
Current Treatment Regimens and Promising Molecular Therapies for Chronic Hepatobiliary Diseases

 ,
,  , , , , and
, , , , and 
Abstract
1. Introduction
1.1. Metabolic Dysfunction-Associated Steatotic Liver Disease
1.2. Hepatitis D Virus (HDV)
1.3. Autoimmune Hepatitis (AIH)
1.4. Primary Sclerosing Cholangitis (PSC)
1.5. Primary Biliary Cholangitis (PBC)
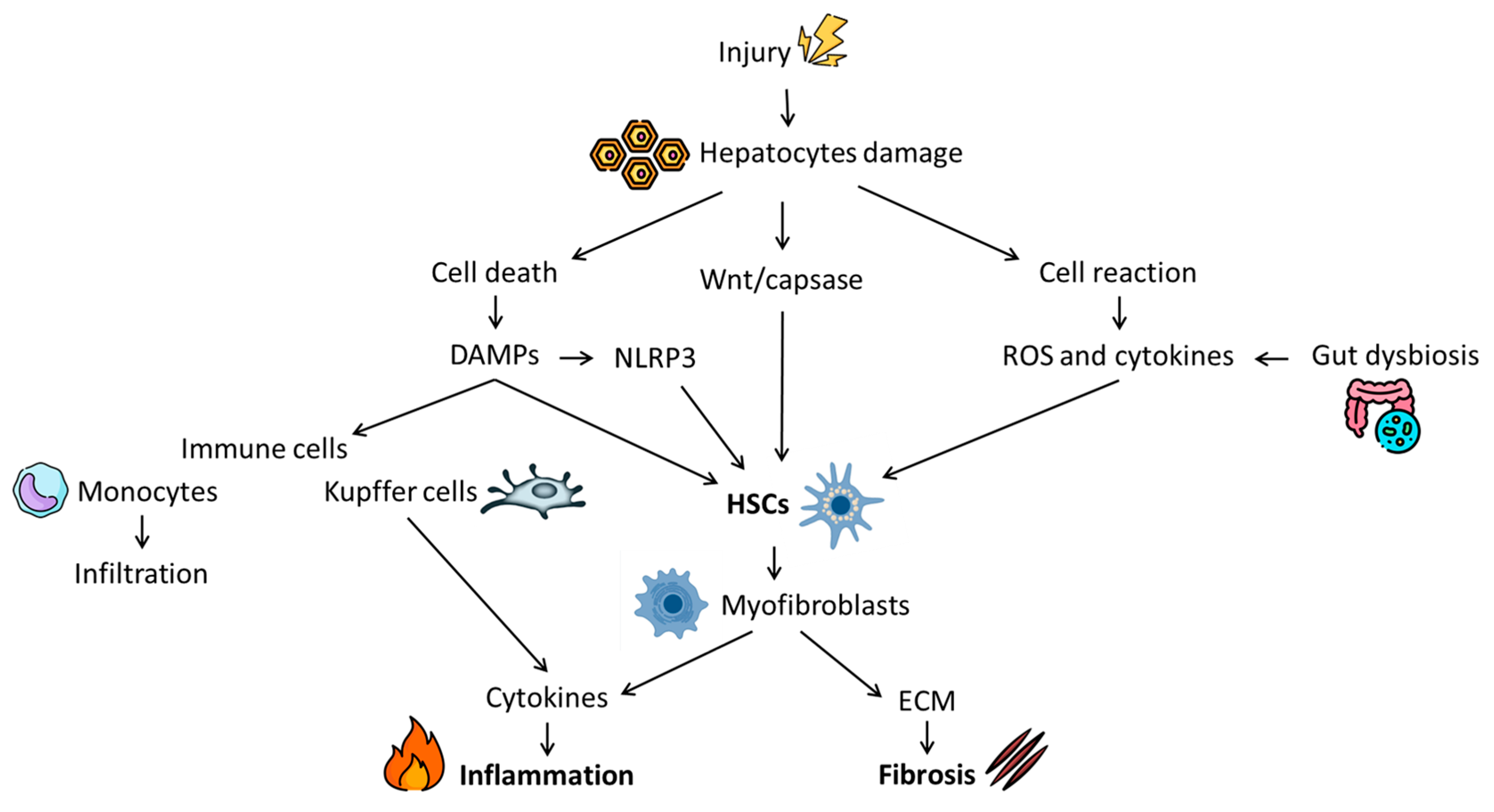
2. Fibrosis: Pathophysiology and Natural History
3. Traditional Therapies
3.1. MASLD
3.1.1. Lifestyle Interventions
3.1.2. Pharmacological Therapy
3.1.3. Surgical and Endoscopic Therapy
3.2. HDV
3.3. AIH
3.4. PSC
3.5. PBC
4. Emerging Therapeutic Approaches
4.1. Inflammation Control
4.2. Hepatic Stellate Cells Inhibition
4.3. Extracellular Matrix Regulation
5. Preclinical CHD Models
5.1. Animal Models
5.1.1. MASLD and MASH
5.1.2. HDV
5.1.3. AIH
5.1.4. Mice Models of PBC and PSC
5.2. Organoids for Liver Diseases Modelling
5.2.1. MASLD
5.2.2. PBC and PSC
6. Promising Targeted Therapies in CHD
6.1. Nucleic Acid-Based Therapeutics
6.1.1. MASLD
6.1.2. HDV
6.1.3. AIH
6.1.4. Cholangiopathies (PBC, PSC)
6.2. Other Potential Molecular Therapies for CHD
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Devarbhavi, H.; Asrani, S.K.; Arab, J.P.; Nartey, Y.A.; Pose, E.; Kamath, P.S. Global burden of liver disease: 2023 update. J. Hepatol. 2023, 79, 516–537. [Google Scholar] [CrossRef] [PubMed]
- Moon, A.M.; Singal, A.G.; Tapper, E.B. Contemporary Epidemiology of Chronic Liver Disease and Cirrhosis. Clin. Gastroenterol. Hepatol. 2020, 18, 2650–2666. [Google Scholar] [CrossRef] [PubMed]
- Ferro, A.; Saccu, G.; Mattivi, S.; Gaido, A.; Herrera Sanchez, M.B.; Haque, S.; Silengo, L.; Altruda, F.; Durazzo, M.; Fagoonee, S. Extracellular Vesicles as Delivery Vehicles for Non-Coding RNAs: Potential Biomarkers for Chronic Liver Diseases. Biomolecules 2024, 14, 277. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, M.; Shigefuku, R.; Eguchi, A.; Tamai, Y.; Takei, Y. Update on blood-based biomarkers for chronic liver diseases prognosis: Literature review and institutional experience. JGH Open 2021, 5, 1250–1256. [Google Scholar] [CrossRef]
- Zheng, M.; Allington, G.; Vilarinho, S. Genomic medicine for liver disease. Hepatology 2022, 76, 860–868. [Google Scholar] [CrossRef]
- Bashir, M.R.; Horowitz, J.M.; Kamel, I.R.; Arif-Tiwari, H.; Asrani, S.K.; Chernyak, V.; Goldstein, A.; Grajo, J.R.; Hindman, N.M.; Kamaya, A.; et al. ACR Appropriateness Criteria® Chronic Liver Disease. J. Am. Coll. Radiol. 2020, 17, S70–S80. [Google Scholar] [CrossRef]
- Jordan, V.C.; Sojoodi, M.; Shroff, S.; Pagan, P.G.; Barrett, S.C.; Wellen, J.; Tanabe, K.K.; Chung, R.T.; Caravan, P.; Gale, E.M. Molecular magnetic resonance imaging of liver inflammation using an oxidatively activated probe. JHEP Rep. 2023, 5, 100850. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): A systematic review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef]
- Riazi, K.; Azhari, H.; Charette, J.H.; Underwood, F.E.; King, J.A.; Afshar, E.E.; Swain, M.G.; Congly, S.E.; Kaplan, G.G.; Shaheen, A.A. The prevalence and incidence of NAFLD worldwide: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2022, 7, 851–861. [Google Scholar] [CrossRef]
- Niro, G.A.; Ferro, A.; Cicerchia, F.; Brascugli, I.; Durazzo, M. Hepatitis delta virus: From infection to new therapeutic strategies. World J. Gastroenterol. 2021, 27, 3530–3542. [Google Scholar] [CrossRef] [PubMed]
- Stockdale, A.J.; Kreuels, B.; Henrion, M.Y.R.; Giorgi, E.; Kyomuhangi, I.; de Martel, C.; Hutin, Y.; Geretti, A.M. The global prevalence of hepatitis D virus infection: Systematic review and meta-analysis. J. Hepatol. 2020, 73, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Gish, R.G.; Wong, R.J.; Di Tanna, G.L.; Kaushik, A.; Kim, C.; Smith, N.J.; Kennedy, P.T.F. Association of hepatitis delta virus with liver morbidity and mortality: A systematic literature review and meta-analysis. Hepatology 2024, 79, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Gatselis, N.K.; Zachou, K.; Koukoulis, G.K.; Dalekos, G.N. Autoimmune hepatitis, one disease with many faces: Etiopathogenetic, clinico-laboratory and histological characteristics. World J. Gastroenterol. 2015, 21, 60–83. [Google Scholar] [CrossRef]
- Trivedi, P.J.; Hirschfield, G.M. Recent advances in clinical practice: Epidemiology of autoimmune liver diseases. Gut 2021, 70, 1989–2003. [Google Scholar] [CrossRef]
- Czaja, A.J. The overlap syndromes of autoimmune hepatitis. Dig. Dis. Sci. 2013, 58, 326–343. [Google Scholar] [CrossRef]
- Gleeson, D. Long-Term Outcomes of Autoimmune Hepatitis. Clin. Liver Dis. 2019, 14, 24–28. [Google Scholar] [CrossRef]
- Chapman, M.H.; Thorburn, D.; Hirschfield, G.M.; Webster, G.G.J.; Rushbrook, S.M.; Alexander, G.; Collier, J.; Dyson, J.K.; Jones, D.E.; Patanwala, I.; et al. British Society of Gastroenterology and UK-PSC guidelines for the diagnosis and management of primary sclerosing cholangitis. Gut 2019, 68, 1356–1378. [Google Scholar] [CrossRef]
- Karlsen, T.H.; Folseraas, T.; Thorburn, D.; Vesterhus, M. Primary sclerosing cholangitis—A comprehensive review. J. Hepatol. 2017, 67, 1298–1323. [Google Scholar] [CrossRef]
- Aune, D.; Sen, A.; Norat, T.; Riboli, E.; Folseraas, T. Primary sclerosing cholangitis and the risk of cancer, cardiovascular disease, and all-cause mortality: A systematic review and meta-analysis of cohort studies. Sci. Rep. 2021, 11, 10646. [Google Scholar] [CrossRef]
- Lindor, K.D.; Bowlus, C.L.; Boyer, J.; Levy, C.; Mayo, M. Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2019, 69, 394–419. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A. Current understanding of primary biliary cholangitis. Clin. Mol. Hepatol. 2021, 27, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Lleo, A.; Jepsen, P.; Morenghi, E.; Carbone, M.; Moroni, L.; Battezzati, P.M.; Podda, M.; Mackay, I.R.; Gershwin, M.E.; Invernizzi, P. Evolving Trends in Female to Male Incidence and Male Mortality of Primary Biliary Cholangitis. Sci. Rep. 2016, 6, 25906. [Google Scholar] [CrossRef]
- Harms, M.H.; van Buuren, H.R.; Corpechot, C.; Thorburn, D.; Janssen, H.L.A.; Lindor, K.D.; Hirschfield, G.M.; Parés, A.; Floreani, A.; Mayo, M.J.; et al. Ursodeoxycholic acid therapy and liver transplant-free survival in patients with primary biliary cholangitis. J. Hepatol. 2019, 71, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, Y.; Tansel, A.; Patel, P.; Emologu, K.; Shukla, R.; Qureshi, Z.; El-Serag, H.B.; Thrift, A.P.; Kanwal, F. Incidence of Hepatocellular Carcinoma in Primary Biliary Cholangitis: A Systematic Review and Meta-Analysis. Dig. Dis. Sci. 2021, 66, 2439–2451. [Google Scholar] [CrossRef]
- Berumen, J.; Baglieri, J.; Kisseleva, T.; Mekeel, K. Liver fibrosis: Pathophysiology and clinical implications. WIREs Mech. Dis. 2021, 13, e1499. [Google Scholar] [CrossRef]
- D’Amico, G.; Morabito, A.; D’Amico, M.; Pasta, L.; Malizia, G.; Rebora, P.; Valsecchi, M.G. New concepts on the clinical course and stratification of compensated and decompensated cirrhosis. Hepatol. Int. 2018, 12, 34–43. [Google Scholar] [CrossRef]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef]
- Mihm, S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. Int. J. Mol. Sci. 2018, 19, 3104. [Google Scholar] [CrossRef]
- Affo, S.; Yu, L.X.; Schwabe, R.F. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu. Rev. Pathol. 2017, 12, 153–186. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, C. The Roles of Liver-Resident Lymphocytes in Liver Diseases. Front. Immunol. 2019, 10, 1582. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, P.; Liu, Y.; Zhang, Y. Efficacy of Probiotics and Synbiotics in Patients with Nonalcoholic Fatty Liver Disease: A Meta-Analysis. Dig. Dis. Sci. 2019, 64, 3402–3412. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [PubMed]
- Berg, T.; DeLanghe, S.; Al Alam, D.; Utley, S.; Estrada, J.; Wang, K.S. β-catenin regulates mesenchymal progenitor cell differentiation during hepatogenesis. J. Surg. Res. 2010, 164, 276–285. [Google Scholar] [CrossRef]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef]
- The French METAVIR Cooperative Study Group. Intraobserver and interobserver variations in liver biopsy interpretation in patients with chronic hepatitis C. Hepatology 1994, 20, 15–20. [Google Scholar] [CrossRef]
- Machado, M.V. MASLD treatment-a shift in the paradigm is imminent. Front. Med. 2023, 10, 1316284. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines on the management of metabolic dysfunction-associated steatotic liver disease (MASLD): Executive Summary. Diabetologia 2024, 67, 2375–2392. [Google Scholar] [CrossRef]
- Li, Y.; Yang, P.; Ye, J.; Xu, Q.; Wu, J.; Wang, Y. Updated mechanisms of MASLD pathogenesis. Lipids Health Dis. 2024, 23, 117. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Nephew, L.D.; Vuppalanchi, R.; Gawrieh, S.; Mladenovic, A.; Pike, F.; Samala, N.; Chalasani, N. High-quality diet, physical activity, and college education are associated with low risk of NAFLD among the US population. Hepatology 2022, 75, 1491–1506. [Google Scholar] [CrossRef]
