
The Therapeutic Potential of Supersulfides in Oxidative Stress-Related Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Occurrence, Biosynthesis, and Metabolism of Supersulfdes in Organisms
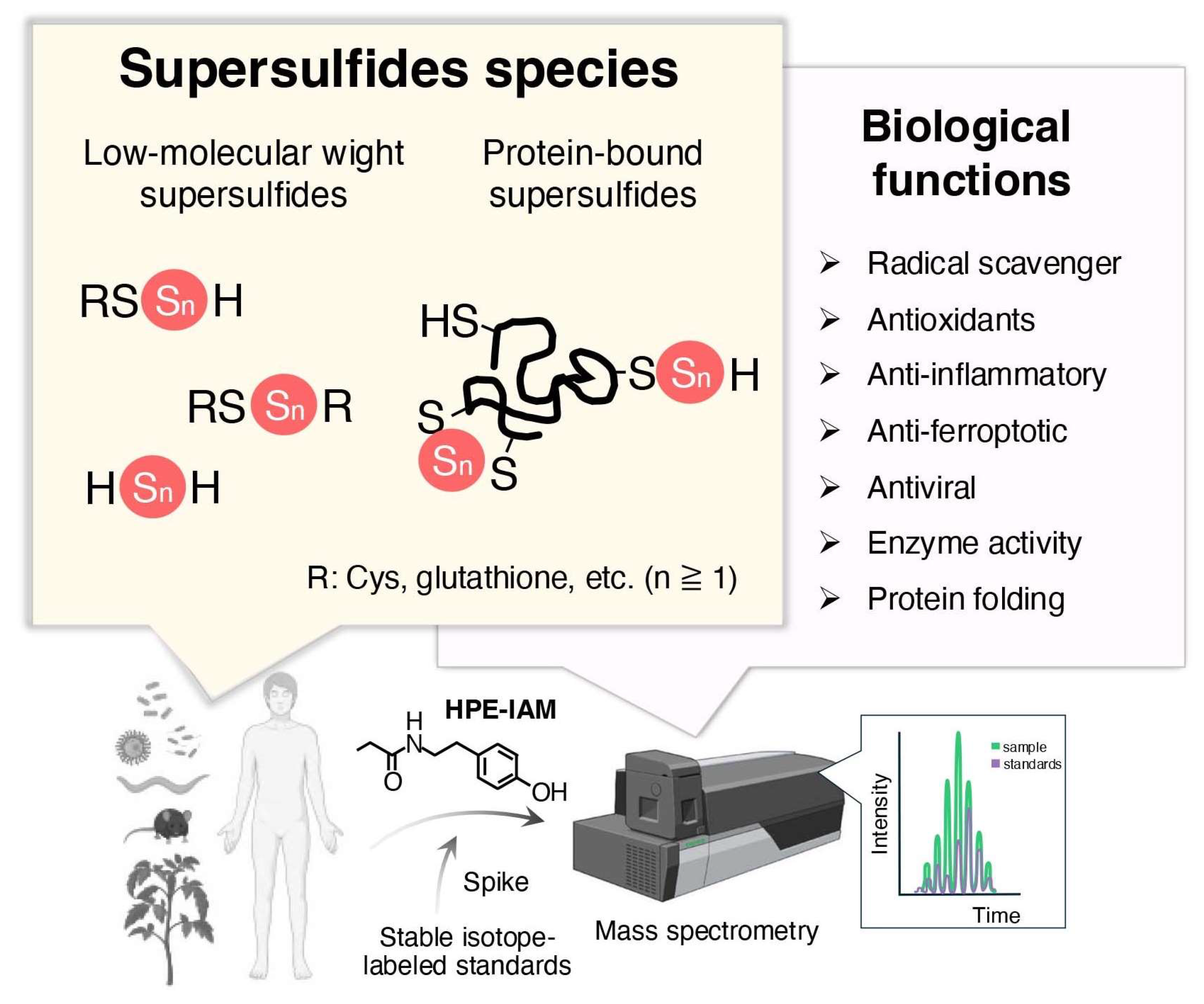
2.1. Occurrence and Quantification
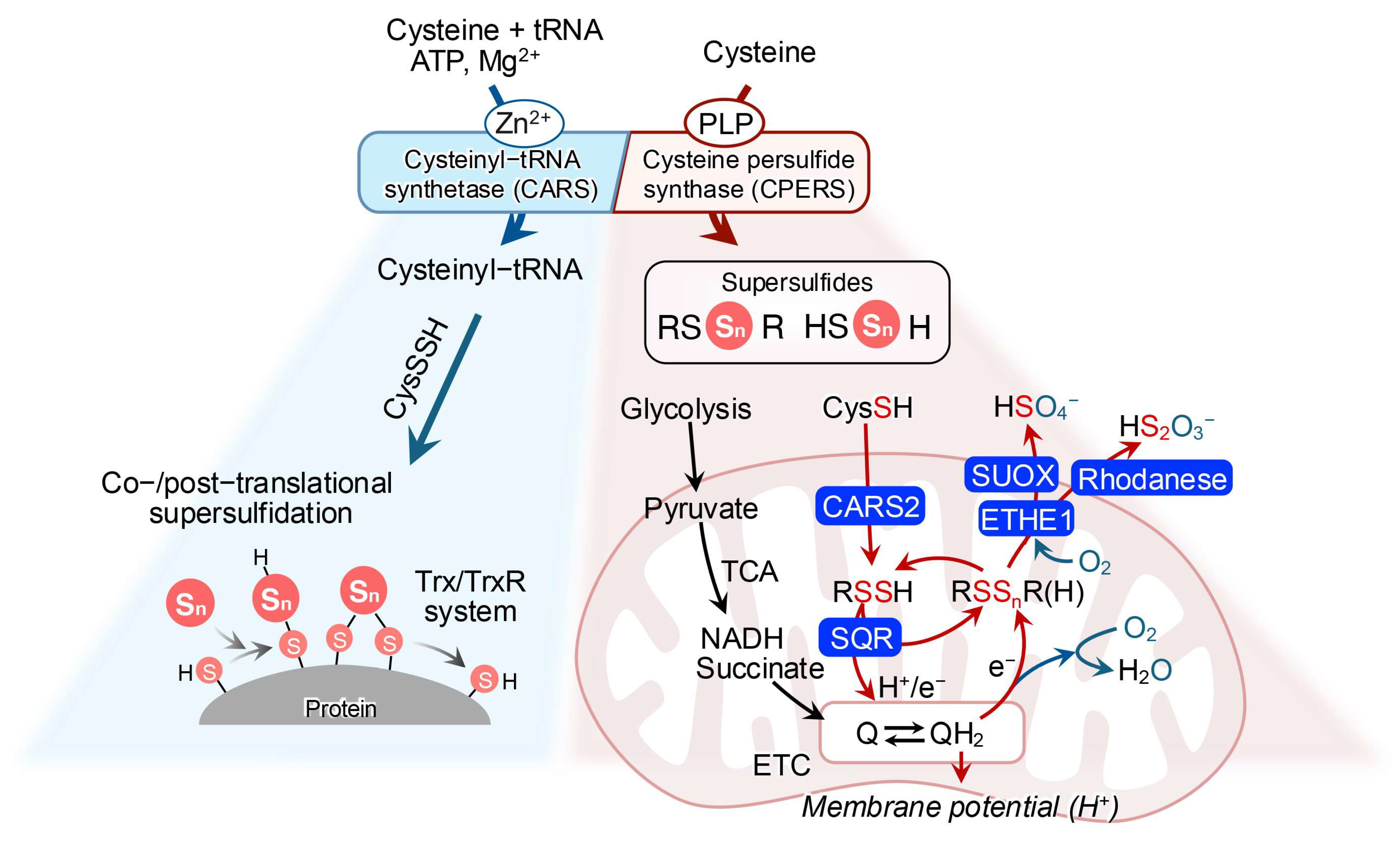
2.2. Biosynthesis
2.3. Catabolism
3. Versatile Physiological Potentials of Supersulfides in Oxidative Distress
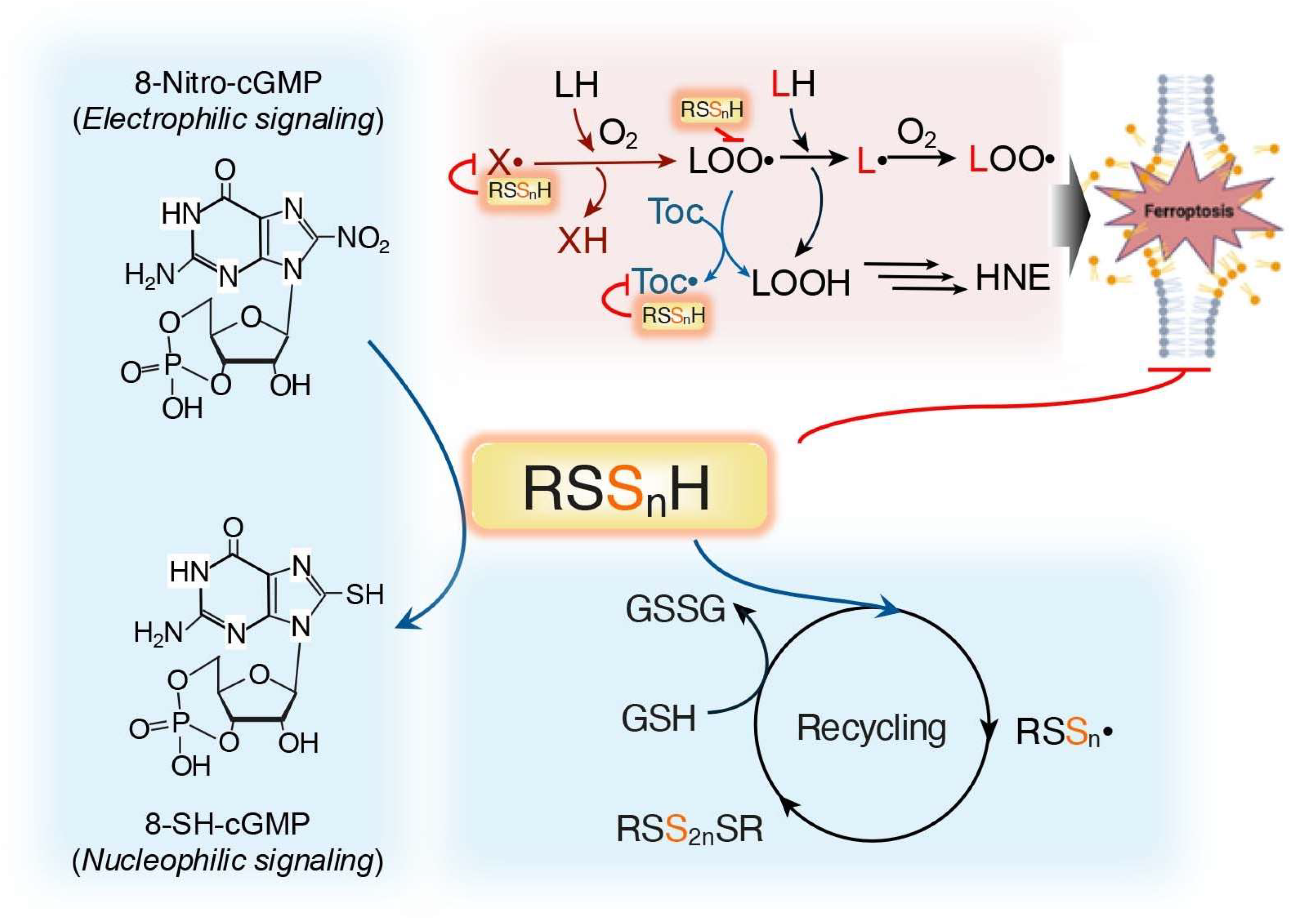
3.1. Supersulfides Regulate Oxidative Distress by Reacting with ROS
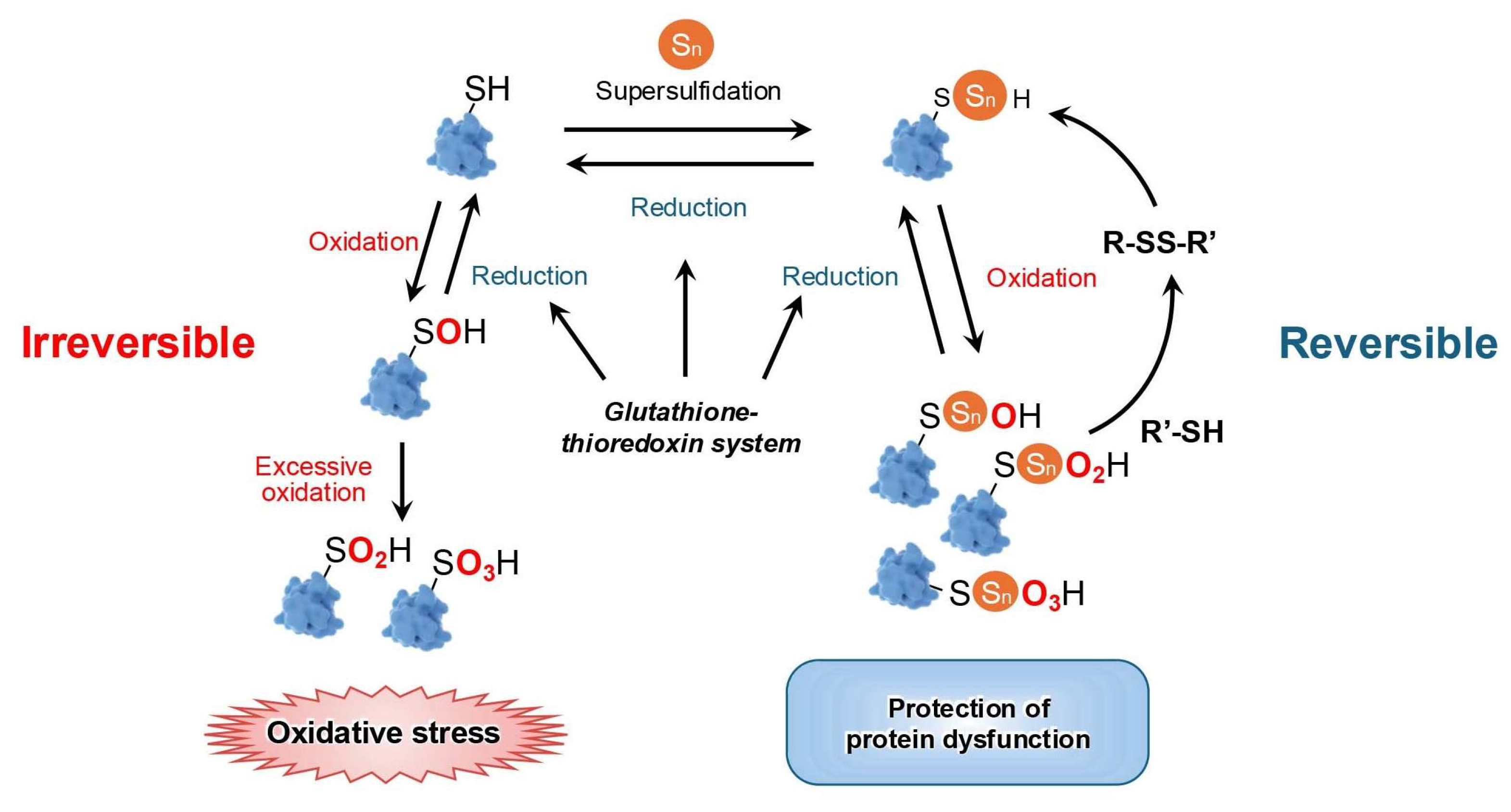
3.2. Supersulfides Regulate Oxidative Distress by Modulating Proteins
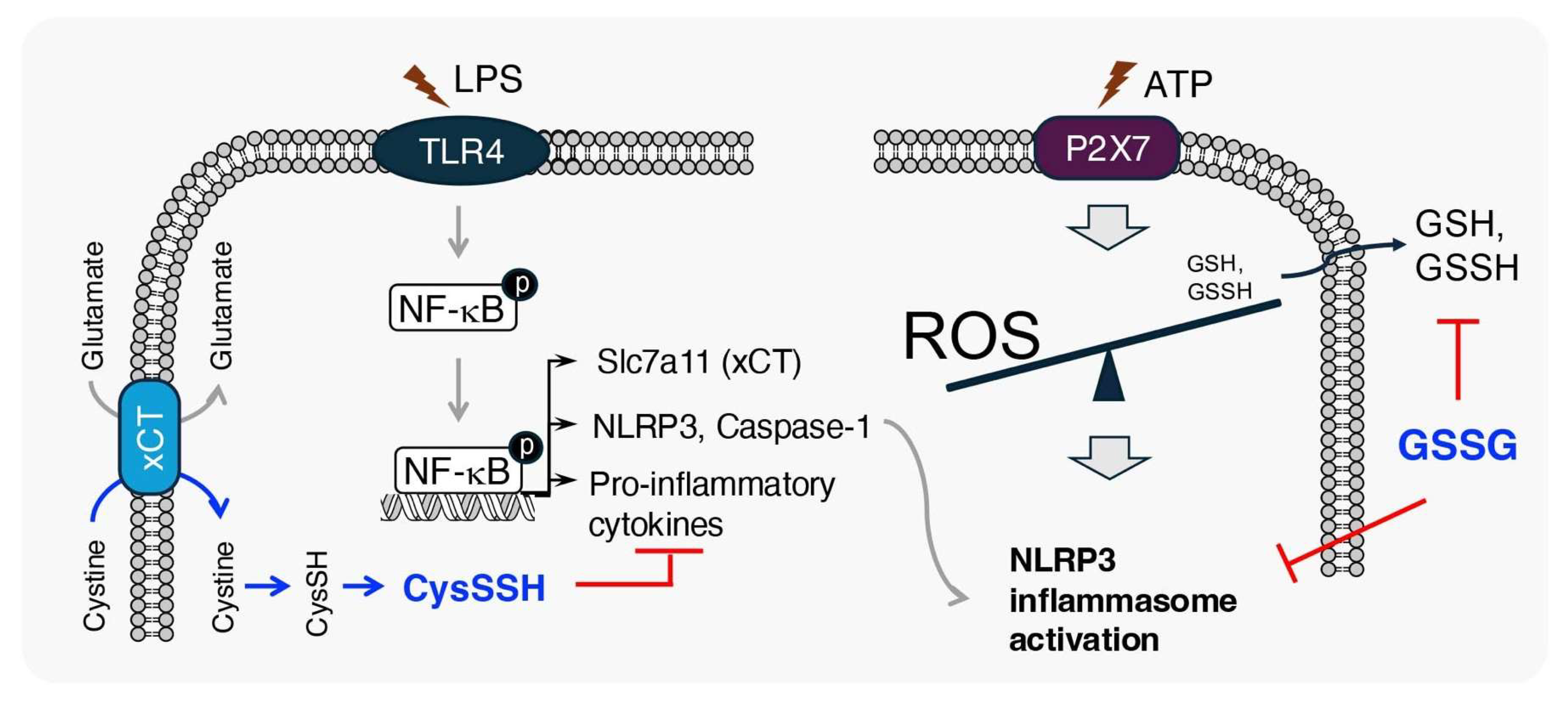
3.3. Anti-Inflammatory Effects of Supersulfides
4. Limitations of Current Antioxidant Therapeutic Agents
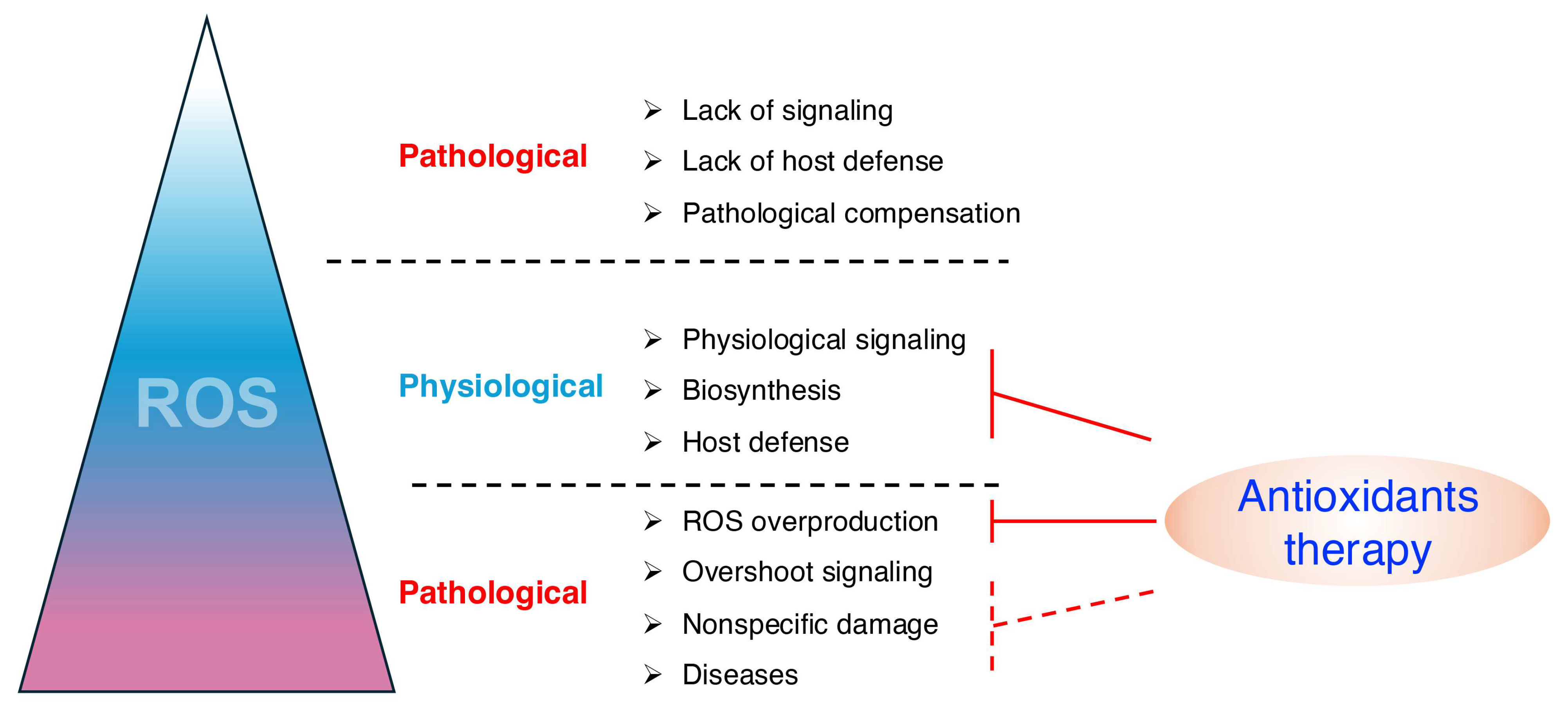
4.1. ROS Play Important Physiological Roles
4.2. Oxidative Stress Is Not the Primary Mediator of Disease Pathology
4.3. Other Limitations
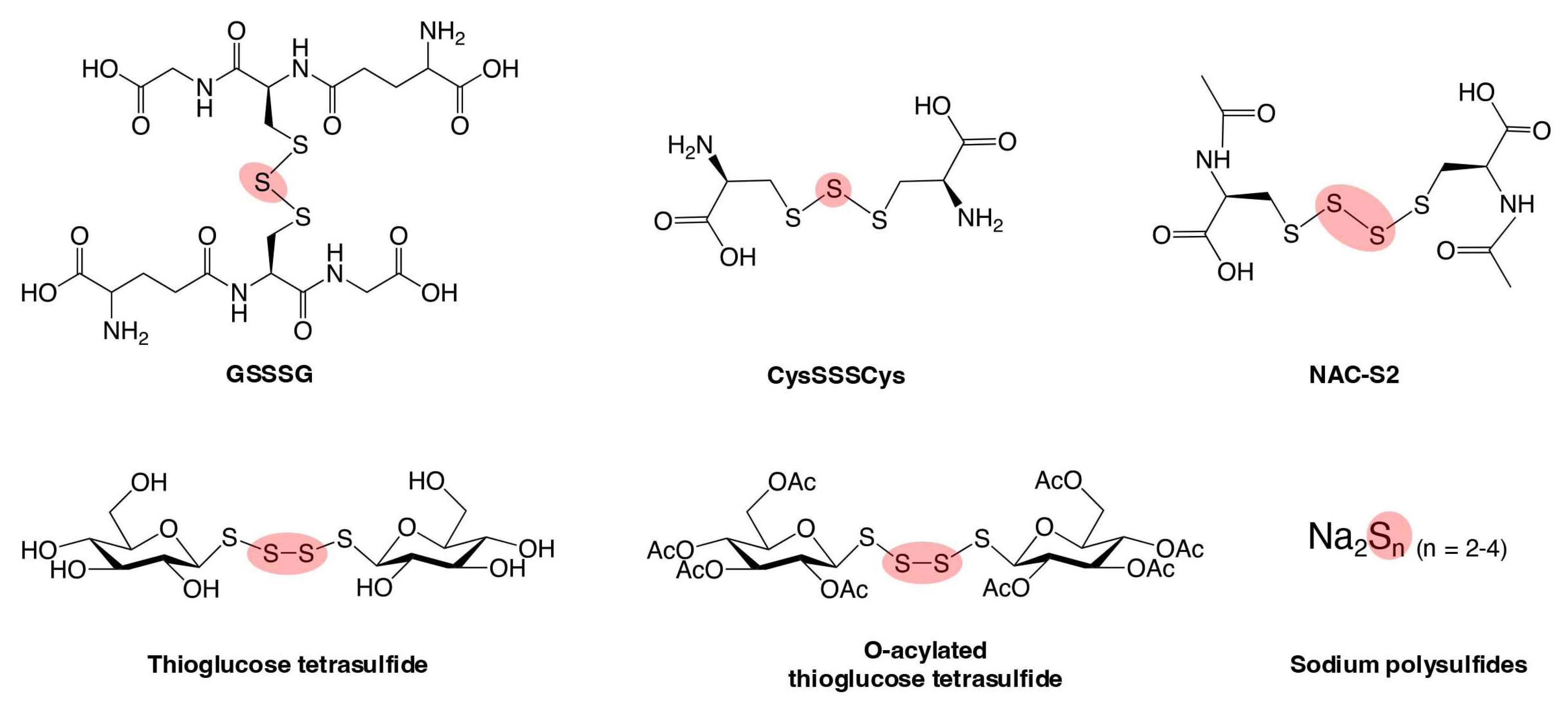
5. Supersulfide Donors as Potential Therapeutic Agents for Oxidative Distress-Related Diseases
5.1. Neurodegenerative Diseases
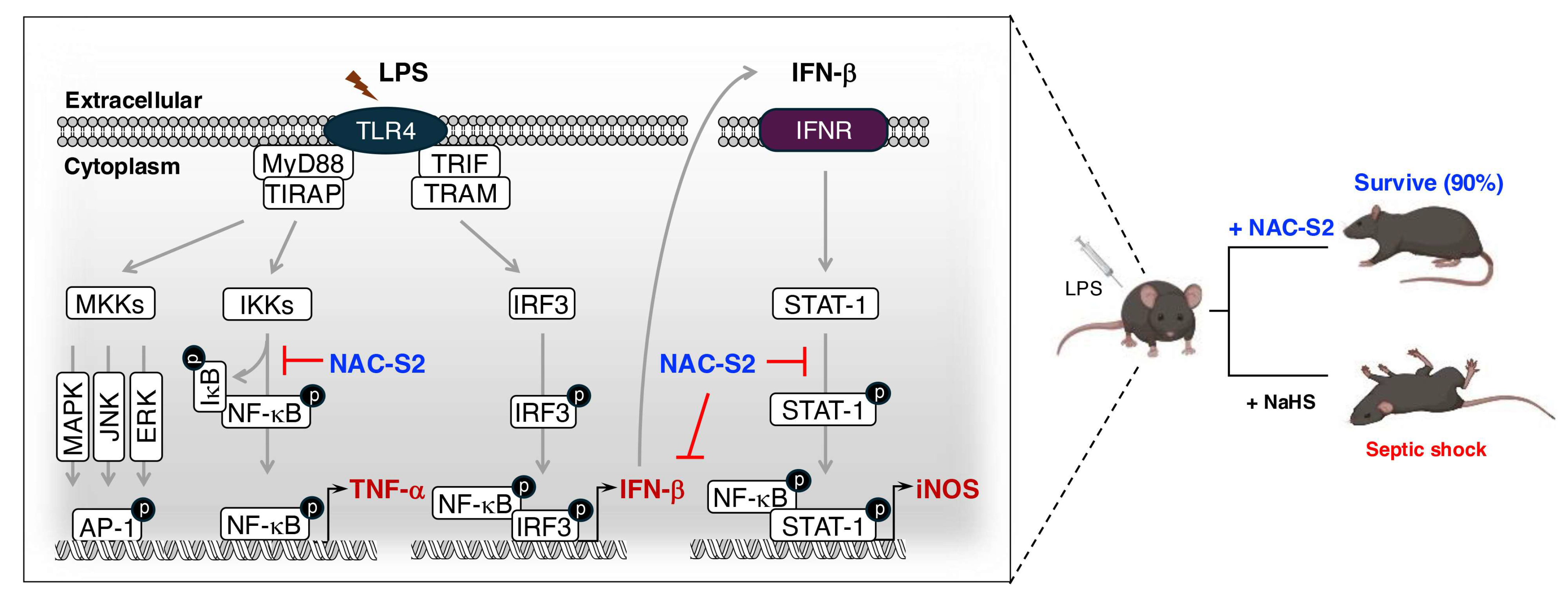
5.2. Systemic Inflammatory Response Syndrome (SIRS)
5.3. Chronic Obstructive Pulmonary Disease (COPD)
6. Limitations and Future Perspectives of Supersulfide-Based Therapy
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sies, H.; Mailloux, R.J.; Jakob, U. Fundamentals of redox regulation in biology. Nat. Rev. Mol. Cell Biol. 2024, 25, 701–719. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.C. Antimicrobial reactive oxygen and nitrogen species: Concepts and controversies. Nat. Rev. Microbiol. 2004, 2, 820–832. [Google Scholar] [CrossRef]
- Mishanina, T.V.; Libiad, M.; Banerjee, R. Biogenesis of reactive sulfur species for signaling by hydrogen sulfide oxidation pathways. Nat. Chem. Biol. 2015, 11, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Cortese-Krott, M.M.; Koning, A.; Kuhnle, G.G.C.; Nagy, P.; Bianco, C.L.; Pasch, A.; Wink, D.A.; Fukuto, J.M.; Jackson, A.A.; van Goor, H.; et al. The Reactive Species Interactome: Evolutionary Emergence, Biological Significance, and Opportunities for Redox Metabolomics and Personalized Medicine. Antioxid. Redox Signal 2017, 27, 684–712. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.; Redman, R. Balancing the generation and elimination of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2005, 102, 3175–3176. [Google Scholar] [CrossRef] [PubMed]
- Sharp, R.E.; Chapman, S.K. Mechanisms for regulating electron transfer in multi-centre redox proteins. Biochim. Biophys. Acta 1999, 1432, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Kohler, A.; Barrientos, A.; Fontanesi, F.; Ott, M. The functional significance of mitochondrial respiratory chain supercomplexes. EMBO Rep. 2023, 24, e57092. [Google Scholar] [CrossRef] [PubMed]
- Lennicke, C.; Cocheme, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 2021, 81, 3691–3707. [Google Scholar] [CrossRef]
- Feelisch, M.; Cortese-Krott, M.M.; Santolini, J.; Wootton, S.A.; Jackson, A.A. Systems redox biology in health and disease. EXCLI J. 2022, 21, 623–646. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I.; Lushchak, O. Interplay between reactive oxygen and nitrogen species in living organisms. Chem. Biol. Interact. 2021, 349, 109680. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. FDA approves first Friedreich’s ataxia drug. Nat. Rev. Drug Discov. 2023, 22, 258. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, E.; Rojas, D.A.; Morales, S.; Miralles, V.; Solari, A. Dual and Opposite Roles of Reactive Oxygen Species (ROS) in Chagas Disease: Beneficial on the Pathogen and Harmful on the Host. Oxid. Med. Cell Longev. 2020, 2020, 8867701. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.J.; Davies, K.J.A.; Davies, M.J.; Dick, T.P.; Finkel, T.; Forman, H.J.; Janssen-Heininger, Y.; et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat. Metab. 2022, 4, 651–662. [Google Scholar] [CrossRef]
