1. Introduction
CLL is characterized by chronic clonal expansion of mature CD19-expressing B-lymphocytes. Recent novel targeted therapies have greatly expanded the treatment choices for many patients, such as Bruton tyrosine kinase and BCL-2 inhibitors (i.e., ibrutinib, acalabrutinib, zanubrutinib, pirtobrutinib, and venetoclax); these therapies have demonstrated potent antitumor activity with prolonged remission. However, CLL remains an incurable disease.
In this context, novel therapeutic approaches including those that potentiate the killing of tumor cells by the immune system’s cell action are needed to improve the eradication of malignant cells. The benefit of CAR-T cells has been observed in some relapsed/refractory patients with CLL [
1]. Recent studies have investigated the efficacy of autologous or allogenic NK-cell-based therapy [
2,
3,
4].
In patients with progressive CLL, NK cell dysfunction is often observed, although little is known about the mechanisms leading to this dysfunction. Reduced effector responses such as direct cytotoxicity [
5,
6] have been described, including reduced production of pro-inflammatory cytokines such as IFNγ [
7]. Various studies have identified some factors that can contribute to NK cell deficiencies. A dysregulated expression of activating/inhibitory surface receptors [
8,
9]—in particular, a decrease in the NKG2D-activating expression—has been reported [
5,
6].
Interestingly, a reduction in fully mature and cytotoxic NK cells [
10], as well as higher percentages of more immunomodulant CD56bright NK cells in patients with CLL with a higher beta 2-microglobulin level [
11], has been observed.
Several mechanisms underlying NK dysfunction are linked to the tumor microenvironment. CLL cells can produce suppressing soluble factors, such as TGF-β1, and express a series of inhibitory ligands that can directly impair NK cells [
6,
12,
13,
14]. Moreover, CLL patients show impaired regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), which suppress NK cells. In addition, novel agents may exert negative effects on the cytotoxicity, proliferation, and survival of NK cells [
14,
15].
Several cytokines, such as IL2, IL15, IL12 IL18, IL21, and their combinations, have been found to effectively stimulate in vitro NK cells in CLL patients and reverse their impairment [
12,
13,
16]. Interestingly, IL15 is not associated with the promotion of immunosuppressive regulatory T cells, in contrast to other NK-activating cytokines such as IL2, IL18, and IL21.
However, the administration of IL15 in clinical settings shows some limitations, including its short half-life and severe adverse events due to the high doses required to reach functional responses. In recent years, new formulations of IL15 and IL15-based constructs have been developed with increased half-life, strength, and potencies of biological activities in order to minimize the risk of toxicity and maximize therapeutic potential. Modifications made to the IL15 molecule include (but are not limited to) empowering mutations; conjugation to receptor α (IL15Rα, sushi domain) or other cytokines (to potentiate activity); linking to the PEG or IgG FC domain (to increase half-life); and conjugation with antibodies targeting the receptors on effectors or target cells [
17,
18,
19].
IL15 and some of its derivatives (i.e., NIZ985; ALT-803; Receptor-Linker-IL15, RLI) has been investigated in different clinical trials, with preliminary data suggesting its role as a single agent, or in cellular treatment approaches for solid and hematological malignancies, including CLL [
17,
18,
19].
A controversial issue regarding the use of IL15-based therapy for treating lymphoproliferative disorders is the pathogenic role of IL15 in these malignancies [
20]. IL15 can promote B-cell proliferation and lymphomagenesis, both as a secreted cytokine or a cytokine trans-presented by the surrounding cells in the germinal centers [
21,
22].
The objective of our work is to demonstrate that, thanks to conjugation to an antibody specific for NK cells subpopulation, the action of IL15 can be specifically targeted, reducing unwanted side effects due to the action of the cytokine on off-target cellular subpopulation or reducing the doses needed to obtain a desired effect on a specific population.
For this purpose, IL15 was conjugated with a recombinant human anti-CD56 antibody through a flexible linker, thus allowing simultaneous binding to CD56 and IL15 receptors on the same cell. Firstly, we tested this bi-functional construct on PBMCs derived from healthy donors and subsequently on PBMCs derived from patients affected by progressive CLL, as an example of a disease that induces phenotypic and functional alterations of NK cells.
We found that this construct was able to direct IL15 activity to the NK subpopulation, potentiate NK cell functionality, and restore NK-mediated cytotoxicity in CLL PBMCs, suggesting its potential application as an agent for sustaining the anti-tumoral activity of NK cells in CLL and promoting NK-cell-based immunotherapies.
2. Materials and Methods
2.1. scFvB1, CD56 Antigen, and Cell Lines
The scFvB1 antibody specific for the CD56 antigen was isolated from our IORISS naive human antibody library; its characterization has been previously described [
23,
24].
The CD56 antigen used for direct ELISA with the immunocytokine was a purified recombinant protein corresponding to the extracellular domain of human CD56 with 6xHis-tag (CD56ecd); it was produced in E. coli by Biologics International Corp, a protein manufacturer (Indianapolis, IN, USA).
Glucose oxidase (GO) antigen was purchased from Sigma-Aldrich (St. Louis, MO, USA).
NK92 is an interleukin-2 (IL-2)-dependent natural killer cell line and was a kind gift of Professor L. Moretta, who obtained the cells from the ATCC repository (ATCC; CRL-2407).
K562 is a human immortalized myelogenous leukemia cell line, which was obtained from the ATCC repository (CCL-243).
The cells were grown under standard conditions for mammalian cell cultures. The basic medium for cell culturing consisted of RPMI-1640 (EuroClone, Milan, Italy) supplemented with 10% heat-inactivated fetal bovine serum (FBS), L-glutamine (2 mM), and penicillin (100 U/mL)/streptomycin (100 U/mL). All of these components were purchased from Euroclone (Milan, Italy). The recombinant human IL-2 for NK-92 culture (50 ng/mL) and IL15 (10 ng/mL) used in some experimental procedures were purchased from Cell Guidance Systems (CellGS, Cambridge, UK).
2.2. PBMC Isolation from HD and Patient Samples
The PBMCs were purified from buffy coats obtained from the Transfusion Center of the “Sapienza” University of Rome, as waste material derived from the procedures of plasma/platelet/red blood isolation from the whole blood of HDs, who provided written informed consent. According to the Italian law (Legislative Decree of the Italian Ministry of Health of 25 January 2001, Gazzetta Ufficiale, 3 April 2001), the donor ages ranged between 21 and 60 years for women and 21 and 65 years for men. All donors were healthy, non-medicated, consenting adults who were prescreened for exposure to infectious agents.
The CLL patient blood samples were obtained from the Institute of Hematology in the Department of Translational and Precision Medicine at the University of Rome, “Sapienza” Italy, after written consent was obtained from the patients. The use of PBMCs from patients was approved by the Ethical Committee (EC) of the Istituto Superiore di Sanità and the EC of the University of Rome “Sapienza”/Azienda Policlinico Umberto I of Rome, Italy.
The patients with CLL were previously untreated and showed progressive disease.
PBMCs were isolated from the heparinized blood samples of the HDs or CLL samples via Ficoll–Hypaque density gradient centrifugation and cultured as previously described for mammalian cells.
