
Clinical Insights in RNA-Binding Protein Motif 20 Cardiomyopathy: A Systematic Review

 , , and
, , and
Abstract
1. Introduction
2. Methods
2.1. Protocol Registration
2.2. Study Plan
2.3. Inclusion and Exclusion Criteria
2.4. Data Extraction
3. Results
3.1. Study Retrieval
3.2. Clinical Features of Patients with RBM20 Cardiomyopathy
3.3. Electrocardiographic Features of Patients with RBM20 Cardiomyopathy
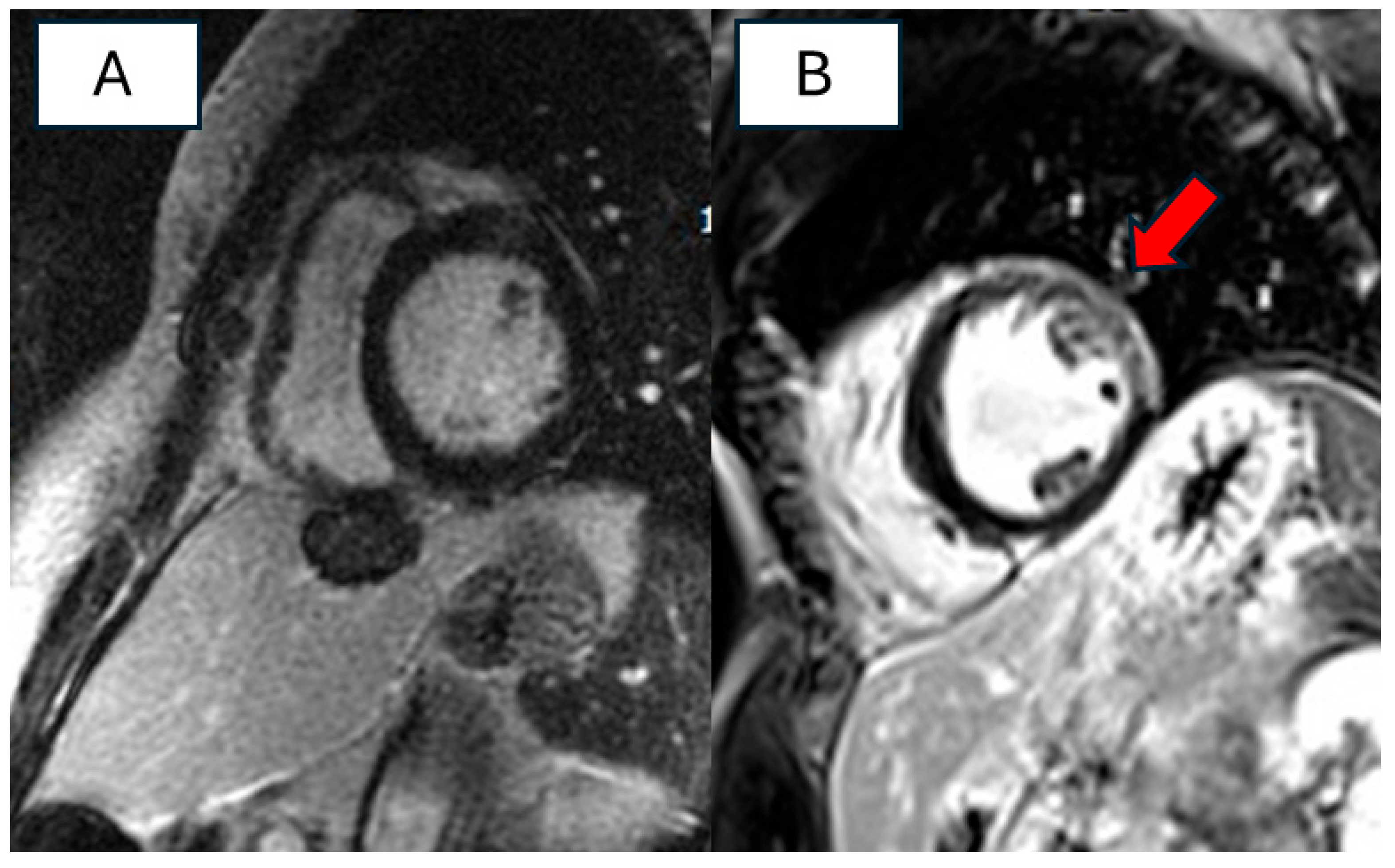
3.4. Imaging Features of Patients with RBM20 Cardiomyopathy
3.5. Outcome Data of Patients with RBM20 Cardiomyopathy
3.6. Sex-Related Disease Expression in RBM20 Cardiomyopathy
4. Discussion
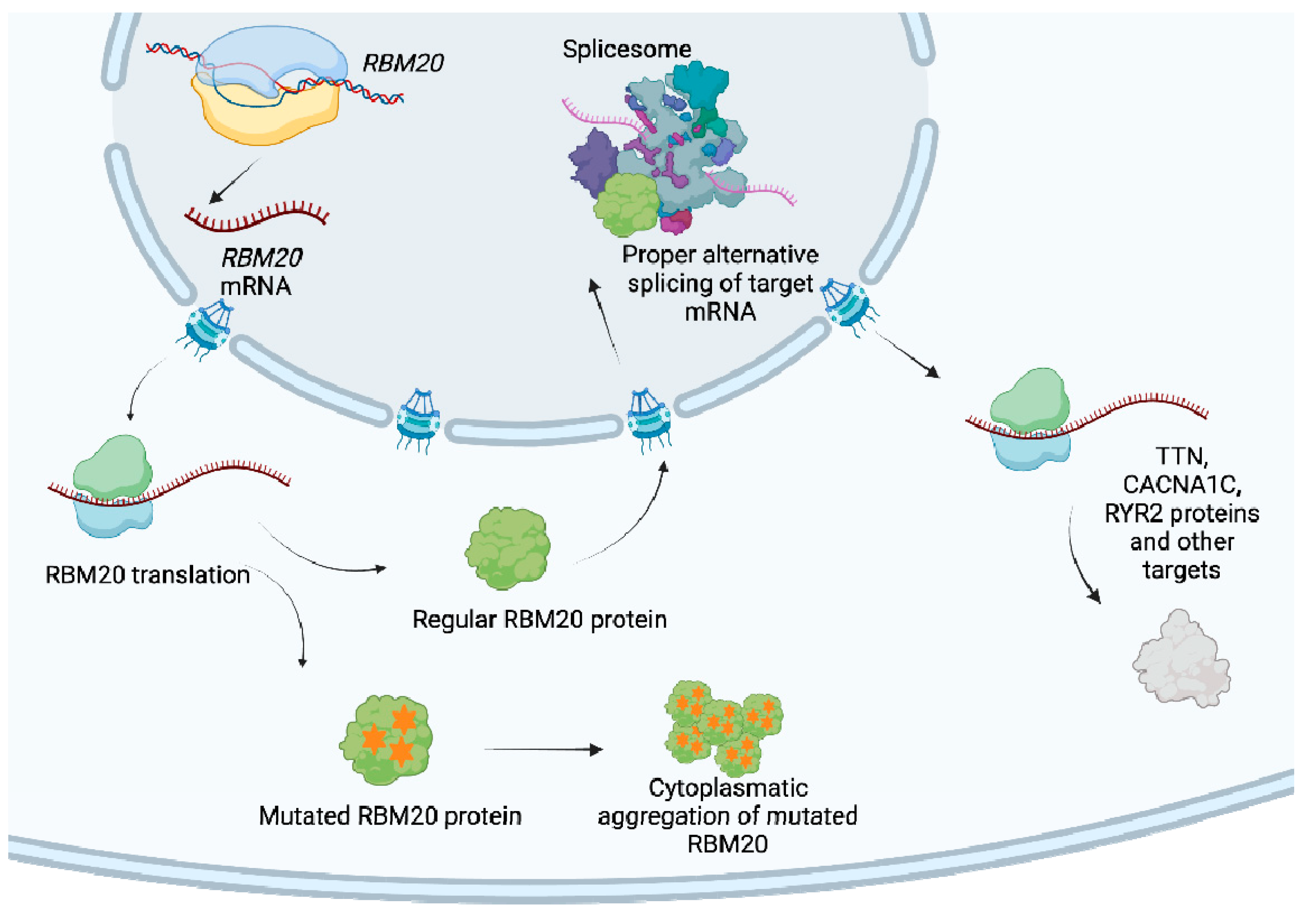
4.1. Genetic Background
4.2. Clinical Features of Patients with RBM20 Cardiomyopathy
4.3. Instrumental Features of RBM20 Cardiomyopathy
4.4. Outcomes Data and Gender Difference in Patients with RBM20 Cardiomyopathy
4.5. Limitation of the Study
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Burkett, E.L.; Hershberger, R.E. Clinical and Genetic Issues in Familial Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2005, 45, 969–981. [Google Scholar] [CrossRef] [PubMed]
- Akinrinade, O.; Ollila, L.; Vattulainen, S.; Tallila, J.; Gentile, M.; Salmenperä, P.; Koillinen, H.; Kaartinen, M.; Nieminen, M.S.; Myllykangas, S.; et al. Genetics and Genotype-Phenotype Correlations in Finnish Patients with Dilated Cardiomyopathy. Eur. Heart J. 2015, 36, 2327–2337. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated Cardiomyopathy: The Complexity of a Diverse Genetic Architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef] [PubMed]
- Brauch, K.M.; Karst, M.L.; Herron, K.J.; de Andrade, M.; Pellikka, P.A.; Rodeheffer, R.J.; Michels, V.V.; Olson, T.M. Mutations in Ribonucleic Acid Binding Protein Gene Cause Familial Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Wyles, S.P.; Li, X.; Hrstka, S.C.; Reyes, S.; Oommen, S.; Beraldi, R.; Edwards, J.; Terzic, A.; Olson, T.M.; Nelson, T.J. Modeling Structural and Functional Deficiencies of RBM20 Familial Dilated Cardiomyopathy Using Human Induced Pluripotent Stem Cells. Hum. Mol. Genet. 2016, 25, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Refaat, M.M.; Lubitz, S.A.; Makino, S.; Islam, Z.; Frangiskakis, J.M.; Mehdi, H.; Gutmann, R.; Zhang, M.L.; Bloom, H.L.; MacRae, C.A.; et al. Genetic Variation in the Alternative Splicing Regulator RBM20 Is Associated with Dilated Cardiomyopathy. Heart Rhythm. 2012, 9, 390–396. [Google Scholar] [CrossRef]