- Li, M. Association of physical activity with MAFLD/MASLD and LF among adults in NHANES, 2017-2020. Wien Klin Wochenschr 2024, 136, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J. Resmetirom: First Approval. Drugs 2024, 84, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Talha, M.; Ali, M.H.; Nadeem, Z.A.; Akram, U.; Saravanan, P.B.; Khalid, M.H.A. Efficacy and safety of resmetirom for the treatment of nonalcoholic steatohepatitis: A GRADE assessed systematic review and meta-analysis. Eur. J. Gastroenterol. Hepatol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Farci, P.; Niro, G.A. Current and Future Management of Chronic Hepatitis D. Gastroenterol. Hepatol. 2018, 14, 342–351. [Google Scholar]
- Pan, C.; Gish, R.; Jacobson, I.M.; Hu, K.Q.; Wedemeyer, H.; Martin, P. Diagnosis and Management of Hepatitis Delta Virus Infection. Dig. Dis. Sci. 2023, 68, 3237–3248. [Google Scholar] [CrossRef]
- Abbas, Z.; Khan, M.A.; Salih, M.; Jafri, W. Interferon alpha for chronic hepatitis D. Cochrane Database Syst. Rev. 2011, 2011, CD006002. [Google Scholar] [CrossRef]
- Brunetto, M.R.; Ricco, G.; Negro, F.; Wedemeyer, H.; Yurdaydin, C.; Asselah, T.; Papatheodoridis, G.; Gheorghe, L.; Agarwal, K.; Farci, P.; et al. EASL Clinical Practice Guidelines on hepatitis delta virus. J. Hepatol. 2023, 79, 433–460. [Google Scholar] [CrossRef]
- Wedemeyer, H.; Aleman, S.; Brunetto, M.R.; Blank, A.; Andreone, P.; Bogomolov, P.; Chulanov, V.; Mamonova, N.; Geyvandova, N.; Morozov, V.; et al. A Phase 3, Randomized Trial of Bulevirtide in Chronic Hepatitis D. N. Engl. J. Med. 2023, 389, 22–32. [Google Scholar] [CrossRef]
- Steinmann, S.; Lohse, A.W. Treatment of autoimmune hepatitis: Budesonide does not solve our problems. Hepatology 2023, 77, 1071–1073. [Google Scholar] [CrossRef]
- Díaz-González, Á.; Hernández-Guerra, M.; Pérez-Medrano, I.; Sapena, V.; Riveiro-Barciela, M.; Barreira-Díaz, A.; Gómez, E.; Morillas, R.M.; Del Barrio, M.; Escudé, L.; et al. Budesonide as first-line treatment in patients with autoimmune hepatitis seems inferior to standard predniso(lo)ne administration. Hepatology 2023, 77, 1095–1105. [Google Scholar] [CrossRef]
- Mack, C.L.; Adams, D.; Assis, D.N.; Kerkar, N.; Manns, M.P.; Mayo, M.J.; Vierling, J.M.; Alsawas, M.; Murad, M.H.; Czaja, A.J. Diagnosis and Management of Autoimmune Hepatitis in Adults and Children: 2019 Practice Guidance and Guidelines from the American Association for the Study of Liver Diseases. Hepatology 2020, 72, 671–722. [Google Scholar] [CrossRef] [PubMed]
- Bacon, B.R.; Treuhaft, W.H.; Goodman, A.M. Azathioprine-induced pancytopenia. Occurrence in two patients with connective-tissue diseases. Arch. Intern. Med. 1981, 141, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Dalekos, G.N.; Arvaniti, P.; Gatselis, N.K.; Gabeta, S.; Samakidou, A.; Giannoulis, G.; Rigopoulou, E.; Koukoulis, G.K.; Zachou, K. Long-term results of mycophenolate mofetil. JHEP Rep. 2022, 4, 100601. [Google Scholar] [CrossRef] [PubMed]
- Snijders, R.J.A.L.; Stoelinga, A.E.C.; Gevers, T.J.G.; Pape, S.; Biewenga, M.; Tushuizen, M.E.; Verdonk, R.C.; de Jonge, H.J.M.; Vrolijk, J.M.; Bakker, S.F.; et al. An open-label randomised-controlled trial of azathioprine vs. mycophenolate mofetil for the induction of remission in treatment-naive autoimmune hepatitis. J. Hepatol. 2024, 80, 576–585. [Google Scholar] [CrossRef]
- Liver, E.A.f.t.S.o.t. EASL Clinical Practice Guidelines on sclerosing cholangitis. J. Hepatol. 2022, 77, 761–806. [Google Scholar] [CrossRef]
- Poupon, R. Ursodeoxycholic acid and bile-acid mimetics as therapeutic agents for cholestatic liver diseases: An overview of their mechanisms of action. Clin. Res. Hepatol. Gastroenterol. 2012, 36 (Suppl. S1), S3–S12. [Google Scholar] [CrossRef]
- Bowlus, C.L.; Arrivé, L.; Bergquist, A.; Deneau, M.; Forman, L.; Ilyas, S.I.; Lunsford, K.E.; Martinez, M.; Sapisochin, G.; Shroff, R.; et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology 2023, 77, 659–702. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Beuers, U.; Corpechot, C.; Invernizzi, P.; Jones, D.; Marzioni, M.; Schramm, C. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J. Hepatol. 2017, 67, 145–172. [Google Scholar] [CrossRef]
- Poupon, R.E.; Lindor, K.D.; Cauch-Dudek, K.; Dickson, E.R.; Poupon, R.; Heathcote, E.J. Combined analysis of randomized controlled trials of ursodeoxycholic acid in primary biliary cirrhosis. Gastroenterology 1997, 113, 884–890. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Mason, A.; Luketic, V.; Lindor, K.; Gordon, S.C.; Mayo, M.; Kowdley, K.V.; Vincent, C.; Bodhenheimer, H.C.; Parés, A.; et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology 2015, 148, 751–761.e758. [Google Scholar] [CrossRef]
- Barreyro, F.J.; Holod, S.; Finocchietto, P.V.; Camino, A.M.; Aquino, J.B.; Avagnina, A.; Carreras, M.C.; Poderoso, J.J.; Gores, G.J. The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. 2015, 35, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Sancho, J.; Manicardi, N.; Ortega-Ribera, M.; Maeso-Díaz, R.; Guixé-Muntet, S.; Fernández-Iglesias, A.; Hide, D.; García-Calderó, H.; Boyer-Díaz, Z.; Contreras, P.C.; et al. Emricasan Ameliorates Portal Hypertension and Liver Fibrosis in Cirrhotic Rats Through a Hepatocyte-Mediated Paracrine Mechanism. Hepatol. Commun. 2019, 3, 987–1000. [Google Scholar] [CrossRef]
- Jie, Y.; Youchun, L.; Jingxiang, C. Emricasan for the treatment of liver cirrhosis: A meta-analysis of randomized controlled trials. Afr. Health Sci. 2023, 23, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Wei, G.; Huang, P.; Matta, H.; Gao, W.; An, P.; Zhao, S.; Lin, Y.; Tan, L.; Vaid, K.; et al. ASK1/ p38 axis inhibition blocks the release of mitochondrial “danger signals” from hepatocytes and suppresses progression to cirrhosis and liver cancer. Hepatology 2024, 80, 346–362. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Johnson, J.D.; Lee, J.J.; Song, J.; Matthews, M.; Hellerstein, M.K.; McWherter, C.A. Seladelpar combined with complementary therapies improves fibrosis, inflammation, and liver injury in a mouse model of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2024, 326, G120–G132. [Google Scholar] [CrossRef]
- Harrison, S.A.; Wong, V.W.; Okanoue, T.; Bzowej, N.; Vuppalanchi, R.; Younes, Z.; Kohli, A.; Sarin, S.; Caldwell, S.H.; Alkhouri, N.; et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: Results from randomized phase III STELLAR trials. J. Hepatol. 2020, 73, 26–39. [Google Scholar] [CrossRef]
- Malandris, K.; Papandreou, S.; Vasilakou, D.; Kakotrichi, P.; Sarakapina, A.; Kalopitas, G.; Karagiannis, T.; Giouleme, O.; Bekiari, E.; Liakos, A.; et al. Efficacy of pharmacologic interventions on magnetic resonance imaging biomarkers in patients with nonalcoholic fatty liver disease: Systematic review and network meta-analysis. J. Gastroenterol. Hepatol. 2024, 39, 1219–1229. [Google Scholar] [CrossRef]
- Wen, H.; Deng, H.; Yang, L.; Li, L.; Lin, J.; Zheng, P.; Bjelakovic, M.; Ji, G. Vitamin E for people with non-alcoholic fatty liver disease. Cochrane Database Syst. Rev. 2024, 10, CD015033. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Jandeleit-Dahm, K.; Szyndralewiez, C.; Török, N.J. Pre-clinical evidence of a dual NADPH oxidase 1/4 inhibitor (setanaxib) in liver, kidney and lung fibrosis. J. Cell. Mol. Med. 2023, 27, 471–481. [Google Scholar] [CrossRef]
- Invernizzi, P.; Carbone, M.; Jones, D.; Levy, C.; Little, N.; Wiesel, P.; Nevens, F.; investigators, s. Setanaxib, a first-in-class selective NADPH oxidase 1/4 inhibitor for primary biliary cholangitis: A randomized, placebo-controlled, phase 2 trial. Liver Int. 2023, 43, 1507–1522. [Google Scholar] [CrossRef]
- Han, S.; Wang, K.; Shen, J.; Xia, H.; Lu, Y.; Zhuge, A.; Li, S.; Qiu, B.; Zhang, S.; Dong, X.; et al. Probiotic. Nutrients 2023, 15, 4864. [Google Scholar] [CrossRef]
- Kim, H.J.; Jeon, H.J.; Kim, D.G.; Kim, J.Y.; Shim, J.J.; Lee, J.H. HY7207 Alleviates Hepatic Steatosis, Inflammation, and Liver Fibrosis in Mice with Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2024, 25, 9870. [Google Scholar] [CrossRef] [PubMed]
- Mohamad Nor, M.H.; Ayob, N.; Mokhtar, N.M.; Raja Ali, R.A.; Tan, G.C.; Wong, Z.; Shafiee, N.H.; Wong, Y.P.; Mustangin, M.; Nawawi, K.N.M. The Effect of Probiotics (MCP® BCMC® Strains) on Hepatic Steatosis, Small Intestinal Mucosal Immune Function, and Intestinal Barrier in Patients with Non-Alcoholic Fatty Liver Disease. Nutrients 2021, 13, 3192. [Google Scholar] [CrossRef] [PubMed]
- Scorletti, E.; Afolabi, P.R.; Miles, E.A.; Smith, D.E.; Almehmadi, A.; Alshathry, A.; Childs, C.E.; Del Fabbro, S.; Bilson, J.; Moyses, H.E.; et al. Synbiotics Alter Fecal Microbiomes, But Not Liver Fat or Fibrosis, in a Randomized Trial of Patients with Nonalcoholic Fatty Liver Disease. Gastroenterology 2020, 158, 1597–1610.e1597. [Google Scholar] [CrossRef]
- Leitner, U.; Brits, A.; Xu, D.; Patil, S.; Sun, J. Efficacy of probiotics on improvement of health outcomes in cirrhotic liver disease patients: A systematic review and meta-analysis of randomised controlled trials. Eur. J. Pharmacol. 2024, 981, 176874. [Google Scholar] [CrossRef]
- van der Heide, D.; Weiskirchen, R.; Bansal, R. Therapeutic Targeting of Hepatic Macrophages for the Treatment of Liver Diseases. Front. Immunol. 2019, 10, 2852. [Google Scholar] [CrossRef]