2.3. Molecular Modeling
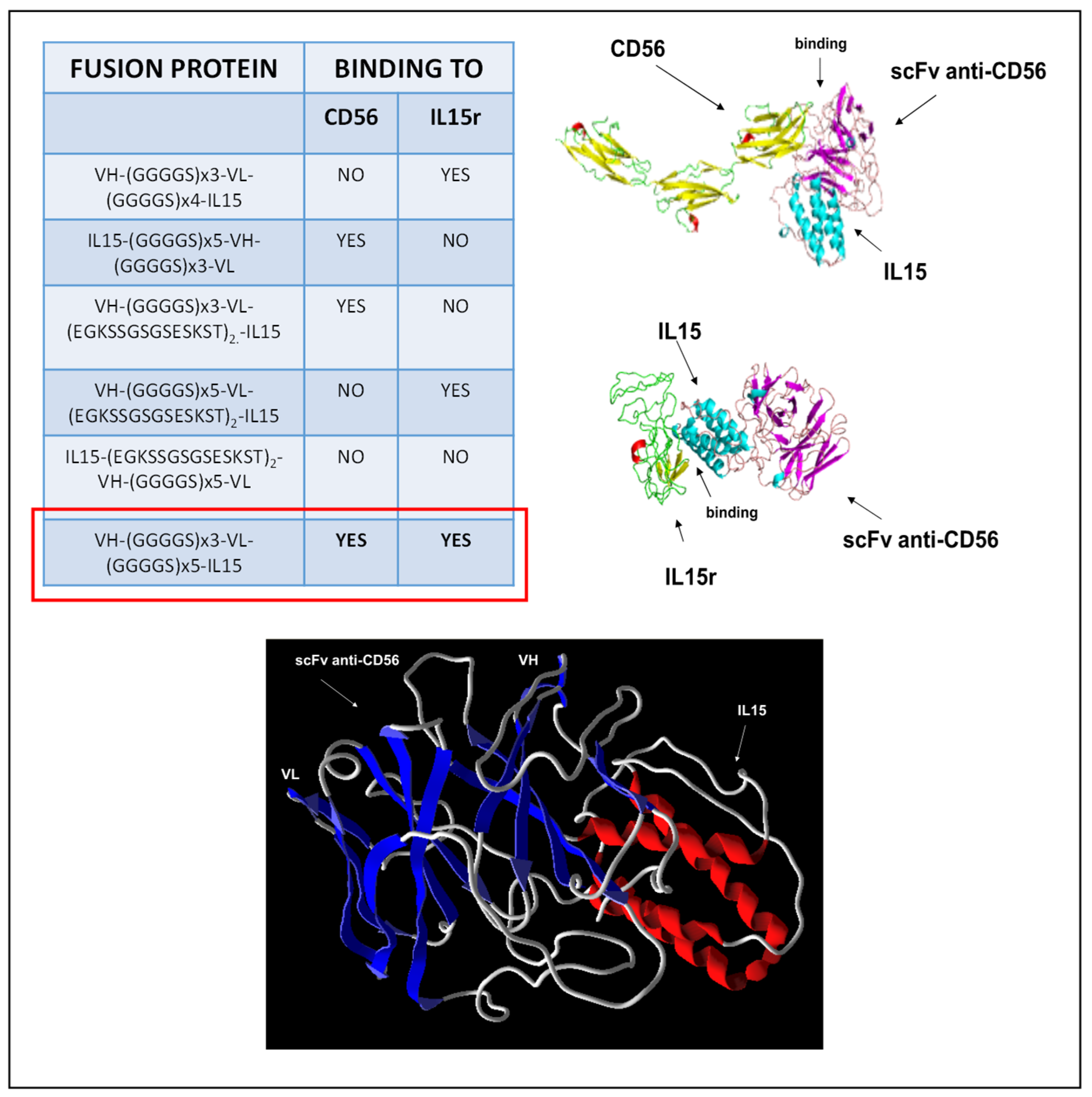
The 3D structural simulation of scFvB1IL15 and the extracellular domain of both human CD56 and IL15R structures were built with the Phyre2 Protein Fold Recognition Server [
25], and the homology model structure was retrieved for analysis. Three-dimensional models of scFvB1IL15 and the extracellular domain of CD56 or IL15R were submitted to the ClusPro protein–protein docking server [
26] to observe the most probable interaction between the two inferred 3D structures and visualized using the Molegro Molecular Viewer software for the coherent contact interface.
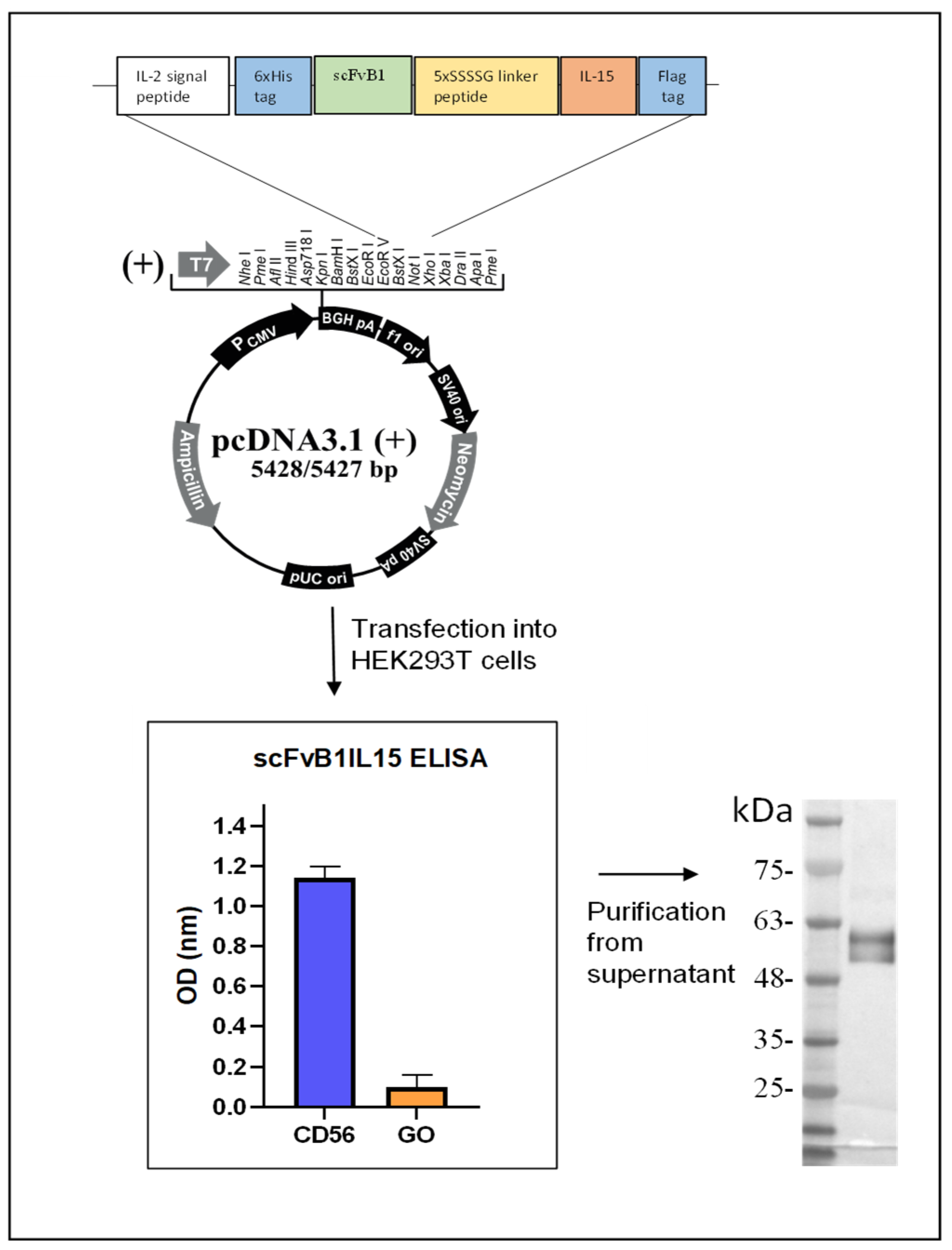
2.4. Genetic Construction, Expression, and Purification of Immunocytokine scFvB1IL15
In order to isolate IL15 cDNA, the PBMCs isolated from the HDs were placed in 24-well plates at 2 × 10
6 cells/mL in the presence of IL2 (50 ng/mL) in RPMI 20% FBS overnight (ON). On the next day, the supernatant was harvested, and adherent macrophages were activated with 100 ng/mL of LPS (Sigma, St. Louis, MO, USA) in RPMI for 24 h. Then, the macrophages were harvested and used for mRNA extraction using a QuickPrep Micro mRNA Purification Kit (Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer’s instructions. The extracted mRNA (1 μg) was reverse-transcribed for double-strand cDNA synthesis using a SmarTer PCR Synthesis kit (Clontech, Takara Bio, Saint-Germain-en-Laye, France). Then, DNA coding for IL15 was amplified by PCR from the total cDNA, while the scFvB1 coding sequence was amplified by PCR from the IORISS phagemide vector [
24] with tailored primers.
The complete immunocytokine gene (1250 bp) was synthetized using DNA recombinant and ligation techniques. The final configuration includes the 6xHis tag, scFvB1 sequence, a 25 aa (GGGGS)5 linker, the IL15 sequence, and the Flag tag. The gene was cloned in pcDNA3.1 vector at the EcoRV/XhoI restriction sites for protein expression in a eukaryotic system already modified with an IL-2 signal peptide sequence for protein secretion in the culture medium, which was kindly provided by Prof. Ferrini (IRCCS A.O.U. San Martino—IST, Istituto Nazionale per la Ricerca sul Cancro, Genova), and cloned at the HindIII/EcoRVrestriction sites.
The construct was transplanted into competent E. coli cells, and the resulting amplified and purified plasmid was used for transfection of 293LentiX cells for the transient production of the fusion protein. In brief, the cells (3.5 × 106) were transfected with 10 µg of plasmid DNA per 10 cm of the tissue culture plate using the jetPRIME® DNA Transfection Reagent (Polyplus-transfection, S.A., Illkirch, France) according to the manufacturer’s instructions. The supernatants were collected 72 h following the transfection to evaluate antibody production and binding activity using ELISA assays. His-tagged protein was purified via immobilized metal affinity chromatography using Ni2+-nitriloacetic acid agarose (Qiagen, Hilden, Germany). The ScFvB1IL15 purified protein was eluted with 250 mM imidazole in PBS 1×, dialyzed against PBS 1×, and stored at −80 °C.
2.5. Western Blotting