- van den Hoogenhof, M.M.G.; Beqqali, A.; Amin, A.S.; van der Made, I.; Aufiero, S.; Khan, M.A.F.; Schumacher, C.A.; Jansweijer, J.A.; van Spaendonck-Zwarts, K.Y.; Remme, C.A.; et al. RBM20 Mutations Induce an Arrhythmogenic Dilated Cardiomyopathy Related to Disturbed Calcium Handling. Circulation 2018, 138, 1330–1342. [Google Scholar] [CrossRef] [PubMed]
- Hey, T.M.; Rasmussen, T.B.; Madsen, T.; Aagaard, M.M.; Harbo, M.; Mølgaard, H.; Møller, J.E.; Eiskjær, H.; Mogensen, J. Pathogenic RBM20-Variants Are Associated With a Severe Disease Expression in Male Patients With Dilated Cardiomyopathy. Circ. Heart Fail. 2019, 12, e005700. [Google Scholar] [CrossRef]
- Parikh, V.N.; Caleshu, C.; Reuter, C.; Lazzeroni, L.C.; Ingles, J.; Garcia, J.; McCaleb, K.; Adesiyun, T.; Sedaghat-Hamedani, F.; Kumar, S.; et al. Regional Variation in RBM20 Causes a Highly Penetrant Arrhythmogenic Cardiomyopathy. Circ. Heart Fail. 2019, 12, e005371. [Google Scholar] [CrossRef]
- Robles-Mezcua, A.; Rodríguez-Miranda, L.; Morcillo-Hidalgo, L.; Jiménez-Navarro, M.; García-Pinilla, J.M. Phenotype and Progression among Patients with Dilated Cardiomyopathy and RBM20 Mutations. Eur. J. Med. Genet. 2021, 64, 104278. [Google Scholar] [CrossRef] [PubMed]
- Cannie, D.E.; Protonotarios, A.; Bakalakos, A.; Syrris, P.; Lorenzini, M.; De Stavola, B.; Bjerregaard, L.; Dybro, A.M.; Hey, T.M.; Hansen, F.G.; et al. Risks of Ventricular Arrhythmia and Heart Failure in Carriers of RBM20 Variants. Circ. Genom. Precis. Med. 2023, 16, 434–441. [Google Scholar] [CrossRef]
- de Frutos, F.; Ochoa, J.P.; Fernández, A.I.; Gallego-Delgado, M.; Navarro-Peñalver, M.; Casas, G.; Basurte, M.T.; Larrañaga-Moreira, J.M.; Mogollón, M.V.; Robles-Mezcua, A.; et al. Late Gadolinium Enhancement Distribution Patterns in Non-Ischaemic Dilated Cardiomyopathy: Genotype-Phenotype Correlation. Eur. Heart J. Cardiovasc. Imaging 2023, 25, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M.; et al. Genetic evaluation of cardiomyopathy: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Schultheiss, H.-P.; Fairweather, D.; Caforio, A.L.P.; Escher, F.; Hershberger, R.E.; Lipshultz, S.E.; Liu, P.P.; Matsumori, A.; Mazzanti, A.; McMurray, J.; et al. Dilated Cardiomyopathy. Nat. Rev. Dis. Primers 2019, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dobreva, G. Epigenetics in LMNA-Related Cardiomyopathy. Cells 2023, 12, 783. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Guo, W.; Dewey, C.N.; Greaser, M.L. Rbm20 Regulates Titin Alternative Splicing as a Splicing Repressor. Nucleic Acids Res. 2013, 41, 2659–2672. [Google Scholar] [CrossRef]
- Guo, W.; Schafer, S.; Greaser, M.L.; Radke, M.H.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, a Gene for Hereditary Cardiomyopathy, Regulates Titin Splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar] [CrossRef]
