
Determining Ligand Binding and Specificity Within the β2-Integrin Family with a Novel Assay Platform

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmids, Cells, Antibodies, and Purified Proteins
2.2. Protein Expression and Purification
2.3. Interaction Analyses
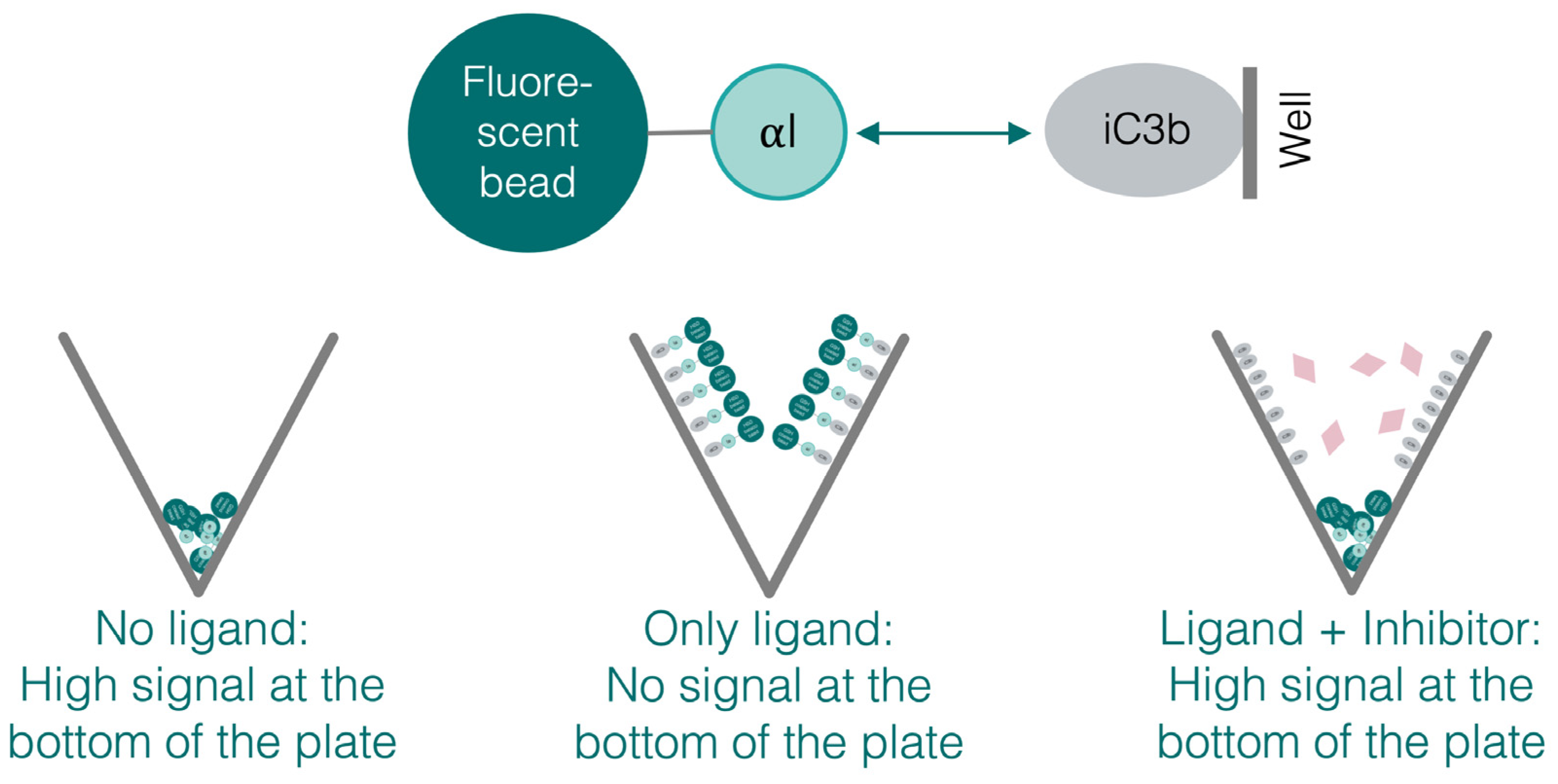
2.4. Bead-Based Adhesion Assay
2.5. Transfected HEK Cell Adhesion Assay
3. Results
3.1. The αI Domains of β2 Integrins Can Be Uniformly Expressed in E. coli
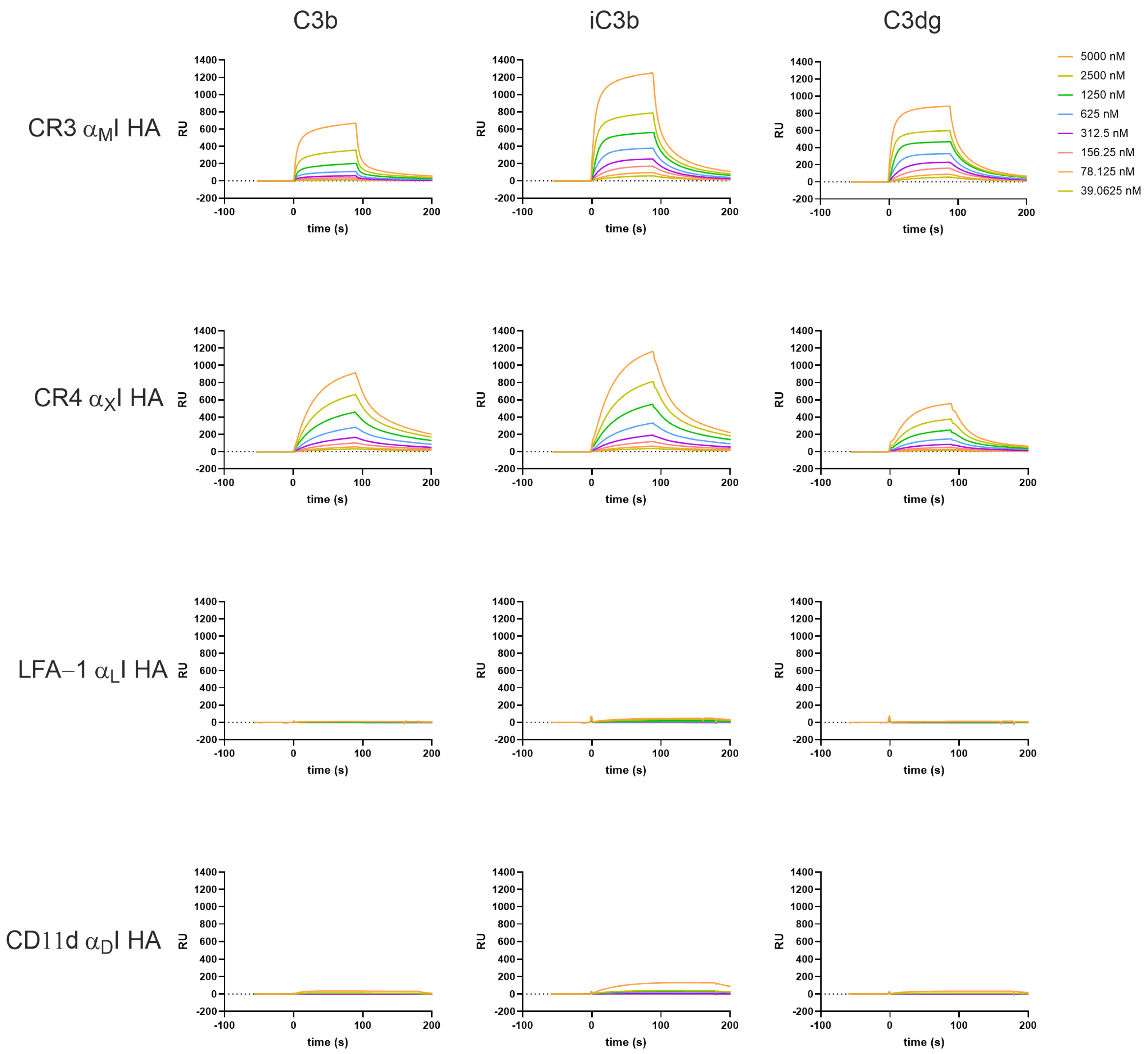
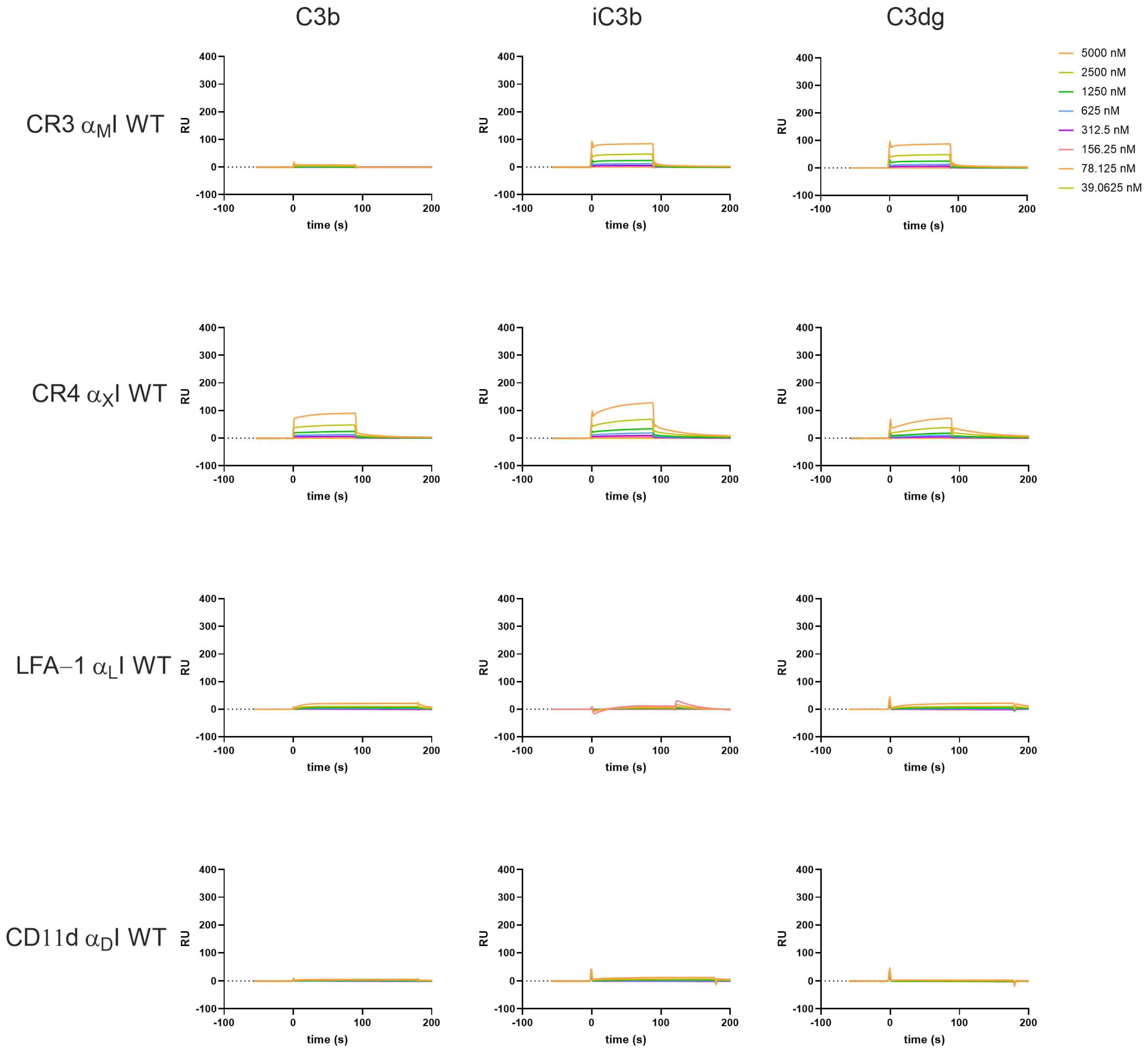
3.2. β2-Integrin I Domains Show Distinct Binding to C3-Derived Opsonins
3.3. Recombinant αI Domains of β2 Integrins Recognize ICAMs
3.4. The Interaction Platform Enables the Characterization of β2-Integrin Modulators
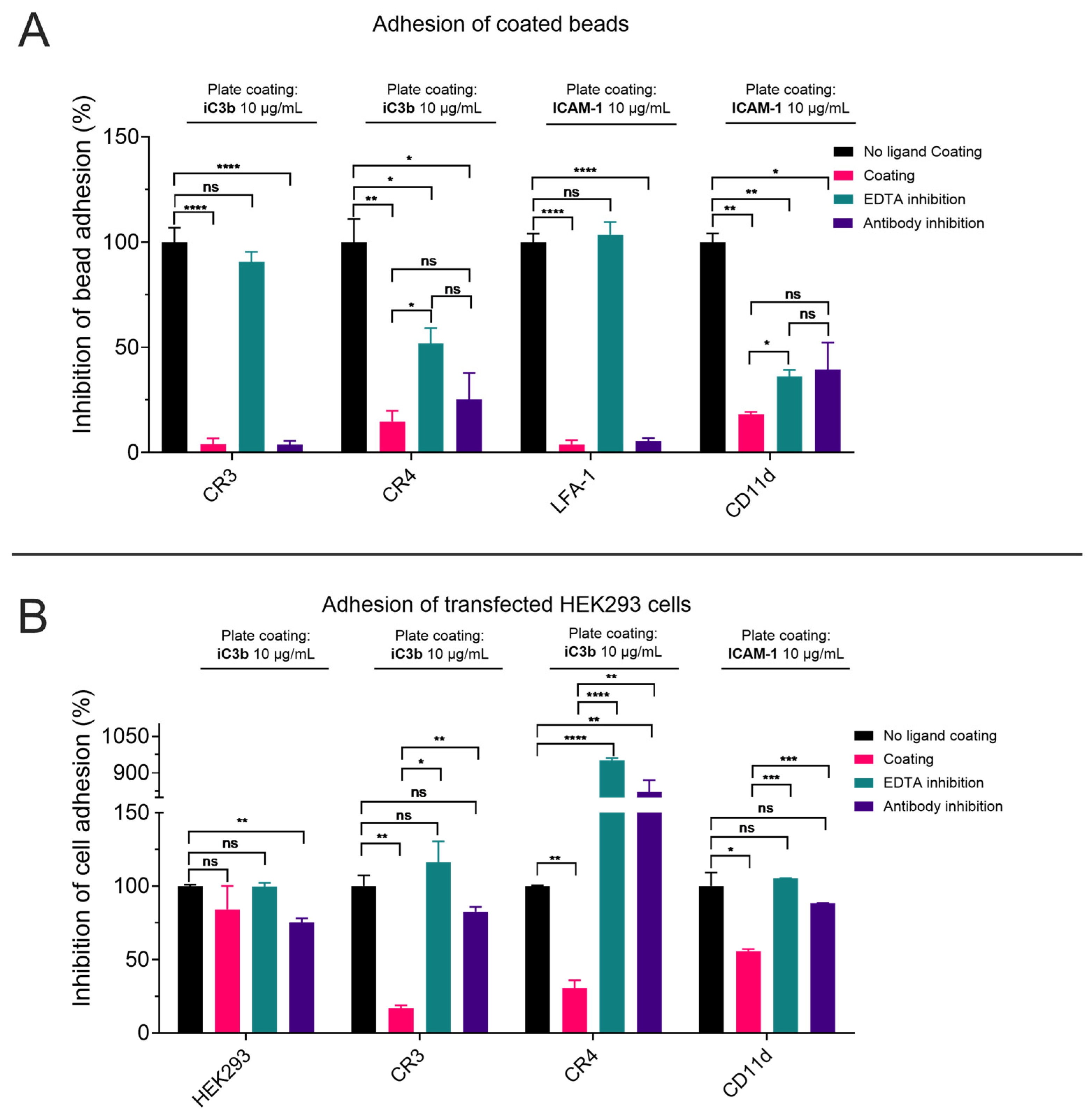
3.5. The Recombinant αI Domains Can Be Used in Functional Adhesion Assays
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMINDAS | adjacent to MIDAS |
| BCECF-AM | 2′,7′-Bis-(2-Carboxyethyl)-5-(and-6)-Carboxyfluorescein, Acetoxymethyl Ester |
| CR1 | Complement receptor 1, CD35 |
| CR3 | Complement receptor 3, macrophage-1antigen, αMβ2, CD11b/CD18 |
| CR4 | Complement receptor 4, p150,95, αXβ2, CD11c/CD18 |
| ECM | Extracellular matrix |
| EDTA | Ethylenediaminetetraacetic acid |
| FB | Complement factor B |
| FBS | Fetal bovine serum |
| FD | Complement factor D |
| FH | Complement Factor H |
| FI | Complement Factor I |
| FITC | Fluorescein isothiocyanate |
| FRET | Förster resonance energy transfer |
| GST | Glutathione-S-Transferase |
| HA | High affinity |
| HEK293 | Human Embryonic Kidney 293 cells |
| HEPES | 2-(4-(2-Hydroxyethyl)-1-piperazinyl)-ethansulfuric acid |
| ICAM | Intercellular adhesion molecule |
| IPTG | Isopropyl β-D-1-thiogalactopyranoside |
| LAD | Leucocyte adhesion deficiency |
| LFA-1 | Lymphocyte function-associated antigen 1, αLβ2, CD11a/CD18 |
| MIDAS | Metal ion-dependent adhesion site |
| nanoDSF | Nano differential scanning fluorimetry |
| rpm | Revolutions per minute |
| RU | Response units |
| SILEN | Socket for isoleucine |
| SPR | Surface plasmon resonance |
| VCAM-1 | Vascular cell adhesion molecule 1 |
| vWFA | Von Willebrand factor type A |
| WT | Wild type |
References
- Bednarczyk, M.; Stege, H.; Grabbe, S.; Bros, M. Beta2 Integrins—Multi-Functional Leukocyte Receptors in Health and Disease. Int. J. Mol. Sci. 2020, 21, 1402. [Google Scholar] [CrossRef]