- Svendsen, P.; Graversen, J.H.; Etzerodt, A.; Hager, H.; Røge, R.; Grønbæk, H.; Christensen, E.I.; Møller, H.J.; Vilstrup, H.; Moestrup, S.K. Antibody-Directed Glucocorticoid Targeting to CD163 in M2-type Macrophages Attenuates Fructose-Induced Liver Inflammatory Changes. Mol. Ther. Methods Clin. Dev. 2017, 4, 50–61. [Google Scholar] [CrossRef]
- Colino, C.I.; Lanao, J.M.; Gutierrez-Millan, C. Targeting of Hepatic Macrophages by Therapeutic Nanoparticles. Front. Immunol. 2020, 11, 218. [Google Scholar] [CrossRef]
- Bartneck, M.; Scheyda, K.M.; Warzecha, K.T.; Rizzo, L.Y.; Hittatiya, K.; Luedde, T.; Storm, G.; Trautwein, C.; Lammers, T.; Tacke, F. Fluorescent cell-traceable dexamethasone-loaded liposomes for the treatment of inflammatory liver diseases. Biomaterials 2015, 37, 367–382. [Google Scholar] [CrossRef]
- Vogel, A.; Brunner, J.S.; Hajto, A.; Sharif, O.; Schabbauer, G. Lipid scavenging macrophages and inflammation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2022, 1867, 159066. [Google Scholar] [CrossRef]
- Qian, Z.; Xiong, W.; Mao, X.; Li, J. Macrophage Perspectives in Liver Diseases: Programmed Death, Related Biomarkers, and Targeted Therapy. Biomolecules 2024, 14, 700. [Google Scholar] [CrossRef] [PubMed]
- Akcora, B.; Storm, G.; Bansal, R. Inhibition of canonical WNT signaling pathway by β-catenin/CBP inhibitor ICG-001 ameliorates liver fibrosis in vivo through suppression of stromal CXCL12. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 804–818. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Fan, S.; Zhao, P.; Li, H.; Cai, C.; Li, X.; Zhou, Y.; Huang, M.; Bi, H. β-catenin/TCF4 inhibitors ICG-001 and LF3 alleviate BDL-induced liver fibrosis by suppressing LECT2 signaling. Chem. Biol. Interact. 2023, 371, 110350. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, Y.; Osawa, Y.; Ohtsuki, T.; Hayashi, Y.; Yamaji, K.; Yamane, D.; Hara, M.; Munekata, K.; Tsukiyama-Kohara, K.; Hishima, T.; et al. Selective inhibitor of Wnt/β-catenin/CBP signaling ameliorates hepatitis C virus-induced liver fibrosis in mouse model. Sci. Rep. 2017, 7, 325. [Google Scholar] [CrossRef]
- Yamaji, K.; Iwabuchi, S.; Tokunaga, Y.; Hashimoto, S.; Yamane, D.; Toyama, S.; Kono, R.; Kitab, B.; Tsukiyama-Kohara, K.; Osawa, Y.; et al. Molecular insights of a CBP/β-catenin-signaling inhibitor on nonalcoholic steatohepatitis-induced liver fibrosis and disorder. Biomed. Pharmacother. 2023, 166, 115379. [Google Scholar] [CrossRef]
- Kimura, M.; Nishikawa, K.; Osawa, Y.; Imamura, J.; Yamaji, K.; Harada, K.; Yatsuhashi, H.; Murata, K.; Miura, K.; Tanaka, A.; et al. Inhibition of CBP/β-catenin signaling ameliorated fibrosis in cholestatic liver disease. Hepatol. Commun. 2022, 6, 2732–2747. [Google Scholar] [CrossRef]
- Kimura, K.; Kanto, T.; Shimoda, S.; Harada, K.; Kimura, M.; Nishikawa, K.; Imamura, J.; Ogawa, E.; Saio, M.; Ikura, Y.; et al. Safety, tolerability, and anti-fibrotic efficacy of the CBP/β-catenin inhibitor PRI-724 in patients with hepatitis C and B virus-induced liver cirrhosis: An investigator-initiated, open-label, non-randomised, multicentre, phase 1/2a study. EBioMedicine 2022, 80, 104069. [Google Scholar] [CrossRef]
- Tian, S.Y.; Chen, S.M.; Pan, C.X.; Li, Y. FXR: Structures, biology, and drug development for NASH and fibrosis diseases. Acta Pharmacol. Sin. 2022, 43, 1120–1132. [Google Scholar] [CrossRef]
- Fiorucci, S.; Antonelli, E.; Rizzo, G.; Renga, B.; Mencarelli, A.; Riccardi, L.; Orlandi, S.; Pellicciari, R.; Morelli, A. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology 2004, 127, 1497–1512. [Google Scholar] [CrossRef]
- Úbeda, M.; Lario, M.; Muñoz, L.; Borrero, M.J.; Rodríguez-Serrano, M.; Sánchez-Díaz, A.M.; Del Campo, R.; Lledó, L.; Pastor, Ó.; García-Bermejo, L.; et al. Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J. Hepatol. 2016, 64, 1049–1057. [Google Scholar] [CrossRef]
- You, H.; Duan, W.; Li, S.; Lv, T.; Chen, S.; Lu, L.; Ma, X.; Han, Y.; Nan, Y.; Xu, X.; et al. Guidelines on the Diagnosis and Management of Primary Biliary Cholangitis (2021). J. Clin. Transl. Hepatol. 2023, 11, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.V.; Tevethia, H.V.; Arab, J.P.; Candia, R.; Premkumar, M.; Kumar, P.; Sharma, M.; Reddy, D.N.; Padaki, N.R. Efficacy and safety of obeticholic acid in liver disease-A systematic review and meta-analysis. Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101675. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Han, N.; Staatz, C.E.; Kwak, J.H.; Baek, I.H. Effect on lipid profile and clinical outcomes of obeticholic acid for the treatment of primary biliary cholangitis and metabolic dysfunction-associated steatohepatitis: A systematic review and meta-analysis. Clin. Res. Hepatol. Gastroenterol. 2023, 47, 102227. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Lopez, P.; Lawitz, E.J.; Lucas, K.J.; Loeffler, J.; Kim, W.; Goh, G.B.B.; Huang, J.F.; Serra, C.; Andreone, P.; et al. Tropifexor for nonalcoholic steatohepatitis: An adaptive, randomized, placebo-controlled phase 2a/b trial. Nat. Med. 2023, 29, 392–400. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Lucas, K.J.; Francque, S.; Abdelmalek, M.F.; Sanyal, A.J.; Ratziu, V.; Gadano, A.C.; Rinella, M.; Charlton, M.; Loomba, R.; et al. Tropifexor plus cenicriviroc combination versus monotherapy in nonalcoholic steatohepatitis: Results from the phase 2b TANDEM study. Hepatology 2023, 78, 1223–1239. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, Y.; Liu, Y.; Wang, W.; Tian, X.; Chen, S.; Lu, Y.; Du, J.; Cai, W. A nonbile acid farnesoid X receptor agonist tropifexor potently inhibits cholestatic liver injury and fibrosis by modulating the gut-liver axis. Liver Int. 2021, 41, 2117–2131. [Google Scholar] [CrossRef]
- Chianelli, D.; Rucker, P.V.; Roland, J.; Tully, D.C.; Nelson, J.; Liu, X.; Bursulaya, B.; Hernandez, E.D.; Wu, J.; Prashad, M.; et al. Nidufexor (LMB763), a Novel FXR Modulator for the Treatment of Nonalcoholic Steatohepatitis. J. Med. Chem. 2020, 63, 3868–3880. [Google Scholar] [CrossRef]
- Nies, V.J.; Sancar, G.; Liu, W.; van Zutphen, T.; Struik, D.; Yu, R.T.; Atkins, A.R.; Evans, R.M.; Jonker, J.W.; Downes, M.R. Fibroblast Growth Factor Signaling in Metabolic Regulation. Front. Endocrinol. 2015, 6, 193. [Google Scholar] [CrossRef]
- Marey, M.M.; Belal, M.; Awad, A.A.; Rabea, E.M.; Hassan, M.A.; Abbas, A.W.; Nashwan, A.J. Efficacy and safety of aldafermin in non-alcoholic steatohepatitis: A systematic review and meta-analysis of randomized controlled trials. Clin. Res. Hepatol. Gastroenterol. 2024, 48, 102357. [Google Scholar] [CrossRef]
- Staiger, H.; Keuper, M.; Berti, L.; Hrabe de Angelis, M.; Häring, H.U. Fibroblast Growth Factor 21-Metabolic Role in Mice and Men. Endocr. Rev. 2017, 38, 468–488. [Google Scholar] [CrossRef]
- Jeong, C.; Han, N.; Jeon, N.; Rhee, S.J.; Staatz, C.E.; Kim, M.S.; Baek, I.H. Efficacy and Safety of Fibroblast Growth Factor-21 Analogs for the Treatment of Metabolic Dysfunction-Associated Steatohepatitis: A Systematic Review and Meta-Analysis. Clin. Pharmacol. Ther. 2024, 116, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, W.; Ge, X. The role of heat shock proteins in the regulation of fibrotic diseases. Biomed. Pharmacother. 2021, 135, 111067. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Nagata, K. Roles of the endoplasmic reticulum-resident, collagen-specific molecular chaperone Hsp47 in vertebrate cells and human disease. J. Biol. Chem. 2019, 294, 2133–2141. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Murase, K.; Kato, J.; Kobune, M.; Sato, T.; Kawano, Y.; Takimoto, R.; Takada, K.; Miyanishi, K.; Matsunaga, T.; et al. Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat. Biotechnol. 2008, 26, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Lawitz, E.J.; Shevell, D.E.; Tirucherai, G.S.; Du, S.; Chen, W.; Kavita, U.; Coste, A.; Poordad, F.; Karsdal, M.; Nielsen, M.; et al. BMS-986263 in patients with advanced hepatic fibrosis: 36-week results from a randomized, placebo-controlled phase 2 trial. Hepatology 2022, 75, 912–923. [Google Scholar] [CrossRef]
- Perepelyuk, M.; Terajima, M.; Wang, A.Y.; Georges, P.C.; Janmey, P.A.; Yamauchi, M.; Wells, R.G. Hepatic stellate cells and portal fibroblasts are the major cellular sources of collagens and lysyl oxidases in normal liver and early after injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G605–G614. [Google Scholar] [CrossRef]
- Zhang, N.; Yang, A.; Zhang, W.; Li, H.; Xu, A.; Yan, X.; Han, Q.; Wang, B.; You, H.; Chen, W. Crosstalk of lysyl oxidase-like 1 and lysyl oxidase prolongs their half-lives and regulates liver fibrosis through Notch signal. Hepatol. Commun. 2024, 8, e0391. [Google Scholar] [CrossRef]
- Ikenaga, N.; Peng, Z.W.; Vaid, K.A.; Liu, S.B.; Yoshida, S.; Sverdlov, D.Y.; Mikels-Vigdal, A.; Smith, V.; Schuppan, D.; Popov, Y.V. Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut 2017, 66, 1697–1708. [Google Scholar] [CrossRef]
- Harrison, S.A.; Abdelmalek, M.F.; Caldwell, S.; Shiffman, M.L.; Diehl, A.M.; Ghalib, R.; Lawitz, E.J.; Rockey, D.C.; Schall, R.A.; Jia, C.; et al. Simtuzumab Is Ineffective for Patients with Bridging Fibrosis or Compensated Cirrhosis Caused by Nonalcoholic Steatohepatitis. Gastroenterology 2018, 155, 1140–1153. [Google Scholar] [CrossRef]