- Halliwell, B. Understanding mechanisms of antioxidant action in health and disease. Nat. Rev. Mol. Cell Biol. 2024, 25, 13–33. [Google Scholar] [CrossRef]
- Meng, J.; Lv, Z.; Zhang, Y.; Wang, Y.; Qiao, X.; Sun, C.; Chen, Y.; Guo, M.; Han, W.; Ye, A.; et al. Precision Redox: The Key for Antioxidant Pharmacology. Antioxid. Redox Signal 2021, 34, 1069–1082. [Google Scholar] [CrossRef]
- Georgieva, M.N.; Little, C.T.S.; Bailey, R.J.; Ball, A.D.; Glover, A.G. Microbial-tubeworm associations in a 440 million year old hydrothermal vent community. Proc. Biol. Sci. 2018, 285, 20182004. [Google Scholar] [CrossRef]
- Atmaca, G. Antioxidant effects of sulfur-containing amino acids. Yonsei Med. J. 2004, 45, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Mukwevho, E.; Ferreira, Z.; Ayeleso, A. Potential role of sulfur-containing antioxidant systems in highly oxidative environments. Molecules 2014, 19, 19376–19389. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, K.; Jakob, U. The role of thiols in antioxidant systems. Free Radic. Biol. Med. 2019, 140, 14–27. [Google Scholar] [CrossRef]
- Kasamatsu, S.; Ida, T.; Koga, T.; Asada, K.; Motohashi, H.; Ihara, H.; Akaike, T. High-Precision Sulfur Metabolomics Innovated by a New Specific Probe for Trapping Reactive Sulfur Species. Antioxid. Redox Signal 2021, 34, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.; Matsunaga, T.; et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [Google Scholar] [CrossRef] [PubMed]
- Akaike, T.; Ida, T.; Wei, F.Y.; Nishida, M.; Kumagai, Y.; Alam, M.M.; Ihara, H.; Sawa, T.; Matsunaga, T.; Kasamatsu, S.; et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 2017, 8, 1177. [Google Scholar] [CrossRef] [PubMed]
- Takata, T.; Jung, M.; Matsunaga, T.; Ida, T.; Morita, M.; Motohashi, H.; Shen, X.; Kevil, C.G.; Fukuto, J.M.; Akaike, T. Methods in sulfide and persulfide research. Nitric Oxide 2021, 116, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Doka, E.; Ida, T.; Dagnell, M.; Abiko, Y.; Luong, N.C.; Balog, N.; Takata, T.; Espinosa, B.; Nishimura, A.; Cheng, Q.; et al. Control of protein function through oxidation and reduction of persulfidated states. Sci. Adv. 2020, 6, eaax8358. [Google Scholar] [CrossRef]
- Barayeu, U.; Sawa, T.; Nishida, M.; Wei, F.Y.; Motohashi, H.; Akaike, T. Supersulfide biology and translational medicine for disease control. Br. J. Pharmacol. 2023. [Google Scholar] [CrossRef]
- Akaike, T.; Morita, M.; Ogata, S.; Yoshitake, J.; Jung, M.; Sekine, H.; Motohashi, H.; Barayeu, U.; Matsunaga, T. New aspects of redox signaling mediated by supersulfides in health and disease. Free Radic. Biol. Med. 2024, 222, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Ogata, S.; Matsunaga, T.; Jung, M.; Barayeu, U.; Morita, M.; Akaike, T. Persulfide Biosynthesis Conserved Evolutionarily in All Organisms. Antioxid. Redox Signal 2023, 39, 983–999. [Google Scholar] [CrossRef] [PubMed]
- Tomaiuolo, M.; Vecchione, G.; Margaglione, M.; Pisanelli, D.; Grandone, E. Stable-isotope dilution LC-ESI-MS/MS techniques for the quantification of total homocysteine in human plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 3292–3299. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Ran, M.; Li, X.; Liu, Y.; Xin, Y.; Liu, H.; Liu, H.; Xia, Y.; Xun, L. The Pathway of Sulfide Oxidation to Octasulfur Globules in the Cytoplasm of Aerobic Bacteria. Appl. Env. Environ. Microbiol. 2022, 88, e0194121. [Google Scholar] [CrossRef] [PubMed]
- Kasamatsu, S.; Nishimura, A.; Alam, M.M.; Morita, M.; Shimoda, K.; Matsunaga, T.; Jung, M.; Ogata, S.; Barayeu, U.; Ida, T.; et al. Supersulfide catalysis for nitric oxide and aldehyde metabolism. Sci. Adv. 2023, 9, eadg8631. [Google Scholar] [CrossRef] [PubMed]