- Springer, T.A. Traffic Signals on Endothelium for Lymphocyte Recirculation and Leukocyte Emigration: The Multistep Paradigm. Annu. Rev. Physiol. 1995, 57, 827–872. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.; Kukkurainen, S.; Hytönen, V.P.; Wehrle-Haller, B. Cell Adhesion by Integrins. Physiol. Rev. 2019, 99, 1655–1699. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, Allosteric Signaling Machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Stanley, P.; Hogg, N. The I Domain of Integrin LFA-1 Interacts with ICAM-1 Domain 1 at Residue Glu-34 but Not Gln-73. J. Biol. Chem. 1998, 273, 3358–3362. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.L.; Lu, J.; Riddle, L.; Kim, K.J.; Presta, L.G.; Bodary, S.C. Identification of the Binding Site in Intercellular Adhesion Molecule 1 for Its Receptor, Leukocyte Function-Associated Antigen 1. Mol. Biol. Cell 1997, 8, 501–515. [Google Scholar] [CrossRef]
- Bailly, P.; Tontti, E.; Hermand, P.; Cartron, J.-P.; Gahmberg, C.G. The Red Cell LW Blood Group Protein Is an Intercellular Adhesion Molecule Which Binds to CD11/CD18 Leukocyte Integrins. Eur. J. Immunol. 1995, 25, 3316–3320. [Google Scholar] [CrossRef]
- Tian, L.; Yoshihara, Y.; Mizuno, T.; Mori, K.; Gahmberg, C.G. The Neuronal Glycoprotein Telencephalin Is a Cellular Ligand for the CD11a/CD18 Leukocyte Integrin. J. Immunol. 1997, 158, 928–936. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Reis, E.S.; Lambris, J.D.; Lindorfer, M.A.; Cook, E.M.; Reis, E.S.; Ricklin, D.; Risitano, A.M.; Lambris, J.D.; Taylor, R.P.; et al. Complement in Disease: A Defence System Turning Offensive. Nat. Rev. Nephrol. 2016, 12, 383–401. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Reis, E.S.; Mastellos, D.C.; Gros, P.; Lambris, J.D. Complement Component C3—The “Swiss Army Knife” of Innate Immunity and Host Defense. Immunol. Rev. 2016, 274, 33–58. [Google Scholar] [CrossRef]
- Bajic, G.; Yatime, L.; Sim, R.B.; Vorup-Jensen, T.; Andersen, G.R. Structural Insight on the Recognition of Surface-Bound Opsonins by the Integrin I Domain of Complement Receptor 3. Proc. Natl. Acad. Sci. USA 2013, 110, 16426–16431. [Google Scholar] [CrossRef] [PubMed]
- Bilsland, C.A.; Diamond, M.S.; Springer, T.A. The Leukocyte Integrin P150,95 (CD11c/CD18) as a Receptor for iC3b. Activation by a Heterologous Beta Subunit and Localization of a Ligand Recognition Site to the I Domain. J. Immunol. 1994, 152, 4582–4589. [Google Scholar] [CrossRef]
- Choi, J.; Buyannemekh, D.; Nham, S.U. Moieties of Complement iC3b Recognized by the I-Domain of Integrin αXβ2. Mol. Cells 2020, 43, 1023–1034. [Google Scholar] [CrossRef]
