
A Cell-Based Evaluation of the Tyrosinase-Mediated Metabolic Activation of Leukoderma-Inducing Phenols, II: The Depletion of Nrf2 Augments the Cytotoxic Effect Evoked by Tyrosinase in Melanogenic Cells

 ,
,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. RNA Interference
2.4. RNA Extraction and Quantitative Real-Time RT-PCR
2.5. Cell Viability and Glutathione Level Assessment
2.6. Western Blotting
2.7. Statistical Analysis
3. Results
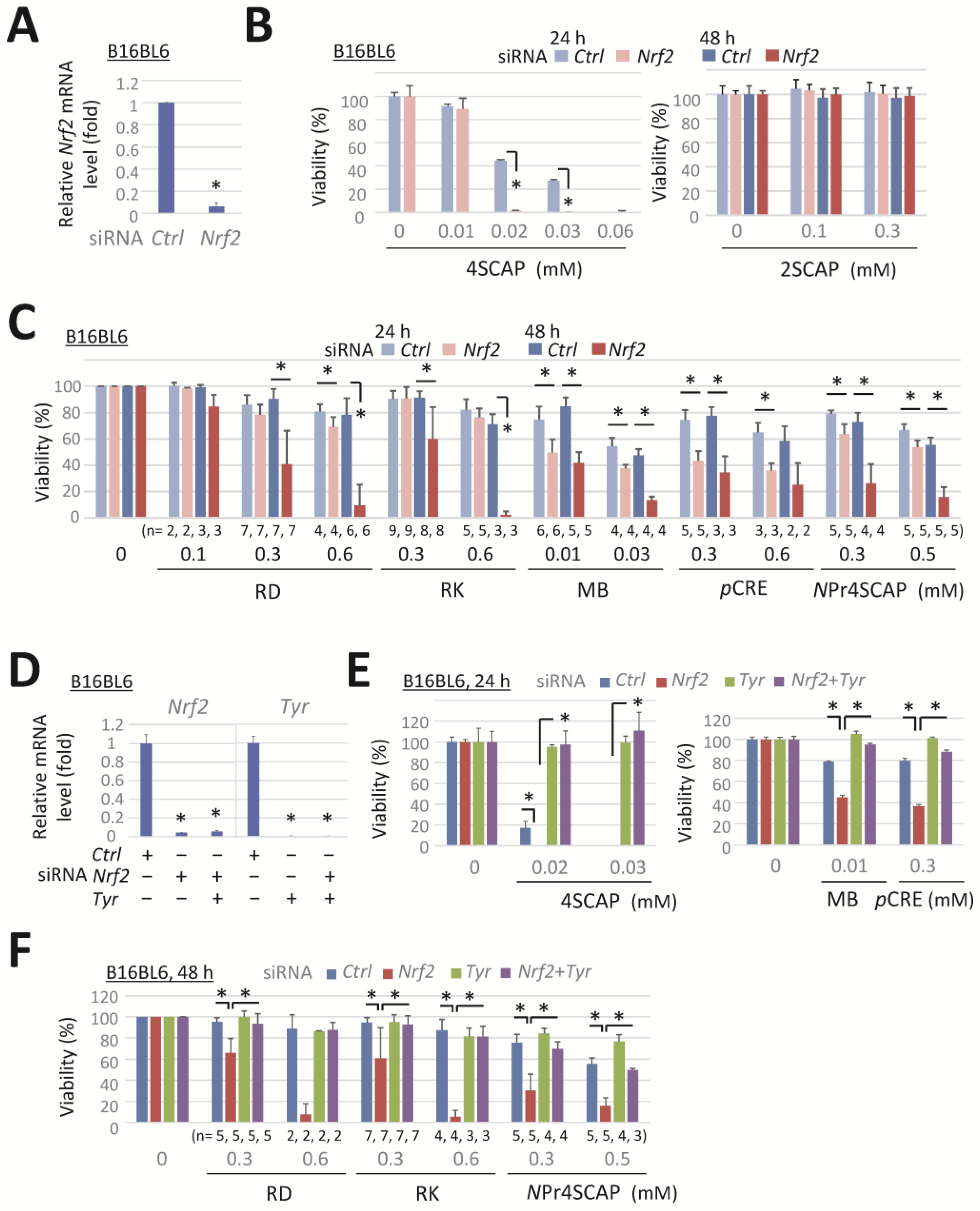
3.1. Knockdown of Nrf2 Enhances the Reduction in Viability of Melanoma Cells in a Tyrosinase-Dependent Manner Following Exposure to Leukoderma-Inducing Phenolic Compounds
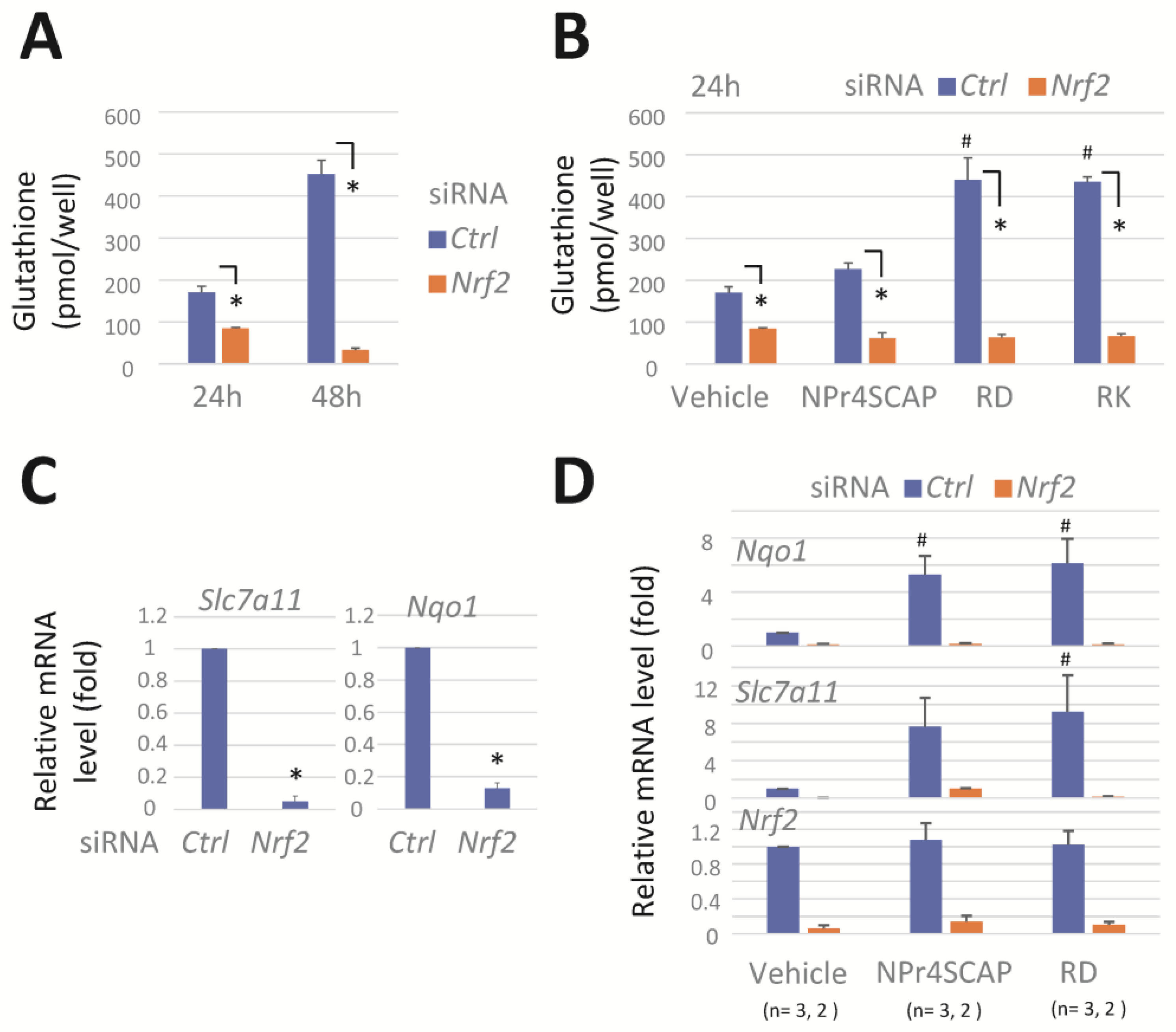
3.2. Nrf2 Knockdown Reduces GSH, Slc7a11, and Nqo1 mRNA Levels and Effectively Prevents Their Induction by Phenolic Compounds
3.3. Slc7a11 Knockdown Enhances the Cytotoxicity Induced by RD, RK, and pCRE, Whereas Nqo1 Knockdown or Inhibition Is Almost Ineffective
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oliver, E.A.; Schwartz, L.; Warren, L.H. Occupational leukoderma. J. Am. Med. Assoc. 1939, 113, 927–928. [Google Scholar] [CrossRef]
- Matsunaga, K.; Suzuki, K.; Ito, A.; Tanemura, A.; Abe, Y.; Suzuki, T.; Yoshikawa, M.; Sumikawa, Y.; Yagami, A.; Masui, Y.; et al. Rhododendrol-induced leukoderma update I: Clinical findings and treatment. J. Dermatol. 2021, 48, 961–968. [Google Scholar] [CrossRef]
- Nishigori, C.; Aoyama, Y.; Ito, A.; Suzuki, K.; Suzuki, T.; Tanemura, A.; Ito, M.; Katayama, I.; Oiso, N.; Kagohashi, Y.; et al. Guide for medical professionals (i.e., dermatologists) for the management of Rhododenol-induced leukoderma. J. Dermatol. 2015, 42, 113–128. [Google Scholar] [CrossRef]
- Fukuda, Y.; Nagano, M.; Futatsuka, M. Occupational leukoderma in workers rngaged in 4-(p-hydroxyphenyl)-2-butanone manufacturing. J. Occup. Health. 1998, 40, 118–122. [Google Scholar] [CrossRef]
- Ito, Y.; Jimbow, K.; Ito, S. Depigmentation of black guinea pig skin by topical application of cysteaminylphenol, cysteinylphenol, and related compounds. J. Investig. Dermatol. 1987, 88, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Ishii-Osai, Y.; Yamashita, T.; Tamura, Y.; Sato, N.; Ito, A.; Honda, H.; Wakamatsu, K.; Ito, S.; Nakayama, E.; Okura, M.; et al. N-propionyl-4-S-cysteaminylphenol induces apoptosis in B16F1 cells and mediates tumor-specific T-cell immune responses in a mouse melanoma model. J. Dermatol. Sci. 2012, 67, 51–60. [Google Scholar] [CrossRef]
