

Alternative Splicing: Emerging Roles in Anti-Aging Strategies

Abstract
1. Introduction
2. The Significant Changes in Alternative Splicing During Aging and Cellular Senescence
3. Alternative Splicing of Genes Associated with Aging and Cellular Senescence
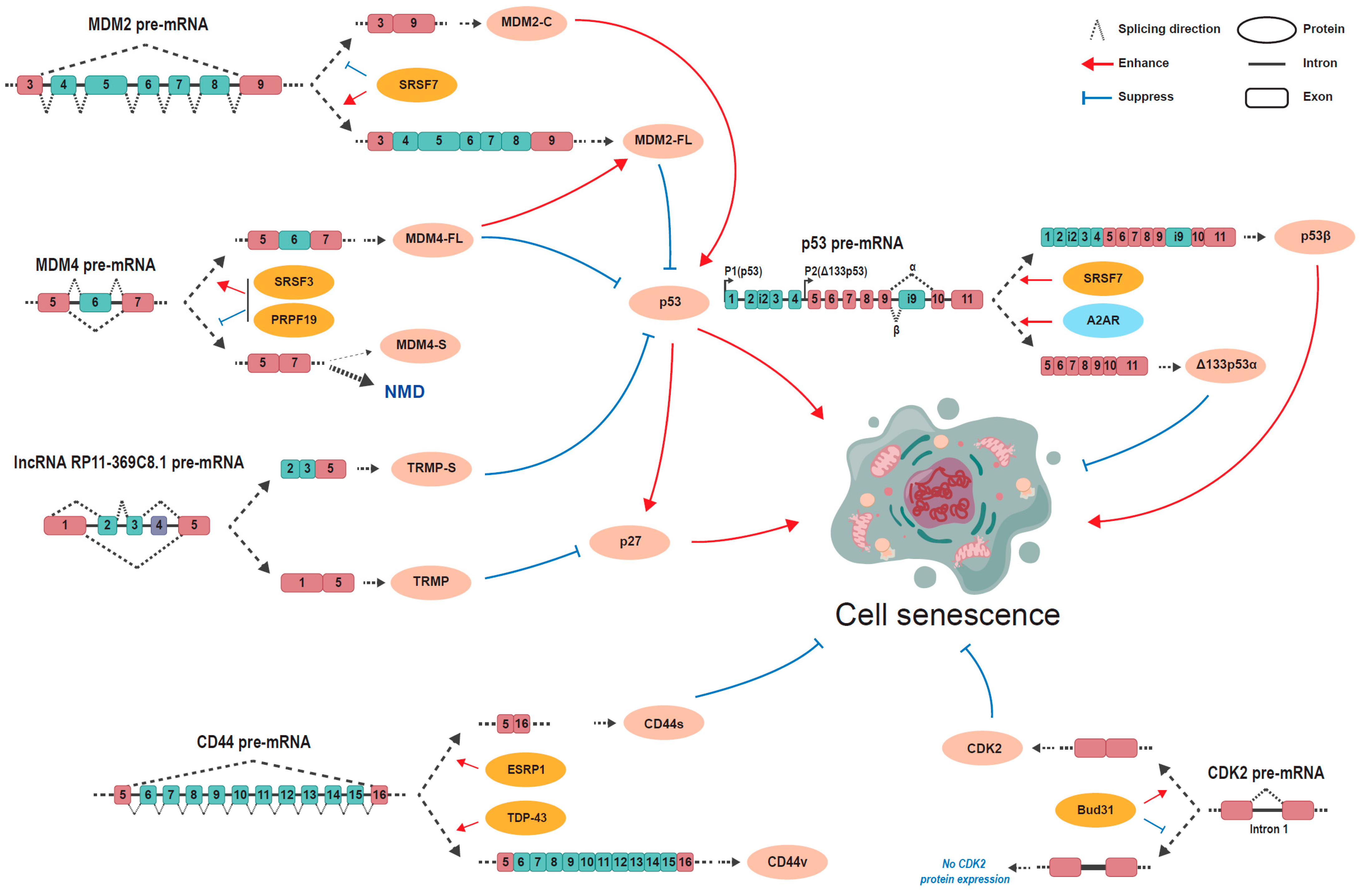
3.1. TP53
3.2. MDM2/MDM4
3.3. lncRNA RP11-369C8.1
3.4. CD44
3.5. CDK2
3.6. SIRT1
4. Controlling Aging or Cellular Senescence by Key Splicing Factors
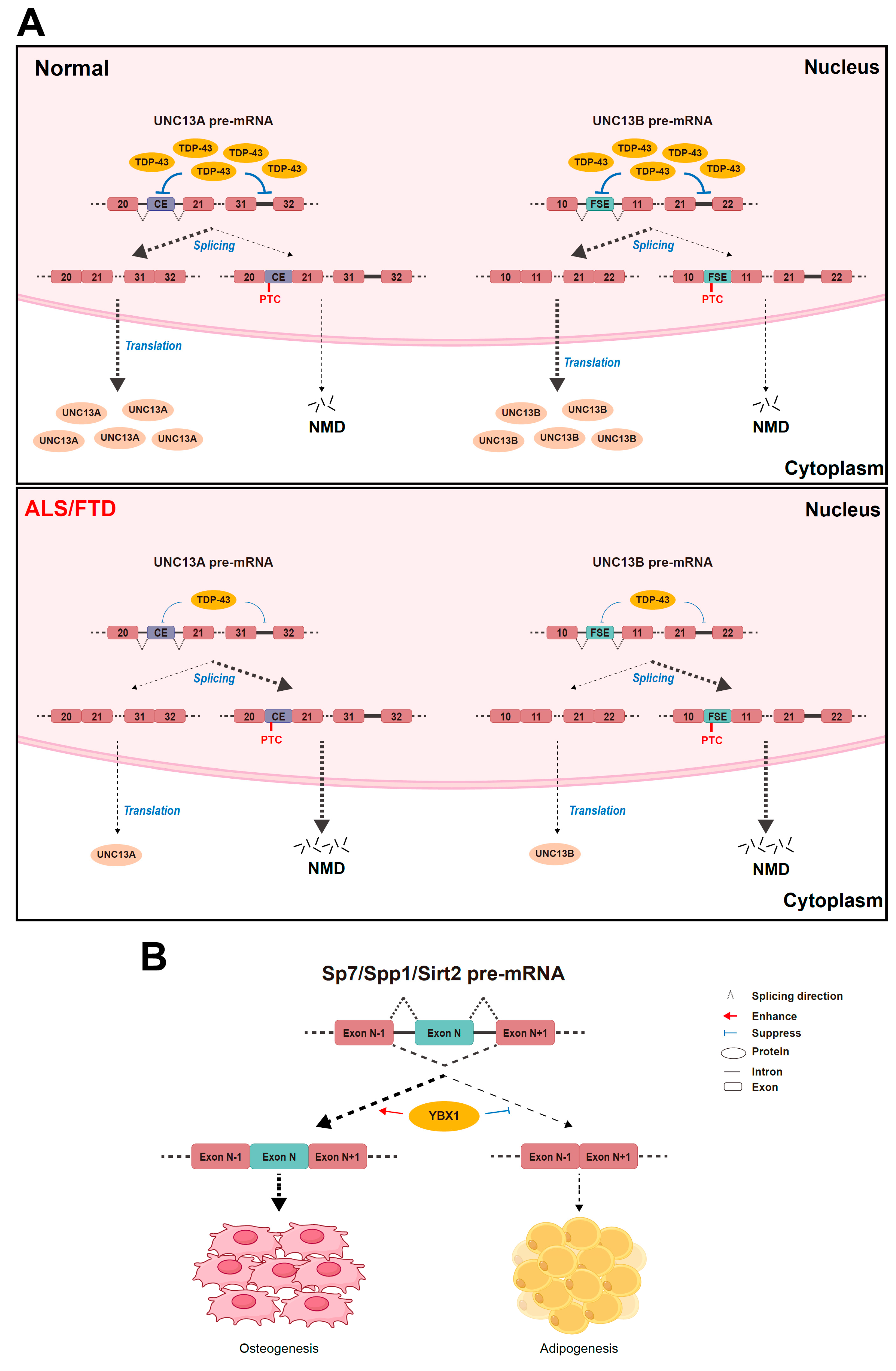
4.1. TDP-43
4.2. YBX1
4.3. Other Splicing Factors Controlling Aging or Cellular Senescence
4.4. Selection of Key Splicing Factors
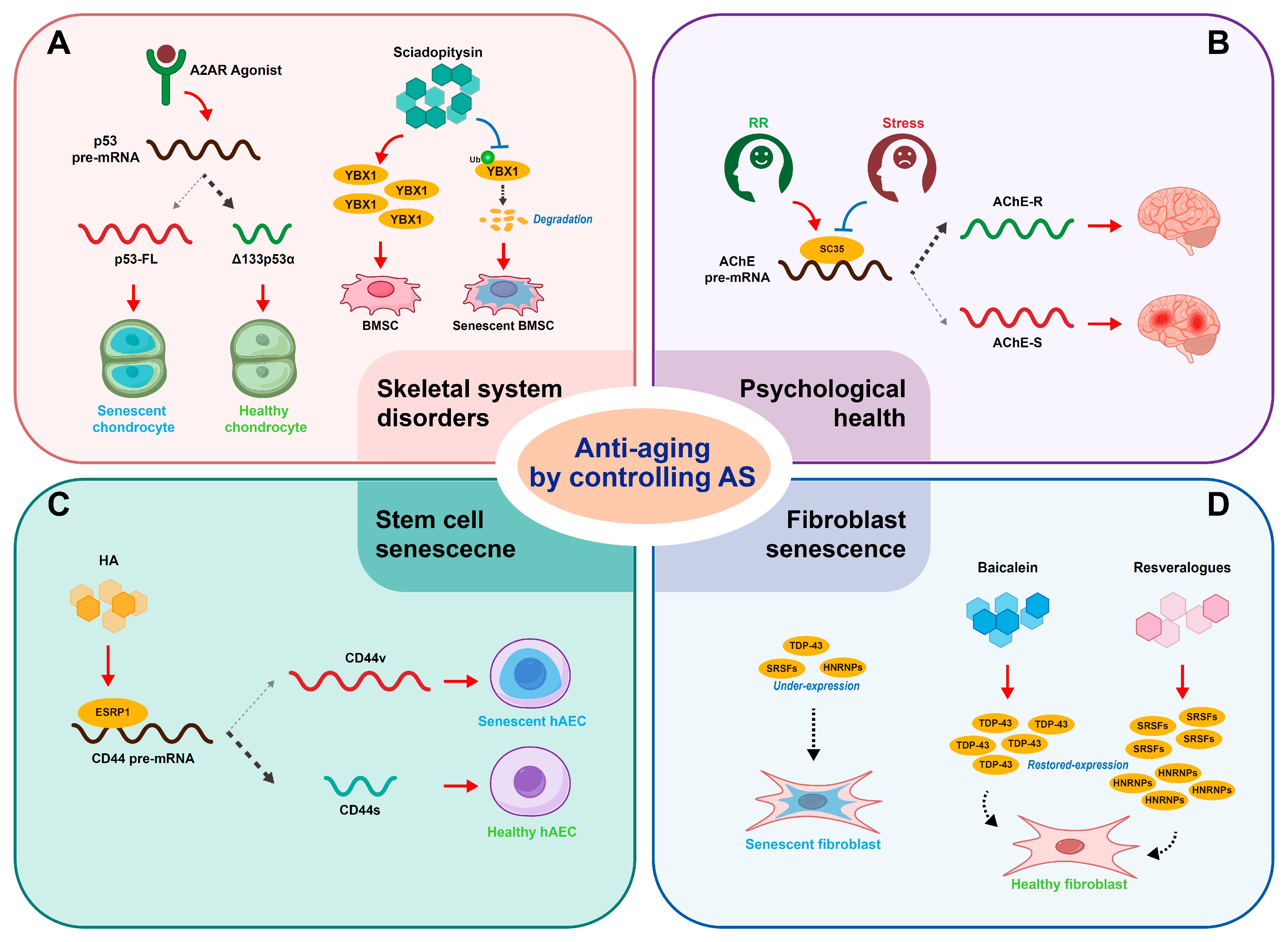
5. Anti-Aging by Controlling Alternative Splicing
5.1. Anti-Aging of Skeletal System
5.2. Anti-Aging of Psychological Health
5.3. Anti-Aging of Stem Cells
5.4. Anti-Aging of Fibroblasts
5.5. Suppressing the Risk of Oncogenesis
5.6. Restoring the Alternative Splicing Profile of Young Cells
6. Conclusions and Remarks
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kirkwood, T.B.; Holliday, R. The evolution of ageing and longevity. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1979, 205, 531–546. [Google Scholar] [CrossRef]
- Bhadra, M.; Howell, P.; Dutta, S.; Heintz, C.; Mair, W.B. Alternative splicing in aging and longevity. Hum. Genet. 2020, 139, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.; Doblhammer, G.; Rau, R.; Vaupel, J.W. Ageing populations: The challenges ahead. Lancet 2009, 374, 1196–1208. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- House, A.E.; Lynch, K.W. Regulation of Alternative Splicing: More than Just the ABCs. J. Biol. Chem. 2008, 283, 1217–1221. [Google Scholar] [CrossRef]
- Black, D.L. Mechanisms of Alternative Pre-Messenger RNA Splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Krainer, A.R. Involvement of SR proteins in mRNA surveillance. Mol. Cell 2004, 16, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.-D.; El Maï, M.; Ladomery, M.; Belali, T.; Leccia, N.; Michiels, J.-F.; Wagner, N. Altered VEGF Splicing Isoform Balance in Tumor Endothelium Involves Activation of Splicing Factors Srpk1 and Srsf1 by the Wilms’ Tumor Suppressor Wt1. Cells 2019, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Stanković, D.; Tain, L.S.; Uhlirova, M. Xrp1 governs the stress response program to spliceosome dysfunction. Nucleic Acids Res. 2024, 52, 2093–2111. [Google Scholar] [CrossRef] [PubMed]
- Salapa, H.E.; Thibault, P.A.; Libner, C.D.; Ding, Y.; Clarke, J.-P.W.E.; Denomy, C.; Hutchinson, C.; Abidullah, H.M.; Austin Hammond, S.; Pastushok, L.; et al. hnRNP A1 dysfunction alters RNA splicing and drives neurodegeneration in multiple sclerosis (MS). Nat. Commun. 2024, 15, 356. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Wei, L.; Zhang, M.Q.; Wang, X. Regulatory RNA binding proteins contribute to the transcriptome-wide splicing alterations in human cellular senescence. Aging 2018, 10, 1489–1505. [Google Scholar] [CrossRef] [PubMed]