- Zivanovic, J.; Kouroussis, E.; Kohl, J.B.; Adhikari, B.; Bursac, B.; Schott-Roux, S.; Petrovic, D.; Miljkovic, J.L.; Thomas-Lopez, D.; Jung, Y.; et al. Selective Persulfide Detection Reveals Evolutionarily Conserved Antiaging Effects of S-Sulfhydration. Cell Metab. 2019, 30, 1152–1170.E13. [Google Scholar] [CrossRef]
- Pedre, B.; Talwar, D.; Barayeu, U.; Schilling, D.; Luzarowski, M.; Sokolowski, M.; Glatt, S.; Dick, T.P. 3-Mercaptopyruvate sulfur transferase is a protein persulfidase. Nat. Chem. Biol. 2023, 19, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Kolluru, G.K.; Shen, X.; Bir, S.C.; Kevil, C.G. Hydrogen sulfide chemical biology: Pathophysiological roles and detection. Nitric Oxide 2013, 35, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Fujii, S.; Matsunaga, T.; Nishimura, A.; Ono, K.; Ida, T.; Ahmed, K.A.; Okamoto, T.; Tsutsuki, H.; Sawa, T.; et al. Reactive Persulfides from Salmonella Typhimurium Downregulate Autophagy-Mediated Innate Immunity in Macrophages by Inhibiting Electrophilic Signaling. Cell Chem. Biol. 2018, 25, 1403–1413.E4. [Google Scholar] [CrossRef] [PubMed]
- Zainol Abidin, Q.H.; Ida, T.; Morita, M.; Matsunaga, T.; Nishimura, A.; Jung, M.; Hassan, N.; Takata, T.; Ishii, I.; Kruger, W.; et al. Synthesis of Sulfides and Persulfides Is Not Impeded by Disruption of Three Canonical Enzymes in Sulfur Metabolism. Antioxidants 2023, 12, 868. [Google Scholar] [CrossRef]
- Guo, M.; Yang, X.L.; Schimmel, P. New functions of aminoacyl-tRNA synthetases beyond translation. Nat. Rev. Mol. Cell Biol. 2010, 11, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Giege, R.; Sissler, M.; Florentz, C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998, 26, 5017–5035. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Kasamatsu, S.; Matsunaga, T.; Akashi, S.; Ono, K.; Nishimura, A.; Morita, M.; Abdul Hamid, H.; Fujii, S.; Kitamura, H.; et al. Protein polysulfidation-dependent persulfide dioxygenase activity of ethylmalonic encephalopathy protein 1. Biochem. Biophys. Res. Commun. 2016, 480, 180–186. [Google Scholar] [CrossRef]
- Alam, M.M.; Kishino, A.; Sung, E.; Sekine, H.; Abe, T.; Murakami, S.; Akaike, T.; Motohashi, H. Contribution of NRF2 to sulfur metabolism and mitochondrial activity. Redox Biol. 2023, 60, 102624. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.; Yoon, S.; Matsunaga, T.; Ida, T.; Jung, M.; Ogata, S.; Morita, M.; Yoshitake, J.; Unno, Y.; Barayeu, U.; et al. Longevity control by supersulfide-mediated mitochondrial respiration and regulation of protein quality. Redox Biol. 2024, 69, 103018. [Google Scholar] [CrossRef] [PubMed]
- Marutani, E.; Morita, M.; Hirai, S.; Kai, S.; Grange, R.M.H.; Miyazaki, Y.; Nagashima, F.; Traeger, L.; Magliocca, A.; Ida, T.; et al. Sulfide catabolism ameliorates hypoxic brain injury. Nat. Commun. 2021, 12, 3108. [Google Scholar] [CrossRef]
- Filipovic, M.R.; Zivanovic, J.; Alvarez, B.; Banerjee, R. Chemical Biology of H2S Signaling through Persulfidation. Chem. Rev. 2018, 118, 1253–1337. [Google Scholar] [CrossRef] [PubMed]
- Doka, E.; Pader, I.; Biro, A.; Johansson, K.; Cheng, Q.; Ballago, K.; Prigge, J.R.; Pastor-Flores, D.; Dick, T.P.; Schmidt, E.E.; et al. A novel persulfide detection method reveals protein persulfide- and polysulfide-reducing functions of thioredoxin and glutathione systems. Sci. Adv. 2016, 2, e1500968. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Maiorino, M.; Ursini, F. Signaling functions of reactive oxygen species. Biochemistry 2010, 49, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef] [PubMed]
