
The Effect of Reduced Fibrinogen on Cerebrovascular Permeability during Traumatic Brain Injury in Fibrinogen Gene Heterozygous Knockout Mice



Abstract
1. Introduction
2. Results
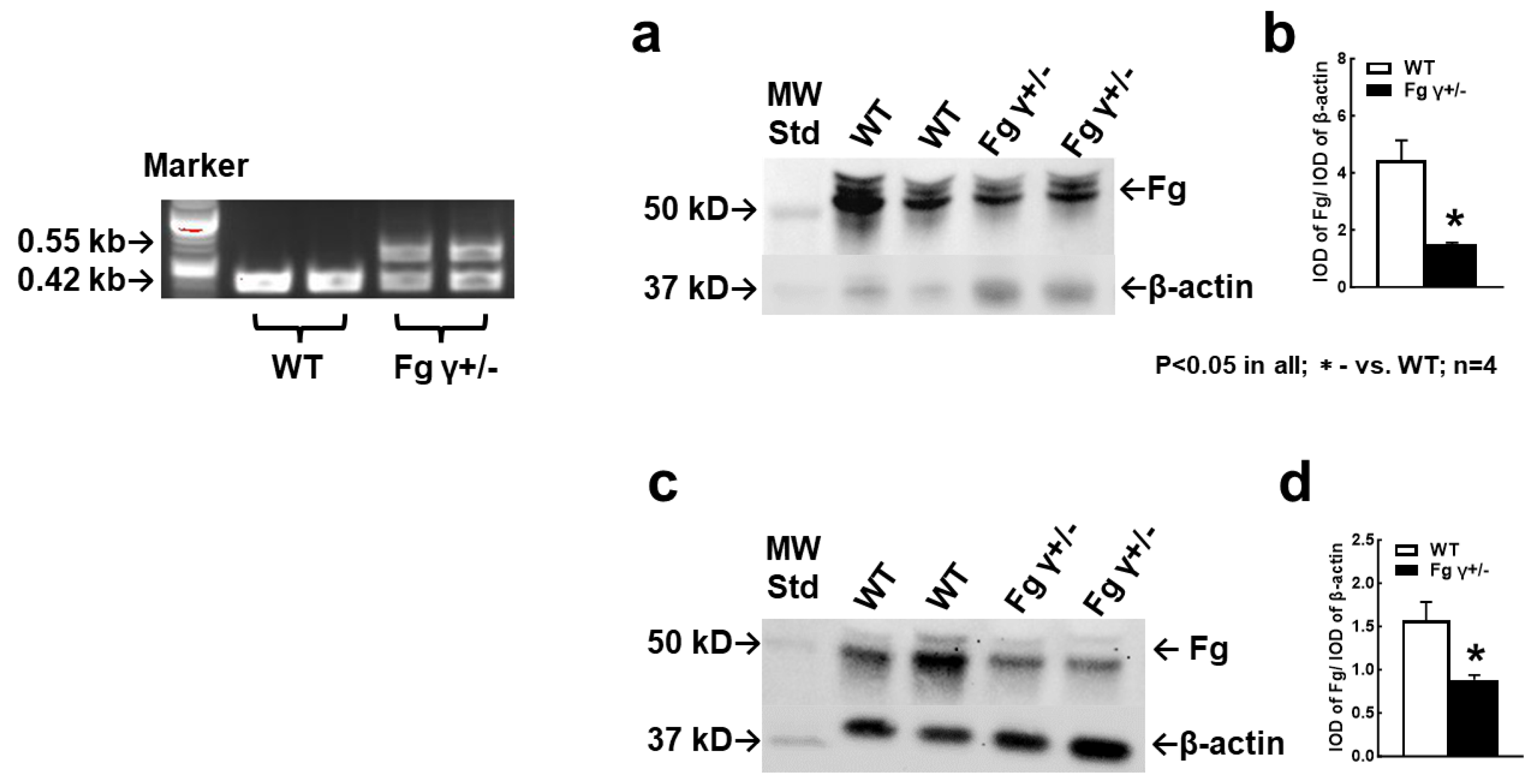
2.1. Blood Level of Fg in the γ+/− Mice
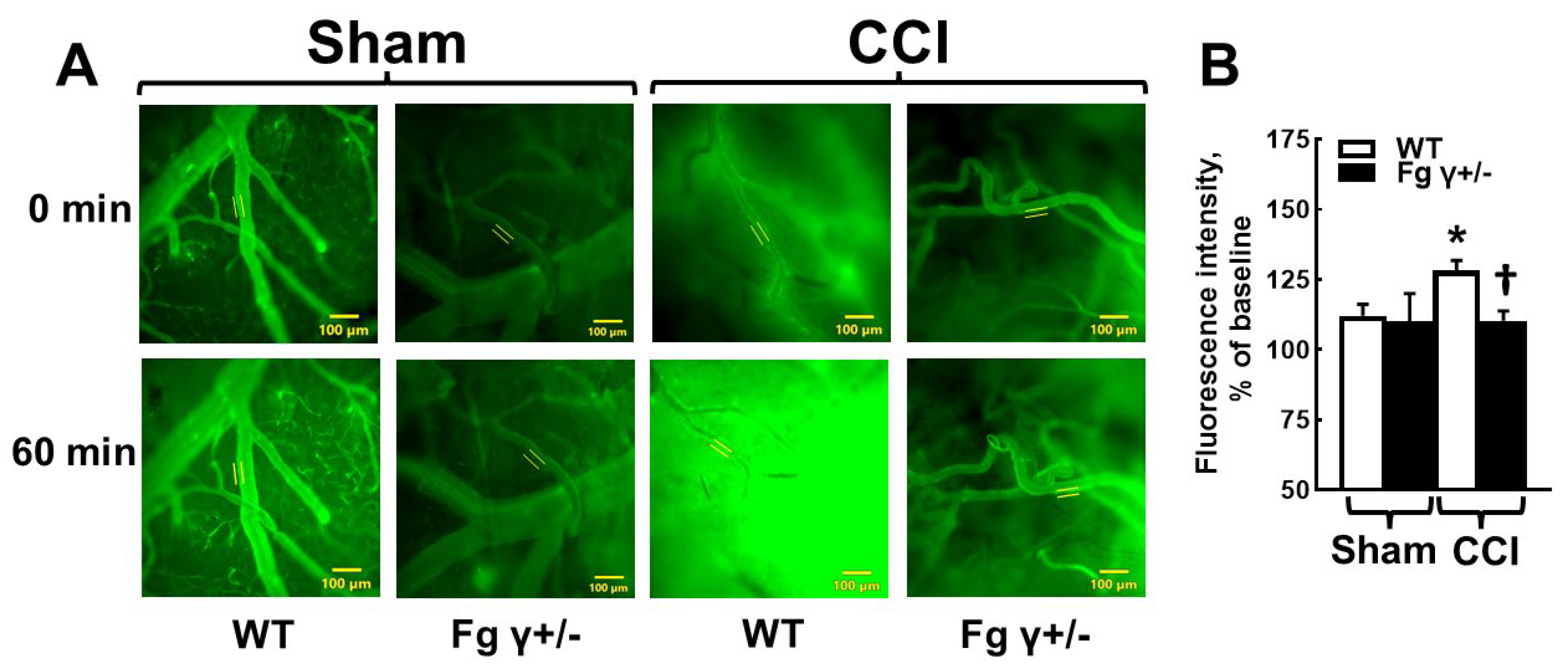
2.2. Cerebrovascular Permeability in Fg γ+/− Mice-
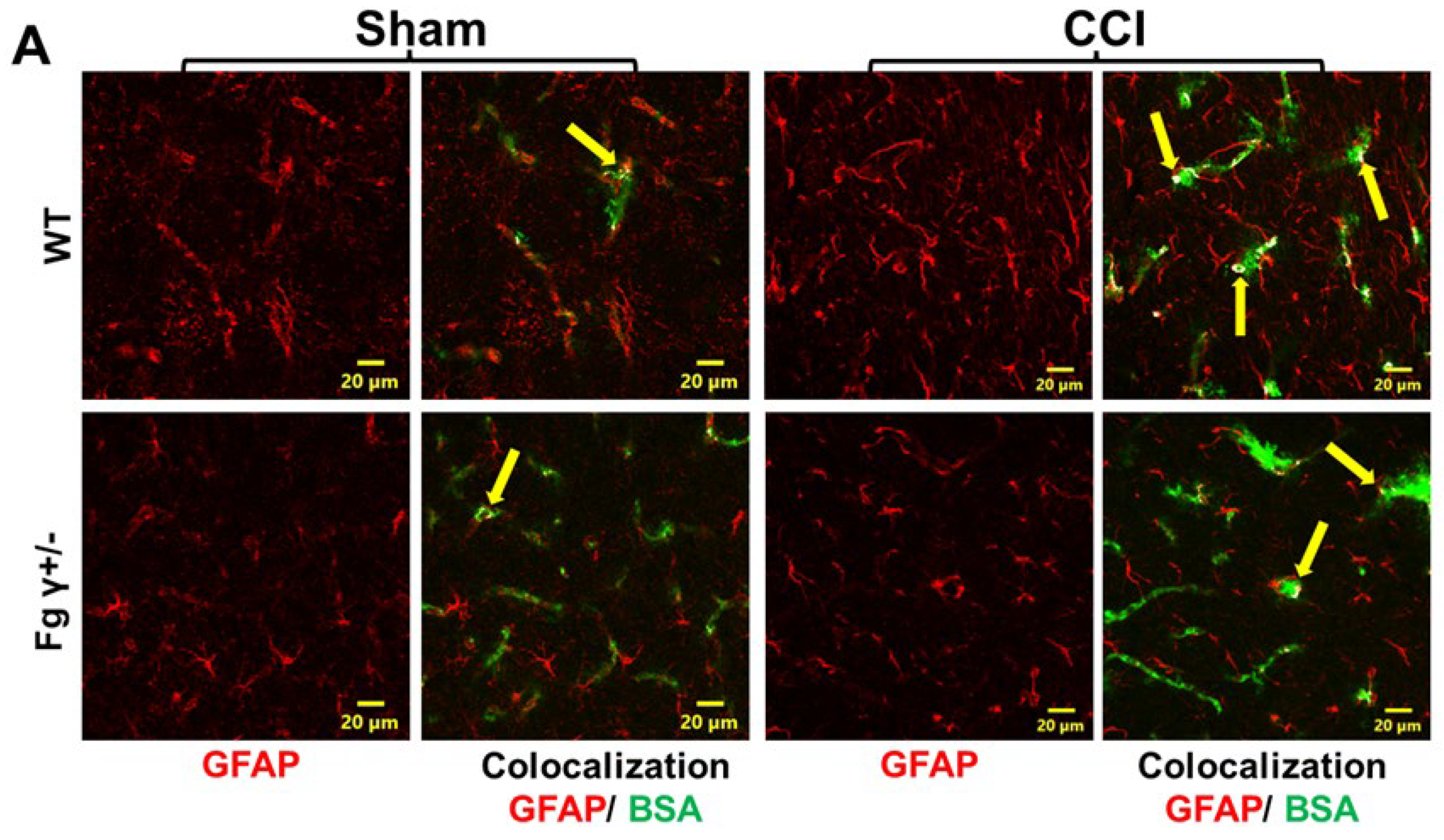
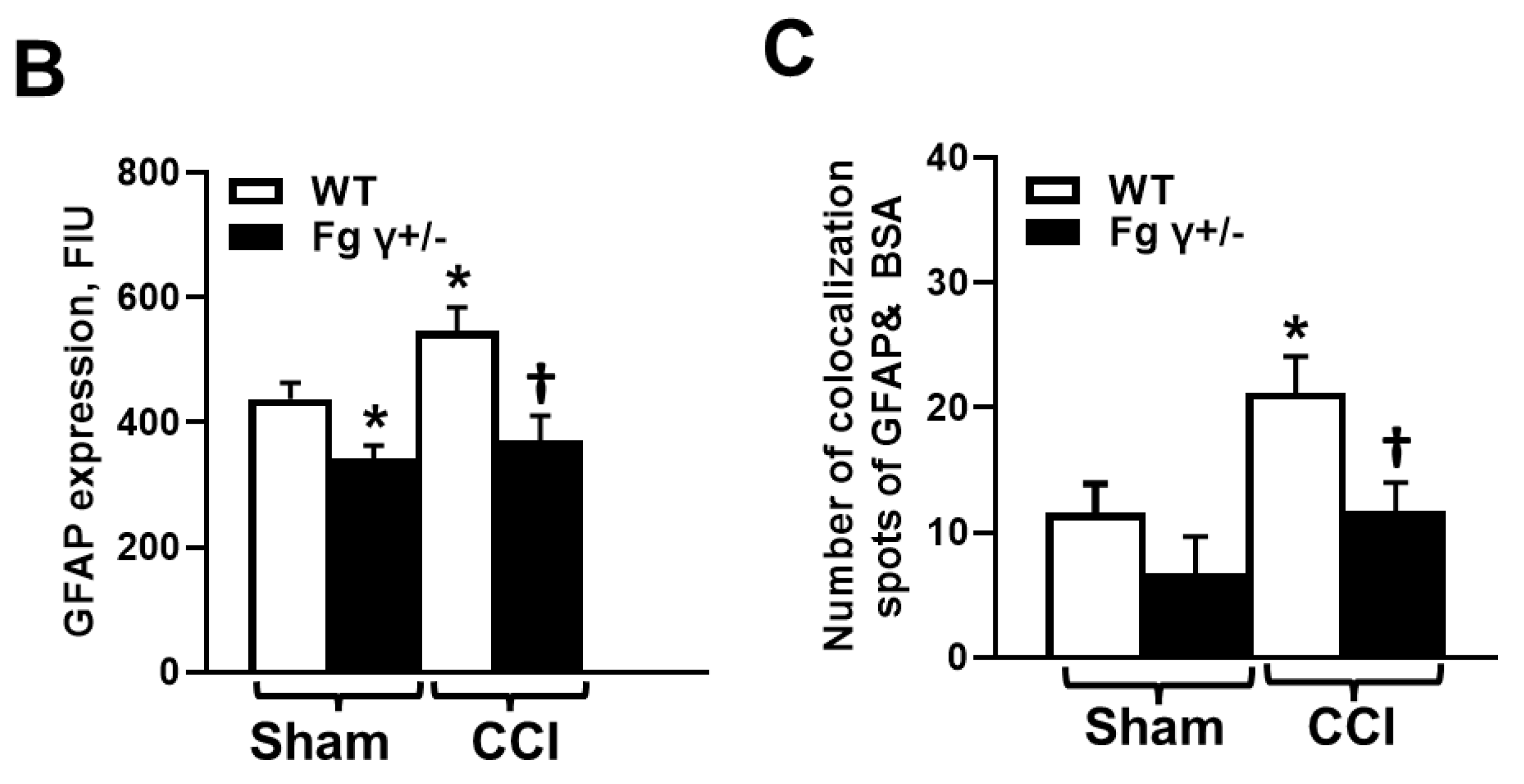
2.3. Extravasation of BSA and Activation of Astrocytes
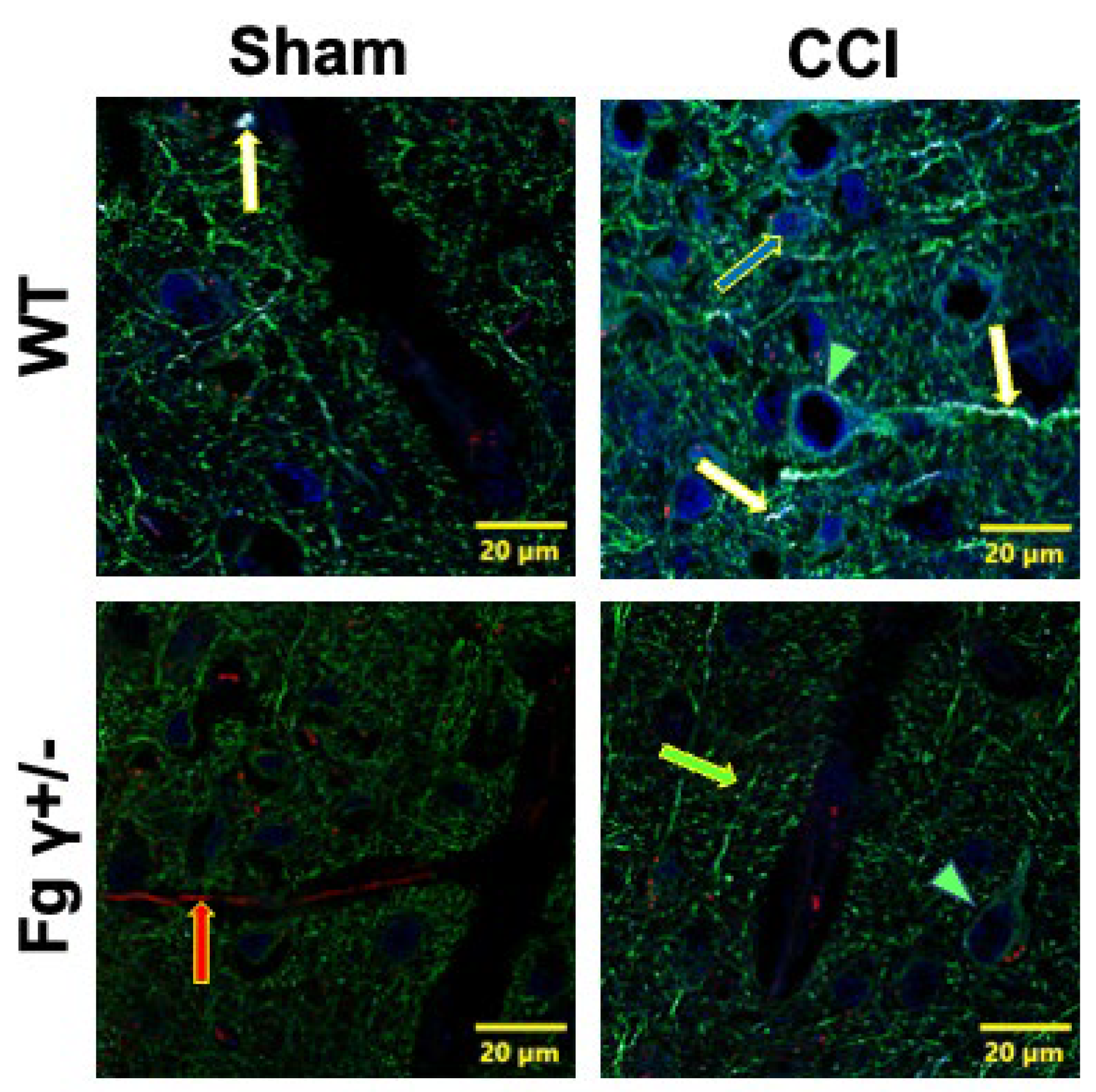