- Vorup-Jensen, T.; Ostermeier, C.; Shimaoka, M.; Hommel, U.; Springer, T.A. Structure and Allosteric Regulation of the αXβ2 Integrin I Domain. Proc. Natl. Acad. Sci. USA 2003, 100, 1873–1878. [Google Scholar] [CrossRef]
- Ross, G. Identification of a C3bi-Specific Membrane Complement Receptor That Is Expressed on Lymphocytes, Monocytes, Neutrophils, and Erythrocytes. J. Exp. Med. 1982, 155, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Ross, G.; Newman, S.L.; Lambris, J.D.; Devery-Pocius, J.E.; Cain, J.A.; Lachmann, P.J. Generation of Three Different Fragments of Bound C3 with Purified Factor I or Serum. II. Location of Binding Sites in the C3 Fragments for Factors B and H, Complement Receptors, and Bovine Conglutinin. J. Exp. Med. 1983, 158, 334–352. [Google Scholar] [CrossRef] [PubMed]
- Wright, B.Y.S.D.; Silverstein, S.C. Tumor-Promoting Phorbol Esters Stimulate C3b and C3b’receptor-Mediated Phagocytosis in Cultured Human Monocytes. J. Exp. Med. 1982, 156, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Yakubenko, V.P.; Yadav, S.P.; Ugarova, T.P. Integrin αDβ2, an Adhesion Receptor up-Regulated on Macrophage Foam Cells, Exhibits Multiligand-Binding Properties. Blood 2006, 107, 1643–1650. [Google Scholar] [CrossRef] [PubMed]
- Blythe, E.N.; Weaver, L.C.; Brown, A.; Dekaban, G.A. B2 Integrin CD11d/CD18: From Expression to an Emerging Role in Staged Leukocyte Migration. Front. Immunol. 2021, 12, 775447. [Google Scholar] [CrossRef]
- Van der Vieren, M.; Le Trong, H.; Wood, C.L.; Moore, P.F.; John, T.S.; Staunton, D.E.; Gallatin, W.M. A Novel Leukointegrin, αDβ2, Binds Preferentially to ICAM-3. Immunity 1995, 3, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.; Plüss, C.J.; Ricklin, D. The Promiscuous Profile of Complement Receptor 3 in Ligand Binding, Immune Modulation, and Pathophysiology. Front. Immunol. 2021, 12, 662164. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; He, X.; Qiu, Z.; Zhang, H.; Xie, R.; Liu, Z.; Gu, Y.; Zhao, N.; Xiang, Q.; Cui, Y. Targeting Integrin Pathways: Mechanisms and Advances in Therapy. Signal Transduct. Target. Ther. 2023, 8, 1. [Google Scholar] [CrossRef]
- Mitroulis, I.; Alexaki, V.; Kourtzelis, I.; Ziogas, A.; Hajishengallis, G.; Chavakis, T. Leukocyte Integrins: Role in Leukocyte Recruitment and as Therapeutic Targets in Inflammatory Disease. Pharmacol. Ther. 2015, 147, 123–135. [Google Scholar] [CrossRef]
