
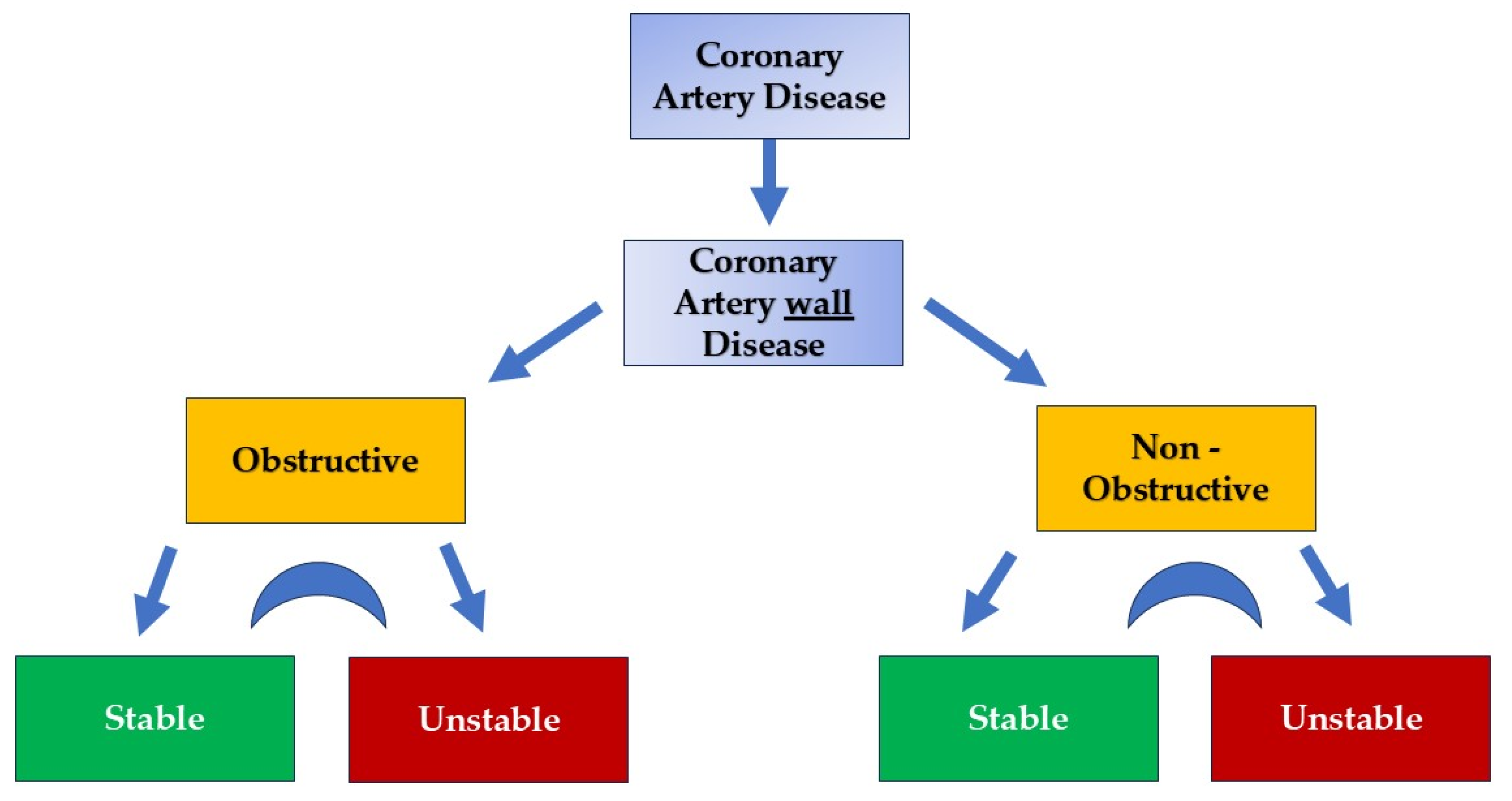
Chronic Coronary Artery Disease: Wall Disease vs. Lumenopathy

Abstract
1. Introduction
2. Role of Coronary Microcirculation in CAD Development and Progression
3. Myocardial Vasculature: Why and What to Open
4. Pathophysiologic Concerns and Questions
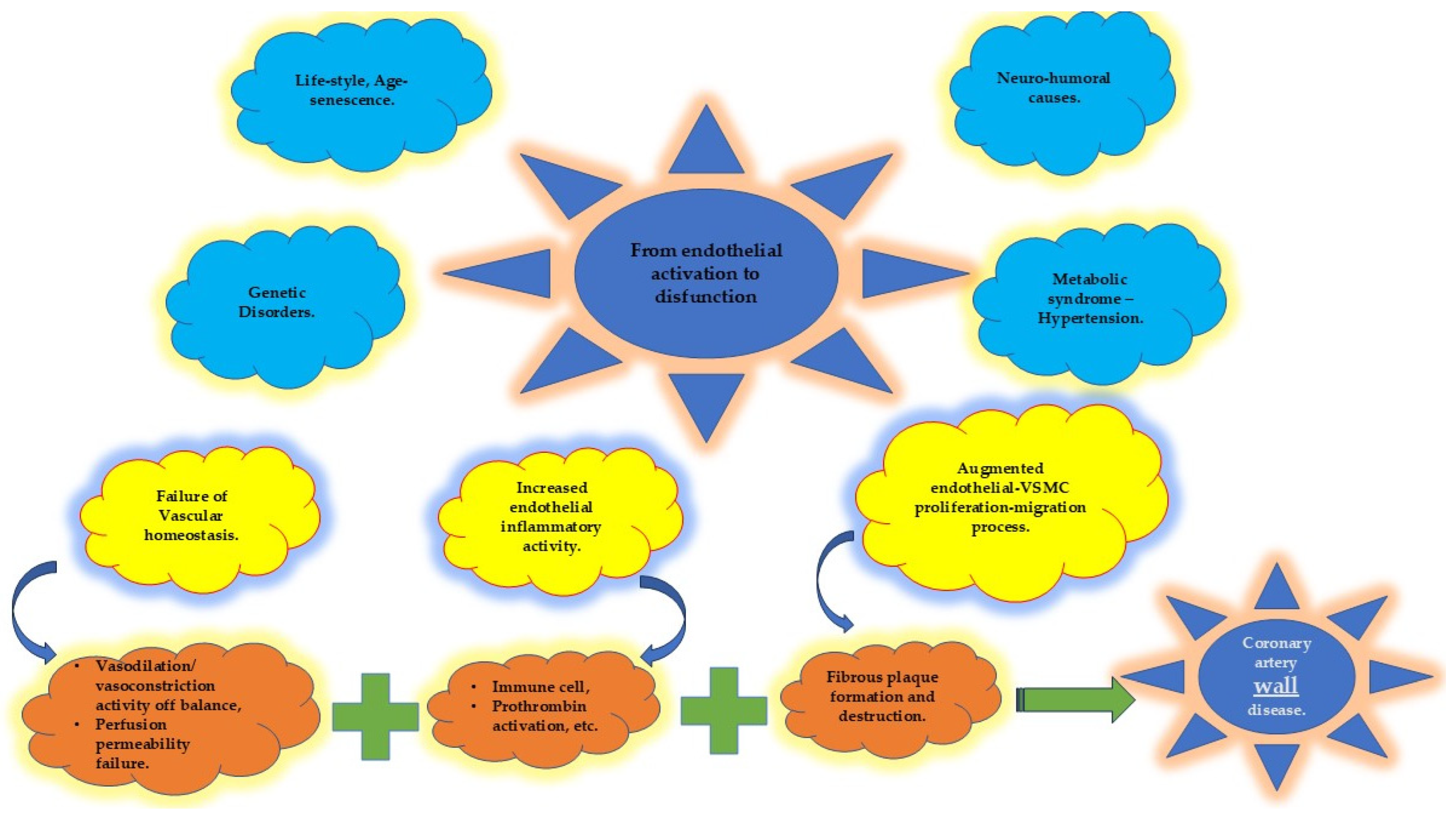
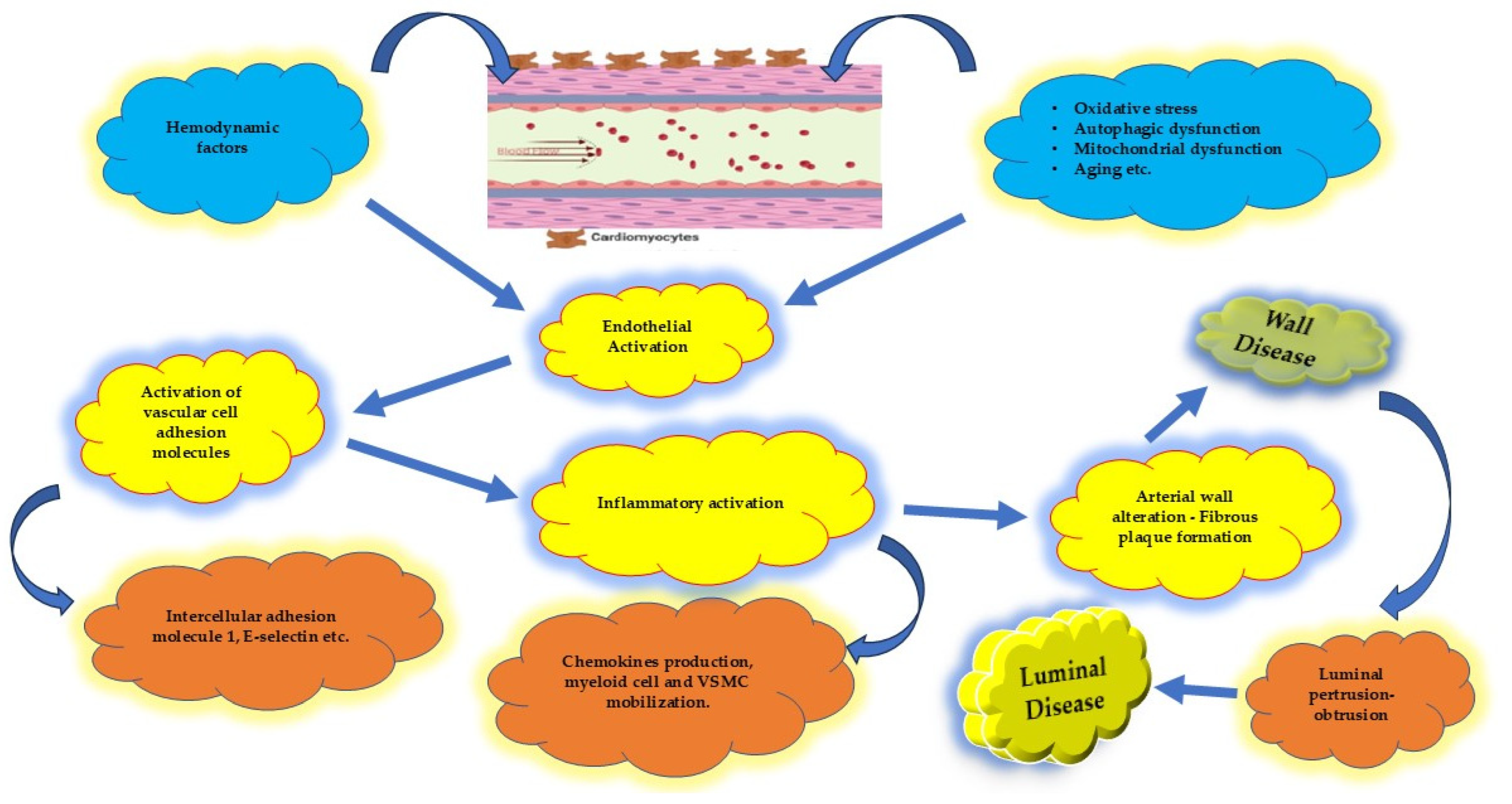
4.1. Endothelium and Coronary Artery Atherosclerosis
4.2. Systemic Inflammatory Disorders in CAD
4.3. Environmental Factors and Genetics in CAD
4.4. Protecting the “Housekeeper”
4.4.1. Vascular Homeostasis
4.4.2. Inflammatory Activity
4.4.3. Vascular Smooth Muscle Cells (VSMCs)
5. Advanced Imaging Techniques in Atherosclerotic Disease
6. Clinical Events
7. Identification of the Source and Therapeutic Actions
7.1. Preventing Inflammation and Beyond
7.2. VSMC Modulation
8. Limitations and Future Directions in Genetic Research in CAD
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vrints, C.; Andreotti, F.; Koskinas, K.C.; Rossello, X.; Adamo, M.; Ainslie, J.; Banning, A.P.; Budaj, A.; Buechel, R.R.; Chiariello, G.A.; et al. ESC 2024 Guidelines for the management of chronic coronary syndromes. Eur. Heart J. 2024, 45, 3415–3537. [Google Scholar] [PubMed]
- Jebari-Benslaiman, S.; Galicia-García, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martín, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef] [PubMed]
- Scarsini, R.; Fezzi, S.; Leone, A.M.; De Maria, G.L.; Pighi, M.; Marcoli, M.; Tavella, D.; Pesarini, G.; Banning, A.P.; Barbato, E.; et al. Functional patterns of coronary disease: Diffuse, focal, and serial lesions. JACC Cardiovasc. Interv. 2022, 15, 2174–2191. [Google Scholar] [CrossRef]
- Maron, D.J.; Hochman, J.S.; Reynolds, H.R.; Bangalore, S.; O’Brien, S.M.; Boden, W.E.; Chaitman, B.R.; Senior, R.; López-Sendón, J.; Alexander, K.P.; et al. Initial invasive or conservative strategy for stable coronary disease. N. Engl. J. Med. 2020, 382, 1395–1407. [Google Scholar] [CrossRef] [PubMed]
- Hochman, J.S.; Anthopolos, R.; Reynolds, H.R.; Bangalore, S.; Xu, Y.; O’Brien, S.M.; Mavromichalis, S.; Chang, M.; Contreras, A.; Rosenberg, Y.; et al. Survival after invasive or conservative management of stable coronary disease. Circulation 2023, 147, 8–19. [Google Scholar] [CrossRef]