A total of 15 µL of the purified scFvB1IL15 was separated using 12% SDS-PAGE and then transferred to a nitrocellulose membrane. After blocking with 2% non-fat milk in PBS (MPBS), the membrane was washed and incubated for 1 h at R.T. with the monoclonal mouse anti-hIL15 antibody (cod. 500-M15, PeproTech, London, UK) in 2% MPBS; after three washings with PBS Tween 0,1%, the membrane was incubated with the secondary HRP-conjugated anti-mouse antibody (1:1000 dilution, cod. P0447, Dako, Glostrup, Denmark) in 2% MPBS. The filter was developed using the DAB (3,3′-diaminobenzidine) Color Development Solution (BioRad, Hercules, CA, USA).
2.6. ELISA Using Recombinant CD56 Protein
ELISA MaxiSorp Nunc 96-well immunoplates (Thermo Fisher Scientific, Rome, Italy) were coated ON with 50 µL/well of 10 µg/mL CD56ecd or GO irrelevant antigen in PBS at 4 °C. On the next day, a blocking solution consisting of 2% MPBS was added, and after 2 h, the plates were washed three times with PBS. The plates were incubated for 2 h at room temperature (RT) with 50 µL of the supernatants containing soluble scFvB1IL15 immunocytokine, anti-Flag M2 antibody (2.5 µg/mL, cod. F3165, Sigma, St. Louis, USA,), and HRP-conjugated anti-mouse antibody (5 μg/mL, cod. P0447, Dako, Denmark). All antibodies were re-suspended in 2% MPBS.
The reaction was visualized with 50 μL/well of 3.3′-5.5′-tetramethylbenzidine (TMB) substrate (Surmodics, Eden Prairie, MN, USA) and stopped by adding 50 µL/well of a liquid stop solution (Surmodics, USA). The reaction was detected using an ELISA reader (Model 680 microplate reader, Biorad, Hercules, CA, USA). The results are expressed as A = A (450 nm)−A (620 nm).
2.7. Determination of Binding Affinity
The determination of antibody affinity was performed via equilibrium saturation analysis using a competitive ELISA assay according to S. Rath et al. [
27], with minor modifications.
A first ELISA step was performed to determine the antibody concentration at which half of the target was present in the bound state; that is, the half maximal effective concentration (EC50). Microtiter plates (Nunc, Roskilde, Denmark) were coated with 50 μL of the CD56ecd antigen (10 μg/mL) in a PBS buffer and incubated ON at 4 °C. The microtiter plates were washed three times with 0.05% Tween PBS (TPBS) and blocked with MPBS for 2 h at RT. The microtiter plates were then washed three times with TPBS and incubated for 2 h at RT with 50 µL of the serial dilutions of scFvB1IL15 immunocytokine in MPBS, together with 10 μL of a freshly prepared mixture consisting of an anti-FLAG M2 (12.5 μg/mL, cod. F3165, Sigma-Aldrich, St. Louis, USA) and an anti-mouse HRP-conjugated antibody (22 μg/mL, cod. P0447, Dako, Germany). After the incubation period, the plates were washed three times with TPBS and three times with PBS. The color development system was added and the color intensities were measured as indicated above. The EC50 was calculated based on obtained absorbance values.
A second ELISA step was performed to determine the ratio of the concentration of free antibody at equilibrium. The wells were coated with 0.5 μg of CD56ecd protein. The day after, the serial dilutions of the same antigen were incubated in a solution with scFvB1IL15 at the determined EC50 dilution point (calculated from the first ELISA step) for 1 h to reach equilibrium.