- Muir, A.J.; Levy, C.; Janssen, H.L.A.; Montano-Loza, A.J.; Shiffman, M.L.; Caldwell, S.; Luketic, V.; Ding, D.; Jia, C.; McColgan, B.J.; et al. Simtuzumab for Primary Sclerosing Cholangitis: Phase 2 Study Results with Insights on the Natural History of the Disease. Hepatology 2019, 69, 684–698. [Google Scholar] [CrossRef]
- Kirsch, R.; Clarkson, V.; Shephard, E.G.; Marais, D.A.; Jaffer, M.A.; Woodburne, V.E.; Kirsch, R.E.; Hall, P.e.L. Rodent nutritional model of non-alcoholic steatohepatitis: Species, strain and sex difference studies. J. Gastroenterol. Hepatol. 2003, 18, 1272–1282. [Google Scholar] [CrossRef] [PubMed]
- Stöppeler, S.; Palmes, D.; Fehr, M.; Hölzen, J.P.; Zibert, A.; Siaj, R.; Schmidt, H.H.; Spiegel, H.U.; Bahde, R. Gender and strain-specific differences in the development of steatosis in rats. Lab. Anim. 2013, 47, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.K.; Zhang, X.; Yu, J. Animal models of non-alcoholic fatty liver disease: Current perspectives and recent advances. J. Pathol. 2017, 241, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Flessa, C.M.; Nasiri-Ansari, N.; Kyrou, I.; Leca, B.M.; Lianou, M.; Chatzigeorgiou, A.; Kaltsas, G.; Kassi, E.; Randeva, H.S. Genetic and Diet-Induced Animal Models for Non-Alcoholic Fatty Liver Disease (NAFLD) Research. Int. J. Mol. Sci. 2022, 23, 15791. [Google Scholar] [CrossRef]
- Ito, M.; Suzuki, J.; Tsujioka, S.; Sasaki, M.; Gomori, A.; Shirakura, T.; Hirose, H.; Ishihara, A.; Iwaasa, H.; Kanatani, A. Longitudinal analysis of murine steatohepatitis model induced by chronic exposure to high-fat diet. Hepatol. Res. 2007, 37, 50–57. [Google Scholar] [CrossRef]
- Lee, J.; Purello, C.; Booth, S.L.; Bennett, B.; Wiley, C.D.; Korstanje, R. Chow diet in mouse aging studies: Nothing regular about it. Geroscience 2023, 45, 2079–2084. [Google Scholar] [CrossRef]
- Velázquez, K.T.; Enos, R.T.; Bader, J.E.; Sougiannis, A.T.; Carson, M.S.; Chatzistamou, I.; Carson, J.A.; Nagarkatti, P.S.; Nagarkatti, M.; Murphy, E.A. Prolonged high-fat-diet feeding promotes non-alcoholic fatty liver disease and alters gut microbiota in mice. World J. Hepatol. 2019, 11, 619–637. [Google Scholar] [CrossRef]
- Larter, C.Z.; Yeh, M.M.; Williams, J.; Bell-Anderson, K.S.; Farrell, G.C. MCD-induced steatohepatitis is associated with hepatic adiponectin resistance and adipogenic transformation of hepatocytes. J. Hepatol. 2008, 49, 407–416. [Google Scholar] [CrossRef]
- Machado, M.V.; Michelotti, G.A.; Xie, G.; Almeida Pereira, T.; Boursier, J.; Bohnic, B.; Guy, C.D.; Diehl, A.M. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS ONE 2015, 10, e0127991. [Google Scholar] [CrossRef]
- Savard, C.; Tartaglione, E.V.; Kuver, R.; Haigh, W.G.; Farrell, G.C.; Subramanian, S.; Chait, A.; Yeh, M.M.; Quinn, L.S.; Ioannou, G.N. Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology 2013, 57, 81–92. [Google Scholar] [CrossRef]
- Gautam, J.; Aggarwal, H.; Kumari, D.; Gupta, S.K.; Kumar, Y.; Dikshit, M. A methionine-choline-deficient diet induces nonalcoholic steatohepatitis and alters the lipidome, metabolome, and gut microbiome profile in the C57BL/6J mouse. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2024, 1869, 159545. [Google Scholar] [CrossRef] [PubMed]
- Ikawa-Yoshida, A.; Matsuo, S.; Kato, A.; Ohmori, Y.; Higashida, A.; Kaneko, E.; Matsumoto, M. Hepatocellular carcinoma in a mouse model fed a choline-deficient, L-amino acid-defined, high-fat diet. Int. J. Exp. Pathol. 2017, 98, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Motiño, O.; Li, S.; Lambertucci, F.; Anagnostopoulos, G.; Montégut, L.; Nogueira-Recalde, U.; Chen, H.; Maiuri, M.C.; Kroemer, G.; Martins, I. A Mouse Model of Hepatocellular Carcinoma Induced by Streptozotocin and High-Fat Diet. Methods Mol. Biol. 2024, 2769, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Lee, Y.A.; Fujiwara, N.; Ybanez, M.; Allen, B.; Martins, S.; Fiel, M.I.; Goossens, N.; Chou, H.I.; Hoshida, Y.; et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J. Hepatol. 2018, 69, 385–395. [Google Scholar] [CrossRef]
- Harris, S.E.; Poolman, T.M.; Arvaniti, A.; Cox, R.D.; Gathercole, L.L.; Tomlinson, J.W. The American lifestyle-induced obesity syndrome diet in male and female rodents recapitulates the clinical and transcriptomic features of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G345–G360. [Google Scholar] [CrossRef]
- Karimkhanloo, H.; Keenan, S.N.; Bayliss, J.; De Nardo, W.; Miotto, P.M.; Devereux, C.J.; Nie, S.; Williamson, N.A.; Ryan, A.; Watt, M.J.; et al. Mouse strain-dependent variation in metabolic associated fatty liver disease (MAFLD): A comprehensive resource tool for pre-clinical studies. Sci. Rep. 2023, 13, 4711. [Google Scholar] [CrossRef]
- Suriano, F.; Vieira-Silva, S.; Falony, G.; Roumain, M.; Paquot, A.; Pelicaen, R.; Régnier, M.; Delzenne, N.M.; Raes, J.; Muccioli, G.G.; et al. Novel insights into the genetically obese (ob/ob) and diabetic (db/db) mice: Two sides of the same coin. Microbiome 2021, 9, 147. [Google Scholar] [CrossRef]
- Oana, F.; Takeda, H.; Hayakawa, K.; Matsuzawa, A.; Akahane, S.; Isaji, M.; Akahane, M. Physiological difference between obese (fa/fa) Zucker rats and lean Zucker rats concerning adiponectin. Metabolism 2005, 54, 995–1001. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Fujita, S.; Yamagishi, A.; Shirai, T.; Maeda, Y.; Suzuki, T.; Kobayashi, K.I.; Inoue, J.; Yamamoto, Y. Brown Rice Inhibits Development of Nonalcoholic Fatty Liver Disease in Obese Zucker (fa/fa) Rats by Increasing Lipid Oxidation Via Activation of Retinoic Acid Synthesis. J. Nutr. 2021, 151, 2705–2713. [Google Scholar] [CrossRef]
- Gibbs, R.A.; Rogers, J.; Katze, M.G.; Bumgarner, R.; Weinstock, G.M.; Mardis, E.R.; Remington, K.A.; Strausberg, R.L.; Venter, J.C.; Wilson, R.K.; et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science 2007, 316, 222–234. [Google Scholar] [CrossRef]
- Qu, P.; Rom, O.; Li, K.; Jia, L.; Gao, X.; Liu, Z.; Ding, S.; Zhao, M.; Wang, H.; Chen, S.; et al. DT-109 ameliorates nonalcoholic steatohepatitis in nonhuman primates. Cell Metab. 2023, 35, 742–757.e710. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, L.; Xiao, R.; Li, Y.; Liao, S.; Zhang, Z.; Yang, W.; Liang, B. Plasma lipidomic signatures of spontaneous obese rhesus monkeys. Lipids Health Dis. 2019, 18, 8. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Liu, Y.; Shang, H.; Zhang, Y.; Ma, D.; Hou, N.; Wang, J.; Sun, X.; Peng, Y.; Pan, L.; et al. Characterization of spontaneously-developed non-alcoholic fatty liver disease in aged rhesus monkeys. Diabetol. Metab. Syndr. 2018, 10, 68. [Google Scholar] [CrossRef]
- Nagarajan, P.; Venkatesan, R.; Kumar, M.; Usmani, A.; Majumdar, S.S. Macaca radiata (bonnet monkey): A spontaneous model of nonalcoholic fatty liver disease. Liver Int. 2008, 28, 856–864. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, R.; Raab, S.; Zheng, W.; Wang, J.; Liu, N.; Zhu, T.; Xue, L.; Song, Z.; Mao, J.; et al. Rhesus macaques develop metabolic syndrome with reversible vascular dysfunction responsive to pioglitazone. Circulation 2011, 124, 77–86. [Google Scholar] [CrossRef]
- Rizzetto, M.; Hoyer, B.; Canese, M.G.; Shih, J.W.; Purcell, R.H.; Gerin, J.L. delta Agent: Association of delta antigen with hepatitis B surface antigen and RNA in serum of delta-infected chimpanzees. Proc. Natl. Acad. Sci. USA 1980, 77, 6124–6128. [Google Scholar] [CrossRef]
- Negro, F.; Shapiro, M.; Satterfield, W.C.; Gerin, J.L.; Purcell, R.H. Reappearance of hepatitis D virus (HDV) replication in chronic hepatitis B virus carrier chimpanzees rechallenged with HDV. J. Infect. Dis. 1989, 160, 567–571. [Google Scholar] [CrossRef]
- Engle, R.E.; De Battista, D.; Danoff, E.J.; Nguyen, H.; Chen, Z.; Lusso, P.; Purcell, R.H.; Farci, P. Distinct Cytokine Profiles Correlate with Disease Severity and Outcome in Longitudinal Studies of Acute Hepatitis B Virus and Hepatitis D Virus Infection in Chimpanzees. mBio 2020, 11. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Barrera, A.; Guerra, B.; Lee, H.; Lanford, R.E. Analysis of host range phenotypes of primate hepadnaviruses by in vitro infections of hepatitis D virus pseudotypes. J. Virol. 2004, 78, 5233–5243. [Google Scholar] [CrossRef]
- Lempp, F.A.; Wiedtke, E.; Qu, B.; Roques, P.; Chemin, I.; Vondran, F.W.R.; Le Grand, R.; Grimm, D.; Urban, S. Sodium taurocholate cotransporting polypeptide is the limiting host factor of hepatitis B virus infection in macaque and pig hepatocytes. Hepatology 2017, 66, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Dandri, M.; Burda, M.R.; Török, E.; Pollok, J.M.; Iwanska, A.; Sommer, G.; Rogiers, X.; Rogler, C.E.; Gupta, S.; Will, H.; et al. Repopulation of mouse liver with human hepatocytes and in vivo infection with hepatitis B virus. Hepatology 2001, 33, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Azuma, H.; Paulk, N.; Ranade, A.; Dorrell, C.; Al-Dhalimy, M.; Ellis, E.; Strom, S.; Kay, M.A.; Finegold, M.; Grompe, M. Robust expansion of human hepatocytes in Fah-/-/Rag2-/-/Il2rg-/- mice. Nat. Biotechnol. 2007, 25, 903–910. [Google Scholar] [CrossRef]
- Bissig, K.D.; Le, T.T.; Woods, N.B.; Verma, I.M. Repopulation of adult and neonatal mice with human hepatocytes: A chimeric animal model. Proc. Natl. Acad. Sci. USA 2007, 104, 20507–20511. [Google Scholar] [CrossRef]