- Schittenhelm, L.; Hilkens, C.M.; Morrison, V.L. Beta2 Integrins As Regulators of Dendritic Cell, Monocyte, and Macrophage Function. Front. Immunol. 2017, 8, 1866. [Google Scholar] [CrossRef] [PubMed]
- Conley, H.E.; Sheats, M.K. Targeting Neutrophil Beta2-Integrins: A Review of Relevant Resources, Tools, and Methods. Biomolecules 2023, 13, 892. [Google Scholar] [CrossRef]
- Fagerholm, S.C.; Guenther, C.; Asens, M.L.; Savinko, T.; Uotila, L.M. Beta2-Integins and Interacting Proteins in Leukocyte Trafficking, Immune Supression, and Immunodeficiency Disease. Front. Immunol. 2019, 10, 254. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T.K.; Hollander, N.; Roberts, T.M.; Anderson, D.C.; Springer, T.A. Heterogeneous Mutations in the β Subunit Common to the LFA-1, Mac-1, and P150,95 Glycoproteins Cause Leukocyte Adhesion Deficiency. Cell 1987, 50, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Fossati-Jimack, L.; Ling, G.S.; Cortini, A.; Szajna, M.; Malik, T.H.; McDonald, J.U.; Pickering, M.C.; Cook, H.T.; Taylor, P.R.; Botto, M. Phagocytosis Is the Main CR3-Mediated Function Affected by the Lupus-Associated Variant of CD11b in Human Myeloid Cells. PLoS ONE 2013, 8, e57082. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, M.; Lek, H.S.; Prescott, A.; Fagerholm, S.C. A Systemic Lupus Erythematosus-Associated R77H Substitution in the CD11b Chain of the Mac-1 Integrin Compromises Leukocyte Adhesion and Phagocytosis. J. Biol. Chem. 2011, 286, 17303–17310. [Google Scholar] [CrossRef]
- Cai, H.Q.; Weickert, T.W.; Catts, V.S.; Balzan, R.; Galletly, C.; Liu, D.; O’Donnell, M.; Shannon Weickert, C. Altered Levels of Immune Cell Adhesion Molecules Are Associated with Memory Impairment in Schizophrenia and Healthy Controls. Brain. Behav. Immun. 2020, 89, 200–208. [Google Scholar] [CrossRef]
- Czirr, E.; Castello, N.A.; Mosher, K.I.; Castellano, J.M.; Hinkson, I.V.; Lucin, K.M.; Baeza-Raja, B.; Ryu, J.K.; Li, L.; Farina, S.N.; et al. Microglial Complement Receptor 3 Regulates Brain Aβ Levels through Secreted Proteolytic Activity. J. Exp. Med. 2017, 214, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Hu, X.; Qian, L.; Chen, S.H.; Zhou, H.; Wilson, B.; Miller, D.S.; Hong, J.S. Microglial MAC1 Receptor and PI3K Are Essential in Mediating β-Amyloid Peptide-Induced Microglial Activation and Subsequent Neurotoxicity. J. Neuroinflamm. 2011, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Pietronigro, E.; Bianca, V.D.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils Promote Alzheimer’s Disease-like Pathology and Cognitive Decline via LFA-1 Integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Celik, E.; Faridi, M.H.; Kumar, V.; Deep, S.; Moy, V.T.; Gupta, V. Agonist Leukadherin-1 Increases CD11b/CD18-Dependent Adhesion via Membrane Tethers. Biophys. J. 2013, 105, 2517–2527. [Google Scholar] [CrossRef]