- D’Autreaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Fukuto, J.M.; Ignarro, L.J.; Nagy, P.; Wink, D.A.; Kevil, C.G.; Feelisch, M.; Cortese-Krott, M.M.; Bianco, C.L.; Kumagai, Y.; Hobbs, A.J.; et al. Biological hydropersulfides and related polysulfides—A new concept and perspective in redox biology. FEBS Lett. 2018, 592, 2140–2152. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Yuan, Z.; Yang, X.; Wang, B. Prodrugs of Persulfides, Sulfur Dioxide, and Carbon Disulfide: Important Tools for Studying Sulfur Signaling at Various Oxidation States. Antioxid. Redox Signal 2020, 33, 1046–1059. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, H.; Chen, Z.; Zhao, R.; Wang, Q.; Ran, M.; Xia, Y.; Hu, X.; Liu, J.; Xian, M.; et al. Using resonance synchronous spectroscopy to characterize the reactivity and electrophilicity of biologically relevant sulfane sulfur. Redox Biol. 2019, 24, 101179. [Google Scholar] [CrossRef] [PubMed]
- Kunikata, H.; Ida, T.; Sato, K.; Aizawa, N.; Sawa, T.; Tawarayama, H.; Murayama, N.; Fujii, S.; Akaike, T.; Nakazawa, T. Metabolomic profiling of reactive persulfides and polysulfides in the aqueous and vitreous humors. Sci. Rep. 2017, 7, 41984. [Google Scholar] [CrossRef]
- Fukuto, J.M.; Carrington, S.J.; Tantillo, D.J.; Harrison, J.G.; Ignarro, L.J.; Freeman, B.A.; Chen, A.; Wink, D.A. Small molecule signaling agents: The integrated chemistry and biochemistry of nitrogen oxides, oxides of carbon, dioxygen, hydrogen sulfide, and their derived species. Chem. Res. Toxicol. 2012, 25, 769–793. [Google Scholar] [CrossRef] [PubMed]
- Everett, S.A.; Wardman, P. Perthiols as antioxidants: Radical-scavenging and prooxidative mechanisms. Methods Enzym. Enzymol. 1995, 251, 55–69. [Google Scholar] [CrossRef]
- Barayeu, U.; Schilling, D.; Eid, M.; Xavier da Silva, T.N.; Schlicker, L.; Mitreska, N.; Zapp, C.; Grater, F.; Miller, A.K.; Kappl, R.; et al. Hydropersulfides inhibit lipid peroxidation and ferroptosis by scavenging radicals. Nat. Chem. Biol. 2023, 19, 28–37. [Google Scholar] [CrossRef]
- Everett, S.A.; Folkes, L.K.; Wardman, P.; Asmus, K.D. Free-radical repair by a novel perthiol: Reversible hydrogen transfer and perthiyl radical formation. Free Radic. Res. 1994, 20, 387–400. [Google Scholar] [CrossRef]
- Ihara, H.; Kasamatsu, S.; Kitamura, A.; Nishimura, A.; Tsutsuki, H.; Ida, T.; Ishizaki, K.; Toyama, T.; Yoshida, E.; Abdul Hamid, H.; et al. Exposure to Electrophiles Impairs Reactive Persulfide-Dependent Redox Signaling in Neuronal Cells. Chem. Res. Toxicol. 2017, 30, 1673–1684. [Google Scholar] [CrossRef]
- Sawa, T.; Zaki, M.H.; Okamoto, T.; Akuta, T.; Tokutomi, Y.; Kim-Mitsuyama, S.; Ihara, H.; Kobayashi, A.; Yamamoto, M.; Fujii, S.; et al. Protein S-guanylation by the biological signal 8-nitroguanosine 3′,5′-cyclic monophosphate. Nat. Chem. Biol. 2007, 3, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Freeman, B.A.; O’Donnell, V.B.; Schopfer, F.J. The discovery of nitro-fatty acids as products of metabolic and inflammatory reactions and mediators of adaptive cell signaling. Nitric Oxide 2018, 77, 106–111. [Google Scholar] [CrossRef]
- Surh, Y.J.; Na, H.K.; Park, J.M.; Lee, H.N.; Kim, W.; Yoon, I.S.; Kim, D.D. 15-Deoxy-Delta12,14-prostaglandin J2, an electrophilic lipid mediator of anti-inflammatory and pro-resolving signaling. Biochem. Pharmacol. 2011, 82, 1335–1351. [Google Scholar] [CrossRef]
- Mol, M.; Regazzoni, L.; Altomare, A.; Degani, G.; Carini, M.; Vistoli, G.; Aldini, G. Enzymatic and non-enzymatic detoxification of 4-hydroxynonenal: Methodological aspects and biological consequences. Free Radic. Biol. Med. 2017, 111, 328–344. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Armstrong, J.S.; Chu, S.H.; Jia-Ling, S.; Wong, B.S.; Cheung, N.S.; Halliwell, B.; Moore, P.K. The novel neuromodulator hydrogen sulfide: An endogenous peroxynitrite ’scavenger’? J. Neurochem. 2004, 90, 765–768. [Google Scholar] [CrossRef]