- Mercer, D.F.; Schiller, D.E.; Elliott, J.F.; Douglas, D.N.; Hao, C.; Rinfret, A.; Addison, W.R.; Fischer, K.P.; Churchill, T.A.; Lakey, J.R.; et al. Hepatitis C virus replication in mice with chimeric human livers. Nat. Med. 2001, 7, 927–933. [Google Scholar] [CrossRef]
- Lütgehetmann, M.; Mancke, L.V.; Volz, T.; Helbig, M.; Allweiss, L.; Bornscheuer, T.; Pollok, J.M.; Lohse, A.W.; Petersen, J.; Urban, S.; et al. Humanized chimeric uPA mouse model for the study of hepatitis B and D virus interactions and preclinical drug evaluation. Hepatology 2012, 55, 685–694. [Google Scholar] [CrossRef]
- Giersch, K.; Allweiss, L.; Volz, T.; Helbig, M.; Bierwolf, J.; Lohse, A.W.; Pollok, J.M.; Petersen, J.; Dandri, M.; Lütgehetmann, M. Hepatitis Delta co-infection in humanized mice leads to pronounced induction of innate immune responses in comparison to HBV mono-infection. J. Hepatol. 2015, 63, 346–353. [Google Scholar] [CrossRef]
- Giersch, K.; Homs, M.; Volz, T.; Helbig, M.; Allweiss, L.; Lohse, A.W.; Petersen, J.; Buti, M.; Pollicino, T.; Sureau, C.; et al. Both interferon alpha and lambda can reduce all intrahepatic HDV infection markers in HBV/HDV infected humanized mice. Sci. Rep. 2017, 7, 3757. [Google Scholar] [CrossRef]
- Washburn, M.L.; Bility, M.T.; Zhang, L.; Kovalev, G.I.; Buntzman, A.; Frelinger, J.A.; Barry, W.; Ploss, A.; Rice, C.M.; Su, L. A humanized mouse model to study hepatitis C virus infection, immune response, and liver disease. Gastroenterology 2011, 140, 1334–1344. [Google Scholar] [CrossRef]
- Bility, M.T.; Cheng, L.; Zhang, Z.; Luan, Y.; Li, F.; Chi, L.; Zhang, L.; Tu, Z.; Gao, Y.; Fu, Y.; et al. Hepatitis B virus infection and immunopathogenesis in a humanized mouse model: Induction of human-specific liver fibrosis and M2-like macrophages. PLoS Pathog. 2014, 10, e1004032. [Google Scholar] [CrossRef]
- Strick-Marchand, H.; Dusséaux, M.; Darche, S.; Huntington, N.D.; Legrand, N.; Masse-Ranson, G.; Corcuff, E.; Ahodantin, J.; Weijer, K.; Spits, H.; et al. A novel mouse model for stable engraftment of a human immune system and human hepatocytes. PLoS ONE 2015, 10, e0119820. [Google Scholar] [CrossRef] [PubMed]
- Gutti, T.L.; Knibbe, J.S.; Makarov, E.; Zhang, J.; Yannam, G.R.; Gorantla, S.; Sun, Y.; Mercer, D.F.; Suemizu, H.; Wisecarver, J.L.; et al. Human hepatocytes and hematolymphoid dual reconstitution in treosulfan-conditioned uPA-NOG mice. Am. J. Pathol. 2014, 184, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.M.; Bial, J.; Tarlow, B.; Bial, G.; Jensen, B.; Greiner, D.L.; Brehm, M.A.; Grompe, M. Extensive double humanization of both liver and hematopoiesis in FRGN mice. Stem Cell Res. 2014, 13, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Berzofsky, J.A.; Pendleton, C.D.; Clerici, M.; Ahlers, J.; Lucey, D.R.; Putney, S.D.; Shearer, G.M. Construction of peptides encompassing multideterminant clusters of human immunodeficiency virus envelope to induce in vitro T cell responses in mice and humans of multiple MHC types. J. Clin. Investig. 1991, 88, 876–884. [Google Scholar] [CrossRef]
- Koboziev, I.; Jones-Hall, Y.; Valentine, J.F.; Webb, C.R.; Furr, K.L.; Grisham, M.B. Use of Humanized Mice to Study the Pathogenesis of Autoimmune and Inflammatory Diseases. Inflamm. Bowel Dis. 2015, 21, 1652–1673. [Google Scholar] [CrossRef]
- Shultz, L.D.; Brehm, M.A.; Garcia-Martinez, J.V.; Greiner, D.L. Humanized mice for immune system investigation: Progress, promise and challenges. Nat. Rev. Immunol. 2012, 12, 786–798. [Google Scholar] [CrossRef]
- Richardson, N.; Ng, S.T.H.; Wraith, D.C. Antigen-Specific Immunotherapy for Treatment of Autoimmune Liver Diseases. Front. Immunol. 2020, 11, 1586. [Google Scholar] [CrossRef]
- Cardon, A.; Conchon, S.; Renand, A. Mechanisms of autoimmune hepatitis. Curr. Opin. Gastroenterol. 2021, 37, 79–85. [Google Scholar] [CrossRef]
- Avci, E.; Abasiyanik, F. Autoimmune hepatitis after SARS-CoV-2 vaccine: New-onset or flare-up? J. Autoimmun. 2021, 125, 102745. [Google Scholar] [CrossRef]
- Mathew, M.; John, S.B.; Sebastian, J.; Ravi, M.D. COVID-19 vaccine triggered autoimmune hepatitis: Case report. Eur. J. Hosp. Pharm. 2023, 30, e27. [Google Scholar] [CrossRef]
- Sergi, C.M. COVID-19 vaccination-related autoimmune hepatitis-a perspective. Front. Pharmacol. 2023, 14, 1190367. [Google Scholar] [CrossRef] [PubMed]
- Christen, U.; Hintermann, E. Animal Models for Autoimmune Hepatitis: Are Current Models Good Enough? Front. Immunol. 2022, 13, 898615. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hao, H.; Hou, T. Concanavalin A-induced autoimmune hepatitis model in mice: Mechanisms and future outlook. Open Life Sci. 2022, 17, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Cheng, X.; Luo, Y.; Li, X. Umbilical Cord-Derived Mesenchymal Stem Cells Attenuate S100-Induced Autoimmune Hepatitis via Modulating Th1 and Th17 Cell Responses in Mice. Stem Cells Int. 2023, 2023, 9992207. [Google Scholar] [CrossRef] [PubMed]
- Limmer, A.; Sacher, T.; Alferink, J.; Kretschmar, M.; Schönrich, G.; Nichterlein, T.; Arnold, B.; Hämmerling, G.J. Failure to induce organ-specific autoimmunity by breaking of tolerance: Importance of the microenvironment. Eur. J. Immunol. 1998, 28, 2395–2406. [Google Scholar] [CrossRef]
- Kido, M.; Watanabe, N.; Okazaki, T.; Akamatsu, T.; Tanaka, J.; Saga, K.; Nishio, A.; Honjo, T.; Chiba, T. Fatal autoimmune hepatitis induced by concurrent loss of naturally arising regulatory T cells and PD-1-mediated signaling. Gastroenterology 2008, 135, 1333–1343. [Google Scholar] [CrossRef]
- Oikawa, T.; Takahashi, H.; Ishikawa, T.; Hokari, A.; Otsuki, N.; Azuma, M.; Zeniya, M.; Tajiri, H. Intrahepatic expression of the co-stimulatory molecules programmed death-1, and its ligands in autoimmune liver disease. Pathol. Int. 2007, 57, 485–492. [Google Scholar] [CrossRef]
- Mühlbauer, M.; Fleck, M.; Schütz, C.; Weiss, T.; Froh, M.; Blank, C.; Schölmerich, J.; Hellerbrand, C. PD-L1 is induced in hepatocytes by viral infection and by interferon-alpha and -gamma and mediates T cell apoptosis. J. Hepatol. 2006, 45, 520–528. [Google Scholar] [CrossRef]
- Bayer, E.M.; Herr, W.; Kanzler, S.; Waldmann, C.; Meyer Zum Büschenfelde, K.H.; Dienes, H.P.; Lohse, A.W. Transforming growth factor-beta1 in autoimmune hepatitis: Correlation of liver tissue expression and serum levels with disease activity. J. Hepatol. 1998, 28, 803–811. [Google Scholar] [CrossRef]
- Mieli-Vergani, G.; Vergani, D.; Czaja, A.J.; Manns, M.P.; Krawitt, E.L.; Vierling, J.M.; Lohse, A.W.; Montano-Loza, A.J. Autoimmune hepatitis. Nat. Rev. Dis. Primers 2018, 4, 18017. [Google Scholar] [CrossRef]
- Yousefi, A.; Zare Bidoki, A.; Shafioyoun, A.; Sadr, M.; Varzaneh, F.N.; Shabani, M.; Motamed, F.; Farahmand, F.; Khodadad, A.; Fallahi, G.; et al. Association of IL-10 and TGF-beta cytokine gene polymorphisms with autoimmune hepatitis. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Guan, Y.; Han, C.; Zhang, Y.; Liu, Q.; Wei, W.; Ma, Y. The pathogenesis, models and therapeutic advances of primary biliary cholangitis. Biomed. Pharmacother. 2021, 140, 111754. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.H.; Ridgway, W.M.; Ueno, Y.; Gershwin, M.E. Animal models of primary biliary cirrhosis. Clin. Liver Dis. 2008, 12, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Arechederra, M.; Fernandez-Barrena, M.; Berasain, C.; Avila, M. Mouse models of primary sclerosing cholangitis: We just can’t get enough. Metab. Target. Organ. Damage 2024, 4, 44. [Google Scholar] [CrossRef]
- Goetz, M.; Lehr, H.A.; Neurath, M.F.; Galle, P.R.; Orth, T. Long-term evaluation of a rat model of chronic cholangitis resembling human primary sclerosing cholangitis. Scand. J. Immunol. 2003, 58, 533–540. [Google Scholar] [CrossRef]
- Fagoonee, S.; Arigoni, M.; Manco, M.; Olivero, M.; Bizzaro, F.; Magagnotti, C.; Andolfo, A.; Miniscalco, B.; Forni, M.; Todeschi, S.; et al. Circulating Extracellular Vesicles Contain Liver-Derived RNA Species as Indicators of Severe Cholestasis-Induced Early Liver Fibrosis in Mice. Antioxid. Redox Signal. 2022, 36, 480–504. [Google Scholar] [CrossRef]
- Pellicano, R.; Ménard, A.; Rizzetto, M.; Mégraud, F. Helicobacter species and liver diseases: Association or causation? Lancet Infect. Dis. 2008, 8, 254–260. [Google Scholar] [CrossRef]
- Verdon, R.; Polianski, J.; Grodet, A.; Garry, L.; Carbon, C. Cryptosporidium parvum biliary tract infection in adult immunocompetent and immunosuppressed mice. J. Med. Microbiol. 1998, 47, 71–77. [Google Scholar] [CrossRef]
- Lukasova, M.; Weinberger, K.; Weiskirchen, R.; Stremmel, W. Onion-skin type of periductular sclerosis in mice with genetic deletion of biliary kindlin-2 as tight junction stabilizer: A pilot experiment indicating a primary sclerosing cholangitis (PSC) phenotype. Metab. Target. Organ. Damage 2024, 4, 36. [Google Scholar] [CrossRef]
- Matsui, T.; Shinozawa, T. Human Organoids for Predictive Toxicology Research and Drug Development. Front. Genet. 2021, 12, 767621. [Google Scholar] [CrossRef]
- Marsee, A.; Roos, F.J.M.; Verstegen, M.M.A.; Gehart, H.; de Koning, E.; Lemaigre, F.; Forbes, S.J.; Peng, W.C.; Huch, M.; Takebe, T.; et al. Building consensus on definition and nomenclature of hepatic, pancreatic, and biliary organoids. Cell Stem Cell 2021, 28, 816–832. [Google Scholar] [CrossRef] [PubMed]