After the blocking step, the solution with the antigen at varying concentrations and the immunocytokine was added to the designated antigen-coated wells, together with a freshly prepared mixture consisting of an anti-FLAG M2 (12.5 μg/mL, cod. F3165, Sigma-Aldrich, USA) and an anti-mouse HR-conjugated antibody (22 μg/mL, cod. P0447, Dako, Germany). The microtiter plates were rinsed thoroughly, the substrate solution applied, and then the enzyme reaction was developed and measured as described above. The unbound immunocytokine was determined based on the ELISA values; the molar inhibitor concentration required for 50% inhibition (IC50) represents an estimate of the average antibody affinity. Analysis of the results was performed using the GraphPrism program.
2.8. ELISA Sandwich for Determination of Purified Protein Concentration
We quantified the scFvB1IL15 purified protein using a sandwich ELISA, with the commercial recombinant human IL15 cytokine from Cell Guidance Systems as a standard, which has an expected specific activity of 2 × 105 units/mg. Since this cytokine was used as the positive control in all functional experiments with PBMCs from the HDs and CLL patients, this quantification method allowed for an accurate comparison of the activity of scFvB1IL15 and free IL15.
The monoclonal mouse anti-hIL15 antibody (cod. 500-M15, PeproTech, London, UK) was diluted in PBS at 10 μg/mL, added to a 96-well plate (Nunc, Roskilde, Denmark), and incubated ON as a coating. Then, the recombinant human IL15 was used as a standard ranging from 1000 to 0.5 ng/mL. After several washes, proteins were detected with 1 μg/mL of biotinylated rabbit anti-hIL15 (500-P15bt, PeproTech, London, UK) and then with HRP-conjugated streptavidin (Dako, Denmark) diluted 1:1000 in 2% MPBS. The reaction was visualized as previously described.
The obtained OD versus commercial IL15 concentrations or scFvB1IL15 dilutions were graphed, and logarithmic curves were extrapolated using GraphPad Prism 10.2.2. We chose the point at 0.3 OD in the linear portion of both curves and calculated the corresponding concentrations of IL15 and scFvB1IL15 dilution according to the equation of the extrapolated curves. Finally, the scFvB1IL15 concentration was calculated as the concentration of IL15 at the 0.3 OD/the scFvB1IL15 dilution at the same OD (
Supplementary Figure S1).
2.9. Construct Binding to Native CD56
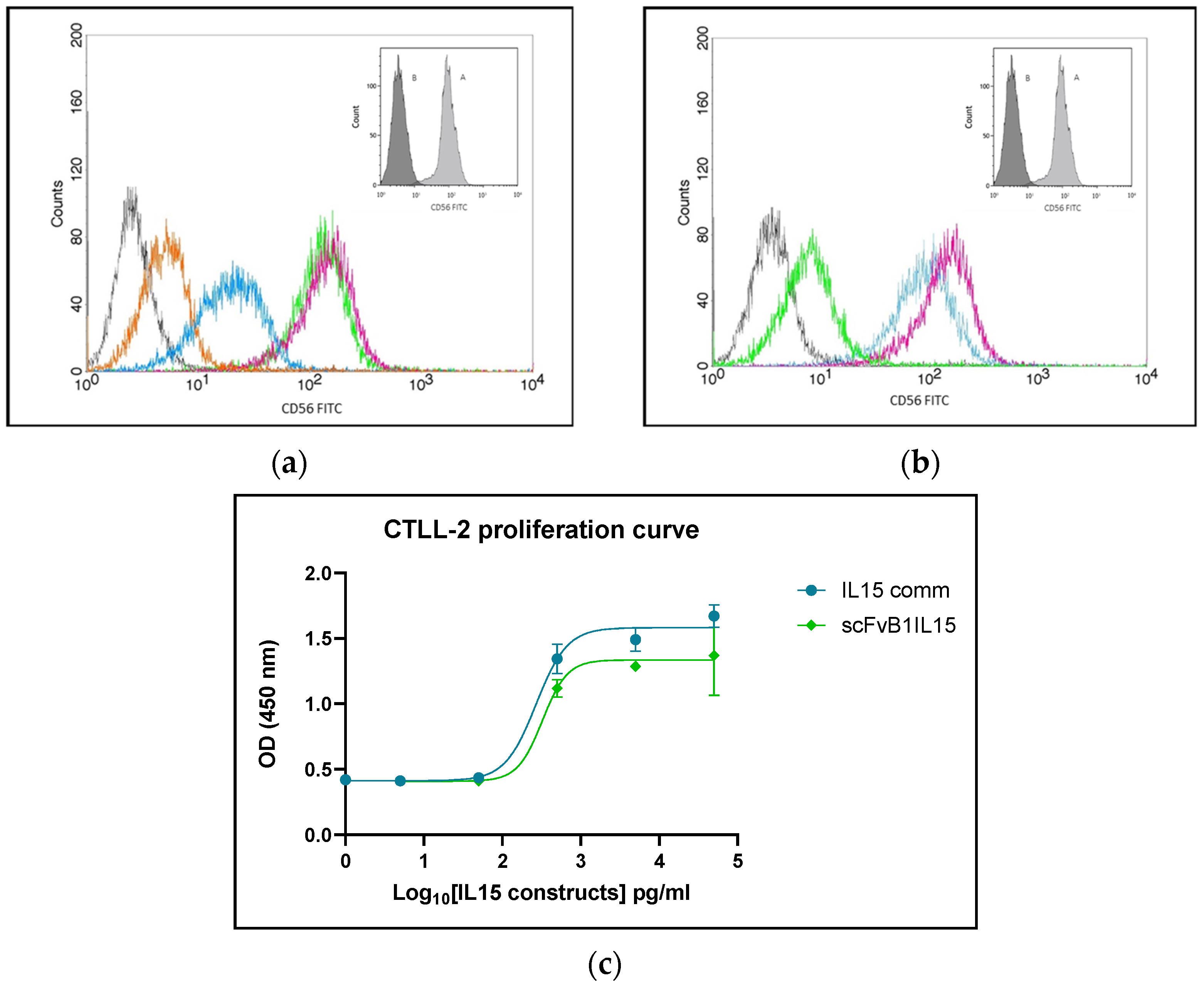
To evaluate the ability of scFvB1IL15 to still recognize CD56 on the cell surface, we performed flow cytometry binding experiments on NK92 cells.
First, cells in the exponential phase of growth were collected and then carefully washed in PBS and pelleted. About 2.5 × 105 cells were re-suspended in PBS containing different concentrations of scFvB1IL15 and incubated for 1 h at RT. After washing, the cells were re-suspended in PBS containing M15 mouse anti-hIL15 (cod. 500-M15, Peptotech, London, UK) diluted 1:1000 in PBS 1× for 30 min at 4 °C. After washing, the cells were incubated with 6 μg/mL of an FITC-goat anti-mouse IgG (cod. 31569, ThermoFischer scientific, Rome, Italy) for 30 min at 4 °C. As a positive control, cells were marked with commercial anti-CD56 FITC-conjugated antibody (Clone NCAM16.2, cod. 340410, Becton Dickinson BD, Franklin Lakes, NJ, USA) for 30 min at 4 °C. After staining, the cell samples were washed, maintained at 4 °C, and immediately analyzed. Fluorescence compensation was determined using the samples stained with anti-glucose oxidase scFv and goat FITC-conjugated anti-mouse secondary antibody.