- Cuevasanta, E.; Moller, M.N.; Alvarez, B. Biological chemistry of hydrogen sulfide and persulfides. Arch. Biochem. Biophys. 2017, 617, 9–25. [Google Scholar] [CrossRef]
- Netto, L.E.; Antunes, F. The Roles of Peroxiredoxin and Thioredoxin in Hydrogen Peroxide Sensing and in Signal Transduction. Mol. Cells 2016, 39, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Delaunay, A.; Pflieger, D.; Barrault, M.B.; Vinh, J.; Toledano, M.B. A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell 2002, 111, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Vignane, T.; Filipovic, M.R. Emerging Chemical Biology of Protein Persulfidation. Antioxid. Redox Signal 2023, 39, 19–39. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Zhang, Z.; Huang, Z.; Duan, X.; Wang, Y.; Cao, J.; Li, L.; He, K.; Nice, E.C.; He, W.; et al. Protein persulfidation: Rewiring the hydrogen sulfide signaling in cell stress response. Biochem. Pharmacol. 2023, 209, 115444. [Google Scholar] [CrossRef] [PubMed]
- Pettinati, I.; Brem, J.; McDonough, M.A.; Schofield, C.J. Crystal structure of human persulfide dioxygenase: Structural basis of ethylmalonic encephalopathy. Hum. Mol. Genet. 2015, 24, 2458–2469. [Google Scholar] [CrossRef] [PubMed]
- Cuevasanta, E.; Reyes, A.M.; Zeida, A.; Mastrogiovanni, M.; De Armas, M.I.; Radi, R.; Alvarez, B.; Trujillo, M. Kinetics of formation and reactivity of the persulfide in the one-cysteine peroxiredoxin from Mycobacterium tuberculosis. J. Biol. Chem. 2019, 294, 13593–13605. [Google Scholar] [CrossRef]
- Garrido Ruiz, D.; Sandoval-Perez, A.; Rangarajan, A.V.; Gunderson, E.L.; Jacobson, M.P. Cysteine Oxidation in Proteins: Structure, Biophysics, and Simulation. Biochemistry 2022, 61, 2165–2176. [Google Scholar] [CrossRef]
- Millikin, R.; Bianco, C.L.; White, C.; Saund, S.S.; Henriquez, S.; Sosa, V.; Akaike, T.; Kumagai, Y.; Soeda, S.; Toscano, J.P.; et al. The chemical biology of protein hydropersulfides: Studies of a possible protective function of biological hydropersulfide generation. Free Radic. Biol. Med. 2016, 97, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Ono, K.; Tsutsuki, H.; Ihara, H.; Islam, W.; Akaike, T.; Sawa, T. Enhanced Cellular Polysulfides Negatively Regulate TLR4 Signaling and Mitigate Lethal Endotoxin Shock. Cell Chem. Biol. 2019, 26, 686–698.E4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Tsutsuki, H.; Islam, W.; Ono, K.; Takeda, K.; Akaike, T.; Sawa, T. ATP exposure stimulates glutathione efflux as a necessary switch for NLRP3 inflammasome activation. Redox Biol. 2021, 41, 101930. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, T.; Sano, H.; Takita, K.; Morita, M.; Yamanaka, S.; Ichikawa, T.; Numakura, T.; Ida, T.; Jung, M.; Ogata, S.; et al. Supersulphides provide airway protection in viral and chronic lung diseases. Nat. Commun. 2023, 14, 4476. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Murakami, S.; Liu, Z.; Sawa, T.; Takahashi, M.; Izumi, Y.; Bamba, T.; Sato, H.; Akaike, T.; Sekine, H.; et al. Sulfur metabolic response in macrophage limits excessive inflammatory response by creating a negative feedback loop. Redox Biol. 2023, 65, 102834. [Google Scholar] [CrossRef] [PubMed]
- Dang, A.T.; Turner, A.W.; Lau, P.; Mohottalage, D.; Stephanie Fong, Y.K.; Eriksson, P.; Folkersen, L.; Matic, L.; Hedin, U.; Soubeyrand, S.; et al. A novel anti-inflammatory role links the CARS2 locus to protection from coronary artery disease. Atherosclerosis 2022, 348, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Nunez, G. The NLRP3 inflammasome: Activation and regulation. Trends Biochem. Sci. 2023, 48, 331–344. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M.; Forman, H.J. Redox homeostasis: The Golden Mean of healthy living. Redox Biol. 2016, 8, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Oxidative stress and stress-activated signaling pathways: A unifying hypothesis of type 2 diabetes. Endocr. Rev. 2002, 23, 599–622. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, Y.; Li, Y.; Ren, X.; Zhang, X.; Hu, D.; Gao, Y.; Xing, Y.; Shang, H. Oxidative Stress-Mediated Atherosclerosis: Mechanisms and Therapies. Front. Physiol. 2017, 8, 600. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020, 33, 101544. [Google Scholar] [CrossRef] [PubMed]