- Linares, L.K.; Hengstermann, A.; Ciechanover, A.; Müller, S.; Scheffner, M. HdmX stimulates Hdm2-mediated ubiquitination and degradation of p53. Proc. Natl. Acad. Sci. USA 2003, 100, 12009–12014. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, P.; Jia, L.; Li, T.; Yang, X.; Liang, Q.; Zeng, Y.; Liu, J.; Wu, T.; Hu, W.; et al. Multi-Omics Analysis Reveals Translational Landscapes and Regulations in Mouse and Human Oocyte Aging. Adv. Sci. 2023, 10, e2301538. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Dong, X.; Yuan, X.; Song, J.; Wang, J.; Liu, B.; Wu, K. Proteomic analysis implicates that postovulatory aging leads to aberrant gene expression, biosynthesis, RNA metabolism and cell cycle in mouse oocytes. J. Ovarian Res. 2022, 15, 112. [Google Scholar] [CrossRef] [PubMed]
- Winsky-Sommerer, R.; King, H.A.; Iadevaia, V.; Möller-Levet, C.; Gerber, A.P. A post-transcriptional regulatory landscape of aging in the female mouse hippocampus. Front. Aging Neurosci. 2023, 15, 1119873. [Google Scholar] [CrossRef]
- Guo, W.; Zhang, Z.; Kang, J.; Gao, Y.; Qian, P.; Xie, G. Single-cell transcriptome profiling highlights the importance of telocyte, kallikrein genes, and alternative splicing in mouse testes aging. Sci. Rep. 2024, 14, 14795. [Google Scholar] [CrossRef] [PubMed]
- Achiro, J.M.; Tao, Y.; Gao, F.; Lin, C.-H.; Watanabe, M.; Neumann, S.; Coppola, G.; Black, D.L.; Martin, K.C. Aging differentially alters the transcriptome and landscape of chromatin accessibility in the male and female mouse hippocampus. Front. Mol. Neurosci. 2024, 17, 1334862. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.; Joo, J.; Choi, S.; Kang, B.-G.; Lee, A.J.; Youn, S.-Y.; Park, S.-H.; Kim, E.-M.; Masliah, E.; Ko, Y.; et al. A novel splicing variant of DJ-1 in Parkinson’s disease induces mitochondrial dysfunction. Heliyon 2023, 9, e14039. [Google Scholar] [CrossRef]
- Sato, N.; Hori, O.; Yamaguchi, A.; Lambert, J.C.; Chartier-Harlin, M.C.; Robinson, P.A.; Delacourte, A.; Schmidt, A.M.; Furuyama, T.; Imaizumi, K.; et al. A novel presenilin-2 splice variant in human Alzheimer’s disease brain tissue. J. Neurochem. 1999, 72, 2498–2505. [Google Scholar] [CrossRef] [PubMed]
- Friedman, B.; Larranaga-Vera, A.; Castro, C.M.; Corciulo, C.; Rabbani, P.; Cronstein, B.N. Adenosine A2A receptor activation reduces chondrocyte senescence. FASEB J. 2023, 37, e22838. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Crutchley, J.; Zhang, D.; Owzar, K.; Kastan, M.B. Identification of a DNA Damage–Induced Alternative Splicing Pathway That Regulates p53 and Cellular Senescence Markers. Cancer Discov. 2017, 7, 766–781. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Mondal, A.M.; Horikawa, I.; Nguyen, G.H.; Kumamoto, K.; Sohn, J.J.; Bowman, E.D.; Mathe, E.A.; Schetter, A.J.; Pine, S.R.; et al. p53 isoforms Δ133p53 and p53β are endogenous regulators of replicative cellular senescence. Nat. Cell Biol. 2009, 11, 1135–1142. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, D.; Qin, X.; Owzar, K.; McCann, J.J.; Kastan, M.B. DNA-Damage-Induced Alternative Splicing of p53. Cancers 2021, 13, 251. [Google Scholar] [CrossRef]