- Lopes, R.D.; Alexander, K.P.; Stevens, S.R.; Reynolds, H.R.; Stone, G.W.; Piña, I.L.; Rockhold, F.W.; Elghamaz, A.; Lopez-Sendon, J.L.; Farsky, P.S.; et al. Initial invasive versus conservative management of stable ischemic heart disease in patients with a history of heart failure or left ventricular dysfunction. Circulation 2020, 142, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Fearon, W.F.; Zimmermann, F.M.; De Bruyne, B.; Piroth, Z.; van Straten, A.H.M.; Szekely, L.; Davidavičius, G.; Kalinauskas, G.; Mansour, S.; Kharbanda, R.; et al. Fractional flow reserve-guided PCI as compared with coronary bypass surgery. N. Engl. J. Med. 2022, 386, 128–137. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Bergmark, B.A.; Murphy, S.A.; O’Gara, P.T.; Smith, P.K.; Serruys, W.; Kappetein, A.P.; Park, S.J.; Park, D.W.; Christiansen, E.H.; et al. Percutaneous coronary intervention with drug-eluting stents versus coronary artery bypass grafting in left main coronary artery disease: An individual patient data meta-analysis. Lancet 2021, 398, 2247–2257. [Google Scholar] [CrossRef]
- Rozanski, A.; Miller, R.J.H.; Gransar, H.; Han, D.; Slomka, P.; Dey, D.; Hayes, S.W.; Friedman, J.D.; Thomson, L.; Berman, D.S. Benefit of early revascularization based on inducible ischemia and left ventricular ejection fraction. J. Am. Coll. Cardiol. 2022, 80, 202–215. [Google Scholar] [CrossRef]
- Reynolds, H.R.; Shaw, L.J.; Min, J.K.; Page, C.B.; Berman, D.S.; Chaitman, B.R.; Picard, M.H.; Kwong, R.Y.; O’Brien, S.M.; Huang, S.; et al. Outcomes in the ISCHEMIA trial based on coronary artery disease and ischemia severity. Circulation 2021, 144, 1024–1038. [Google Scholar] [CrossRef]
- Safiri, S.; Karamzad, N.; Singh, K.; Carson-Chahhoud, K.; Adams, C.; Nejadghaderi, S.A.; Almasi-Hashiani, A.; Sullman, M.J.M.; Mansournia, M.A.; Bragazzi, N.L. Burden of ischemic heart disease and its attributable risk factors in 204 countries and territories, 1990–2019. Eur. J. Prev. Cardiol. 2022, 29, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, F.; Crea, F.; Sechtem, U. Diagnoses and outcomes in patients with suspected angina: What are they trying to tell us? Eur. Heart J. 2019, 40, 1436–1439. [Google Scholar] [CrossRef]
- Juarez-Orozco, L.E.; Saraste, A.; Capodanno, D.; Prescott, E.; Ballo, H.; Bax, J.J.; Wijns, W.; Knuuti, J. Impact of a decreasing pre-test probability on the performance of diagnostic tests for coronary artery disease. Eur. Heart J. Cardiovasc. Imaging 2019, 20, 1198–1207. [Google Scholar] [CrossRef]