- Mutations of the Cardiac Ryanodine Receptor (RyR2) Gene in Familial Polymorphic Ventricular Tachycardia—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/11157710/ (accessed on 17 March 2024).
- Zhang, M.; Gao, H.; Liu, D.; Zhong, X.; Shi, X.; Yu, P.; Jin, L.; Liu, Y.; Tang, Y.; Song, Y.; et al. CaMKII-Δ9 Promotes Cardiomyopathy through Disrupting UBE2T-Dependent DNA Repair. Nat. Cell Biol. 2019, 21, 1152–1163. [Google Scholar] [CrossRef]
- Murayama, R.; Kimura-Asami, M.; Togo-Ohno, M.; Yamasaki-Kato, Y.; Naruse, T.K.; Yamamoto, T.; Hayashi, T.; Ai, T.; Spoonamore, K.G.; Kovacs, R.J.; et al. Phosphorylation of the RSRSP Stretch Is Critical for Splicing Regulation by RNA-Binding Motif Protein 20 (RBM20) through Nuclear Localization. Sci. Rep. 2018, 8, 8970. [Google Scholar] [CrossRef]
- Ihara, K.; Sasano, T.; Hiraoka, Y.; Togo-Ohno, M.; Soejima, Y.; Sawabe, M.; Tsuchiya, M.; Ogawa, H.; Furukawa, T.; Kuroyanagi, H. A Missense Mutation in the RSRSP Stretch of Rbm20 Causes Dilated Cardiomyopathy and Atrial Fibrillation in Mice. Sci. Rep. 2020, 10, 17894. [Google Scholar] [CrossRef]
- Gaertner, A.; Klauke, B.; Felski, E.; Kassner, A.; Brodehl, A.; Gerdes, D.; Stanasiuk, C.; Ebbinghaus, H.; Schulz, U.; Dubowy, K.-O.; et al. Cardiomyopathy-Associated Mutations in the RS Domain Affect Nuclear Localization of RBM20. Hum. Mutat. 2020, 41, 1931–1943. [Google Scholar] [CrossRef]
- Schneider, J.W.; Oommen, S.; Qureshi, M.Y.; Goetsch, S.C.; Pease, D.R.; Sundsbak, R.S.; Guo, W.; Sun, M.; Sun, H.; Kuroyanagi, H.; et al. Dysregulated Ribonucleoprotein Granules Promote Cardiomyopathy in RBM20 Gene-Edited Pigs. Nat. Med. 2020, 26, 1788–1800. [Google Scholar] [CrossRef] [PubMed]
- Pantou, M.P.; Gourzi, P.; Gkouziouta, A.; Tsiapras, D.; Zygouri, C.; Constantoulakis, P.; Adamopoulos, S.; Degiannis, D. Phenotypic Heterogeneity within Members of a Family Carrying the Same RBM20 Mutation R634W. Cardiology 2018, 141, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Dec, G.W.; Fuster, V. Idiopathic Dilated Cardiomyopathy. N. Engl. J. Med. 1994, 331, 1564–1575. [Google Scholar] [CrossRef] [PubMed]
- Ollila, L.; Nikus, K.; Holmström, M.; Jalanko, M.; Jurkko, R.; Kaartinen, M.; Koskenvuo, J.; Kuusisto, J.; Kärkkäinen, S.; Palojoki, E.; et al. Clinical Disease Presentation and ECG Characteristics of LMNA Mutation Carriers. Open Heart 2017, 4, e000474. [Google Scholar] [CrossRef]