- Horikawa, I.; Park, K.; Isogaya, K.; Hiyoshi, Y.; Li, H.; Anami, K.; Robles, A.I.; Mondal, A.M.; Fujita, K.; Serrano, M.; et al. Δ133p53 represses p53-inducible senescence genes and enhances the generation of human induced pluripotent stem cells. Cell Death Differ. 2017, 24, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Min, S.; Yoon, G.; Lim, S.B. SRSF7 downregulation induces cellular senescence through generation of MDM2 variants. Aging 2023, 15, 14591–14606. [Google Scholar] [CrossRef] [PubMed]
- Dewaele, M.; Tabaglio, T.; Willekens, K.; Bezzi, M.; Teo, S.X.; Low, D.H.P.; Koh, C.M.; Rambow, F.; Fiers, M.; Rogiers, A.; et al. Antisense oligonucleotide–mediated MDM4 exon 6 skipping impairs tumor growth. J. Clin. Investig. 2015, 126, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Yano, K.; Takahashi, R.-U.; Shiotani, B.; Abe, J.; Shidooka, T.; Sudo, Y.; Yamamoto, Y.; Kan, S.; Sakagami, H.; Tahara, H. PRPF19 regulates p53-dependent cellular senescence by modulating alternative splicing of MDM4 mRNA. J. Biol. Chem. 2021, 297, 100882. [Google Scholar] [CrossRef]
- Shuai, T.; Khan, M.R.; Zhang, X.D.; Li, J.; Thorne, R.F.; Wu, M.; Shao, F. lncRNA TRMP-S directs dual mechanisms to regulate p27-mediated cellular senescence. Mol. Ther. Nucleic Acids 2021, 24, 971–985. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, C.; Zhao, K. TRMP, a p53-inducible long noncoding RNA, regulates G1/S cell cycle progression by modulating IRES-dependent p27 translation. Cell Death Dis. 2018, 9, 886. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Ke, H.; Zhang, H.; Zou, L.; Yang, Q.; Lu, X.; Zhao, L.; Jiao, B. TDP43 promotes stemness of breast cancer stem cells through CD44 variant splicing isoforms. Cell Death Dis. 2022, 13, 428. [Google Scholar] [CrossRef]
- Yu, C.; Yuan, H.; Xu, Y.; Luo, Y.; Wu, Z.-H.; Zhong, J.-J.; Xiao, J.-H. Hyaluronan delays human amniotic epithelial stem cell senescence by regulating CD44 isoform switch to activate AKT/mTOR signals. Biomed. Pharmacother. 2024, 170, 116100. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.Q.; Wang, J.; Jiang, S.T.; Yuan, L.Q.; Ma, H.Y.; Hu, Y.M.; Han, X.M.; Tan, L.M.; Wang, Z.X. SIRT7-Induced PHF5A Decrotonylation Regulates Aging Progress Through Alternative Splicing-Mediated Downregulation of CDK2. Front. Cell Dev. Biol. 2021, 9, 710479. [Google Scholar] [CrossRef]
- Qin, J.; Huang, T.; Wang, Z.; Zhang, X.; Wang, J.; Dang, Q.; Cui, D.; Wang, X.; Zhai, Y.; Zhao, L.; et al. Bud31-mediated alternative splicing is required for spermatogonial stem cell self-renewal and differentiation. Cell Death Differ. 2023, 30, 184–194. [Google Scholar] [CrossRef]