- Tandon, M.; Thomas, P.D.; Shokravi, M.; Singh, S.; Samra, S.; Chang, D.; Jimbow, K. Synthesis and antitumour effect of the melanogenesis-based antimelanoma agent N-propionyl-4-S-cysteaminylphenol. Biochem. Pharmacol. 1998, 55, 2023–2029. [Google Scholar] [CrossRef]
- Shelley, W.B. p-Cresol: Cause of ink-induced hair depigmentation in mice. Br. J. Dermatol. 1974, 90, 169–174. [Google Scholar] [CrossRef]
- Riley, P.A. Hydroxyanisole depigmentation: In-vivo studies. J. Pathol. 1969, 97, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Boissy, R.E.; Manga, P. On the etiology of contact/occupational vitiligo. Pigment. Cell Res. 2004, 17, 208–214. [Google Scholar] [CrossRef]
- Cummings, M.; Nordlund, J. Chemical leukoderma: Fact or fancy. Am. J. Contact Dermat. 1995, 6, 122–126. [Google Scholar] [CrossRef]
- Manini, P.; Napolitano, A.; Westerhof, W.; Riley, P.A.; d’Ischia, M. A reactive ortho-quinone generated by tyrosinase-catalyzed oxidation of the skin depigmenting agent monobenzone: Self-coupling and thiol-conjugation reactions and possible implications for melanocyte toxicity. Chem. Res. Toxicol. 2009, 22, 1398–1405. [Google Scholar] [CrossRef]
- Van den Boorn, J.G.; Picavet, D.I.; van Swieten, P.F.; van Veen, H.A.; Konijnenberg, D.; van Veelen, P.A.; van Capel, T.; Jong, E.C.; Reits, E.A.; Drijfhout, J.W.; et al. Skin-depigmenting agent monobenzone induces potent T-cell autoimmunity toward pigmented cells by tyrosinase haptenation and melanosome autophagy. J. Investig. Dermatol. 2011, 131, 1240–1251. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Ojika, M.; Yamashita, T.; Wakamatsu, K. Tyrosinase-catalyzed oxidation of rhododendrol produces 2-methylchromane-6,7-dione, the putative ultimate toxic metabolite: Implications for melanocyte toxicity. Pigment. Cell Melanoma Res. 2014, 27, 744–753. [Google Scholar] [CrossRef]
- Ito, S.; Hinoshita, M.; Suzuki, E.; Ojika, M.; Wakamatsu, K. Tyrosinase-catalyzed oxidation of the leukoderma-inducing agent raspberry ketone produces (E)-4-(3-oxo-1-butenyl)-1,2-benzoquinone: Implications for melanocyte toxicity. Chem. Res. Toxicol. 2017, 30, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Ito, S.; Inoue, S.; Wakamatsu, K.; Ozeki, H.; Ishiguro, I. Dihydro-1,4-benzothiazine-6,7-dione, the ultimate toxic metabolite of 4-S-cysteaminylphenol and 4-S-cysteaminylcatechol. Biochem. Pharmacol. 1997, 53, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Nishigaki, A.; Ishii-Osai, Y.; Ojika, M.; Wakamatsu, K.; Yamashita, T.; Tamura, Y.; Ito, A.; Honda, H.; Nakayama, E.; et al. Mechanism of putative neo-antigen formation from N-propionyl-4-S-cysteaminylphenol, a tyrosinase substrate, in melanoma models. Biochem. Pharmacol. 