To confirm that the binding we observed with scFvB1IL15 was specific and attributable to the antibody portion, we also performed a competition experiment where NK92 cells were pre-incubated with scFvB1 at 250 μg/mL or an irrelevant anti-glucose oxidase scFv (scFvGO) [
28] together with scFvB1IL15 at 1 μg/mL. Finally, the cells were marked with anti-hIL15 and FITC-conjugated anti-mouse secondary antibodies, as described before.
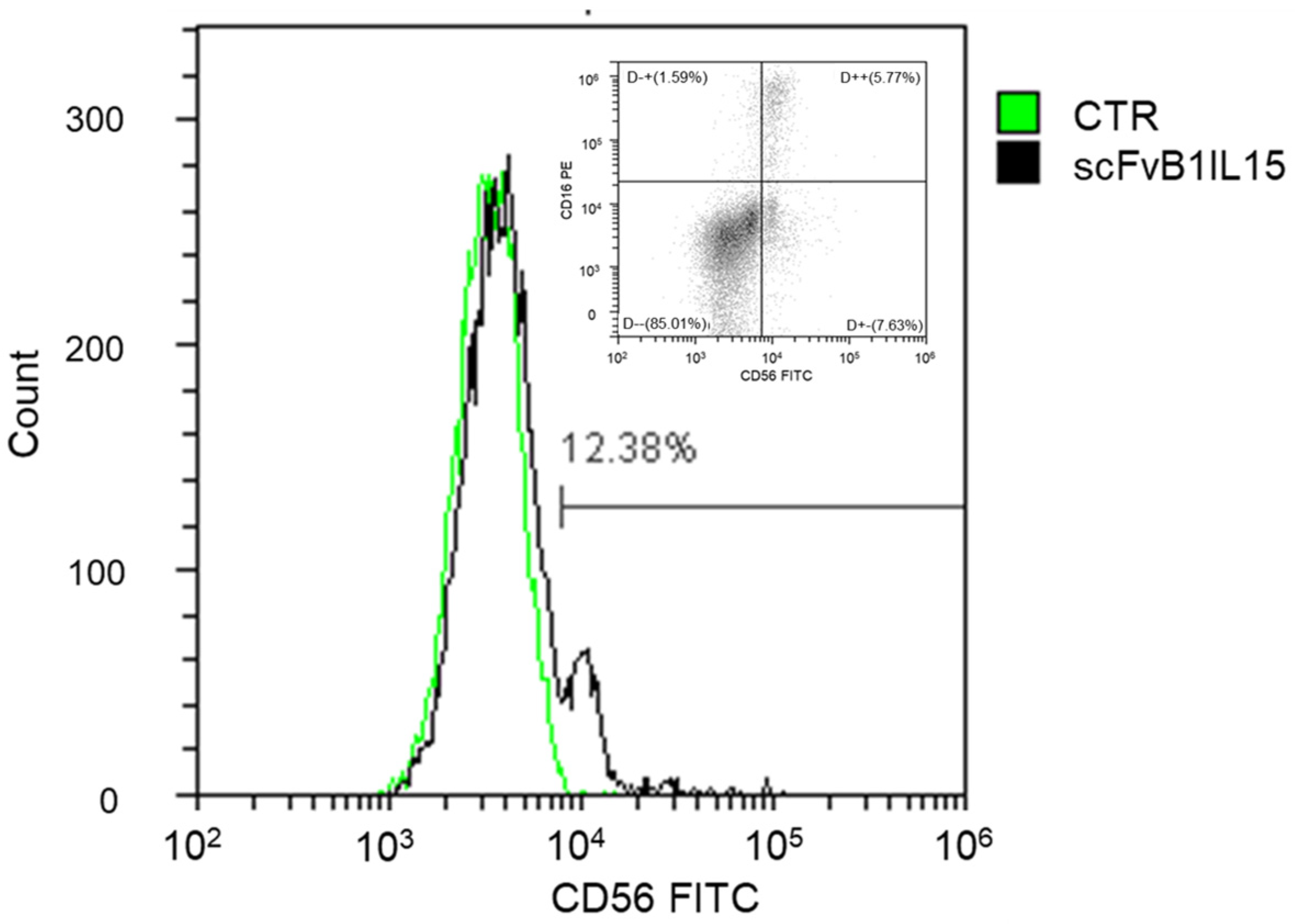
To verify that scFvB1IL15 was able to recognize NK cells in a mixed population under physiological conditions, PBMCs at 1 × 10
6 cells/sample were collected, washed with PBS, and marked with the LIVE/DEAD™ Fixable Aqua Dead Cell Stain reagent (Invitrogen, Waltham, MA, USA) in order to exclude dead cells during the FACS analysis. The cells were then washed, re-suspended in PBS containing 1 μg/mL of scFvB1IL15, and incubated for 1 h at 4 °C to avoid IL15R-mediated internalization of the construct [
29]. After washing, the cells were re-suspended in PBS containing anti-His (MCA1396, BioRad, CA, USA) diluted 1:1000 in PBS 1× for 30 min at 4 °C. After washing, the cells were incubated with 6 μg/mL of an FITC-goat anti-mouse IgG (ThermoFischer scientific, 31569) for 30 min at 4 °C. As a positive control, PBMCs were marked with commercial anti-CD16-PE (Clone B73.1, cod. 561313) and anti-CD56-FITC (Clone NCAM16.2, cod. 340410) (Becton Dickinson BD, NJ, USA) for 30 min at 4 °C. After staining, the cell samples were washed, maintained at 4 °C, and immediately analyzed.
2.10. CTLL-2 Growth Curve
The determination of cytokine functionality was performed based on the growth curve of the CTLL-2 cell line in 96-well plates and by determining the relative EC50. In particular, the cells were seeded at a density of 104 cells per well in the 96-well plates in 200 of RMPI 10% FBS, cultured in the presence of different concentrations of scFvB1IL15 or the commercial IL15 (CellGS, Cambridge, UK) ranging from 50 to 0 ng/mL, and incubated at 37 °C with 5% CO2. After 48 h, Premix WST-1 (Takara) (20 μL/well) was added, and the plates were incubated for 90 min at 37 °C with 5% CO2. Absorbance at 450 nm was measured using a Model 680 microplate reader, Biorad (CA, USA). Data analysis and EC50 calculation were carried out using the GraphPad Prism 10.2.2 program.