- Zhang, X.; Ameer, F.S.; Azhar, G.; Wei, J.Y. Alternative Splicing Increases Sirtuin Gene Family Diversity and Modulates Their Subcellular Localization and Function. Int. J. Mol. Sci. 2021, 22, 473. [Google Scholar] [CrossRef]
- Shlomi, S.; Toledano, R.; Nitzan, K.; Shahaf, S.D.; Break, E.P.; Frenkel, D.; Doron, R. Imbalance in Sirt1 Alternative Splicing in Response to Chronic Stress during the Adolescence Period in Female Mice. Int. J. Mol. Sci. 2022, 23, 4945. [Google Scholar] [CrossRef] [PubMed]
- Zu, Y.; Liu, L.; Lee, M.Y.K.; Xu, C.; Liang, Y.; Man, R.Y.; Vanhoutte, P.M.; Wang, Y. SIRT1 promotes proliferation and prevents senescence through targeting LKB1 in primary porcine aortic endothelial cells. Circ. Res. 2010, 106, 1384–1393. [Google Scholar] [CrossRef]
- Hu, W. The Role of p53 Gene Family in Reproduction. Cold Spring Harb. Perspect. Biol. 2009, 1, a001073. [Google Scholar] [CrossRef]
- Lane, D.; Levine, A. p53 Research: The Past Thirty Years and the Next Thirty Years. Cold Spring Harb. Perspect. Biol. 2010, 2, a000893. [Google Scholar] [CrossRef] [PubMed]
- Joruiz, S.M.; Bourdon, J.-C. p53 Isoforms: Key Regulators of the Cell Fate Decision. Cold Spring Harb. Perspect. Med. 2016, 6, a026039. [Google Scholar] [CrossRef]
- Bourdon, J.-C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef]
- Moll, U.M.; Petrenko, O. The MDM2-p53 interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar] [PubMed]
- Rosso, M.; Okoro, D.E.; Bargonetti, J. Splice Variants of MDM2 in Oncogenesis. In Mutant p53 and MDM2 in Cancer; Deb, S.P., Deb, S., Eds.; Springer: Dordrecht, The Netherlands, 2014; pp. 247–261. [Google Scholar]
- Ponta, H.; Wainwright, D.; Herrlich, P. Molecules in focus The CD44 protein family. Int. J. Biochem. Cell Biol. 1998, 30, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Gomari, M.M.; Farsimadan, M.; Rostami, N.; Mahmoudi, Z.; Fadaie, M.; Farhani, I.; Tarighi, P. CD44 polymorphisms and its variants, as an inconsistent marker in cancer investigations. Mutat. Res./Rev. Mutat. Res. 2021, 787, 108374. [Google Scholar] [CrossRef]
- Stamenkovic, I.; Amiot, M.; Pesando, J.M.; Seed, B. A lymphocyte molecule implicated in lymph node homing is a member of the cartilage link protein family. Cell 1989, 56, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- Screaton, G.R.; Bell, M.V.; Jackson, D.G.; Cornelis, F.B.; Gerth, U.; Bell, J.I. Genomic structure of DNA encoding the lymphocyte homing receptor CD44 reveals at least 12 alternatively spliced exons. Proc. Natl. Acad. Sci. USA 1992, 89, 12160–12164. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.N.; Chandavarkar, V.; Sharma, R.; Bhargava, D. Structure, function and role of CD44 in neoplasia. J. Oral Maxillofac. Pathol. 2019, 23, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Bei, Y.; Cheng, N.; Chen, T.; Shu, Y.; Yang, Y.; Yang, N.; Zhou, X.; Liu, B.; Wei, J.; Liu, Q.; et al. CDK5 Inhibition Abrogates TNBC Stem-Cell Property and Enhances Anti-PD-1 Therapy. Adv. Sci. 2020, 7, 2001417. [Google Scholar] [CrossRef]