- Alam, S.; Pepine, C.J. Physiology and functional significance of the coronary microcircu-lation: An overview of its implications in health and disease. Am. Heart J. Plus. 2024, 40, 100381. [Google Scholar]
- Pries, A.R.; Reglin, B. Coronary microcirculatory pathophysiology: Can we afford it to remain a black box? Eur. Heart J. 2017, 38, 478–488. [Google Scholar] [CrossRef]
- Del Buono, M.G.; Montone, R.A.; Camilli, M.; Carbone, S.; Narula, J.; Lavie, C.J.; Niccoli, G.; Crea, F. Coronary Microvascular Dysfunction Across the Spectrum of Cardiovascu-lar Diseases: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 78, 1352–1371. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, B.; Ling, H.; Li, Y.; Fu, S.; Xu, M.; Li, B.; Liu, X.; Wang, Q.; Li, A.; et al. Navigating the Landscape of Coronary Microvascular Research: Trends, Triumphs, and Challenges Ahead. Rev. Cardiovasc. Med. 2024, 25, 288. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, T.; Galiero, R.; Caturano, A.; Vetrano, E.; Loffredo, G.; Rinaldi, L.; Catalini, C.; Gjeloshi, K.; Albanese, G.; Di Martino, A.; et al. Coronary Microvascular Dysfunction in Diabetes Mellitus: Pathogenetic Mechanisms and Potential Therapeutic Options. Biomedicines 2022, 10, 2274. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Wu, X.; Liu, H.; Zheng, D.; Xia, L. Index of microcirculatory resistance: State-of-the-art and potential applications in computational simulation of coronary artery disease. J. Zhejiang Univ. Sci. B 2022, 23, 123–140. (In English) [Google Scholar] [CrossRef] [PubMed]
- Travieso, A.; Jeronimo-Baza, A.; Faria, D.; Shabbir, A.; Mejia-Rentería, H.; Escaned, J. Invasive evaluation of coronary microvascular dysfunction. J. Nucl. Cardiol. 2022, 29, 2474–2486. [Google Scholar] [CrossRef]
- Ciaramella, L.; Di Serafino, L.; Mitrano, L.; De Rosa, M.L.; Carbone, C.; Rea, F.S.; Monaco, S.; Scalamogna, M.; Cirillo, P.; Esposito, G. Invasive Assessment of Coronary Microcirculation: A State-of-the-Art Review. Diagnostics 2023, 14, 86. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Tuzcu, E.M.; Sipahi, I. The Role of Inflammation in Atherosclerosis. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK549786/ (accessed on 23 January 2025).