2012, 84, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Nishimaki-Mogami, T.; Ito, S.; Cui, H.; Akiyama, T.; Tamehiro, N.; Adachi, R.; Wakamatsu, K.; Ikarashi, Y.; Kondo, K. A cell-based evaluation of human tyrosinase-mediated metabolic activation of leukoderma-inducing phenolic compounds. J. Dermatol. Sci. 2022, 108, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Wakamatsu, K. A convenient screening method to differentiate phenolic skin whitening tyrosinase inhibitors from leukoderma-inducing phenols. J. Dermatol. Sci. 2015, 80, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Thörneby-Andersson, K.; Sterner, O.; Hansson, C. Tyrosinase-mediated formation of a reactive quinone from the depigmenting agents, 4-tert-butylphenol and 4-tert-butylcatechol. Pigment. Cell Res. 2000, 13, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Okura, M.; Yamashita, T.; Ishii-Osai, Y.; Yoshikawa, M.; Sumikawa, Y.; Wakamatsu, K.; Ito, S. Effects of rhododendrol and its metabolic products on melanocytic cell growth. J. Dermatol. Sci. 2015, 80, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Miyaji, A.; Gabe, Y.; Kohno, M.; Baba, T. Generation of hydroxyl radicals and singlet oxygen during oxidation of rhododendrol and rhododendrol-catechol. J. Clin. Biochem. Nutr. 2017, 60, 86–92. [Google Scholar] [CrossRef] [PubMed]
- van den Boorn, J.G.; Melief, C.J.; Luiten, R.M. Monobenzone-induced depigmentation: From enzymatic blockade to autoimmunity. Pigment. Cell Melanoma Res. 2011, 24, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Kato, T.; Ishikawa, K.; Kasuga, T.; Jimbow, K. Mechanism of selective toxicity of 4-S-cysteinylphenol and 4-S-cysteaminylphenol to melanocytes. Biochem. Pharmacol. 1987, 36, 2007–2011. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Kondo, M.; Sato, K.; Umeda, M.; Kawabata, K.; Takahashi, Y.; Suzuki, T.; Matsunaga, K.; Inoue, S. Rhododendrol, a depigmentation-inducing phenolic compound, exerts melanocyte cytotoxicity via a tyrosinase-dependent mechanism. Pigment. Cell Melanoma Res. 2014, 27, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Katayama, I.; Suzuki, T.; Tanemura, A.; Ito, S.; Abe, Y.; Sumikawa, Y.; Yoshikawa, M.; Suzuki, K.; Yagami, A.; et al. Rhododendrol-induced leukoderma update II: Pathophysiology, mechanisms, risk evaluation, and possible mechanism-based treatments in comparison with vitiligo. J. Dermatol. 2021, 48, 969–978. [Google Scholar] [CrossRef]