2.11. Intracellular Cytokine Staining
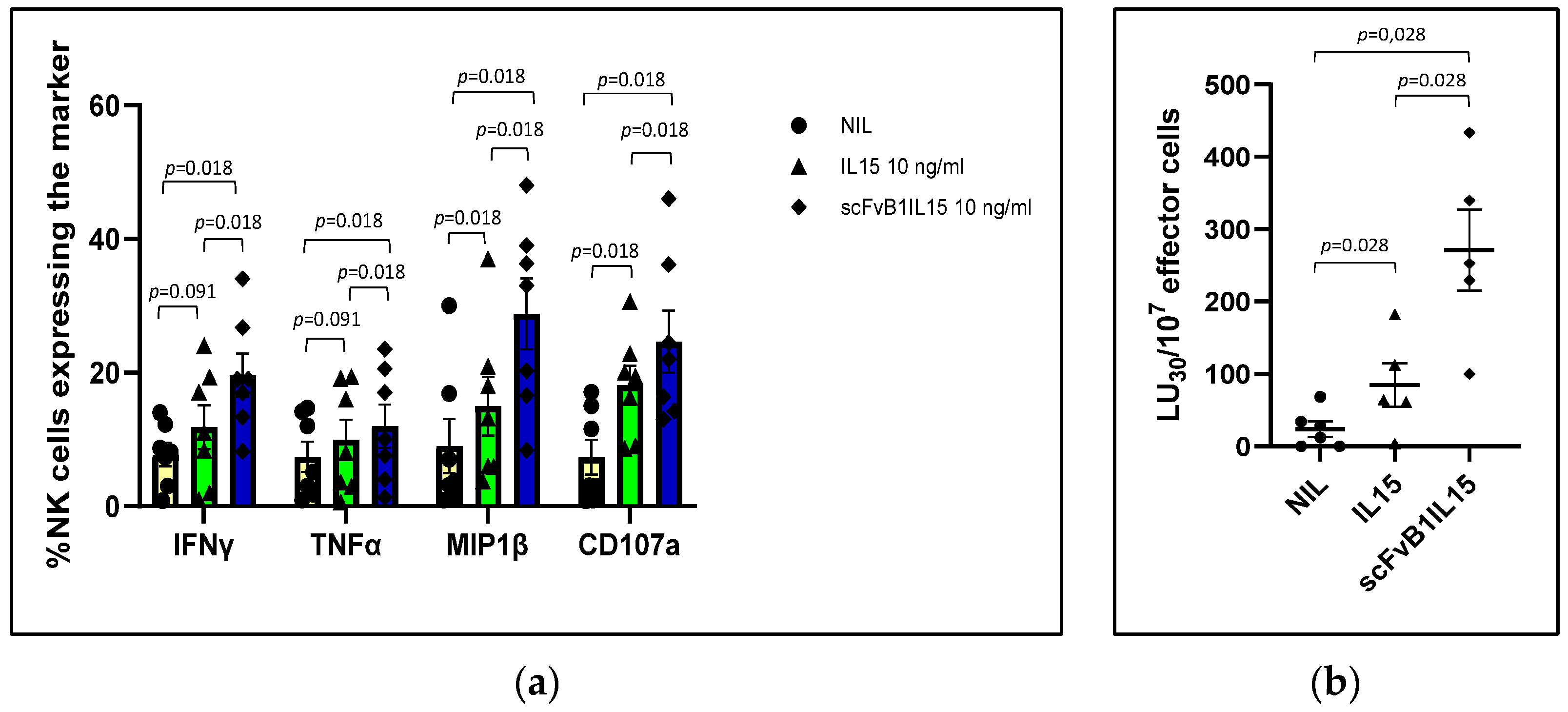
PBMCs were isolated from the HDs’ buffy-coat samples and the CLL patients’ blood samples via Lympholyte-H (Cedarlane, Burlington, ON, Canada) density centrifugation and incubated ON (37 °C, 5% CO2) at a concentration of 2 million/mL in RPMI 10% FBS without any treatment (null) or in the presence of IL15 or scFvB1IL15 at 10 ng/mL. K562 cells were marked with CFSE (Invitrogen, Waltham, USA) according to the manufacturer’s instruction and incubated ON in RPMI 10% FBS. On the next day, the PBMCs were plated at 250,000 cells/150 µL/well in 96-well plates with RPMI 10% FBS in the presence or absence of commercial IL15 or immunocytokines at 10 ng/mL (4 replicates per condition), together with CD107aPerCP-Cy5.5 (Clone H4A3, cod. 328616, Biolegend, San Diego, CA, USA). The plates were incubated for 1 h; then, 50 µL of K562 cells were added at 10,000 cells/patient and the plates were further incubated at 37 °C for 1 h. GolgiStop (1:1500, Becton Dickinson BD, Franklin Lakes, NJ, USA) and GolgiPlug (1:1000, BD Biosciences) were added, and the plates were further incubated for 4 h. After incubation, the cells were harvested, washed, and treated with the LIVE/DEAD™ Fixable Aqua Dead Cell Stain reagent (Invitrogen) in order to exclude dead cells during the FACS analysis. Then, the cells were stained for surface antigens with CD19PE (Clone HIB19, cod. 555413, BD, Franklin Lakes, NJ, USA), CD14PE (Clone M5E2, cod. 555398, BD, Franklin Lakes, NJ, USA), and CD3APC-Cy7 (Clone SK7, cod. 557832, BD, Franklin Lakes, NJ, USA) and permeabilized and stained for intracellular antigens with MIP1b-PeCy7 (Clone D21-1351, cod. 560687, BD, Franklin Lakes, NJ, USA), IFNγBV421 (Clone B27, cod. 562988, BD, Franklin Lakes, NJ, USA), and TNFαPECF594 (Clone MAb11, cod. 562784, BD, Franklin Lakes, NJ, USA). The NK population was identified excluding CD19/CD14/CD3+ cells, as direct staining with CD56 antibody was not possible due to the partial or total loss of binding after the scFvB1IL15 treatment.
2.12. NK-Specific Surface Receptor Staining
PBMCs from the HD and CLL patient samples were isolated and incubated ON (37 °C, 5% CO2) at a concentration of 2 million/mL in RPMI 10% FBS without any treatment (null) or with supplementation with IL15 (CellGS, Cambridge, UK) or scFvB1IL15 at 10 ng/mL. K562 cells were marked with CFSE (Invitrogen) according to the manufacturer’s instruction and incubated ON in RPMI 10% FBS. On the next day, the PBMCs were plated at 250,000 cells/150 µL/patient in 96-well plates with RPMI 10% FBS in the presence or absence of commercial IL15 or immunocytokines at 10 ng/mL (4 replicates per condition); then, 50 µL of the K562 cells were added at 10,000 cells/patient and the plates were incubated at 37 °C for 1 h. After incubation, the cells were harvested, washed, and treated with the LIVE/DEAD™ Fixable Aqua Dead Cell Stain reagent (Invitrogen) in order to exclude dead cells during the FACS analysis. Then, the cells were stained for surface antigens with CD19/CD14PE, CD3APC-Cy7, and NKp30 BV421 (Clone p30-15, cod. 563385, BD, Franklin Lakes, NJ, USA); NKG2D APC (Clone 1D11, cod. 558071, BD, Franklin Lakes, NJ, USA); and NKp46 PeCy7 (Clone 9E2, cod. 562101, BD, Franklin Lakes, NJ, USA). The NK population was identified excluding CD19/CD14/CD3+ cells, since direct staining with CD56 antibody was not possible because of the partial or total loss of binding after the scFvB1IL15 treatment.