- Cipriani, A.; Bauce, B.; De Lazzari, M.; Rigato, I.; Bariani, R.; Meneghin, S.; Pilichou, K.; Motta, R.; Aliberti, C.; Thiene, G.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy: Characterization of Left Ventricular Phenotype and Differential Diagnosis With Dilated Cardiomyopathy. J. Am. Heart Assoc. 2020, 9, e014628. [Google Scholar] [CrossRef]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the Management of Cardiomyopathies. Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef]
- Gregorich, Z.R.; Zhang, Y.; Kamp, T.J.; Granzier, H.L.; Guo, W. Mechanisms of RBM20 Cardiomyopathy: Insights From Model Systems. Circ. Genom. Precis. Med. 2024, 17, e004355. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Reference | Aim and Design of the Study | Study Population | Main Results | Pts with RBM20 Variant | Conclusions |
|---|---|---|---|---|---|
| Brauch KM, J Am Coll Cardiol. 2009 [5] | Authors conducted a genetic linkage analysis in two large families with autosomal dominant DCM to map a disease locus, eventually leading to the discovery of a variant hotspot in RNA-binding protein. | 280 unrelated probands with DCM | RBM20 variant carriers exhibit an earlier onset of disease compared to other familial DCM cases. RBM20 cardiomyopathy is characterized by a high arrhythmic burden and the development of end-stage HF. | 39 patients | RBM20 accounted for 3% of DCM genetic variants and 13% of cases with history of SCD. |
| Refaat MM, Heart Rhythm. 2012 Mar [7] | To determine the prevalence of RBM20 variants in a large, multiracial cohort of DCM pts. To analyse the clinical features and outcomes of variant carriers. | A total of 1465 subjects belonging to the GRADE database, including 283 patients with DCM | The prevalence of RBM20 variants was 2.7% in individuals of European ancestry and 3.9% in individuals of African American ancestry. RBM20 variant carriers have an increased risk of atrial fibrillation. Variant carriers had similar survival rates, HTx rates and frequencies of ICD therapy compared to non-variant carriers.” | 8 patients | In this cohort of DCM patients, 3% of patients had an RBM20 variant. RBM20 variants were not associated with adverse survival in terms of ventricular arrhythmias. |
| Van den Hoogenhof, Circulation. 2018 [8] | To investigate the underlying mechanism of arrhythmias in RBM20 variant carriers. | 18 RBM20 variant carriers 22 TTN variant carriers | No differences between the two groups regarding ECG parameters and LV chamber dimension and function. Despite exhibiting similar levels of LV remodelling, RBM20 variant carriers experienced a higher incidence of major ventricular arrhythmias. There was a significant presence of SCD family history in the RBM20 variant carriers group. | 18 patients | RBM20 variant carriers have an increased risk of major ventricular arrhythmias compared to TTN variant carriers, suggesting that dysregulation of TTN constitutes not the sole mechanism in RBM20 cardiomyopathy. |