- Zoneff, E.; Wang, Y.; Jackson, C.; Smith, O.; Duchi, S.; Onofrillo, C.; Farrugia, B.; Moulton, S.E.; Williams, R.; Parish, C.; et al. Controlled oxygen delivery to power tissue regeneration. Nat. Commun. 2024, 15, 4361. [Google Scholar] [CrossRef] [PubMed]
- Boden, W.E.; O’Rourke, R.A.; Teo, K.K.; Hartigan, P.M.; Maron, D.J.; Kostuk, W.J.; Knudtson, M.; Dada, M.; Casperson, P.; Harris, C.L.; et al. Optimal Medical Therapy with or without PCI for Stable Coronary Disease. N. Engl. J. Med. 2007, 356, 1503–1516. [Google Scholar] [CrossRef]
- Fezzi, S.; Ding, D.; Mahfoud, F.; Huang, J.; Lansky, A.J.; Tu, S.; Wijns, W. Illusion of revascularization: Does anyone achieve optimal revascularization during percutaneous coronary intervention? Nat. Rev. Cardiol. 2024, 21, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Collet, C.; Munhoz, D.; Mizukami, T.; Sonck, J.; Matsuo, H.; Shinke, T.; Ando, H.; Ko, B.; Biscaglia, S.; Rivero, F.; et al. Influence of Pathophysiologic Patterns of Coronary Artery Disease on Immediate Percutaneous Coronary Intervention Outcomes. Circulation 2024, 150, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.; Koo, B.K.; Zhang, J.; Park, J.; Yang, S.; Kim, M.; Yun, J.P.; Lee, J.M.; Nam, C.W.; Shin, E.S.; et al. Prognostic implications of fractional flow reserve after coronary stenting: A systematic review and meta-analysis. JAMA Netw. Open 2022, 5, e2232842. [Google Scholar] [CrossRef]
- Collison, D.; Copt, S.; Mizukami, T.; Collet, C.; McLaren, R.; Didagelos, M.; Aetesam-Ur-Rahman, M.; McCartney, P.; Ford, T.J.; Lindsay, M.; et al. Angina after percutaneous coronary intervention: Patient and procedural predictors. Circ. Cardiovasc. Interv. 2023, 16, e012511. [Google Scholar] [CrossRef]
- Sternheim, D.; Power, D.A.; Samtani, R.; Kini, A.; Fuster, V.; Sharma, S. Myocardial bridging: Diagnosis, functional assessment, and management: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2021, 78, 2196–2212. [Google Scholar] [CrossRef]
- Xing, Y.; Shi, J.; Yan, Y.; Liu, Y.; Chen, Y.; Kong, D.; Shu, X.; Pan, C. Subclinical myocardial dysfunction in coronary slow flow phenomenon: Identification by speckle tracking echocardiography. Microcirculation 2019, 26, e12509. [Google Scholar] [CrossRef]
- Zhu, O.; Wang, S.; Huang, X.; Zhao, C.; Wang, Y.; Li, X.; Jia, D.; Ma, C. Understanding the pathogenesis of coronary slow flow: Recent advances. Trends Cardiovasc. Med. 2024, 34, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.B.; Kelsey, S.F.; Matthews, K.; Shaw, L.J.; Sharaf, B.L.; Pohost, G.M.; Cornell, C.E.; McGorray, S.P.; Vido, D.; Bairey Merz, C.N. Symptoms, myocardial ischaemia and quality of life in women. Eur. Heart J. 2003, 24, 1506–1514. [Google Scholar] [CrossRef]
- Reynolds, H.R.; Picard, M.H.; Spertus, J.A.; Peteiro, J.; Sendon, J.L.L.; Senior, R.; El-Hajjar, M.C.; Celutkiene, J.; Shapiro, M.D.; Pellikka, P.A.; et al. Natural History of Patients with Ischemia and No Obstructive Coronary Artery Disease: The CIAO-ISCHEMIA Study. Circulation 2021, 144, 1008–1023. [Google Scholar] [CrossRef]
- Mancini, G.B.J.; Hartigan, P.M.; Shaw, L.J.; Berman, D.S.; Hayes, S.W.; Bates, E.R.; Maron, D.J.; Teo, K.; Sedlis, S.P.; Chaitman, B.R.; et al. Predicting outcome in the COURAGE trial (Clinical Outcomes Utilizing Revascularization and Aggressive Drug Evaluation): Coronary anatomy versus ischemia. JACC Cardiovasc. Interv. 2014, 7, 195–201. [Google Scholar] [CrossRef]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef]
- Ohba, K.; Sugiyama, S.; Sumida, H.; Nozaki, T.; Matsubara, J.; Matsuzawa, Y.; Konishi, M.; Akiyama, E.; Kurokawa, H.; Maeda, H.; et al. Microvascular coronary artery spasm presents distinctive clinical features with endothelial dysfunction as nonobstructive coronary artery disease. J. Am. Heart Assoc. 2012, 1, e002485. [Google Scholar] [CrossRef]
- Hong, Y.M. Atherosclerotic cardiovascular disease beginning in childhood. Korean Circ. J. 2010, 40, 1–9. [Google Scholar] [CrossRef]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, indicators, risk factors and new hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar] [PubMed]
- Beverly, J.K.; Budoff, M.J. Atherosclerosis: Pathophysiology of insulin resistance, hyperglycemia, hyperlipidemia, and inflammation. J. Diabetes 2020, 12, 102–104. [Google Scholar] [CrossRef]
- Singh, R.B.; Mengi, S.A.; Xu, Y.J.; Arneja, A.S.; Dhalla, N.S. Pathogenesis of atherosclerosis: A multifactorial process. Exp. Clin. Cardiol. 2002, 7, 40–53. [Google Scholar] [PubMed]
- Xu, X.; Wang, B.; Ren, C.; Hu, J.; Greenberg, D.A.; Chen, T.; Xie, L.; Jin, K. Age-related Impairment of Vascular Structure and Functions. Aging Dis. 2017, 8, 590–610. [Google Scholar] [CrossRef]
- Zhang, B.; Gu, J.; Qian, M.; Niu, L.; Zhou, H.; Ghista, D. Correlation between quantitative analysis of wall shear stress and intima-media thickness in atherosclerosis development in carotid arteries. Biomed. Eng. Online 2017, 16, 137. [Google Scholar] [CrossRef]
- Campinho, P.; Vilfan, A.; Vermot, J. Blood Flow Forces in Shaping the Vascular System: A Focus on Endothelial Cell Behavior. Front. Physiol. 2020, 11, 552. [Google Scholar] [CrossRef] [PubMed]
- Gordon, E.; Schimmel, L.; Frye, M. The Importance of Mechanical Forces for in vitro Endothelial Cell Biology. Front. Physiol. 2020, 11, 684. [Google Scholar] [CrossRef] [PubMed]