- Redon, J.; Oliva, M.R.; Tormos, C.; Giner, V.; Chaves, J.; Iradi, A.; Saez, G.T. Antioxidant activities and oxidative stress byproducts in human hypertension. Hypertension 2003, 41, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.L.; Takano, H.; Rizvi, A.; Turrens, J.F.; Qiu, Y.; Wu, W.J.; Zhang, Q.; Bolli, R. Oxidant species trigger late preconditioning against myocardial stunning in conscious rabbits. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H281–H291. [Google Scholar] [CrossRef] [PubMed]
- Hultqvist, M.; Olsson, L.M.; Gelderman, K.A.; Holmdahl, R. The protective role of ROS in autoimmune disease. Trends Immunol. 2009, 30, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef] [PubMed]
- Jena, A.B.; Samal, R.R.; Bhol, N.K.; Duttaroy, A.K. Cellular Red-Ox system in health and disease: The latest update. Biomed. Pharmacother. 2023, 162, 114606. [Google Scholar] [CrossRef] [PubMed]
- Pratico, D. Evidence of oxidative stress in Alzheimer’s disease brain and antioxidant therapy: Lights and shadows. Ann. N. Y Acad. Sci. 2008, 1147, 70–78. [Google Scholar] [CrossRef]
- Witting, P.K.; Mohr, D.; Stocker, R. Assessment of prooxidant activity of vitamin E in human low-density lipoprotein and plasma. Methods Enzym. Enzymol. 1999, 299, 362–375. [Google Scholar] [CrossRef]
- Neuhouser, M.L.; Patterson, R.E.; Thornquist, M.D.; Omenn, G.S.; King, I.B.; Goodman, G.E. Fruits and vegetables are associated with lower lung cancer risk only in the placebo arm of the beta-carotene and retinol efficacy trial (CARET). Cancer Epidemiol. Biomark. Prev. 2003, 12, 350–358. [Google Scholar]
- Bohn, T.; de Lera, A.R.; Landrier, J.F.; Ruhl, R. Carotenoid metabolites, their tissue and blood concentrations in humans and further bioactivity via retinoid receptor-mediated signalling. Nutr. Res. Rev. 2023, 36, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Reflections of an aging free radical. Free Radic. Biol. Med. 2020, 161, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, S.; Shieh, M.; Zhang, T.; Guo, C.; Robinson, J.R.; Sawa, T.; Xian, M. Thioglucose-derived tetrasulfide, a unique polysulfide model compound. Redox Biol. 2024, 70, 103045. [Google Scholar] [CrossRef]
- Tsutsuki, H.; Zhang, T.; Akaike, T.; Sawa, T. Regulation of innate immune and inflammatory responses by supersulfides. Int. Immunol. 2024, 36, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Teleanu, D.M.; Niculescu, A.G.; Lungu; Radu, C.I.; Vladacenco, O.; Roza, E.; Costachescu, B.; Grumezescu, A.M.; Teleanu, R.I. An Overview of Oxidative Stress, Neuroinflammation, and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 5938. [Google Scholar] [CrossRef]
- Berndt, C.; Alborzinia, H.; Amen, V.S.; Ayton, S.; Barayeu, U.; Bartelt, A.; Bayir, H.; Bebber, C.M.; Birsoy, K.; Bottcher, J.P.; et al. Ferroptosis in health and disease. Redox Biol. 2024, 75, 103211. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Cai, R.; Volchuk, A.; Steinberg, B.E.; Saito, Y.; Matsuzawa, A.; Grinstein, S.; Freeman, S.A. Lipid peroxidation increases membrane tension, Piezo1 gating, and cation permeability to execute ferroptosis. Curr. Biol. 2023, 33, 1282–1294.e5. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Kagan, V.E.; Bayir, H.; Pagnussat, G.C.; Head, B.; Traber, M.G.; Stockwell, B.R. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes. Dev. 2018, 32, 602–619. [Google Scholar] [CrossRef] [PubMed]