- Schmid, M.C.; Khan, S.Q.; Kaneda, M.M.; Pathria, P.; Shepard, R.; Louis, T.L.; Anand, S.; Woo, G.; Leem, C.; Faridi, M.H.; et al. Integrin CD11b Activation Drives Anti-Tumor Innate Immunity. Nat. Commun. 2018, 9, 5379. [Google Scholar] [CrossRef]
- Park, E.J.; Yuki, Y.; Kiyono, H.; Shimaoka, M. Structural Basis of Blocking Integrin Activation and Deactivation for Anti-Inflammation. J. Biomed. Sci. 2015, 22, 51. [Google Scholar] [CrossRef]
- Arnaout, M.A. Biology and Structure of Leukocyte Beta2 Integrins and Their Role in Inflammation. F1000Research 2016, 5, 2433. [Google Scholar] [CrossRef]
- Xiong, J.P.; Li, R.; Essafi, M.; Stehle, T.; Arnaout, M.A. An Isoleucine-Based Allosteric Switch Controls Affinity and Shape Shifting in Integrin CD11b A-Domain. J. Biol. Chem. 2000, 275, 38762–38767. [Google Scholar] [CrossRef]
- Rosetti, F.; Mayadas, T.N. The Many Faces of Mac-1 in Autoimmune Disease. Immunol. Rev. 2016, 269, 175–193. [Google Scholar] [CrossRef]
- Luo, B.H.; Carman, C.V.; Springer, T.A. Structural Basis of Integrin Regulation and Signaling. Annu. Rev. Immunol. 2007, 25, 619–647. [Google Scholar] [CrossRef]
- Li, J.; Jo, M.H.; Yan, J.; Hall, T.; Lee, J.; López-Sánchez, U.; Yan, S.; Ha, T.; Springer, T.A. Ligand Binding Initiates Single-Molecule Integrin Conformational Activation. Cell 2024, 187, 2990–3005.e17. [Google Scholar] [CrossRef]
- Lin, Z.; Schmidt, C.Q.; Koutsogiannaki, S.; Ricci, P.; Risitano, A.M.; Lambris, J.D.; Ricklin, D. Complement C3dg-Mediated Erythrophagocytosis: Implications for Paroxysmal Nocturnal Hemoglobinuria. Blood 2015, 126, 891–894. [Google Scholar] [CrossRef] [PubMed]
- Tham, W.H.; Schmidt, C.Q.; Hauhart, R.E.; Guariento, M.; Tetteh-Quarcoo, P.B.; Lopaticki, S.; Atkinson, J.P.; Barlow, P.N.; Cowman, A.F. Plasmodium Falciparum Uses a Key Functional Site in Complement Receptor Type-1 for Invasion of Human Erythrocytes. Blood 2011, 118, 1923–1933. [Google Scholar] [CrossRef]
- Shimaoka, M.; Lu, C.; Palframan, R.T.; von Andrian, U.H.; McCormack, A.; Takagi, J.; Springer, T.A. Reversibly Locking a Protein Fold in an Active Conformation with a Disulfid Bond: Integrin AlphaLI Domains with High Affinity and Antagonistic Activity in Vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 6009–6014. [Google Scholar] [CrossRef]