- Sampaziotis, F.; Muraro, D.; Tysoe, O.C.; Sawiak, S.; Beach, T.E.; Godfrey, E.M.; Upponi, S.S.; Brevini, T.; Wesley, B.T.; Garcia-Bernardo, J.; et al. Cholangiocyte organoids can repair bile ducts after transplantation in the human liver. Science 2021, 371, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Tysoe, O.C.; Justin, A.W.; Brevini, T.; Chen, S.E.; Mahbubani, K.T.; Frank, A.K.; Zedira, H.; Melum, E.; Saeb-Parsy, K.; Markaki, A.E.; et al. Isolation and propagation of primary human cholangiocyte organoids for the generation of bioengineered biliary tissue. Nat. Protoc. 2019, 14, 1884–1925. [Google Scholar] [CrossRef] [PubMed]
- Sampaziotis, F.; Justin, A.W.; Tysoe, O.C.; Sawiak, S.; Godfrey, E.M.; Upponi, S.S.; Gieseck, R.L.; de Brito, M.C.; Berntsen, N.L.; Gómez-Vázquez, M.J.; et al. Reconstruction of the mouse extrahepatic biliary tree using primary human extrahepatic cholangiocyte organoids. Nat. Med. 2017, 23, 954–963. [Google Scholar] [CrossRef]
- Schutgens, F.; Clevers, H. Human Organoids: Tools for Understanding Biology and Treating Diseases. Annu. Rev. Pathol. 2020, 15, 211–234. [Google Scholar] [CrossRef]
- Günther, C.; Winner, B.; Neurath, M.F.; Stappenbeck, T.S. Organoids in gastrointestinal diseases: From experimental models to clinical translation. Gut 2022, 71, 1892–1908. [Google Scholar] [CrossRef]
- McCracken, K.W.; Catá, E.M.; Crawford, C.M.; Sinagoga, K.L.; Schumacher, M.; Rockich, B.E.; Tsai, Y.H.; Mayhew, C.N.; Spence, J.R.; Zavros, Y.; et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature 2014, 516, 400–404. [Google Scholar] [CrossRef]
- Hu, H.; Gehart, H.; Artegiani, B.; LÖpez-Iglesias, C.; Dekkers, F.; Basak, O.; van Es, J.; Chuva de Sousa Lopes, S.M.; Begthel, H.; Korving, J.; et al. Long-Term Expansion of Functional Mouse and Human Hepatocytes as 3D Organoids. Cell 2018, 175, 1591–1606.e1519. [Google Scholar] [CrossRef]
- Soroka, C.J.; Assis, D.N.; Alrabadi, L.S.; Roberts, S.; Cusack, L.; Jaffe, A.B.; Boyer, J.L. Bile-Derived Organoids From Patients With Primary Sclerosing Cholangitis Recapitulate Their Inflammatory Immune Profile. Hepatology 2019, 70, 871–882. [Google Scholar] [CrossRef]
- Zhou, Y.; Shen, J.X.; Lauschke, V.M. Comprehensive Evaluation of Organotypic and Microphysiological Liver Models for Prediction of Drug-Induced Liver Injury. Front. Pharmacol. 2019, 10, 1093. [Google Scholar] [CrossRef]
- Shinozawa, T.; Kimura, M.; Cai, Y.; Saiki, N.; Yoneyama, Y.; Ouchi, R.; Koike, H.; Maezawa, M.; Zhang, R.R.; Dunn, A.; et al. High-Fidelity Drug-Induced Liver Injury Screen Using Human Pluripotent Stem Cell-Derived Organoids. Gastroenterology 2021, 160, 831–846.e810. [Google Scholar] [CrossRef] [PubMed]
- Mun, S.J.; Ryu, J.S.; Lee, M.O.; Son, Y.S.; Oh, S.J.; Cho, H.S.; Son, M.Y.; Kim, D.S.; Kim, S.J.; Yoo, H.J.; et al. Generation of expandable human pluripotent stem cell-derived hepatocyte-like liver organoids. J. Hepatol. 2019, 71, 970–985. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Jiang, D.; Yang, Y.; Li, S.; Ding, Q. Modeling drug-induced liver injury and screening for anti-hepatofibrotic compounds using human PSC-derived organoids. Cell Regen. 2023, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Zeilinger, K.; Freyer, N.; Damm, G.; Seehofer, D.; Knöspel, F. Cell sources for in vitro human liver cell culture models. Exp. Biol. Med. (Maywood) 2016, 241, 1684–1698. [Google Scholar] [CrossRef]
- Lee, H.J.; Mun, S.J.; Jung, C.R.; Kang, H.M.; Kwon, J.E.; Ryu, J.S.; Ahn, H.S.; Kwon, O.S.; Ahn, J.; Moon, K.S.; et al. In vitro modeling of liver fibrosis with 3D co-culture system using a novel human hepatic stellate cell line. Biotechnol. Bioeng. 2023, 120, 1241–1253. [Google Scholar] [CrossRef]
- Török, G.; Erdei, Z.; Lilienberg, J.; Apáti, Á.; Homolya, L. The importance of transporters and cell polarization for the evaluation of human stem cell-derived hepatic cells. PLoS ONE 2020, 15, e0227751. [Google Scholar] [CrossRef]
- Wang, Z.; Faria, J.; van der Laan, L.J.W.; Penning, L.C.; Masereeuw, R.; Spee, B. Human Cholangiocytes Form a Polarized and Functional Bile Duct on Hollow Fiber Membranes. Front. Bioeng. Biotechnol. 2022, 10, 868857. [Google Scholar] [CrossRef]
- Dalsbecker, P.; Beck Adiels, C.; Goksör, M. Liver-on-a-chip devices: The pros and cons of complexity. Am. J. Physiol. Gastrointest. Liver Physiol. 2022, 323, G188–G204. [Google Scholar] [CrossRef]
- Lasli, S.; Kim, H.J.; Lee, K.; Suurmond, C.E.; Goudie, M.; Bandaru, P.; Sun, W.; Zhang, S.; Zhang, N.; Ahadian, S.; et al. A Human Liver-on-a-Chip Platform for Modeling Nonalcoholic Fatty Liver Disease. Adv. Biosyst. 2019, 3, e1900104. [Google Scholar] [CrossRef]
- Cast, A.; Kumbaji, M.; D’Souza, A.; Rodriguez, K.; Gupta, A.; Karns, R.; Timchenko, L.; Timchenko, N. Liver Proliferation Is an Essential Driver of Fibrosis in Mouse Models of Nonalcoholic Fatty Liver Disease. Hepatol. Commun. 2019, 3, 1036–1049. [Google Scholar] [CrossRef]
- Hansen, H.H.; Feigh, M.; Veidal, S.S.; Rigbolt, K.T.; Vrang, N.; Fosgerau, K. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov. Today 2017, 22, 1707–1718. [Google Scholar] [CrossRef] [PubMed]
- Paradiso, A.; Volpi, M.; Rinoldi, C.; Celikkin, N.; Contessi Negrini, N.; Bilgen, M.; Dallera, G.; Pierini, F.; Costantini, M.; Święszkowski, W.; et al. functional models for human liver diseases and drug screening: Beyond animal testing. Biomater. Sci. 2023, 11, 2988–3015. [Google Scholar] [CrossRef] [PubMed]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Animal Models of Fibrosis in Nonalcoholic Steatohepatitis: Do They Reflect Human Disease? Adv. Nutr. 2020, 11, 1696–1711. [Google Scholar] [CrossRef] [PubMed]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical models of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, R.; Togo, S.; Kimura, M.; Shinozawa, T.; Koido, M.; Koike, H.; Thompson, W.; Karns, R.A.; Mayhew, C.N.; McGrath, P.S.; et al. Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metab. 2019, 30, 374–384.e376. [Google Scholar] [CrossRef]
- McCarron, S.; Bathon, B.; Conlon, D.M.; Abbey, D.; Rader, D.J.; Gawronski, K.; Brown, C.D.; Olthoff, K.M.; Shaked, A.; Raabe, T.D. Functional Characterization of Organoids Derived from Irreversibly Damaged Liver of Patients with NASH. Hepatology 2021, 74, 1825–1844. [Google Scholar] [CrossRef]
- Duriez, M.; Jacquet, A.; Hoet, L.; Roche, S.; Bock, M.D.; Rocher, C.; Haussy, G.; Vigé, X.; Bocskei, Z.; Slavnic, T.; et al. A 3D Human Liver Model of Nonalcoholic Steatohepatitis. J. Clin. Transl. Hepatol. 2020, 8, 359–370. [Google Scholar] [CrossRef]
- Sampaziotis, F.; de Brito, M.C.; Madrigal, P.; Bertero, A.; Saeb-Parsy, K.; Soares, F.A.C.; Schrumpf, E.; Melum, E.; Karlsen, T.H.; Bradley, J.A.; et al. Cholangiocytes derived from human induced pluripotent stem cells for disease modeling and drug validation. Nat. Biotechnol. 2015, 33, 845–852. [Google Scholar] [CrossRef]
- Rimland, C.A.; Tilson, S.G.; Morell, C.M.; Tomaz, R.A.; Lu, W.Y.; Adams, S.E.; Georgakopoulos, N.; Otaizo-Carrasquero, F.; Myers, T.G.; Ferdinand, J.R.; et al. Regional Differences in Human Biliary Tissues and Corresponding In Vitro-Derived Organoids. Hepatology 2021, 73, 247–267. [Google Scholar] [CrossRef]
- Loarca, L.; De Assuncao, T.M.; Jalan-Sakrikar, N.; Bronk, S.; Krishnan, A.; Huang, B.; Morton, L.; Trussoni, C.; Bonilla, L.M.; Krueger, E.; et al. Development and characterization of cholangioids from normal and diseased human cholangiocytes as an in vitro model to study primary sclerosing cholangitis. Lab. Investig. 2017, 97, 1385–1396. [Google Scholar] [CrossRef]
- Willemse, J.; van der Laan, L.J.W.; de Jonge, J.; Verstegen, M.M.A. Design by Nature: Emerging Applications of Native Liver Extracellular Matrix for Cholangiocyte Organoid-Based Regenerative Medicine. Bioengineering 2022, 9, 110. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Gehart, H.; van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.; Ellis, E.; van Wenum, M.; Fuchs, S.A.; de Ligt, J.; et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jin, Y.; Guo, Y.; Tan, Z.; Zhang, X.; Ye, D.; Yu, Y.; Peng, S.; Zheng, L.; Li, J. Conversion Therapy of Intrahepatic Cholangiocarcinoma Is Associated with Improved Prognosis and Verified by a Case of Patient-Derived Organoid. Cancers 2021, 13, 1179. [Google Scholar] [CrossRef]
- Bansal, R.; Nagórniewicz, B.; Prakash, J. Clinical Advancements in the Targeted Therapies against Liver Fibrosis. Mediat. Inflamm. 2016, 2016, 7629724. [Google Scholar] [CrossRef]
- Gardin, A.; Remih, K.; Gonzales, E.; Andersson, E.R.; Strnad, P. Modern therapeutic approaches to liver-related disorders. J. Hepatol. 2022, 76, 1392–1409. [Google Scholar] [CrossRef]
- Dana, H.; Chalbatani, G.M.; Mahmoodzadeh, H.; Karimloo, R.; Rezaiean, O.; Moradzadeh, A.; Mehmandoost, N.; Moazzen, F.; Mazraeh, A.; Marmari, V.; et al. Molecular Mechanisms and Biological Functions of siRNA. Int. J. Biomed. Sci. 2017, 13, 48–57. [Google Scholar] [CrossRef]
- Arjunan, P.; Kathirvelu, D.; Mahalingam, G.; Goel, A.K.; Zacharaiah, U.G.; Srivastava, A.; Marepally, S. Lipid-nanoparticle-enabled nucleic acid therapeutics for liver disorders. Acta Pharm. Sin. B 2024, 14, 2885–2900. [Google Scholar] [CrossRef]
- Fagoonee, S.; Weiskirchen, R. MicroRNAs and RNA-Binding Protein-Based Regulation of Bone Metastasis from Hepatobiliary Cancers and Potential Therapeutic Strategies. Cells 2024, 13, 1935. [Google Scholar] [CrossRef]