2.4. Association of Extravascularly Deposited Fg with Neurons after CCI
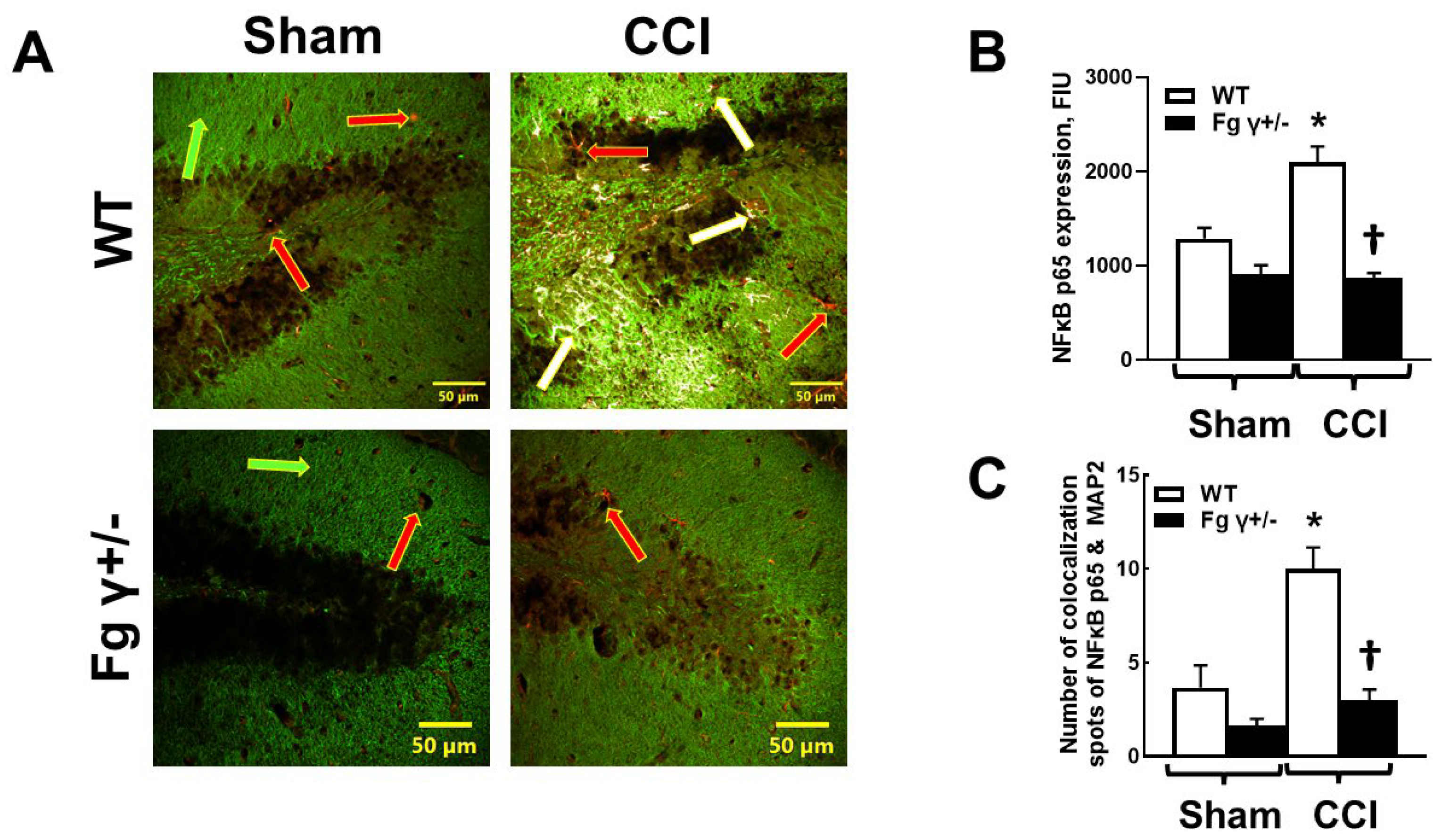
2.5. Expression of Nuclear Factor-кBp65 (NF-кBp65) and Its Colocalization with Neurons in the Brain after CCI
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Permeability Study
4.3. Western Blot
4.4. Immunofluorescence Staining
4.5. Confocal Microscopy
4.6. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hay, J.R.; Johnson, V.E.; Young, A.M.; Smith, D.H.; Stewart, W. Blood-brain barrier disruption is an early event that may persist for many years after traumatic brain injury in humans. J. Neuropathol. Exp. Neurol. 2015, 74, 1147–1157. [Google Scholar]