- Souilhol, C.; Serbanovic-Canic, J.; Fragiadaki, M.; Chico, T.J.; Ridger, V.; Roddie, H.; Evans, P.C. Endothelial responses to shear stress in atherosclerosis: A novel role for developmental genes. Nat. Rev. Cardiol. 2020, 17, 52–63. [Google Scholar] [CrossRef]
- Hachuła, M.; Basiak, M.; Kosowski, M.; Okopień, B. Effect of GLP-1RA Treatment on Adhesion Molecules and Monocyte Chemoattractant Protein-1 in Diabetic Patients with Atherosclerosis. Life 2024, 14, 690. [Google Scholar] [CrossRef]
- Li, G.; Gao, J.; Ding, P.; Gao, Y. The role of endothelial cell-pericyte interactions in vascularization and diseases. J. Adv. Res. 2025, 67, 269–288. [Google Scholar] [CrossRef]
- Baratchi, S.; Khoshmanesh, K.; Woodman, O.L.; Potocnik, S.; Peter, K.; McIntyre, P. Molecular Sensors of Blood Flow in Endothelial Cells. Trends Mol. Med. 2017, 23, 850–868. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, B. Endothelial dysfunction: Molecular mechanisms and clinical implications. Medcomm 2024, 5, e651. [Google Scholar] [CrossRef] [PubMed]
- Naderi-Meshkin, H.; Setyaningsih, W.A.W. Endothelial Cell Dysfunction: Onset, Progression, and Consequences. Front. Biosci. (Landmark Ed.) 2024, 29, 223. [Google Scholar] [CrossRef] [PubMed]
- Mengozzi, A.; Masi, S.; Virdis, A. Obesity-Related Endothelial Dysfunction: Moving from classical to emerging mechanisms. Endocr. Metab. Sci. 2020, 1, 100063. [Google Scholar] [CrossRef]
- Abu-Saleh, N.; Yaseen, H.; Kinaneh, S.; Khamaisi, M.; Abassi, Z. Combination of hyperglycaemia and hyperlipidaemia induces endothelial dysfunction: Role of the endothelin and nitric oxide systems. J. Cell. Mol. Med. 2021, 25, 1884–1895. [Google Scholar] [CrossRef] [PubMed]
- Gallo, G.; Volpe, M.; Savoia, C. Endothelial Dysfunction in Hypertension: Current Concepts and Clinical Implications. Front. Med. 2022, 8, 798958. [Google Scholar] [CrossRef]
- Shaito, A.; Aramouni, K.; Assaf, R.; Parenti, A.; Orekhov, A.; El Yazbi, A.; Pintus, G.; Eid, A.H. Oxidative Stress-Induced Endothelial Dysfunction in Cardiovascular Diseases. Front. Biosci. 2022, 27, 105. [Google Scholar] [CrossRef]
- Bjorkegren, J.L.M.; Lusis, A.J. Atherosclerosis: Recent developments. Cell 2022, 185, 1630–1645. [Google Scholar] [CrossRef]
- Döring, Y.; van der Vorst, E.P.C.; Weber, C. Targeting immune recruitment in atherosclerosis. Nat. Rev. Cardiol. 2024, 21, 824–840. [Google Scholar] [CrossRef]
- McLaren, J.; Nunes de Alencar, J.; Aslanger, E.K.; Meyers, P.H.; Smith, S.W. From ST-Segment Elevation MI to Occlusion MI. The New Paradigm Shift in Acute Myocardial Infarction. JACC Adv. 2024, 3, 101314. [Google Scholar] [CrossRef] [PubMed]
- Raffin, C.; Vo, L.T.; Bluestone, J.A. Treg cell-based therapies: Challenges and perspectives. Nat. Rev. Immunol. 2020, 20, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Poredos, P.; Poredos, A.V.; Gregoric, I. Endothelial dysfunction and its clinical implications. Angiology 2021, 72, 604–615. [Google Scholar] [CrossRef]
- Patel, N.H.; Dey, A.K.; Sorokin, A.V.; Teklu, M.; Petrole, R.; Zhou, W.; Mehta, N.N. Chronic inflammatory diseases and coronary heart disease: Insights from cardiovascular CT. J. Cardiovasc. Comput. Tomogr. 2022, 16, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Abou-Raya, A.; Abou-Raya, S. Inflammation: A pivotal link between autoimmune diseases and atherosclerosis. Autoimmun. Rev. 2006, 5, 331–337. [Google Scholar] [CrossRef]
- Asenjo-Lobos, C.; González, L.; Bulnes, J.F.; Roque, M.; Muñoz Venturelli, P.; Rodríguez, G.M. Cardiovascular events risk in patients with systemic autoimmune diseases: A prognostic systematic review and meta-analysis. Clin. Res. Cardiol. 2024, 113, 246–259. [Google Scholar] [CrossRef]
- Wang, Z.; Nakayama, T. Inflammation, a link between obesity and cardiovascular disease. Mediat. Inflamm. 2010, 2010, 535918. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Suárez, V.J.; Redondo-Flórez, L.; Beltrán-Velasco, A.I.; Martín-Rodríguez, A.; Martínez-Guardado, I.; Navarro-Jiménez, E.; Laborde-Cárdenas, C.C.; Tornero-Aguilera, J.F. The Role of Adipokines in Health and Disease. Biomedicines 2023, 11, 1290. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, J.; Rodrigues, C.F.; Sharopov, F.; Docea, A.O.; Can Karaca, A.; Sharifi-Rad, M.; Kahveci Karıncaoglu, D.; Gülseren, G.; Şenol, E.; Demircan, E.; et al. Diet, Lifestyle and Cardiovascular Diseases: Linking Pathophysiology to Cardioprotective Effects of Natural Bioactive Compounds. Int. J. Environ. Res. Public Health 2020, 17, 2326. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Mutlu, G.M. Particulate Matter Air Pollution: Effects on the Cardiovascular System. Front. Endocrinol. 2018, 9, 680. [Google Scholar] [CrossRef]
- Huang, H.B.; Chen, G.W.; Wang, C.J.; Lin, Y.Y.; Liou, S.H.; Lai, C.H.; Wang, S.L. Exposure to heavy metals and polycyclic aromatic hydrocarbons and DNA damage in taiwanese traffic conductors. Cancer Epidemiol. Biomarkers Prev. 2013, 22, 102–108. [Google Scholar] [CrossRef]