- Zhang, H.; Brown, R.L.; Wei, Y.; Zhao, P.; Liu, S.; Liu, X.; Deng, Y.; Hu, X.; Zhang, J.; Gao, X.D.; et al. CD44 splice isoform switching determines breast cancer stem cell state. Genes Dev. 2019, 33, 166–179. [Google Scholar] [CrossRef]
- Faber, E.B.; Wang, N.; Georg, G.I. Review of rationale and progress toward targeting cyclin-dependent kinase 2 (CDK2) for male contraception. Biol. Reprod. 2020, 103, 357–367. [Google Scholar] [CrossRef]
- Kwon, T.K.; Buchholz, M.A.; Jun, D.Y.; Kim, Y.H.; Nordin, A.A. The Differential Catalytic Activity of Alternatively Spliced cdk2α and cdk2β in the G1/S Transition and Early S Phase. Exp. Cell Res. 1998, 238, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.G.; Rogina, B.; Lavu, S.; Howitz, K.; Helfand, S.L.; Tatar, M.; Sinclair, D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 2004, 430, 686–689. [Google Scholar] [CrossRef]
- Yamamoto, H.; Schoonjans, K.; Auwerx, J. Sirtuin functions in health and disease. Mol. Endocrinol. 2007, 21, 1745–1755. [Google Scholar] [CrossRef]
- Di Lorenzo, R.; Falanga, D.; Ricci, L.; Colantuono, A.; Greco, G.; Angelillo, M.; Nugnes, F.; Di Serio, T.; Costa, D.; Tito, A.; et al. NAD-Driven Sirtuin Activation by Cordyceps sinensis Extract: Exploring the Adaptogenic Potential to Promote Skin Longevity. Int. J. Mol. Sci. 2024, 25, 4282. [Google Scholar] [CrossRef]
- Ling, J.P.; Pletnikova, O.; Troncoso, J.C.; Wong, P.C. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 2015, 349, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.R.; Prudencio, M.; Koike, Y.; Vatsavayai, S.C.; Kim, G.; Harbinski, F.; Briner, A.; Rodriguez, C.M.; Guo, C.; Akiyama, T.; et al. TDP-43 represses cryptic exon inclusion in the FTD–ALS gene UNC13A. Nature 2022, 603, 124–130. [Google Scholar] [CrossRef]
- Brown, A.-L.; Wilkins, O.G.; Keuss, M.J.; Hill, S.E.; Zanovello, M.; Lee, W.C.; Bampton, A.; Lee, F.C.Y.; Masino, L.; Qi, Y.A.; et al. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature 2022, 603, 131–137. [Google Scholar] [CrossRef]
- Koyama, A.; Sugai, A.; Kato, T.; Ishihara, T.; Shiga, A.; Toyoshima, Y.; Koyama, M.; Konno, T.; Hirokawa, S.; Yokoseki, A.; et al. Increased cytoplasmic TARDBP mRNA in affected spinal motor neurons in ALS caused by abnormal autoregulation of TDP-43. Nucleic Acids Res. 2016, 44, 5820–5836. [Google Scholar] [CrossRef] [PubMed]
- Koike, Y. Molecular mechanisms linking loss of TDP-43 function to amyotrophic lateral sclerosis/frontotemporal dementia-related genes. Neurosci. Res. 2024, 208, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Didier, D.K.; Schiffenbauer, J.; Woulfe, S.L.; Zacheis, M.; Schwartz, B.D. Characterization of the cDNA encoding a protein binding to the major histocompatibility complex class II Y box. Proc. Natl. Acad. Sci. USA 1988, 85, 7322–7326. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.