- Pahatouridis, D.; Alexiou, G.; Zigouris, A.; Mihos, E.; Drosos, D.; Voulgaris, S. Coagulopathy in moderate head injury. The role of early administration of low molecular weight heparin. Brain Inj. 2010, 24, 1189–1192. [Google Scholar] [CrossRef]
- Aono, Y.; Ohkubo, T.; Kikuya, M.; Hara, A.; Kondo, T.; Obara, T.; Metoki, H.; Inoue, R.; Asayama, K.; Shintani, Y.; et al. Plasma fibrinogen, ambulatory blood pressure, and silent cerebrovascular lesions: The Ohasama study. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 963–968. [Google Scholar] [CrossRef]
- Jenkins, D.R.; Craner, M.J.; Esiri, M.M.; DeLuca, G.C. The contribution of fibrinogen to inflammation and neuronal density in human traumatic brain injury. J. Neurotrauma 2018, 35, 2259–2271. [Google Scholar] [CrossRef]
- Redman, C.M.; Xia, H.U.I. Fibrinogen biosynthesis. Ann. N. Y. Acad. Sci. 2001, 936, 480–495. [Google Scholar] [CrossRef]
- Tennent, G.A.; Brennan, S.O.; Stangou, A.J.; O’Grady, J.; Hawkins, P.N.; Pepys, M.B. Human plasma fibrinogen is synthesized in the liver. Blood 2007, 109, 1971–1974. [Google Scholar] [CrossRef]