- Zilka, O.; Shah, R.; Li, B.; Angeli, J.P.F.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Angeli, J.P.F.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Jenkins, N.L.; James, S.A.; Salim, A.; Sumardy, F.; Speed, T.P.; Conrad, M.; Richardson, D.R.; Bush, A.I.; McColl, G. Changes in ferrous iron and glutathione promote ferroptosis and frailty in aging Caenorhabditis elegans. Elife 2020, 9, e56580. [Google Scholar] [CrossRef]
- Wu, Z.; Barayeu, U.; Schilling, D.; Dick, T.P.; Pratt, D.A. Emergence of (hydro)persulfides as suppressors of lipid peroxidation and ferroptotic cell death. Curr. Opin. Chem. Biol. 2023, 76, 102353. [Google Scholar] [CrossRef]
- Kaneko, T.; Mita, Y.; Nozawa-Kumada, K.; Yazaki, M.; Arisawa, M.; Niki, E.; Noguchi, N.; Saito, Y. Antioxidant action of persulfides and polysulfides against free radical-mediated lipid peroxidation. Free Radic. Res. 2022, 56, 677–690. [Google Scholar] [CrossRef]
- Wu, Z.; Khodade, V.S.; Chauvin, J.R.; Rodriguez, D.; Toscano, J.P.; Pratt, D.A. Hydropersulfides Inhibit Lipid Peroxidation and Protect Cells from Ferroptosis. J. Am. Chem. Soc. 2022, 144, 15825–15837. [Google Scholar] [CrossRef]
- Kanemaru, E.; Miyazaki, Y.; Marutani, E.; Ezaka, M.; Goto, S.; Ohshima, E.; Bloch, D.B.; Ichinose, F. Intranasal administration of polysulfide prevents neurodegeneration in spinal cord and rescues mice from delayed paraplegia after spinal cord ischemia. Redox Biol. 2023, 60, 102620. [Google Scholar] [CrossRef]
- Takahashi, S.; Hisatsune, A.; Kurauchi, Y.; Seki, T.; Katsuki, H. Polysulfide protects midbrain dopaminergic neurons from MPP+-induced degeneration via enhancement of glutathione biosynthesis. J. Pharmacol. Sci. 2018, 137, 47–54. [Google Scholar] [CrossRef]
- Lei, G.; Zhuang, L.; Gan, B. The roles of ferroptosis in cancer: Tumor suppression, tumor microenvironment, and therapeutic interventions. Cancer Cell 2024, 42, 513–534. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.K.; Burns, B. Systemic Inflammatory Response Syndrome; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Kuzmich, N.N.; Sivak, K.V.; Chubarev, V.N.; Porozov, Y.B.; Savateeva-Lyubimova, T.N.; Peri, F. TLR4 Signaling Pathway Modulators as Potential Therapeutics in Inflammation and Sepsis. Vaccines 2017, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Ma, C.; Zhang, Z.; Zhang, H.; Hu, H. NF-kappaB signaling in inflammation and cancer. MedComm 2021, 2, 618–653. [Google Scholar] [CrossRef]
- Guo, Q.; Jin, Y.; Chen, X.; Ye, X.; Shen, X.; Lin, M.; Zeng, C.; Zhou, T.; Zhang, J. NF-kappaB in biology and targeted therapy: New insights and translational implications. Signal Transduct. Target. Ther. 2024, 9, 53. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr. STATs and gene regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Mazewski, C.; Perez, R.E.; Fish, E.N.; Platanias, L.C. Type I Interferon (IFN)-Regulated Activation of Canonical and Non-Canonical Signaling Pathways. Front. Immunol. 2020, 11, 606456. [Google Scholar] [CrossRef] [PubMed]
- Tawarayama, H.; Umeki, K.; Inoue-Yanagimachi, M.; Takahashi, N.; Hasegawa, H.; Himori, N.; Tsuda, S.; Kunikata, H.; Akaike, T.; Nakazawa, T. Glutathione trisulfide prevents lipopolysaccharide-induced retinal inflammation via inhibition of proinflammatory cytokine production in glial cells. Sci. Rep. 2023, 13, 11513. [Google Scholar] [CrossRef]
- Alonso de Vega, J.M.; Diaz, J.; Serrano, E.; Carbonell, L.F. Oxidative stress in critically ill patients with systemic inflammatory response syndrome. Crit. Care Med. 2002, 30, 1782–1786. [Google Scholar] [CrossRef]
- Takeda, K.; Shimada, Y.; Amano, M.; Sakai, T.; Okada, T.; Yoshiya, I. Plasma lipid peroxides and alpha-tocopherol in critically ill patients. Crit. Care Med. 1984, 12, 957–959. [Google Scholar] [CrossRef]