-J.; Mu, S.-R.; Heiner, M.; Fu, X.; Cao, L.-J.; Gong, X.-F.; Bindereif, A.; Hui, J. YB-1 binds to CAUC motifs and stimulates exon inclusion by enhancing the recruitment of U2AF to weak polypyrimidine tracts. Nucleic Acids Res. 2012, 40, 8622–8636. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-J.; Zhu, H.; Mu, S.-R.; Wei, W.-J.; Yuan, X.; Wang, M.; Liu, Y.; Hui, J.; Huang, Y. Crystal structure of a Y-box binding protein 1 (YB-1)-RNA complex reveals key features and residues interacting with RNA. J. Biol. Chem. 2019, 294, 10998–11010. [Google Scholar] [CrossRef]
- Kohno, K.; Izumi, H.; Uchiumi, T.; Ashizuka, M.; Kuwano, M. The pleiotropic functions of the Y-box-binding protein, YB-1. BioEssays 2003, 25, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Lyabin, D.N.; Eliseeva, I.A.; Ovchinnikov, L.P. YB-1 protein: Functions and regulation. WIREs RNA 2014, 5, 95–110. [Google Scholar] [CrossRef]
- Xiao, Y.; Cai, G.-P.; Feng, X.; Li, Y.-J.; Guo, W.-H.; Guo, Q.; Huang, Y.; Su, T.; Li, C.-J.; Luo, X.-H.; et al. Splicing factor YBX1 regulates bone marrow stromal cell fate during aging. EMBO J. 2023, 42, e111762. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Horikawa, I.; Ajiro, M.; Robles, A.I.; Fujita, K.; Mondal, A.M.; Stauffer, J.K.; Zheng, Z.-M.; Harris, C.C. Downregulation of splicing factor SRSF3 induces p53β, an alternatively spliced isoform of p53 that promotes cellular senescence. Oncogene 2013, 32, 2792–2798. [Google Scholar] [CrossRef]
- Raihan, O.; Brishti, A.; Li, Q.; Zhang, Q.; Li, D.; Li, X.; Zhang, Q.; Xie, Z.; Li, J.; Zhang, J.; et al. SFRS11 Loss Leads to Aging-Associated Cognitive Decline by Modulating LRP8 and ApoE. Cell Rep. 2019, 28, 78–90.e6. [Google Scholar] [CrossRef] [PubMed]
- Grillari, J.; Ajuh, P.; Stadler, G.; Löscher, M.; Voglauer, R.; Ernst, W.; Chusainow, J.; Eisenhaber, F.; Pokar, M.; Fortschegger, K.; et al. SNEV is an evolutionarily conserved splicing factor whose oligomerization is necessary for spliceosome assembly. Nucleic Acids Res. 2005, 33, 6868–6883. [Google Scholar] [CrossRef]
- Voglauer, R.; Chang, M.W.-F.; Dampier, B.; Wieser, M.; Baumann, K.; Sterovsky, T.; Schreiber, M.; Katinger, H.; Grillari, J. SNEV overexpression extends the life span of human endothelial cells. Exp. Cell Res. 2006, 312, 746–759. [Google Scholar] [CrossRef]
- Wang, Z.; Peng, Q.; Zhang, Z.; You, X.; Duan, H.; Sha, R.; Yuan, N.; Li, Z.; Xie, Z.; Han, J.; et al. SRSF1 Is Crucial for Maintaining Satellite Cell Homeostasis During Skeletal Muscle Growth and Regeneration. J. Cachexia Sarcopenia Muscle 2024, 15, 2629–2641. [Google Scholar] [CrossRef]
- Fregoso, O.I.; Das, S.; Akerman, M.; Krainer, A.R. Splicing-factor oncoprotein SRSF1 stabilizes p53 via RPL5 and induces cellular senescence. Mol. Cell 2013, 50, 56–66. [Google Scholar] [CrossRef]