| Hey TM, Circ Heart Fail. 2019 [9] | To further explore the phenotypic expression of patients carrying pathogenetic variants in the RBM20 gene. | 80 individuals carrying pathogenic RBM20 variants across 15 families 30 familiar DCM patients with unknown genetic cause | The penetrance of RBM20 cardiomyopathy was 66%. Asymptomatic family members were diagnosed with DCM at a younger age than the probands. A total of 30% of RBM20 variant carriers experienced a life-threatening event. Male patients exhibited a more severe disease expression, but no differences were observed in arrhythmic outcomes. RBM20 male patients had a shorter event-free survival compared to males with DCM of unknown genetic causes. | 80 patients | The disease expression is severe, particularly in males, with earlier onset and ventricular dysfunction. The penetrance of the disease is also severe in family members, necessitating careful follow-up. |
| Parikh VN, Circ Heart Fail. 2019 [10] | To characterize clinical features of RBM20 variant carriers. | Patients carrying different disease gene variants were analysed: A total of 74 patients with RBM20 variant from an international registry (43 index cases) 633 patients affected with DCM 83 patients with TTN cardiomyopathy 87 patients with lamin (LMNA) cardiomyopathy | RBM20 index patients had a higher incidence of DCM and SCD compared to the general DCM cohort and the Titin (TTN) cohort. The rate of ventricular arrhythmias did not differ compared to patients with LMNA cardiomyopathy. | 74 patients | Cardiomyopathy associated with RBM20 has a severe phenotypic expression with early onset and high arrhythmic burden, sharing features similar to LMNA cardiomyopathy. |
| Robles-Mezcua A, Eur J Med Genet. 2021 [11] | To investigate the genotype-phenotype correlation in patients with RBM20 gene variants. | 8 patients carrying RBM20 genetic variants | The main phenotype observed was DCM, except for one patient who was diagnosed with HCM. Overall, 62.5% had a history of cardiomyopathy among first-degree relatives, and 37.5% had a family history of SCD. No significant decline in systolic function was observed during the follow-up. | 8 patients | RBM20 cardiomyopathy is a highly penetrant disease with early onset and a frequent family history of DCM and SCD. |