- Schorah, C.J.; Downing, C.; Piripitsi, A.; Gallivan, L.; Al-Hazaa, A.H.; Sanderson, M.J.; Bodenham, A. Total vitamin C, ascorbic acid, and dehydroascorbic acid concentrations in plasma of critically ill patients. Am. J. Clin. Nutr. 1996, 63, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Lyons, J.; Rauh-Pfeiffer, A.; Ming-Yu, Y.; Lu, X.M.; Zurakowski, D.; Curley, M.; Collier, S.; Duggan, C.; Nurko, S.; Thompson, J.; et al. Cysteine metabolism and whole blood glutathione synthesis in septic pediatric patients. Crit. Care Med. 2001, 29, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef] [PubMed]
- Montuschi, P.; Collins, J.V.; Ciabattoni, G.; Lazzeri, N.; Corradi, M.; Kharitonov, S.A.; Barnes, P.J. Exhaled 8-isoprostane as an in vivo biomarker of lung oxidative stress in patients with COPD and healthy smokers. Am. J. Respir. Crit. Care Med. 2000, 162, 1175–1177. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; van Schadewijk, A.A.; Crowther, A.J.; Hiemstra, P.S.; Stolk, J.; MacNee, W.; De Boer, W.I. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2002, 166, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Igishi, T.; Hitsuda, Y.; Kato, K.; Sako, T.; Burioka, N.; Yasuda, K.; Sano, H.; Shigeoka, Y.; Nakanishi, H.; Shimizu, E. Elevated urinary 8-hydroxydeoxyguanosine, a biomarker of oxidative stress, and lack of association with antioxidant vitamins in chronic obstructive pulmonary disease. Respirology 2003, 8, 455–460. [Google Scholar] [CrossRef]
- Dey, S.; Eapen, M.S.; Chia, C.; Gaikwad, A.V.; Wark, P.A.B.; Sohal, S.S. Pathogenesis, clinical features of asthma COPD overlap, and therapeutic modalities. Am. J. Physiol. Lung Cell Mol. Physiol. 2022, 322, L64–L83. [Google Scholar] [CrossRef]
- Guo, P.; Li, R.; Piao, T.H.; Wang, C.L.; Wu, X.L.; Cai, H.Y. Pathological Mechanism and Targeted Drugs of COPD. Int. J. Chron. Obs. Pulmon Dis. 2022, 17, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Brusselle, G.G.; Joos, G.F.; Bracke, K.R. New insights into the immunology of chronic obstructive pulmonary disease. Lancet 2011, 378, 1015–1026. [Google Scholar] [CrossRef]
- Uwagboe, I.; Adcock, I.M.; Lo Bello, F.; Caramori, G.; Mumby, S. New drugs under development for COPD. Minerva Med. 2022, 113, 471–496. [Google Scholar] [CrossRef]
- Numakura, T.; Sugiura, H.; Akaike, T.; Ida, T.; Fujii, S.; Koarai, A.; Yamada, M.; Onodera, K.; Hashimoto, Y.; Tanaka, R.; et al. Production of reactive persulfide species in chronic obstructive pulmonary disease. Thorax 2017, 72, 1074–1083. [Google Scholar] [CrossRef]
- Tabas, I.; Glass, C.K. Anti-inflammatory therapy in chronic disease: Challenges and opportunities. Science 2013, 339, 166–172. [Google Scholar] [CrossRef]
- Agrahari, V.; Burnouf, P.A.; Burnouf, T.; Agrahari, V. Nanoformulation properties, characterization, and behavior in complex biological matrices: Challenges and opportunities for brain-targeted drug delivery applications and enhanced translational potential. Adv. Drug Deliv. Rev. 2019, 148, 146–180. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, Y.; Matsunaga, T.; Zhang, T.; Akaike, T. The Therapeutic Potential of Supersulfides in Oxidative Stress-Related Diseases. Biomolecules 2025, 15, 172. https://doi.org/10.3390/biom15020172
Pan Y, Matsunaga T, Zhang T, Akaike T. The Therapeutic Potential of Supersulfides in Oxidative Stress-Related Diseases. Biomolecules. 2025; 15(2):172. https://doi.org/10.3390/biom15020172
Chicago/Turabian StylePan, Yuexuan, Tetsuro Matsunaga, Tianli Zhang, and Takaaki Akaike. 2025. "The Therapeutic Potential of Supersulfides in Oxidative Stress-Related Diseases" Biomolecules 15, no. 2: 172. https://doi.org/10.3390/biom15020172
APA StylePan, Y., Matsunaga, T., Zhang, T., & Akaike, T. (2025). The Therapeutic Potential of Supersulfides in Oxidative Stress-Related Diseases. Biomolecules, 15(2), 172. https://doi.org/10.3390/biom15020172