2.13. Direct NK Cytotoxicity Assay
K562 cells were marked with Cell Trace Violet (CTV, Invitrogen) according to the manufacturer’s instruction and incubated ON in RPMI 10% FBS. PBMCs from the HD and CLL patient samples were isolated and incubated ON (37 °C, 5% CO2) in RPMI 10% FBS without any treatment (null) or with supplementation with IL15 or scFvB1IL15 at 10 ng/mL. On the next day, the PBMCs were co-cultured with the K562 cells at 50:1 (250,000 PBMCs/well with 5000 K562 cells/well), 25:1, 12.5:1, 6.25:1, and 3.12:1 ratios in the presence or absence of IL15 or immunocytokines at 10 ng/mL in 96-well plates (4 replicates for each condition) and incubated for 5 h.
After incubation, the cells were harvested, washed, and then re-suspended in PBS with 1 μg/mL of 7-aminoactinomycin D (7AAD, Thermo Fisher Scientific, Rome, Italy).
The gating strategy was performed as previously described [
30]. The specific lysis of target cells measured at different effector versus target ratios allows for the calculation of lytic units as LU
30/10
7 cells. The percentage of specific lysis was plotted as a function of the actual E:T ratio. A linear regression was calculated from the plot, and the equation of the trendline was used to calculate the E:T ratio required to lyse 30% of K562 cells. The lytic activity is defined as the number of lytic units contained in 10
7 effector cells: LU
30/10
7 = 10
7/T × Xp, where T is the number of target cells; p is the reference lysis level (30%); and Xp is the E:T ratio required to lyse 30% of the targets [
31].
2.14. Flow Cytometer Acquisition and Analysis
The samples from the binding experiments using NK92 cells were acquired with a FACScan (BD, Franklin Lakes, NJ, USA) equipped with a 15 mW, 488 nm argon laser.
The samples from the ex vivo experiments using the PBMCs from the HDs and CLL patients were analyzed using a Gallios flow cytometry system (Beckman Coulter, Brea, CA, United States) with multi-lasers (emitting light at 405, 488, and 640 nm) and multiple detectors. The data were processed with the Kaluza analysis software 2.1 (Beckman Coulter, Brea, CA, USA).
2.15. Statistical Analysis
Statistical analysis of the experimental results was performed using SPSS Statistics version 28. As the collected data were generally not normally distributed, we report the medians and ranges for the parameter values. Non-parametric statistical methods were used to analyze the data.
The Wilcoxon test was used for comparison between the experimental and control conditions; the Mann–Whitney test was used for comparison between the CLL patients and HDs; correlations were analyzed using Spearman’s rank correlations. Significance was assessed at the p = 0.05 level.
4. Discussion
Herein, we describe the construction and functional characterization of an immunocytokine, scFvB1IL15, designed to target NK cells through the CD56-specific scFvB1 antibody arm and activate the IL15 receptor on the same cell, with the aim of inducing targeted—and, therefore, improved—NK stimulation, as compared to free IL15.
Our results confirm that the antibody arm has the right affinity and specificity to target NK cells in a mixed population of cells, such as PBMCs, even when NK cells are present in a small percentage, as in patients with progressive CLL.
As expected, the immunocytokine was also able to activate NK cells from the HD PBMCs better than free IL15 in terms of both direct cytotoxicity and production of cytokines and chemokines, thus underlining its potential for enhancing a receptor-mediated biological action in a particular cell subpopulation. Interestingly, when purified NK cells were used in cytotoxicity assays, the difference between the immunocytokine and the free IL15 was cancelled, since in this case no targeting effect was necessary, as there was no mixed population (
Supplementary Figure S6). This fact further underlines how the enhancement of IL15 activity was not due to new or stronger functions acquired thanks to IL15 conjugation to the antibody in itself but only to a targeting effect on the specific subpopulation.
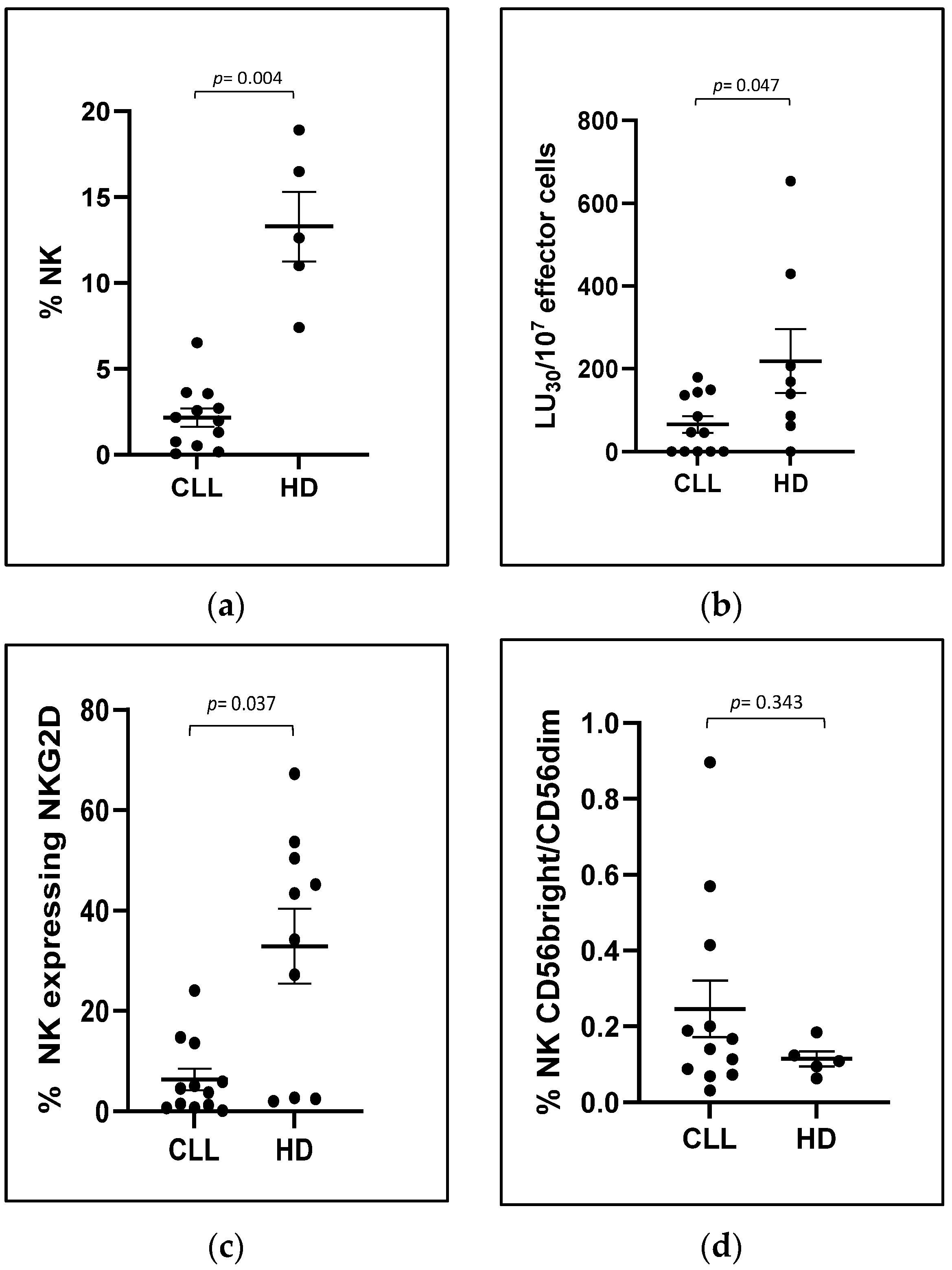
The same potentiated effect was observed on PBMCs derived from CLL patients, which, in line with previously published data, showed a dysfunctionality of CLL-NK cells with a reduction in NK-mediated direct cytotoxicity (specifically measured with K562 target population) and expression of the activating receptor NKG2D in NK cells.