- Carpi, S.; Daniele, S.; de Almeida, J.F.M.; Gabbia, D. Recent Advances in miRNA-Based Therapy for MASLD/MASH and MASH-Associated HCC. Int. J. Mol. Sci. 2024, 25, 12229. [Google Scholar] [CrossRef]
- Clarke, J.D.; Sharapova, T.; Lake, A.D.; Blomme, E.; Maher, J.; Cherrington, N.J. Circulating microRNA 122 in the methionine and choline-deficient mouse model of non-alcoholic steatohepatitis. J. Appl. Toxicol. 2014, 34, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.H.; Wang, B.; Kota, J.; Yu, J.; Costinean, S.; Kutay, H.; Yu, L.; Bai, S.; La Perle, K.; Chivukula, R.R.; et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J. Clin. Investig. 2012, 122, 2871–2883. [Google Scholar] [CrossRef] [PubMed]
- Padgett, K.A.; Lan, R.Y.; Leung, P.C.; Lleo, A.; Dawson, K.; Pfeiff, J.; Mao, T.K.; Coppel, R.L.; Ansari, A.A.; Gershwin, M.E. Primary biliary cirrhosis is associated with altered hepatic microRNA expression. J. Autoimmun. 2009, 32, 246–253. [Google Scholar] [CrossRef]
- Tan, Y.; Pan, T.; Ye, Y.; Ge, G.; Chen, L.; Wen, D.; Zou, S. Serum microRNAs as potential biomarkers of primary biliary cirrhosis. PLoS ONE 2014, 9, e111424. [Google Scholar] [CrossRef]
- Li, S.; Xiong, F.; Zhang, S.; Liu, J.; Gao, G.; Xie, J.; Wang, Y. Oligonucleotide therapies for nonalcoholic steatohepatitis. Mol. Ther. Nucleic Acids 2024, 35, 102184. [Google Scholar] [CrossRef]
- Yuan, L.; Terrrault, N.A. PNPLA3 and nonalcoholic fatty liver disease: Towards personalized medicine for fatty liver. Hepatobiliary Surg. Nutr. 2020, 9, 353–356. [Google Scholar] [CrossRef]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef]
- Su, W.; Wang, Y.; Jia, X.; Wu, W.; Li, L.; Tian, X.; Li, S.; Wang, C.; Xu, H.; Cao, J.; et al. Comparative proteomic study reveals 17β-HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2014, 111, 11437–11442. [Google Scholar] [CrossRef]
- Mak, L.Y.; Gane, E.; Schwabe, C.; Yoon, K.T.; Heo, J.; Scott, R.; Lee, J.H.; Lee, J.I.; Kweon, Y.O.; Weltman, M.; et al. A phase I/II study of ARO-HSD, an RNA interference therapeutic, for the treatment of non-alcoholic steatohepatitis. J. Hepatol. 2023, 78, 684–692. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, J.; Yang, Q.; Li, X.; Qiu, Y.; Zhang, Y.; Liu, M.; Zhu, A.J. Therapeutic siRNA targeting PLIN2 ameliorates steatosis, inflammation, and fibrosis in steatotic liver disease models. J. Lipid Res. 2024, 65, 100635. [Google Scholar] [CrossRef]
- Lopez-Yus, M.; Frendo-Cumbo, S.; Del Moral-Bergos, R.; Garcia-Sobreviela, M.P.; Bernal-Monterde, V.; Rydén, M.; Lorente-Cebrian, S.; Arbones-Mainar, J.M. CRISPR/Cas9-mediated deletion of adipocyte genes associated with NAFLD alters adipocyte lipid handling and reduces steatosis in hepatocytes in vitro. Am. J. Physiol. Cell Physiol. 2023, 325, C1178–C1189. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, D.; Brouwers, J.F.; Hamer, K.; Geurts, M.H.; Luciana, L.; Massalini, S.; López-Iglesias, C.; Peters, P.J.; Rodríguez-Colman, M.J.; Chuva de Sousa Lopes, S.; et al. Engineered human hepatocyte organoids enable CRISPR-based target discovery and drug screening for steatosis. Nat. Biotechnol. 2023, 41, 1567–1581. [Google Scholar] [CrossRef] [PubMed]
- Vogt, A.; Wohlfart, S.; Urban, S.; Mier, W. Medical Advances in Hepatitis D Therapy: Molecular Targets. Int. J. Mol. Sci. 2022, 23, 10817. [Google Scholar] [CrossRef] [PubMed]
- Roehl, I.; Seiffert, S.; Brikh, C.; Quinet, J.; Jamard, C.; Dorfler, N.; Lockridge, J.A.; Cova, L.; Vaillant, A. Nucleic Acid Polymers with Accelerated Plasma and Tissue Clearance for Chronic Hepatitis B Therapy. Mol. Ther. Nucleic Acids 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Bazinet, M.; Pântea, V.; Cebotarescu, V.; Cojuhari, L.; Jimbei, P.; Albrecht, J.; Schmid, P.; Le Gal, F.; Gordien, E.; Krawczyk, A.; et al. Safety and efficacy of REP 2139 and pegylated interferon alfa-2a for treatment-naive patients with chronic hepatitis B virus and hepatitis D virus co-infection (REP 301 and REP 301-LTF): A non-randomised, open-label, phase 2 trial. Lancet Gastroenterol. Hepatol. 2017, 2, 877–889. [Google Scholar] [CrossRef]
- Sneller, L.; Lin, C.; Price, A.; Kottilil, S.; Chua, J.V. RNA Interference Therapeutics for Chronic Hepatitis B: Progress, Challenges, and Future Prospects. Microorganisms 2024, 12, 599. [Google Scholar] [CrossRef]
- Wooddell, C.I.; Rozema, D.B.; Hossbach, M.; John, M.; Hamilton, H.L.; Chu, Q.; Hegge, J.O.; Klein, J.J.; Wakefield, D.H.; Oropeza, C.E.; et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol. Ther. 2013, 21, 973–985. [Google Scholar] [CrossRef]
- Nair, J.K.; Attarwala, H.; Sehgal, A.; Wang, Q.; Aluri, K.; Zhang, X.; Gao, M.; Liu, J.; Indrakanti, R.; Schofield, S.; et al. Impact of enhanced metabolic stability on pharmacokinetics and pharmacodynamics of GalNAc-siRNA conjugates. Nucleic Acids Res. 2017, 45, 10969–10977. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, X.; Steer, C.J.; Song, G.; Niu, J. Efficient silencing of hepatitis B virus S gene through CRISPR-mediated base editing. Hepatol. Commun. 2022, 6, 1652–1663. [Google Scholar] [CrossRef]
- Billioud, G.; Kruse, R.L.; Carrillo, M.; Whitten-Bauer, C.; Gao, D.; Kim, A.; Chen, L.; McCaleb, M.L.; Crosby, J.R.; Hamatake, R.; et al. In vivo reduction of hepatitis B virus antigenemia and viremia by antisense oligonucleotides. J. Hepatol. 2016, 64, 781–789. [Google Scholar] [CrossRef]
- Yuen, M.F.; Heo, J.; Jang, J.W.; Yoon, J.H.; Kweon, Y.O.; Park, S.J.; Tami, Y.; You, S.; Yates, P.; Tao, Y.; et al. Safety, tolerability and antiviral activity of the antisense oligonucleotide bepirovirsen in patients with chronic hepatitis B: A phase 2 randomized controlled trial. Nat. Med. 2021, 27, 1725–1734. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Hyunsoon, K.; Nie, Y.; Pandey, R.; Cortez, J.; Rajwanshi, V.; Smith, D.; Blatt, L.; Beigelman, L.; Symons, J.; et al. Combination drug interactions of hepatitis B virus (HBV) small interfering RNA (siRNA) and antisense oligonucleotides (ASO) in vitro and in vivo. J. Hepatol. 2021, 75, S720. [Google Scholar]
- Muratori, L.; Longhi, M.S. The interplay between regulatory and effector T cells in autoimmune hepatitis: Implications for innovative treatment strategies. J. Autoimmun. 2013, 46, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, M.; Sun, Q.; Cheng, S.; Chi, Y.; Zhang, J.; Wang, B.; Zhou, L.; Zhao, J. Engineering M2 type macrophage-derived exosomes for autoimmune hepatitis immunotherapy via loading siRIPK3. Biomed. Pharmacother. 2024, 171, 116161. [Google Scholar] [CrossRef] [PubMed]
- Isaac, R.; Bandyopadhyay, G.; Rohm, T.V.; Kang, S.; Wang, J.; Pokhrel, N.; Sakane, S.; Zapata, R.; Libster, A.M.; Vinik, Y.; et al. TM7SF3 controls TEAD1 splicing to prevent MASH-induced liver fibrosis. Cell Metab. 2024, 36, 1030–1043.e1037. [Google Scholar] [CrossRef]
- Lai, D.; Ho, K.C.; Hao, Y.; Yang, X. Taxol resistance in breast cancer cells is mediated by the hippo pathway component TAZ and its downstream transcriptional targets Cyr61 and CTGF. Cancer Res. 2011, 71, 2728–2738. [Google Scholar] [CrossRef]
- Lipson, K.E.; Wong, C.; Teng, Y.; Spong, S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair. 2012, 5, S24. [Google Scholar] [CrossRef]
- Mooring, M.; Yeung, G.A.; Luukkonen, P.; Liu, S.; Akbar, M.W.; Zhang, G.J.; Balogun, O.; Yu, X.; Mo, R.; Nejak-Bowen, K.; et al. Hepatocyte CYR61 polarizes profibrotic macrophages to orchestrate NASH fibrosis. Sci. Transl. Med. 2023, 15, eade3157. [Google Scholar] [CrossRef]
- Banales, J.M.; Sáez, E.; Uriz, M.; Sarvide, S.; Urribarri, A.D.; Splinter, P.; Tietz Bogert, P.S.; Bujanda, L.; Prieto, J.; Medina, J.F.; et al. Up-regulation of microRNA 506 leads to decreased Cl-/HCO3- anion exchanger 2 expression in biliary epithelium of patients with primary biliary cirrhosis. Hepatology 2012, 56, 687–697. [Google Scholar] [CrossRef]
- Erice, O.; Munoz-Garrido, P.; Vaquero, J.; Perugorria, M.J.; Fernandez-Barrena, M.G.; Saez, E.; Santos-Laso, A.; Arbelaiz, A.; Jimenez-Agüero, R.; Fernandez-Irigoyen, J.; et al. MicroRNA-506 promotes primary biliary cholangitis-like features in cholangiocytes and immune activation. Hepatology 2018, 67, 1420–1440. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, M.; Sun, B.D.; Liu, F.B.; Zhang, X.D.; Ye, L.H. MiR-506 suppresses proliferation of hepatoma cells through targeting YAP mRNA 3′UTR. Acta Pharmacol. Sin. 2014, 35, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Cardus, D.F.; Smith, M.T.; Vernaza, A.; Smith, J.L.; Del Buono, B.; Parajuli, A.; Lewis, E.G.; Mesa-Diaz, N.; Du, L. Systematic Analysis of miR-506-3p Target Genes Identified Key Mediators of Its Differentiation-Inducing Function. Genes 2024, 15, 1268. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; He, X.; Rojas, M.; Leung, P.S.C.; Gao, L. Mechanism-based target therapy in primary biliary cholangitis: Opportunities before liver cirrhosis? Front. Immunol. 2023, 14, 1184252. [Google Scholar] [CrossRef] [PubMed]
- Ahn, I.; Kang, C.S.; Han, J. Where should siRNAs go: Applicable organs for siRNA drugs. Exp. Mol. Med. 2023, 55, 1283–1292. [Google Scholar] [CrossRef]
- Lu, K.; Fan, Q.; Zou, X. Antisense oligonucleotide is a promising intervention for liver diseases. Front. Pharmacol. 2022, 13, 1061842. [Google Scholar] [CrossRef]