- Lamers, C.; Xue, X.; Smieško, M.; van Son, H.; Wagner, B.; Berger, N.; Sfyroera, G.; Gros, P.; Lambris, J.D.; Ricklin, D. Insight into Mode-of-Action and Structural Determinants of the Compstatin Family of Clinical Complement Inhibitors. Nat. Commun. 2022, 13, 5519. [Google Scholar] [CrossRef] [PubMed]
- Weetall, M.; Hugo, R.; Friedman, C.; Maida, S.; West, S.; Wattanasin, S.; Bouhel, R.; Weitz-Schmidt, G.; Lake, P. A Homogeneous Fluorometric Assay for Measuring Cell Adhesion to Immobilized Ligand Using V-Well Microtiter Plates. Anal. Biochem. 2001, 293, 277–287. [Google Scholar] [CrossRef]
- Shimaoka, M.; Xiao, T.; Liu, J.H.; Yang, Y.; Dong, Y.; Jun, C.D.; McCormack, A.; Zhang, R.; Joachimiak, A.; Takagi, J.; et al. Structures of the αL I Domain and Its Complex with ICAM-1 Reveal a Shape-Shifting Pathway for Integrin Regulation. Cell 2003, 112, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.D.; Rao, P.E.; Van Voorhis, W.C.; Craigmyle, L.S.; Iida, K.; Talle, M.A.; Westberg, E.F.; Goldstein, G.; Silverstein, S.C. Identification of the C3bi Receptor of Human Monocytes and Macrophages by Using Monoclonal Antibodies. Proc. Natl. Acad. Sci. USA 1983, 80, 5699–5703. [Google Scholar] [CrossRef]
- Ye, F.; Kim, C.; Ginsberg, M.H. Reconstruction of Integrin Activation. Blood 2012, 119, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Fernández, F.J.; Santos-López, J.; Martínez-Barricarte, R.; Querol-García, J.; Martín-Merinero, H.; Navas-Yuste, S.; Savko, M.; Shepard, W.E.; Rodríguez de Córdoba, S.; Vega, M.C. The Crystal Structure of iC3b-CR3 αI Reveals a Modular Recognition of the Main Opsonin iC3b by the CR3 Integrin Receptor. Nat. Commun. 2022, 13, 1955. [Google Scholar] [CrossRef]
- Jensen, R.K.; Bajic, G.; Sen, M.; Springer, T.A.; Vorup-Jensen, T.; Andersen, G.R. Complement Receptor 3 Forms a Compact High-Affinity Complex with IC3b. J. Immunol. 2021, 206, 3032–3042. [Google Scholar] [CrossRef]
- Vorup-Jensen, T.; Jensen, R.K. Structural Immunology of Complement Receptors 3 and 4. Front. Immunol. 2018, 9, 2716. [Google Scholar] [CrossRef] [PubMed]
- Ustinov, V.A.; Plow, E.F. Identity of the Amino Acid Residues Involved in C3bi Binding to the I-Domain Supports a Mosaic Model to Explain the Broad Ligand Repertoire of Integrin αMβ2. Biochemistry 2005, 44, 4357–4364. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, Y.; Mi, L.-Z.; Walz, T.; Springer, T.A. Molecular Basis for Complement Recognition by Integrin αXβ2. Proc. Natl. Acad. Sci. USA 2012, 109, 4586–4591. [Google Scholar] [CrossRef]