- Choi, S.; Cho, N.; Kim, E.-M.; Kim, K.K. The role of alternative pre-mRNA splicing in cancer progression. Cancer Cell Int. 2023, 23, 249. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.; Wempe, F.; Bahr, I.; Zopf, D. Proteasomal degradation of the multifunctional regulator YB-1 is mediated by an F-Box protein induced during programmed cell death. FEBS Lett. 2006, 580, 3921–3930. [Google Scholar] [CrossRef] [PubMed]
- Polsky, L.R.; Rentscher, K.E.; Carroll, J.E. Stress-induced biological aging: A review and guide for research priorities. Brain Behav. Immun. 2022, 104, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Meerson, A.; Cacheaux, L.; Goosens, K.A.; Sapolsky, R.M.; Soreq, H.; Kaufer, D. Changes in Brain MicroRNAs Contribute to Cholinergic Stress Reactions. J. Mol. Neurosci. 2010, 40, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Meshorer, E.; Bryk, B.; Toiber, D.; Cohen, J.; Podoly, E.; Dori, A.; Soreq, H. SC35 promotes sustainable stress-induced alternative splicing of neuronal acetylcholinesterase mRNA. Mol. Psychiatry 2005, 10, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Dal Lin, C.; Marinova, M.; Brugnolo, L.; Rubino, G.; Plebani, M.; Iliceto, S.; Tona, F. Rapid changes of miRNAs-20, -30, -410, -515, -134, and -183 and telomerase with psychological activity: A one year study on the relaxation response and epistemological considerations. J. Tradit. Complement. Med. 2021, 11, 409–418. [Google Scholar] [CrossRef]
- Latorre, E.; Birar, V.C.; Sheerin, A.N.; Jeynes, J.C.C.; Hooper, A.; Dawe, H.R.; Melzer, D.; Cox, L.S.; Faragher, R.G.A.; Ostler, E.L.; et al. Small molecule modulation of splicing factor expression is associated with rescue from cellular senescence. BMC Cell Biol. 2017, 18, 31. [Google Scholar] [CrossRef]
- Latorre, E.; Ostler, E.L.; Faragher, R.G.A.; Harries, L.W. FOXO1 and ETV6 genes may represent novel regulators of splicing factor expression in cellular senescence. FASEB J. 2019, 33, 1086–1097. [Google Scholar] [CrossRef]
- Bramwell, L.R.; Harries, L.W. Senescence, regulators of alternative splicing and effects of trametinib treatment in progeroid syndromes. Geroscience 2023, 46, 1861–1879. [Google Scholar] [CrossRef]
- Chang, H.-Y.; Wang, I.-F. Restoring functional TDP-43 oligomers in ALS and laminopathic cellular models through baicalein-induced reconfiguration of TDP-43 aggregates. Sci. Rep. 2024, 14, 4620. [Google Scholar] [CrossRef]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Varley, J.M.; McGown, G.; Thorncroft, M.; Santibanez-Koref, M.F.; Kelsey, A.M.; Tricker, K.J.; Evans, D.G.; Birch, J.M. Germ-line mutations of TP53 in Li-Fraumeni families: An extended study of 39 families. Cancer Res. 1997, 57, 3245–3252. [Google Scholar] [PubMed]
- Deschênes, M.; Chabot, B. The emerging role of alternative splicing in senescence and aging. Aging Cell 2017, 16, 918–933. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Splice Variants | Exon Location | Splicing Events | Roles in Aging | Regulatory Mechanism | Refs. |