- Scott, L.J.; Keam, S.J. Lumasiran: First Approval. Drugs 2021, 81, 277–282. [Google Scholar] [CrossRef]
- Eom, Y.W.; Kang, S.H.; Kim, M.Y.; Lee, J.I.; Baik, S.K. Mesenchymal stem cells to treat liver diseases. Ann. Transl. Med. 2020, 8, 563. [Google Scholar] [CrossRef]
- Fagoonee, S. Stem Cell Delivery Routes: From Preclinical Models to Clinical Applications; Bentham Science Publishers: Sharjah, United Arab Emirates, 2021. [Google Scholar]
- Han, H.T.; Jin, W.L.; Li, X. Mesenchymal stem cells-based therapy in liver diseases. Mol. Biomed. 2022, 3, 23. [Google Scholar] [CrossRef]
- Hu, J.; Li, S.; Zhong, X.; Wei, Y.; Sun, Q.; Zhong, L. Human umbilical cord mesenchymal stem cells attenuate diet-induced obesity and NASH-related fibrosis in mice. Heliyon 2024, 10, e25460. [Google Scholar] [CrossRef]
- Kholodenko, I.V.; Yarygin, K.N. Hepatic Macrophages as Targets for the MSC-Based Cell Therapy in Non-Alcoholic Steatohepatitis. Biomedicines 2023, 11, 3056. [Google Scholar] [CrossRef]
- Lightner, A.L.; Dadgar, N.; Vaidya, A.; Simon, R.; Fulmer, C.; Siddiki, H.; Narayanan Menon, K.V.; Liu, P.; Matthew Walsh, R. Mesenchymal stem cells: A novel treatment option for primary sclerosing cholangitis. Cell Biol. Int. 2023, 47, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Han, Q.; Chen, H.; Wang, K.; Shan, G.L.; Kong, F.; Yang, Y.J.; Li, Y.Z.; Zhang, X.; Dong, F.; et al. Allogeneic bone marrow mesenchymal stem cell transplantation in patients with UDCA-resistant primary biliary cirrhosis. Stem Cells Dev. 2014, 23, 2482–2489. [Google Scholar] [CrossRef] [PubMed]
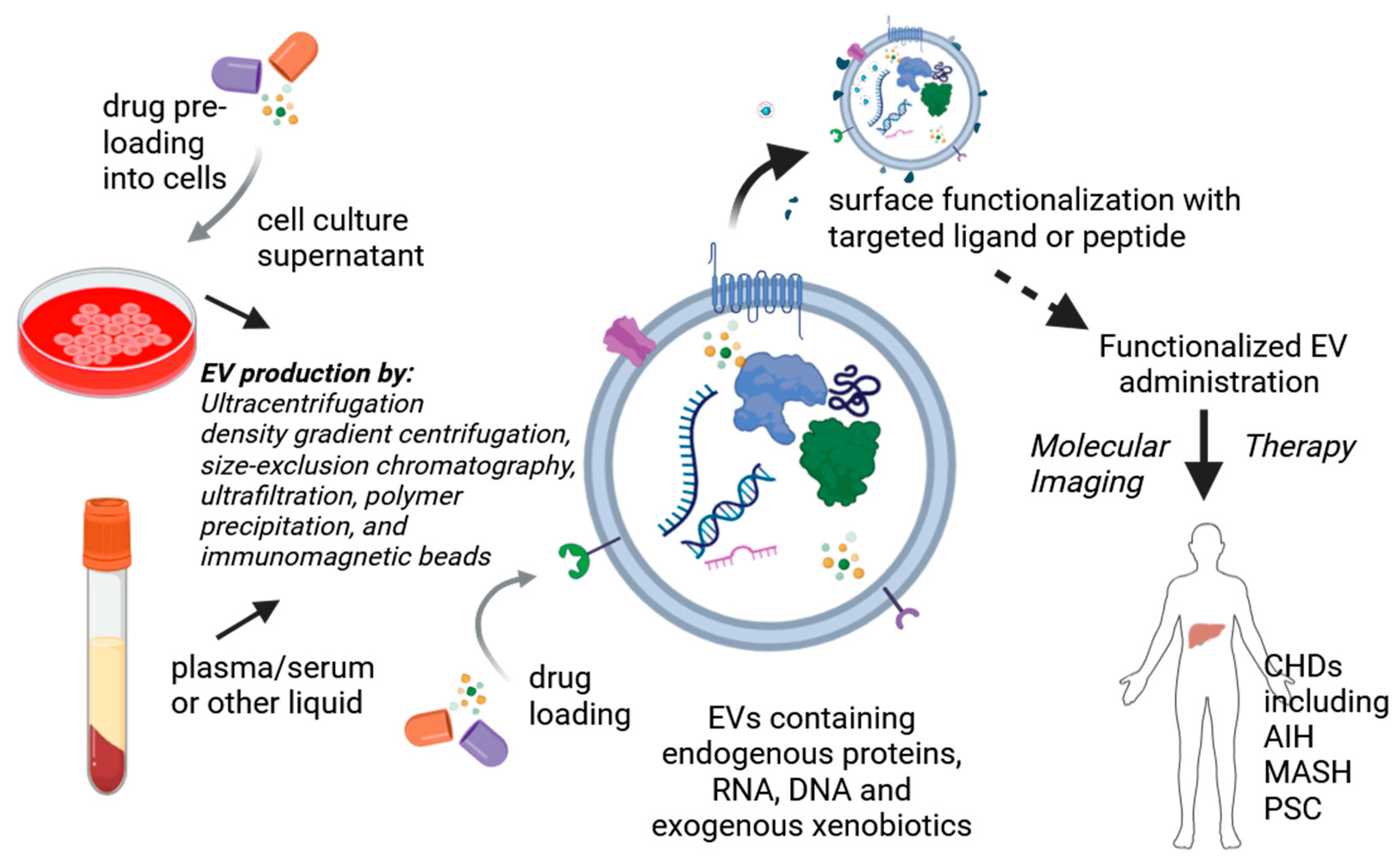
- Kumar, M.A.; Baba, S.K.; Sadida, H.Q.; Marzooqi, S.A.; Jerobin, J.; Altemani, F.H.; Algehainy, N.; Alanazi, M.A.; Abou-Samra, A.B.; Kumar, R.; et al. Extracellular vesicles as tools and targets in therapy for diseases. Signal Transduct. Target. Ther. 2024, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Greenberg, Z.F.; Serafim, M.F.; Ali, S.; Jamieson, J.C.; Traktuev, D.O.; March, K.; He, M. Understanding molecular characteristics of extracellular vesicles derived from different types of mesenchymal stem cells for therapeutic translation. Extracell. Vesicle 2024, 3, 100034. [Google Scholar] [CrossRef]
- Rosenkrans, Z.T.; Thickens, A.S.; Kink, J.A.; Aluicio-Sarduy, E.; Engle, J.W.; Hematti, P.; Hernandez, R. Investigating the In Vivo Biodistribution of Extracellular Vesicles Isolated from Various Human Cell Sources Using Positron Emission Tomography. Mol. Pharm. 2024, 21, 4324–4335. [Google Scholar] [CrossRef]
- Luo, N.; Li, J.; Dong, R.; Lu, J. Exosome-Based Theranostics for Liver Diseases. Dis. Markers 2022, 2022, 7888906. [Google Scholar] [CrossRef]
- Chen, L.; Lu, F.B.; Chen, D.Z.; Wu, J.L.; Hu, E.D.; Xu, L.M.; Zheng, M.H.; Li, H.; Huang, Y.; Jin, X.Y.; et al. BMSCs-derived miR-223-containing exosomes contribute to liver protection in experimental autoimmune hepatitis. Mol. Immunol. 2018, 93, 38–46. [Google Scholar] [CrossRef]
- Lu, F.B.; Chen, D.Z.; Chen, L.; Hu, E.D.; Wu, J.L.; Li, H.; Gong, Y.W.; Lin, Z.; Wang, X.D.; Li, J.; et al. Attenuation of Experimental Autoimmune Hepatitis in Mice with Bone Mesenchymal Stem Cell-Derived Exosomes Carrying MicroRNA-223-3p. Mol. Cells 2019, 42, 906–918. [Google Scholar] [CrossRef]
- Shi, Y.; Yang, X.; Wang, S.; Wu, Y.; Zheng, L.; Tang, Y.; Gao, Y.; Niu, J. Human umbilical cord mesenchymal stromal cell-derived exosomes protect against MCD-induced NASH in a mouse model. Stem Cell Res. Ther. 2022, 13, 517. [Google Scholar] [CrossRef]
- Angioni, R.; Calì, B.; Vigneswara, V.; Crescenzi, M.; Merino, A.; Sánchez-Rodríguez, R.; Liboni, C.; Hoogduijn, M.J.; Newsome, P.N.; Muraca, M.; et al. Administration of Human MSC-Derived Extracellular Vesicles for the Treatment of Primary Sclerosing Cholangitis: Preclinical Data in MDR2 Knockout Mice. Int. J. Mol. Sci. 2020, 21, 8874. [Google Scholar] [CrossRef]
- Zheng, L.; Gong, H.; Zhang, J.; Guo, L.; Zhai, Z.; Xia, S.; Hu, Z.; Chang, J.; Jiang, Y.; Huang, X.; et al. Strategies to improve the therapeutic efficacy of mesenchymal stem cell-derived extracellular vesicle (MSC-EV): A promising cell-free therapy for liver disease. Front. Bioeng. Biotechnol. 2023, 11, 1322514. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, P.; Yu, C.; Shi, Q.; Wei, S.; Li, Y.; Qi, H.; Cao, Q.; Guo, C.; Wu, X.; et al. Hypoxic bone marrow mesenchymal stromal cells-derived exosomal miR-182-5p promotes liver regeneration via FOXO1-mediated macrophage polarization. FASEB J. 2022, 36, e22553. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Pasquino, C.; Herrera Sanchez, M.B.; Tapparo, M.; Figliolini, F.; Grange, C.; Chiabotto, G.; Cedrino, M.; Deregibus, M.C.; Tetta, C.; et al. HLSC-Derived Extracellular Vesicles Attenuate Liver Fibrosis and Inflammation in a Murine Model of Non-alcoholic Steatohepatitis. Mol. Ther. 2020, 28, 479–489. [Google Scholar] [CrossRef]
- Athanasopoulou, F.; Manolakakis, M.; Vernia, S.; Kamaly, N. Nanodrug delivery systems for metabolic chronic liver diseases: Advances and perspectives. Nanomedicine 2023, 18, 67–84. [Google Scholar] [CrossRef]
- Ghosh, S.; Ghosh, I.; Chakrabarti, M.; Mukherjee, A. Genotoxicity and biocompatibility of superparamagnetic iron oxide nanoparticles: Influence of surface modification on biodistribution, retention, DNA damage and oxidative stress. Food Chem. Toxicol. 2020, 136, 110989. [Google Scholar] [CrossRef]
- Zhao, H.Y.; Liu, S.; He, J.; Pan, C.C.; Li, H.; Zhou, Z.Y.; Ding, Y.; Huo, D.; Hu, Y. Synthesis and application of strawberry-like Fe3O4-Au nanoparticles as CT-MR dual-modality contrast agents in accurate detection of the progressive liver disease. Biomaterials 2015, 51, 194–207. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Disease | Etiology | Characteristics | Traditional Treatment | Emerging Treatment | Future Targeted Treatment |
|---|---|---|---|---|---|
| MASLD | Dysmetabolic | Presence of liver steatosis (≥5%) +T2DM, obesity or other metabolic abnormalities | Lifestyle, GLP1-RA, Resmetirom | Emirican, Selonsertib, Vitamin D, Setanaxib, probiotics, PRI-724, OCA, Tropifexor, Aldafermin, Simtuzumab | miR-122, miR-132-3p, miR-10b-5p, miR-22-5p, miR-103a-3p, miR-107, AZD2693, ARO-HSD, SOCS3, DUSP1, SIK1 |
| HDV | Viral | Infection is possible only in HBV+ patients | PEG-IFNα, Bulevirtide | PRI-724 | REP 2139, ATI-2173, AB-729, Bepirovirsen |
| AIH | Autoimmune | Frequently associated with other immune-related disease | Prednisone and AZA | - | M2 Exos/siRIPK3 |
| PSC | Unknown | Progressive inflammation and fibrosis of biliary ducts | UDCA | Probiotics, ICG-001, PRI-724, OCA, Tropifexor, Simtuzumab | ASO 56, STP707 |
| PBC | Autoimmune | Immuno-mediated destruction of biliary ducts | UDCA, OCA | Setanaxib, ICG-001, PRI-724, Tropifexor | ASO 56, miR-506 |
| MSC Source | Disease | Type of Intervention/Results | Dose/Duration/Administration Route | Aim/Expected Outcome | Study Number/Country |
|---|---|---|---|---|---|
| Umbilical cord | AIH and PBC | Interventional/Phase 1/2/results N/A | 1 million cells/kg body weight for 12 weeks/I.V. once per 4 week | Evaluation of efficacy and safety/Liver Histology change | NCT01661842/NCT01662973 China |
| Bone marrow/allogeneic | PBC | Interventional/Phase 1/results N/A | 5–50 million cells /kg body weight/24 months if first dose functional/I.V. | Evaluate efficacy and safety of MSCs/improvement in symptoms, survival and side effects | NCT01440309/China |
| Umbilical cord | AIH and PSC | Adaptive, single arm, multi-centre, phase IIa/active | 0.5, 1.0, 2.5 million cells/kg body weight/I.V. single dose | Reduce inflammation/Dose safety and treatment activity determination | NCT02997878/UK |
| Umbilical cord | PSC | Interventional/Phase 1/2/Results N/A | N/A/Infusion at day 0, 7, 14, and 21/Intra-arterial | Evaluate adverse effects/changes in biliary lesions and inflammation | NCT03516006/China |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Durazzo, M.; Ferro, A.; Navarro-Tableros, V.M.; Gaido, A.; Fornengo, P.; Altruda, F.; Romagnoli, R.; Moestrup, S.K.; Calvo, P.L.; Fagoonee, S. Current Treatment Regimens and Promising Molecular Therapies for Chronic Hepatobiliary Diseases. Biomolecules 2025, 15, 121. https://doi.org/10.3390/biom15010121
Durazzo M, Ferro A, Navarro-Tableros VM, Gaido A, Fornengo P, Altruda F, Romagnoli R, Moestrup SK, Calvo PL, Fagoonee S. Current Treatment Regimens and Promising Molecular Therapies for Chronic Hepatobiliary Diseases. Biomolecules. 2025; 15(1):121. https://doi.org/10.3390/biom15010121
Chicago/Turabian StyleDurazzo, Marilena, Arianna Ferro, Victor Manuel Navarro-Tableros, Andrea Gaido, Paolo Fornengo, Fiorella Altruda, Renato Romagnoli, Søren K. Moestrup, Pier Luigi Calvo, and Sharmila Fagoonee. 2025. "Current Treatment Regimens and Promising Molecular Therapies for Chronic Hepatobiliary Diseases" Biomolecules 15, no. 1: 121. https://doi.org/10.3390/biom15010121
APA StyleDurazzo, M., Ferro, A., Navarro-Tableros, V. M., Gaido, A., Fornengo, P., Altruda, F., Romagnoli, R., Moestrup, S. K., Calvo, P. L., & Fagoonee, S. (2025). Current Treatment Regimens and Promising Molecular Therapies for Chronic Hepatobiliary Diseases. Biomolecules, 15(1), 121. https://doi.org/10.3390/biom15010121