.; Dey, P.; Seshacharyulu, P.; et al. Mucin (Muc) Expression during Pancreatic Cancer Progression in Spontaneous Mouse Model: Potential Implications for Diagnosis and Therapy. J. Hematol. Oncol. 2012, 5, 68. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.K.; Khan, P.; Natarajan, G.; Atri, P.; Aithal, A.; Ganti, A.K.; Batra, S.K.; Nasser, M.W.; Jain, M. Mucins as Potential Biomarkers for Early Detection of Cancer. Cancers 2023, 15, 1640. [Google Scholar] [CrossRef]
- Li, N.S.; Lin, W.L.; Hsu, Y.P.; Chen, Y.T.; Shiue, Y.L.; Yang, H.W. Combined Detection of CA19-9 and MUC1 Using a Colorimetric Immunosensor Based on Magnetic Gold Nanorods for Ultrasensitive Risk Assessment of Pancreatic Cancer. ACS Appl. Bio Mater. 2019, 2, 4847–4855. [Google Scholar] [CrossRef]
- Gao, T.; Cen, Q.; Lei, H. A Review on Development of MUC1-Based Cancer Vaccine. Biomed. Pharmacother. 2020, 132, 110888. [Google Scholar] [CrossRef] [PubMed]
- Pinkhasov, J.; Alvarez, M.L.; Pathangey, L.B.; Tinder, T.L.; Mason, H.S.; Walmsley, A.M.; Gendler, S.J.; Mukherjee, P. Analysis of a Cholera Toxin B Subunit (CTB) and Human Mucin 1 (MUC1) Conjugate Protein in a MUC1-Tolerant Mouse Model. Cancer Immunol. Immunother. 2010, 59, 1801–1811. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; Lee, K.M.; McKolanis, J.; Hitbold, E.; Schraut, W.; Moser, A.J.; Warnick, E.; Whiteside, T.; Osborne, J.; Kim, H.; et al. Phase I Study of a MUC1 Vaccine Composed of Different Doses of MUC1 Peptide with SB-AS2 Adjuvant in Resected and Locally Advanced Pancreatic Cancer. Cancer Immunol. Immunother. 2005, 54, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Barratt-Boyes, S.M.; Vlad, A.; Finn, O.J. Immunization of Chimpanzees with Tumor Antigen MUC1 Mucin Tandem Repeat Peptide Elicits Both Helper and Cytotoxic T-Cell Responses. Clin. Cancer Res. 1999, 5, 1918–1924. [Google Scholar] [PubMed]
- Li, M.; Wang, Z.; Yan, B.; Yin, X.; Zhao, Y.; Yu, F.; Meng, M.; Liu, Y.; Zhao, W. Design of a MUC1-Based Tricomponent Vaccine Adjuvanted with FSL-1 for Cancer Immunotherapy. Medchemcomm 2019, 10, 2073–2077. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdanifar, M.; Das Roy, L.; Whilding, L.M.; Gavrill, A.; Maher, J.; Mukherjee, P. CAR T Cells Targeting the Tumor MUC1 Glycoprotein Reduce Triple-Negative Breast Cancer Growth. Front. Immunol. 2019, 10, 1149. [Google Scholar] [CrossRef] [PubMed]
- Yazdanifar, M.; Zhou, R.; Grover, P.; Williams, C.; Bose, M.; Moore, L.J.; Wu, S.T.; Maher, J.; Dreau, D.; Mukherjee, P. Overcoming Immunological Resistance Enhances the Efficacy of a Novel Anti-TMUC1-CAR T Cell Treatment against Pancreatic Ductal Adenocarcinoma. Cells 2019, 8, 1070. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.; Farrukh, H.; Chittepu, V.C.S.R.; Xu, H.; Pan, C.X.; Zhu, Z. CAR Race to Cancer Immunotherapy: From CAR T, CAR NK to CAR Macrophage Therapy. J. Exp. Clin. Cancer Res. 2022, 41, 119. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Jin, G.; Wang, L.; Li, M.; He, C.; Guo, X.; Zhu, Q. The Role of PAM4 in the Management of Pancreatic Cancer: Diagnosis, Radioimmunodetection, and Radioimmunotherapy. J. Immunol. Res. 2014, 2014, 268479. [Google Scholar] [CrossRef]
- Yao, Y.; Fan, D. Advances in MUC1 Resistance to Chemotherapy in Pancreatic Cancer. J. Chemother. 2023, 36, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Mujumdar, N.; Dudeja, V.; Mackenzie, T.; Krosch, T.K.; Sangwan, V.; Vickers, S.M.; Saluja, A.K. MUC1 Regulates Cell Survival in Pancreatic Cancer by Preventing Lysosomal Permeabilization. PLoS ONE 2012, 7, e43020. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dieli, R.; Lioy, R.; Crispo, F.; Cascelli, N.; Martinelli, M.; Lerose, R.; Telesca, D.; Milella, M.R.; Colella, M.; Loperte, S.; et al. The Oncoprotein Mucin 1 in Pancreatic Cancer Onset and Progression: Potential Clinical Implications. Biomolecules 2025, 15, 275. https://doi.org/10.3390/biom15020275
Dieli R, Lioy R, Crispo F, Cascelli N, Martinelli M, Lerose R, Telesca D, Milella MR, Colella M, Loperte S, et al. The Oncoprotein Mucin 1 in Pancreatic Cancer Onset and Progression: Potential Clinical Implications. Biomolecules. 2025; 15(2):275. https://doi.org/10.3390/biom15020275
Chicago/Turabian StyleDieli, Rosalia, Rosa Lioy, Fabiana Crispo, Nicoletta Cascelli, Mara Martinelli, Rosa Lerose, Donatella Telesca, Maria Rita Milella, Marco Colella, Simona Loperte, and et al. 2025. "The Oncoprotein Mucin 1 in Pancreatic Cancer Onset and Progression: Potential Clinical Implications" Biomolecules 15, no. 2: 275. https://doi.org/10.3390/biom15020275
APA StyleDieli, R., Lioy, R., Crispo, F., Cascelli, N., Martinelli, M., Lerose, R., Telesca, D., Milella, M. R., Colella, M., Loperte, S., & Mazzoccoli, C. (2025). The Oncoprotein Mucin 1 in Pancreatic Cancer Onset and Progression: Potential Clinical Implications. Biomolecules, 15(2), 275. https://doi.org/10.3390/biom15020275