- Tazaki, A.; Nishadhi, D.; Li, A.; Zhang, L.; Maw, T.H.; Kondo-Ida, L.; Yanagisawa, K.; Kato, M. Progression from in vivo validation to in vitro screening in hazard assessment for leukoderma-inducible chemicals. Environ. Pollut. 2024, 356, 124508. [Google Scholar] [CrossRef] [PubMed]
- Prezioso, J.A.; Epperly, M.W.; Wang, N.; Bloomer, W.D. Effects of tyrosinase activity on the cytotoxicity of 4-S-cysteaminylphenol and N-acetyl-4-S-cysteaminylphenol in melanoma cells. Cancer Lett. 1992, 63, 73–79. [Google Scholar] [CrossRef]
- Simonova, M.; Wall, A.; Weissleder, R.; Bogdanov, A., Jr. Tyrosinase mutants are capable of prodrug activation in transfected nonmelanotic cells. Cancer Res. 2000, 60, 6656–6662. [Google Scholar] [PubMed]
- Kasamatsu, S.; Hachiya, A.; Nakamura, S.; Yasuda, Y.; Fujimori, T.; Takano, K.; Moriwaki, S.; Hase, T.; Suzuki, T.; Matsunaga, K. Depigmentation caused by application of the active brightening material, rhododendrol, is related to tyrosinase activity at a certain threshold. J. Dermatol. Sci. 2014, 76, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Kondo, M.; Kawabata, K.; Sato, K.; Yamaguchi, S.; Hachiya, A.; Takahashi, Y.; Inoue, S. Glutathione maintenance is crucial for survival of melanocytes after exposure to rhododendrol. Pigment. Cell Melanoma Res. 2016, 29, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Kusano, Y.; Okazaki, K.; Akaike, T.; Motohashi, H. NRF2 signalling in cytoprotection and metabolism. Br. J. Pharmacol. 2023. [Google Scholar] [CrossRef]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes. Cells 2011, 16, 123–140. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, E.L.; Becker, A.L.; Indra, A.K. NRF2 and key transcriptional targets in melanoma redox manipulation. Cancers 2022, 14, 1531. [Google Scholar] [CrossRef]
- Feng, Q.; Xu, X.; Zhang, S. Nrf2 protein in melanoma progression, as a new means of treatment. Pigment. Cell Melanoma Res. 2024, 37, 247–258. [Google Scholar] [CrossRef]
- Arowojolu, O.A.; Orlow, S.J.; Elbuluk, N.; Manga, P. The nuclear factor (erythroid-derived 2)-like 2 (NRF2) antioxidant response promotes melanocyte viability and reduces toxicity of the vitiligo-inducing phenol monobenzone. Exp. Dermatol. 