- Kessler, T.; Schunkert, H. Coronary Artery Disease Genetics Enlightened by Genome-Wide Association Studies. JACC Basic Transl. Sci. 2021, 6, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Aragam, K.G.; Jiang, T.; Goel, A.; Kanoni, S.; Wolford, B.N.; Atri, D.S.; Weeks, E.M.; Wang, M.; Hindy, G.; Zhou, W.; et al. Discovery and systematic characterization of risk variants and genes for coronary artery disease in over a million participants. Nat. Genet. 2022, 54, 1803–1815. [Google Scholar] [CrossRef]
- Zarkasi, K.A.; Abdullah, N.; Abdul Murad, N.A.; Ahmad, N.; Jamal, R. Genetic Factors for Coronary Heart Disease and Their Mechanisms: A Meta-Analysis and Comprehensive Review of Common Variants from Genome-Wide Association Studies. Diagnostics 2022, 12, 2561. [Google Scholar] [CrossRef]
- Toraño, E.G.; García, M.G.; Fernández-Morera, J.L.; Niño-García, P.; Fernández, A.F. The Impact of External Factors on the Epigenome: In Utero and over Lifetime. Biomed. Res. Int. 2016, 2016, 2568635. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.; Shymka, M.; Ng, T.; Phulka, J.S.; Safabakhsh, S.; Laksman, Z. Polygenic Risk Score Implementation into Clinical Practice for Primary Prevention of Cardiometabolic Disease. Genes 2024, 15, 1581. [Google Scholar] [CrossRef] [PubMed]
- Little, P.J.; Askew, C.D.; Xu, S.; Kamato, D. Endothelial Dysfunction and Cardiovascular Disease: History and Analysis of the Clinical Utility of the Relationship. Biomedicines 2021, 9, 699. [Google Scholar] [CrossRef] [PubMed]
- Alexander, Y.; Osto, E.; Schmidt-Trucksäss, A.; Shechter, M.; Trifunovic, D.; Duncker, D.J.; Aboyans, V.; Bäck, M.; Badimon, L.; Cosentino, F.; et al. Endothelial function in cardiovascular medicine: A consensus paper of the European Society of Cardiology Working Groups on Atherosclerosis and Vascular Biology, Aorta and Peripheral Vascular Diseases, Coronary Pathophysiology and Microcirculation, and Thrombosis. Cardiovasc. Res. 2021, 117, 29–42. [Google Scholar]
- van Trier, T.J.; Mohammadnia, N.; Snaterse, M.; Peters, R.J.G.; Jørstad, H.T.; Bax, W.A. Lifestyle management to prevent atherosclerotic cardiovascular disease: Evidence and challenges. Neth. Heart J. 2022, 30, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Presa, M.; Bustos, C.; Ortego, M.; Tuñon, J.; Renedo, G.; Ruiz-Ortega, M.; Egido, J. Angiotensin-converting enzyme inhibition prevents arterial nuclear factor-kappa B activation, monocyte chemoattractant protein-1 expression, and macrophage infiltration in a rabbit model of early accelerated atherosclerosis. Circulation 1997, 95, 1532–1541. [Google Scholar] [CrossRef]
- Silva, G.M.; França-Falcão, M.S.; Calzerra, N.T.M.; Luz, M.S.; Gadelha, D.D.A.; Balarini, C.M.; Queiroz, T.M. Role of Renin-Angiotensin System Components in Atherosclerosis: Focus on Ang-II, ACE2, and Ang-1-7. Front. Physiol. 2020, 11, 1067. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M.; Gao, Y. Beta blockers, nitric oxide, and cardiovascular disease. Curr. Opin. Pharmacol. 2013, 13, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Almeida, S.O.; Budoff, M. Effect of statins on atherosclerotic plaque. Trends Cardiovasc. Med. 2019, 29, 451–455. [Google Scholar] [CrossRef]
- Xu, J.; Hirai, T.; Koya, D.; Kitada, M. Effects of SGLT2 Inhibitors on Atherosclerosis: Lessons from Cardiovascular Clinical Outcomes in Type 2 Diabetic Patients and Basic Researches. J. Clin. Med. 2021, 11, 137. [Google Scholar] [CrossRef]
- Kang, K.T. Endothelium-derived Relaxing Factors of Small Resistance Arteries in Hypertension. Toxicol. Res. 2014, 30, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F.; Fiore, A.; Masiglat, J.; Cavuoti, T.; Romandini, M.; Nappi, P.; Avtaar Singh, S.S.; Couetil, J.P. Endothelium-Derived Relaxing Factors and Endothelial Function: A Systematic Review. Biomedicines 2022, 10, 2884. [Google Scholar] [CrossRef]
- Sandoo, A.; van Zanten, J.J.; Metsios, G.S.; Carroll, D.; Kitas, G.D. The endothelium and its role in regulating vascular tone. Open Cardiovasc. Med. J. 2010, 4, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Wazir, M.; Olanrewaju, O.A.; Yahya, M.; Kumari, J.; Kumar, N.; Singh, J.; Abbas Al-Itbi, A.Y.; Kumari, K.; Ahmed, A.; Islam, T.; et al. Lipid Disorders and Cardiovascular Risk: A Comprehensive Analysis of Current Perspectives. Cureus 2023, 15, e51395. [Google Scholar] [CrossRef] [PubMed]
- Farah, C.; Michel, L.; Balligand, J.L. Nitric oxide signaling in cardiovascular health and disease. Nat. Rev. Cardiol. 2018, 15, 292–316. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, P.; Sun, Y.; Shao, Y.; Yang, W.Y.; Jiang, X.; Wang, H.; Yang, X. Anti-inflammatory cytokines IL-35 and IL-10 block atherogenic lysophosphatidylcholine—induced, mitochondrial ROS-mediated innate immune activation, but spare innate immune memory signature in endothelial cells. Redox Biol. 2020, 28, 101373. [Google Scholar] [CrossRef]
- Paraskevaidis, I.; Farmakis, D.; Papingiotis, G.; Tsougos, E. Inflammation and Heart Failure: Searching for the Enemy—Reaching the Entelechy. J. Cardiovasc. Dev. Dis. 2023, 10, 19. [Google Scholar] [CrossRef]
- Lu, L.; Jang, S.; Zhu, J.; Qin, Q.; Sun, L.; Sun, J. Nur77 mitigates endothelial dysfunction through activation of both nitric oxide production and anti-oxidant pathways. Redox Biol. 2024, 70, 103056. [Google Scholar] [CrossRef]