| Cannie DE, Circ Genom Precis Med. 2023 [12] | To detail the natural history of RBM20 gene carriers in comparison with a group of gene-elusive patients showing LV systolic dysfunction. | 149 patients carrying a RBM20 variant, including 32 probands 238 patients with gene-elusive systolic dysfunction | Men exhibited an earlier onset of cardiac disease with a greater degree of ventricular dilation and dysfunction compared to women. Among men and women with the RBM20 variants, there was no difference in arrhythmic outcomes, while men more frequently experienced HF. In comparison to patients with variant-elusive systolic dysfunction, RBM20 variant carriers had a 6-fold increase in the risk of ventricular arrhythmias and HF. | 149 patients | RBM20 cardiomyopathy exhibits high penetrance, even among relatives. Men display more severe phenotypes and a poorer prognosis, yet the arrhythmic burden is similar in both sexes. In RBM20 variant carriers, ICD implantation should be considered for LVEF ≤45%. |
| De Frutos F, Eur Heart J Cardiovasc Imaging. 2023 [13] | To describe late LGE patterns according to genotypes in a cohort of DCM patients and to analyze the risk of major ventricular arrhythmias based on these patterns. | 577 patients affected with DCM: 219 with P/LP variants; 358 gene elusive pts | LGE was absent or very rare in RBM20 variant carriers. There was no significant association between the presence of LGE and lower LVEF. RBM20 variant carriers experienced MVA even in absence of LGE. | 22 patients | LGE is rare in patients with RBM20 cardiomyopathy, and it does not correlate with the degree of electrical instability. |
| Reference | HGVSc | HGVSp | Exon | ACMG | gnomAD |
|---|---|---|---|---|---|
| Brauch KM, J Am Coll Cardiol. 2009 [5] | c.1913C > T | p.(Pro638Leu) | 9 | P | 0.00000354 |
| c.1901G > A | p.(Arg634Gln) | 9 | P | 0.00000712 | |
| c.1906C > A | p.(Arg636Ser) | 9 | P | NA | |
| c.1907G > A | p.(Arg634Gln) | 9 | P | 0.00000712 | |
| c.1909A > G | p.(Ser637Gly) | 9 | P | NA | |
| Refaat MM, Heart Rhythm. 2012 Mar [7] | c.247C > A | p.(Leu83Ile) | 2 | LB | 0.00000645 |
| c.1364C > T | p.(Ser455Leu) | 4 | B | 0.0056 | |
| c.1913C > T | p.(Arg636Ser) | 9 | P | NA | |
| c.2109G > C | p.(Arg703Ser) | 9 | VUS | NA | |
| c.2662G > A | p.(Asp888Asn) | 11 | B | 0.00387 | |
| c.3091G > T | p.(Gly1031Ter) | 11 | LP | NA | |
| c.3242C > G | p.(Pro1081Arg) | 11 | LB | NA | |
| c.3616G > A | p.(Glu1206Lys) | 14 | B | 0.0000467 | |
| van den Hoogenhof, Circulation. 2018 [8] | c.769A > G | p.(Thr257Ala) | 2 | LB | NA |
| c.1175G > A | p.(Arg392Gln) | 2 | LB | 0.0000129 | |
| c.1494C > A | p.(Ser498Arg) | 5 | LB | 0.000013 | |
| c.1760T > A | p.(Leu587His) | 7 | VUS | NA | |
| c.1900C > T | p.(Arg634Trp) | 9 | P | NA | |
| c.1913C > T | p.(Pro638Leu) | 9 | P | NA | |