|---|---|---|---|---|---|---|
| TP53 | p53β | Exon i9 | Alternative 3′ splice site | Promoting cellular senescence | RPL26–SRSF7 promotes the p53β isoform | [20,21,22,23,24] |
| Δ133p53α | P2 promoter | Alternative promoter | Inhibiting cellular senescence | A2AR promotes the Δ133p53α isoform | ||
| MDM2 | MDM2-C | Exons 4–8 | Exon skipping | Promoting senescence | SRSF7 inhibits the skipping of exons 4–8 | [25] |
| MDM4 | MDM4-FL and MDM4-S | Exon 6 | Exon skipping | MDM4-FL suppresses senescence | SRSF3 and PRPF19 inhibit the skipping of exon 6 | [26,27] |
| lncRNA RP11-369C8.1 | TRMP | Exons 2–4 | Exon skipping | Promoting cell proliferation and inhibiting cellular senescence | N/A | [28,29] |
| TRMP-S | Exons 1 and 4 | Exon skipping | ||||
| CD44 | CD44s | exons 1–5 and 16–20 | Standard exons | Inhibiting hAECs senescence | ESRP1 upregulates the CD44s and downregulates the CD44v TDP-43 promotes the inclusion of variable exons | [30,31] |
| CD44v | exons 6–15 | Variable exons | Promoting hAECs senescence | |||
| CDK2 | N/A | Intron 1 | Intron retention | Promoting senescence | PHF5A K25 decrotonylation and the depletion of Bud31 promote the retention of intron 1 | [32,33] |
| SIRT1 | SIRT1-v1 | N/A | N/A | Inhibiting senescence and improving longevity | N/A | [34,35,36] |
| SIRT1-v2 | Exons 1 and 3 | Exon skipping | Promoting senescence | |||
| Exon 1′ | Exon inclusion | |||||
| SIRT1-v3 | Exons 1, 2, and 3 | Exon skipping | ||||
| Exon 4′ | Exon inclusion |
| Aging-Related Disorders | Splicing Events or Splicing Regulators | Anti-Aging Methods | Refs. | |
|---|---|---|---|---|
| Skeletal system | Senescence of chondrocytes and osteoarthritis | Decrease in full-length p53 Increase in Δ133p53α | A2AR agonist | [20] |
| Senescence of BMSCs and age-related osteoporosis | Exon skipping of BMSC osteogenesis-related and senescence-related genes | Sciadopitysin | [65] | |
| Psychological health | Increase in SC35 | Relaxation response | [81] | |
| Stem cells | Senescence of hAECs | Increase in CD44s and decrease in the CD44v regulated by ESRP1 | Hyaluronic acid | [31] |
| Fibroblast | Increase in SRSFs and HNRNPs | Resveralogues, trametinib, and SH-6 | [78,79] | |
| TDP-43-mediated exon skipping | Baicalein | [75,77] | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, L.; Jia, R. Alternative Splicing: Emerging Roles in Anti-Aging Strategies. Biomolecules 2025, 15, 131. https://doi.org/10.3390/biom15010131
Gao L, Jia R. Alternative Splicing: Emerging Roles in Anti-Aging Strategies. Biomolecules. 2025; 15(1):131. https://doi.org/10.3390/biom15010131
Chicago/Turabian StyleGao, Lingyue, and Rong Jia. 2025. "Alternative Splicing: Emerging Roles in Anti-Aging Strategies" Biomolecules 15, no. 1: 131. https://doi.org/10.3390/biom15010131
APA StyleGao, L., & Jia, R. (2025). Alternative Splicing: Emerging Roles in Anti-Aging Strategies. Biomolecules, 15(1), 131. https://doi.org/10.3390/biom15010131