2017, 26, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Okubo, A.; Yasuhira, S.; Shibazaki, M.; Takahashi, K.; Akasaka, T.; Masuda, T.; Maesawa, C. NAD(P)H dehydrogenase, quinone 1 (NQO1), protects melanin-producing cells from cytotoxicity of rhododendrol. Pigment. Cell Melanoma Res. 2016, 29, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yang, F.; Teng, L.; Katayama, I. 6-Shogaol protects human melanocytes against oxidative stress through activation of the Nrf2-antioxidant response element signaling pathway. Int. J. Mol. Sci. 2020, 21, 3537. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Ham, W.; Kim, J. Roles of NAD(P)H:quinone oxidoreductase 1 in diverse diseases. Life 2021, 11, 1301. [Google Scholar] [CrossRef] [PubMed]
- Radjendirane, V.; Joseph, P.; Lee, Y.H.; Kimura, S.; Klein-Szanto, A.J.; Gonzalez, F.J.; Jaiswal, A.K. Disruption of the DT diaphorase (NQO1) gene in mice leads to increased menadione toxicity. J. Biol. Chem. 1998, 273, 7382–7389. [Google Scholar] [CrossRef]
- Siegel, D.; Yan, C.; Ross, D. NAD(P)H:quinone oxidoreductase 1 (NQO1) in the sensitivity and resistance to antitumor quinones. Biochem. Pharmacol. 2012, 83, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, N.; Okubo, A.; Yasuhira, S.; Takahashi, K.; Amano, H.; Akasaka, T.; Masuda, T.; Shibazaki, M.; Maesawa, C. Carnosic acid, an inducer of NAD(P)H quinone oxidoreductase 1, enhances the cytotoxicity of β-lapachone in melanoma cell lines. Oncol. Lett. 2018, 15, 2393–2400. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Sato, H.; Kuriyama-Matsumura, K.; Sato, K.; Maebara, K.; Wang, H.; Tamba, M.; Itoh, K.; Yamamoto, M.; Bannai, S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 2002, 277, 44765–44771. [Google Scholar] [CrossRef] [PubMed]
- Chintala, S.; Li, W.; Lamoreux, M.L.; Ito, S.; Wakamatsu, K.; Sviderskaya, E.V.; Bennett, D.C.; Park, Y.M.; Gahl, W.A.; Huizing, M.; et al. Slc7a11 gene controls production of pheomelanin pigment and proliferation of cultured cells. Proc. Natl. Acad. Sci. USA 2005, 102, 10964–10969. [Google Scholar] [CrossRef]
- Danciu, C.; Oprean, C.; Coricovac, D.E.; Andreea, C.; Cimpean, A.; Radeke, H.; Soica, C.; Dehelean, C. Behaviour of four different B16 murine melanoma cell sublines: C57BL/6J skin. Int. J. Exp. Pathol. 2015, 96, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Dehn, D.L.; Siegel, D.; Swann, E.; Moody, C.J.; Ross, D. Biochemical, cytotoxic, and genotoxic effects of ES936, a mechanism-based inhibitor of NAD(P)H:quinone oxidoreductase 1, in cellular systems. Mol. Pharmacol. 2003, 64, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Winski, S.L.; Faig, M.; Bianchet, M.A.; Siegel, D.; Swann, E.; Fung, K.; Duncan, M.W.; Moody, C.J.; Amzel, L.M.; Ross, D. Characterization of a mechanism-based inhibitor of NAD(P)H:quinone oxidoreductase 1 by biochemical, X-ray crystallographic, and mass spectrometric approaches. Biochemistry 2001, 40, 15135–15142. [Google Scholar] [CrossRef]