- Xu, S.; Wang, J.; Wang, J.-H.; Springer, T.A. Distinct Recognition of Complement iC3b by Integrins αXβ2 and αMβ2. Proc. Natl. Acad. Sci. USA 2017, 114, 3403–3408. [Google Scholar] [CrossRef]
- Kallen, J.; Welzenbach, K.; Ramage, P.; Geyl, D.; Kriwacki, R.; Legge, G.; Cottens, S.; Weitz-Schmidt, G.; Hommel, U. Structural Basis for LFA-1 Inhibition upon Lovastatin Binding to the CD11a I-Domain. J. Mol. Biol. 1999, 292, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Weitz-Schmidt, G.; Welzenbach, K.; Brinkmann, V.; Kamata, T.; Kallen, J.; Bruns, C.; Cottens, S.; Takada, Y.; Hommel, U. Statins Selectively Inhibit Leukocyte Function Antigen-1 by Binding to a Novel Regulatory Integrin Site. Nat. Med. 2001, 7, 687–692. [Google Scholar] [CrossRef]
- Jensen, M.R.; Bajic, G.; Zhang, X.; Laustsen, A.K.; Koldsø, H.; Skeby, K.K.; Schiøtt, B.; Andersen, G.R.; Vorup-Jensen, T. Structural Basis for Simvastatin Competitive Antagonism of Complement Receptor 3. J. Biol. Chem. 2016, 291, 16963–16976. [Google Scholar] [CrossRef]
- Weitz-Schmidt, G.; Chreng, S. Cell Adhesion Assays. Methods Mol. Biol. 2012, 757, 15–30. [Google Scholar]
- Yakubenko, V.P.; Lishko, V.K.; Lam, S.C.T.; Ugarova, T.P. A Molecular Basis for Integrin αMβ2 Ligand Binding Promiscuity. J. Biol. Chem. 2002, 277, 48635–48642. [Google Scholar] [CrossRef]
- Sadhu, C.; Hendrickson, L.; Dick, K.O.; Potter, T.G.; Staunton, D.E. Novel Tools for Functional Analysis of CD11c: Activation-Specific, Activation-Independent, and Activating Antibodies. J. Immunoass. Immunochem. 2008, 29, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Cui, K.; Podolnikova, N.P.; Bailey, W.; Szmuc, E.; Podrez, E.A.; Byzova, T.V.; Yakubenko, V.P. Inhibition of Integrin αDβ2-Mediated Macrophage Adhesion to End Product of Docosahexaenoic Acid (DHA) Oxidation Prevents Macrophage Accumulation during Inflammation. J. Biol. Chem. 2019, 294, 14370–14382. [Google Scholar] [CrossRef]
- Podolnikova, N.P.; Kushchayeva, Y.S.; Wu, Y.F.; Faust, J.; Ugarova, T.P. The Role of Integrins αMβ2 (Mac-1, CD11b/CD18) and αDβ2 (CD11d/CD18) in Macrophage Fusion. Am. J. Pathol. 2016, 186, 2105–2116. [Google Scholar] [CrossRef]
- Koivunen, E.; Madhavan, S.; Bermudez-Garrido, L.; Grönholm, M.; Kaprio, T.; Haglund, C.; Andersson, L.C.; Gahmberg, C.G. Hypoxia Favors Tumor Growth in Colorectal Cancer in an Integrin αDβ1/Hemoglobin δ-Dependent Manner. Life Sci. Alliance 2025, 8, 1–13. [Google Scholar] [CrossRef]