| c.2042A > G | p.(Tyr681Cys) | 9 | B | 0.000135 | |
| c.3115C > T | p.(Pro1039Ser) | 11 | B | 0.00021 | |
| Hey TM, Circ Heart Fail. 2019 [9] | c.1901G > A | p.(Arg634Gln) | 9 | P | 0.00000712 |
| c.1907G > A | p.(Arg634Gln) | 9 | P | 0.00000712 | |
| c.1906C > A | p.(Arg636Ser) | 9 | P | NA | |
| c.1913C > T | p.(Arg636Ser) | 9 | P | NA | |
| c.2737G > A | p.(Glu913Lys) | 11 | P | NA | |
| c.586A > G | p.(Met196Val) | 2 | LB | 0 | |
| c.1174C > T | p.(Arg392Trp) | 2 | VUS | 0.0000129 | |
| c.2021A > G | p.(Asp674Gly) | 9 | LB | 0.00000644 | |
| c.3115C > T | p.(Pro1039Ser) | 11 | B | 0.00021 | |
| Parikh VN, Circ Heart Fail. 2019 [10] | c.2721–2760 region | p.907–920 region | 11 | NA | NA |
| c.1881–1920 region | p.627–640 region | 9 | NA | NA | |
| Robles-Mezcua A, Eur J Med Genet. 2021 [11] | c.1793A > C | p.(Gln598Pro) | 7 | VUS | NA |
| c.776G > T | p.(Gly259Val) | 2 | LB | 0.000013 | |
| c.3169C > T | p.(Arg1057Trp) | 11 | B | 0.000102 | |
| c.580A > G | p.(Met194Val) | 2 | LB | NA | |
| c.529A > T | p.(Thr177Ser) | 2 | B | 0.0000896 | |
| c.154C > A | p.(Pro52Thr) | 1 | LB | NA | |
| Cannie DE, Circ Genom Precis Med. 2023 [12] | c.1906C > A | p.(Arg636Ser) | 9 | P | NA |
| c.1907G > A | p.(Arg634Gln) | 9 | P | 0.00000712 | |
| c.1901G > A | p.(Arg634Gln) | 9 | P | 0.00000712 | |
| c.2737G > A | p.(Glu913Lys) | 11 | P | NA | |
| c.1913C > T | p.(Arg636Ser) | 9 | P | NA | |
| c.1900C > T | p.(Arg634Trp) | 9 | P | NA | |
| c.2746G > A | p.(Glu916Lys) | 11 | VUS | NA | |
| c.2723T > C | p.(Leu908Pro) | 11 | VUS | NA | |
| De Frutos F, Eur Heart J Cardiovasc Imaging. 2023 [13] | NA | NA | NA | NA | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martini, M.; Bueno Marinas, M.; Rigato, I.; Pilichou, K.; Bauce, B. Clinical Insights in RNA-Binding Protein Motif 20 Cardiomyopathy: A Systematic Review. Biomolecules 2024, 14, 702. https://doi.org/10.3390/biom14060702
Martini M, Bueno Marinas M, Rigato I, Pilichou K, Bauce B. Clinical Insights in RNA-Binding Protein Motif 20 Cardiomyopathy: A Systematic Review. Biomolecules. 2024; 14(6):702. https://doi.org/10.3390/biom14060702
Chicago/Turabian StyleMartini, Marika, Maria Bueno Marinas, Ilaria Rigato, Kalliopi Pilichou, and Barbara Bauce. 2024. "Clinical Insights in RNA-Binding Protein Motif 20 Cardiomyopathy: A Systematic Review" Biomolecules 14, no. 6: 702. https://doi.org/10.3390/biom14060702
APA StyleMartini, M., Bueno Marinas, M., Rigato, I., Pilichou, K., & Bauce, B. (2024). Clinical Insights in RNA-Binding Protein Motif 20 Cardiomyopathy: A Systematic Review. Biomolecules, 14(6), 702. https://doi.org/10.3390/biom14060702