- Choi, T.Y.; Sohn, K.C.; Kim, J.H.; Kim, S.M.; Kim, C.H.; Hwang, J.S.; Lee, J.H.; Kim, C.D.; Yoon, T.J. Impact of NAD(P)H:quinone oxidoreductase-1 on pigmentation. J. Investig. Dermatol. 2010, 130, 784–792. [Google Scholar] [CrossRef]
- Sheppard, J.R.; Lester, B.; Doll, J.; Buscarino, C.; Gonzales, E.; Corwin, S.; Greig, R.; Poste, G. Biochemical regulation of adenylate cyclase in murine melanoma clones with different metastatic properties. Int. J. Cancer 1986, 37, 713–722. [Google Scholar] [CrossRef]
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 2004, 84, 1155–1228. [Google Scholar] [CrossRef] [PubMed]
- Vachtenheim, J.; Borovanský, J. “Transcription physiology” of pigment formation in melanocytes: Central role of MITF. Exp. Dermatol. 2010, 19, 617–627. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishimaki-Mogami, T.; Ito, S.; Wakamatsu, K.; Akiyama, T.; Tamehiro, N.; Shibata, N. A Cell-Based Evaluation of the Tyrosinase-Mediated Metabolic Activation of Leukoderma-Inducing Phenols, II: The Depletion of Nrf2 Augments the Cytotoxic Effect Evoked by Tyrosinase in Melanogenic Cells. Biomolecules 2025, 15, 114. https://doi.org/10.3390/biom15010114
Nishimaki-Mogami T, Ito S, Wakamatsu K, Akiyama T, Tamehiro N, Shibata N. A Cell-Based Evaluation of the Tyrosinase-Mediated Metabolic Activation of Leukoderma-Inducing Phenols, II: The Depletion of Nrf2 Augments the Cytotoxic Effect Evoked by Tyrosinase in Melanogenic Cells. Biomolecules. 2025; 15(1):114. https://doi.org/10.3390/biom15010114
Chicago/Turabian StyleNishimaki-Mogami, Tomoko, Shosuke Ito, Kazumasa Wakamatsu, Takumi Akiyama, Norimasa Tamehiro, and Norihito Shibata. 2025. "A Cell-Based Evaluation of the Tyrosinase-Mediated Metabolic Activation of Leukoderma-Inducing Phenols, II: The Depletion of Nrf2 Augments the Cytotoxic Effect Evoked by Tyrosinase in Melanogenic Cells" Biomolecules 15, no. 1: 114. https://doi.org/10.3390/biom15010114
APA StyleNishimaki-Mogami, T., Ito, S., Wakamatsu, K., Akiyama, T., Tamehiro, N., & Shibata, N. (2025). A Cell-Based Evaluation of the Tyrosinase-Mediated Metabolic Activation of Leukoderma-Inducing Phenols, II: The Depletion of Nrf2 Augments the Cytotoxic Effect Evoked by Tyrosinase in Melanogenic Cells. Biomolecules, 15(1), 114. https://doi.org/10.3390/biom15010114