- Vorup-Jensen, T. Surface Plasmon Resonance Biosensing in Studies of the Binding Between Beta2 Integrin I Domains and Their Ligands. Methods Mol. Biol. 2011, 757, 55–71. [Google Scholar]
- Errington, W.J.; Bruncsics, B.; Sarkar, C.A. Mechanisms of Noncanonical Binding Dynamics in Multivalent Protein–Protein Interactions. Proc. Natl. Acad. Sci. USA 2019, 116, 25659–25667. [Google Scholar] [CrossRef] [PubMed]
- Welzenbach, K.A. Differential Effects of Low Molecular Weight Inhibitors on the Conformation and the Immunologic Function of the Adhesion Receptor LFA-1. Doctoral Dissertation, University of Basel, Basel, Switzerland, 2004. [Google Scholar]
- Zhou, L.; Lee, D.H.S.; Plescia, J.; Lau, C.Y.; Altieri, D.C. Differential Ligand Binding Specificities of Recombinant CD11b/CD18 Integrin I-Domain. J. Biol. Chem. 1994, 269, 17075–17079. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| αMI | αXI | α LI | αDI | |||||
|---|---|---|---|---|---|---|---|---|
| HA [µM] | WT [µM] * | HA [µM] | WT [µM] * | HA | WT | HA | WT | |
| C3b | 11.6 ± 9.8 | n.b. | 2.2 ± 1.5 | 55.5 ± 27.1 | n.b. | n.b. | n.b. | n.b. |
| iC3b | 1.8 ± 1.0 | 15.0 ± 9.5 | 2.6 ± 1.6 | 33.0 ± 17.8 | n.b. | n.b. | n.b. | n.b. |
| C3dg | 1.4 ± 0.2 | 16.1 ± 10.2 | 3.8 ± 2.4 | 30.4 ± 12.0 | n.b. | n.b. | n.b. | n.b. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sommer-Plüss, C.J.; Leiggener, C.; Nikci, E.; Mancuso, R.V.; Rabbani, S.; Lamers, C.; Ricklin, D. Determining Ligand Binding and Specificity Within the β2-Integrin Family with a Novel Assay Platform. Biomolecules 2025, 15, 238. https://doi.org/10.3390/biom15020238
Sommer-Plüss CJ, Leiggener C, Nikci E, Mancuso RV, Rabbani S, Lamers C, Ricklin D. Determining Ligand Binding and Specificity Within the β2-Integrin Family with a Novel Assay Platform. Biomolecules. 2025; 15(2):238. https://doi.org/10.3390/biom15020238
Chicago/Turabian StyleSommer-Plüss, Carla Johanna, Céline Leiggener, Elira Nikci, Riccardo Vincenzo Mancuso, Said Rabbani, Christina Lamers, and Daniel Ricklin. 2025. "Determining Ligand Binding and Specificity Within the β2-Integrin Family with a Novel Assay Platform" Biomolecules 15, no. 2: 238. https://doi.org/10.3390/biom15020238
APA StyleSommer-Plüss, C. J., Leiggener, C., Nikci, E., Mancuso, R. V., Rabbani, S., Lamers, C., & Ricklin, D. (2025). Determining Ligand Binding and Specificity Within the β2-Integrin Family with a Novel Assay Platform. Biomolecules, 15(2), 238. https://doi.org/10.3390/biom15020238