- Yahagi, K.; Kolodgie, Y.K.; Otsuka, F.; Finn, A.V.; Davis, H.R.; Joner, M.; Virmani, R. Pathophysiology of native coronary, vein graft, and in-stent atherosclerosis. Nat. Rev. Cardiol. 2016, 13, 79–98. [Google Scholar] [CrossRef]
- Grootaert, M.O.J.; Bennett, M.R. Vascular smooth muscle cells in atherosclerosis: Time for a re-assessment. Cardiovasc. Res. 2021, 117, 2326–2339. [Google Scholar] [CrossRef] [PubMed]
- Basatemur, G.L.; Helle FJorgensen, H.F.; Clarke, M.C.H.; Bennett, M.R.; Mallat, Z. Vascular smooth muscle cells in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 727–744. [Google Scholar] [PubMed]
- Virmani, R.; Kolodgie, F.D.; Burke, A.P.; Farb, A.; Schwartz, S.M. Lessons from sudden coronary death. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1262–1275. [Google Scholar] [CrossRef] [PubMed]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of Intimal Smooth Muscle Cells to Cholesterol Accumulation and Macrophage-Like Cells in Human Atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Skålén, K.; Gustafsson, M.; Rydberg, E.K.; Hultén, L.M.; Wiklund, P.; Innerarity, T.L.; Borén, J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 2002, 417, 750–754. [Google Scholar] [CrossRef]
- Nagao, M.; Lyu, Q.; Zhao, Q.; Wirka, R.C.; Bagga, J.; Nguyen, T.; Cheng, P.; Kim, J.B.; Pjanic, M.; Miano, J.M.; et al. Coronary disease-associated gene TCF21 inhibits smooth muscle cell differentiation by blocking the myocardin-serum response factor pathway. Circ. Res. 2020, 126, 517–529. [Google Scholar] [CrossRef]
- Sarwar, M.; Adedokun, S.; Narayanan, M.A. Role of intravascular ultrasound and optical coherence tomography in intracoronary imaging for coronary artery disease: A systematic review. J. Geriatr. Cardiol. 2024, 21, 104–129. [Google Scholar] [CrossRef] [PubMed]
- Usui, E.; Yonetsu, T.; Ohmori, M.; Kanno, Y.; Nakao, M.; Niida, T.; Matsuda, Y.; Matsuda, J.; Umemoto, T.; Misawa, T.; et al. Predictors of Near-Infrared Spectroscopy-Detected Lipid-Rich Plaques by Optical Coherence Tomography-Defined Morphological Features in Patients with Acute Coronary Syndrome. Front. Cardiovasc. Med. 2022, 9, 842914. [Google Scholar] [CrossRef] [PubMed]
- Kwiecinski, J.; Tzolos, E.; Williams, M.C.; Dey, D.; Berman, D.; Slomka, P.; Newby, D.E.; Dweck, M.R. Noninvasive Coronary Atherosclerotic Plaque Imaging. JACC Cardiovasc. Imaging 2023, 16, 1608–1622. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, J.; de Almeida, J.; Mendes, P.L.; Ferreira, M.J.; Gonçalves, L. Advancements in non-invasive imaging of atherosclerosis: Future perspectives. J. Clin. Lipidol. 2024, 18, e142–e152. [Google Scholar] [CrossRef] [PubMed]
- Bing, R.; Loganath, K.; Adamson, P.; Newby, D.; Moss, A. Non-invasive imaging of high-risk coronary plaque: The role of computed tomography and positron emission tomography. Br. J. Radiol. 2020, 93, 20190740. [Google Scholar] [CrossRef] [PubMed]
- Jansen, K.; Wu, M.; van der Steen, A.F.; van Soest, G. Photoacoustic imaging of human coronary atherosclerosis in two spectral bands. Photoacoustics 2013, 2, 12–20. [Google Scholar] [CrossRef]
- Schneider, M.K.; Wang, J.; Kare, A.; Adkar, S.S.; Salmi, D.; Bell, C.F.; Alsaigh, T.; Wagh, D.; Coller, J.; Mayer, A.; et al. Combined near infrared photoacoustic imaging and ultrasound detects vulnerable atherosclerotic plaque. Biomaterials 2023, 302, 122314. [Google Scholar] [CrossRef] [PubMed]
- Wildgruber, M.; Swirski, F.K.; Zernecke, A. Molecular imaging of inflammation in atherosclerosis. Theranostics 2013, 3, 865–884. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Chen, Y.; Hu, Y.; Wang, H.; Chen, W.; Zhou, Q. Molecular imaging research in atherosclerosis: A 23-year scientometric and visual analysis. Front. Bioeng. Biotechnol. 2023, 11, 1152067. [Google Scholar] [CrossRef]
- Pasterkamp, G.; den Ruijter, H.M.; Libby, P. Temporal shifts in clinical presentation and underlying mechanisms of atherosclerotic disease. Nat. Rev. Cardiol. 2017, 14, 21–29. [Google Scholar] [CrossRef]
- Papafaklis, M.I.; Koros, R.; Tsigkas, G.; Karanasos, A.; Moulias, A.; Davlouros, P. Reversal of Atherosclerotic Plaque Growth and Vulnerability: Effects of Lipid-Modifying and Anti-Inflammatory Therapeutic Agents. Biomedicines 2024, 12, 2435. [Google Scholar] [CrossRef]
- Thim, T.; Hagensen, M.K.; Wallace-Bradley, D.; Granada, J.F.; Kaluza, G.L.; Drouet, L.; Paaske, W.P.; Botker, H.E.; Falk, E. Unreliable assessment of necrotic core by virtual histology intravascular ultrasound in porcine coronary artery disease. Circ. Cardiovasc. Imaging 2010, 3, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Kłosowicz, M.; Leksa, D.; Bartusik-Aebisher, D.; Mysliwiec, A.; Dynarowicz, K.; Aebisher, D. Biomarkers That Seem to Have the Greatest Impact on Promoting the Formation of Atherosclerotic Plaque in Current Scientific Research. Curr. Issues Mol. Biol. 2024, 46, 9503–9522. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Masuda, D.; Matsuzawa, Y. Pemafibrate, a New Selective PPAR alpha Modulator: Drug Concept and Its Clinical Applications for Dyslipidemia and Metabolic Diseases. Curr. Atheroscler. Rep. 2020, 22, 5. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.C.; Waters, D.D.; et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, K.; Arnott, C.; Martinez, G.J.; Ng, B.; McCormack, S.; Sullivan, D.R.; Celermajer, D.S.; Patel, S. Colchicine Therapy and Plaque Stabilization in Patients with Acute Coronary Syndrome: A CT Coronary Angiography Study. JACC Cardiovasc. Imaging 2018, 11, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Yang, Y.; Dong, S.L.; Zhao, C.; Yang, F.; Yuan, Y.F.; Liao, Y.H.; He, S.L.; Liu, K.; Wei, F.; et al. Effect of Colchicine on Coronary Plaque Stability in Acute Coronary Syndrome as Assessed by Optical Coherence Tomography: The COLOCT Randomized Clinical Trial. Circulation 2024, 150, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Hergenhan, S.; Holtkamp, S.; Scheiermann, C. Molecular interactions between components of the circadian clock and the immune system. J. Mol. Biol. 2020, 432, 3700–3713. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Herrero-Fernandez, B.; Almarcha, C.B.; Gomez Bris, R.; Zorita, V.; Sáez, A.; Maas, S.L.; Pérez-Olivares, L.; Herrero-Cervera, A.; Lemnitzer, P.; et al. Time-restricted feeding enhances early atherosclerosis in hypercholesterolemic mice. Circulation 2023, 147, 774–777. [Google Scholar] [CrossRef]
- Panhuis, W.I.H.; Schönke, M.; Modder, M.; Tom, H.E.; Lalai, R.A.; Pronk, A.C.; Streefland, T.C.; van Kerkhof, L.W.; Dollé, M.E.; Depuydt, M.A.; et al. Time-restricted feeding attenuates hypercholesterolaemia and atherosclerosis development during circadian disturbance in APOE *3-Leiden CETP Mice. EBioMedicine 2023, 93, 104680. [Google Scholar]
- Batinac, T.; Batičić, L.; Kršek, A.; Knežević, D.; Marcucci, E.; Sotošek, V.; Ćurko-Cofek, B. Endothelial Dysfunction and Cardiovascular Disease: Hyperbaric Oxygen Therapy as an Emerging Therapeutic Modality? J. Cardiovasc. Dev. Dis. 2024, 11, 408. [Google Scholar] [CrossRef] [PubMed]
- Papp, M.; Ince, C.; Bakker, J.; Molnar, Z. Endothelial Protection and Improved Micro- and Macrocirculation with Hemoadsorption in Critically Ill Patients. J. Clin. Med. 2024, 13, 7044. [Google Scholar] [CrossRef]
- Back, M.; Yurdagul, A., Jr.; Tabas, I.; Oorni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef]
- Irfan, A.; Haider, S.H.; Nasir, A.; Larik, M.O.; Naz, T. Assessing the efficacy of omega-3 fatty acids + statins vs. statins only on cardiovascular outcomes: A systematic review and meta-analysis of 40,991 patients. Curr. Probl. Cardiol. 2024, 49, 102245. [Google Scholar] [CrossRef] [PubMed]
- Lhoëst, M.L.C.; Martinez, A.; Claudi, L.; Garcia, E.; Benitez-Amaro, A.; Polishchuk, A.; Piñero, J.; Vilades, D.; Guerra, J.M.; Sanz, F.; et al. Mechanisms modulating foam cell formation in the arterial intima: Exploring new therapeutic opportunities in atherosclerosis. Front. Cardiovasc. Med. 2024, 11, 1381520. [Google Scholar]
- Linna-Kuosmanen, S.; Bosch, V.T.; Moreau, P.R.; Bouvy-Liivrand, M.; Niskanen, H.; Kansanen, E.; Kivela, A.; Hartikainen, J.; Hippela¨inen, M.; Kokki, H.; et al. NRF2 is a key regulator of endothelial microRNA expression under proatherogenic stimuli. Cardiovasc. Res. 2021, 117, 1339–1357. [Google Scholar] [CrossRef]
- LeBlanc, M.; Zuber, V.; Andreassen, B.K.; Witoelar, A.; Zeng, L.; Bettella, F.; Wang, Y.; McEvoy, L.K.; Thompson, W.K.; Schork, A.J.; et al. Identifying Novel Gene Variants in Coronary Artery Disease and Shared Genes with Several Cardiovascular Risk Factors. Circ. Res. 2016, 118, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Bargieł, W.; Cierpiszewska, K.; Maruszczak, K.; Pakuła, A.; Szwankowska, D.; Wrzesińska, A.; Gutowski, Ł.; Formanowicz, D. Recognized and Potentially New Biomarkers-Their Role in Diagnosis and Prognosis of Cardiovascular Disease. Medicina 2021, 57, 701. [Google Scholar] [CrossRef]
- Quazi, S. Artificial intelligence and machine learning in precision and genomic medicine. Med. Oncol. 2022, 39, 120. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Underlying Mechanism in CAD | Pharmacological Strategy |
|---|---|
| Endothelial dysfunction | ACE inhibitors, ARBs, statins, and NO donors |
| Inflammation | Anti-inflammatory therapies including IL-1β inhibitors, TNF-α blockers, etc. |
| Oxidative stress | Antioxidants including vitamin E, Nrf2 activators, etc. |
| Vascular smooth muscle cells dysfunction | Drugs targeting VSMC phenotype switching including TNF-β inhibitors, microRNA modulators, etc. |
| Plaque instability | MMP inhibitors and PCSK9 inhibitors |
| Microvascular dysfunction | Endothelial protective drugs, vasodilators, and Ca blockers |
| Lipid accumulation and foam cell formation | Statins, PCSK9 inhibitors, and therapies reducing LDL oxidation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paraskevaidis, I.; Kourek, C.; Tsougos, E. Chronic Coronary Artery Disease: Wall Disease vs. Lumenopathy. Biomolecules 2025, 15, 201. https://doi.org/10.3390/biom15020201
Paraskevaidis I, Kourek C, Tsougos E. Chronic Coronary Artery Disease: Wall Disease vs. Lumenopathy. Biomolecules. 2025; 15(2):201. https://doi.org/10.3390/biom15020201
Chicago/Turabian StyleParaskevaidis, Ioannis, Christos Kourek, and Elias Tsougos. 2025. "Chronic Coronary Artery Disease: Wall Disease vs. Lumenopathy" Biomolecules 15, no. 2: 201. https://doi.org/10.3390/biom15020201
APA StyleParaskevaidis, I., Kourek, C., & Tsougos, E. (2025). Chronic Coronary Artery Disease: Wall Disease vs. Lumenopathy. Biomolecules, 15(2), 201. https://doi.org/10.3390/biom15020201