In particular, when PBMCs from patients with progressive CLL were cultured with IL15 or scFvB1IL15, we observed the restoration of the direct NK cytotoxic activity to HDs levels (even exceeding them) in both conditions; however, the scFvB1IL15 immunocytokine was more highly effective in increasing the NK-mediated direct cytotoxicity in CLL patients. This observation suggests that the restoration of NK-mediated cytotoxicity in CLL patients might be obtained using a lower concentration of conjugated IL15, thus further reducing off-target effects.
The increase in cytotoxicity to HD levels (calculated considering the effective number of NK cells and E:T in the well) was not due to an NK proliferation (
Supplementary Figure S9b,c) but to a real restoration of NK cells functions.
In our experiment setting, one factor contributing to IL15-mediated restoration was the cumulative increase in NK-activating receptors. Specifically, we observed a significant increase in NKG2D expression in the NK cells from CLL patients when their PBMCs were cultured with scFvB1IL15 or IL15 compared to the non-stimulated condition. This enhancement contributed to reducing the difference in NKGD expression between the PBMCs from the HDs and CLL patients, which was no longer statistically significant after treatment. However, higher levels of NKGD expression were maintained in the PBMCs from the HDs. Interestingly, at the same time, both IL15 and scFvB1IL15 were able to increase the expression of the NKp30 receptor, another key receptor participating in NK-mediated direct cytotoxicity [
33,
34]. The correlation between the increase in the NKp30 expression and the direct cytotoxicity percentages induced by the IL15 or scFvB1IL15 treatment suggests a contribution of the NKp30 receptor in the restoration of NK-mediated cytotoxicity, when the recovery of the NKG2D receptor after both the IL15 and scFvB1IL15 treatments was not complete when compared to the HD levels. This is an interesting finding, as NKG2D expression was impaired in the CLL patients compared to the HDs at baseline while NKp30 expression was not, suggesting that compensation among different NK receptors can take place in order to restore the impaired NK function.
Another IL15-mediated effect underlying the enhancement of CLL NK functionality was the increase in other factors such as the degranulation marker CD107a along with pro-inflammatory cytokines and chemokines, such as IFNℽ, TNFα, and MIP1β [
35,
36].
Clearly, other mechanisms and factors, in addition to those explored by us, may be involved in the ability of IL15 to restore NK cell functions; nevertheless, our observations show that IL15 and scFvB1IL15 have the same mode of action, but the effects induced by scFvB1IL15 were always statistically higher than those obtained with free IL-15.
It would be interesting to establish if the potentiated activity of scFvB1IL15 could also translate into overcoming the possible intrinsic resistance of CLL cells to NK-mediated direct killing [
12,
37]; a limitation of our study design is, in fact, the absence of experiments evaluating the effect of NK activation using leukemic cells as the target, leaving this aspect unexplored.
On the other hand, using autologous CLL cells as the target instead of the K562 cell line could have introduced high variability in the obtained cytotoxic assay outcomes. Moreover, considering the need to use total PBMCs to demonstrate the potency of scFvB1-mediated targeting, the use of K562 cells allowed us to investigate the specific activation and function of NK cells and to reduce the impact of CD8 T-cell-mediated direct cytotoxicity [
38].
What we can affirm with our experiments is that the treatment with scFvB1IL15 seems to be able to overcome the immunosuppressive soluble factors that may be released by leukemic cells present in the cell cultures.
Further studies need to be carried out to evaluate the effect of the scFvB1IL15 treatment on the killing of CLL cells, alone or in combination with molecules that can overcome the intrinsic resistance of CLL cells to direct killing [
36]. Moreover, the IL15 antibody-mediated stimulation of NK cells should be further investigated in the context of CLL pathology, including evaluation of whether leukemic cells are preserved by the IL15-mediated proliferating action [
21], and in the context of CAR-NK cellular therapy for CLL.
Taken together, our results provide evidence for the ability of antibody-mediated targeting to maximize the activity of IL15 in a particular cell subset and for the efficacy of this approach in potentiating dysfunctional NK cells.
Overall, the scFvB1IL15 immunocytokine joins the group of IL15-containing con-structs developed to overcome the limitations of IL15 as a single immunotherapeutic agent that we previously mentioned in the Introduction section (
Section 1); some of these constructs are being investigated in clinical trials or are in the preclinical stages [
17,
18,
19,
39]. Some constructs enhance the activity of IL15 via conjugation to IL15Rα, an-ti-PDL1/PD1 mAb construct, or NKG2D extracellular domain (N809, anti-PD1-IL15 mutein, and dsNKG2D-IL15). Multifunctional heteromeric fusion protein complexes (HFPCs, such as HCW) containing IL12, IL15, and IL18 have also been developed to exploit the synergy of cytokines. Other constructs (HcW9107, IL15 trike) direct the activity of IL15 specifically to NK cells via conjugation to an anti-CD16 scFv. Finally, another group of constructs (anti-GD2RLI, 2B8T2M, IL15 trike) exploits the targeting mediated by a specific antibody for a tumor-associated antigen (TSA) to direct the activity of effector cells activated by the IL15/IL15Rα sushi domain complex to tumor cells.
The scFvB1IL15 immunocytokine, described in this manuscript, directs IL15 to NK cells through specific binding to the CD56 antigen and restores the NK population in CLL patients to its physiological functions in a targeted way. In this regard, we cannot exclude that, in vivo, the immunocytokine can also act on the small subset of more cytotoxic CD8+ T and NKT cells which also express CD56 and IL15R [
40,
41], but considering the percentages of CD8+ CD56+ T cells (less than 5%) and of NKT cells (less than 1%) in peripheral blood, we can state that the targeting of IL15 by an anti-CD56 antibody can greatly reduce the binding of IL15 to the large variety of cells that express IL15R and are responsive to IL15.
Additionally, being antigen-independent, scFvB1IL15 could also be explored in other disorders that involve dysfunction of cytotoxic NK cells or in adoptive transfer protocols to improve and sustain NK cell expansion and activation.




 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}