- Drury, D.R.; McMaster, P.D. The liver as the source of fibrinogen. J. Exp. Med. 1929, 50, 569–578. [Google Scholar] [CrossRef]
- Golanov, E.V.; Sharpe, M.A.; Regnier-Golanov, A.S.; Del Zoppo, G.J.; Baskin, D.S.; Britz, G.W. Fibrinogen Chains Intrinsic to the Brain. Front. Neurosci. 2019, 13, 541. [Google Scholar] [CrossRef]
- Muradashvili, N.; Tyagi, R.; Lominadze, D. A dual-tracer method for differentiating transendothelial transport from paracellular leakage in vivo and in vitro. Front. Physiol. 2012, 3, 166–172. [Google Scholar] [CrossRef]
- Muradashvili, N.; Tyagi, S.C.; Lominadze, D. Localization of fibrinogen in the vasculo-astrocyte interface after cortical contusion injury in mice. Brain Sci. 2017, 7, 77. [Google Scholar] [CrossRef]
- Pleasant, J.; Carlson, S.; Mao, H.; Scheff, S.; Yang, K.; Saatman, K. Rate of neurodegeneration in the mouse controlled cortical impact model is influenced by impactor tip shape: Implications for mechanistic and therapeutic studies. J. Neurotrauma 2011, 28, 2245–2262. [Google Scholar] [CrossRef]
- Johnson, V.E.; Weber, M.T.; Xiao, R.; Cullen, D.K.; Meaney, D.F.; Stewart, W.; Smith, D.H. Mechanical disruption of the blood–brain barrier following experimental concussion. Acta Neuropathol. 2018, 135, 711–726. [Google Scholar] [CrossRef]
- Kossmann, T.; Hans, V.H.; Imhof, H.G.; Stocker, R.; Grob, P.; Trentz, O.; Morganti-Kossmann, C. Intrathecal and serum interleukin-6 and the acute-phase response in patients with severe traumatic brain injuries. Shock 1995, 4, 311–317. [Google Scholar] [CrossRef]
- Muradashvili, N.; Tyagi, R.; Tyagi, N.; Tyagi, S.C.; Lominadze, D. Cerebrovascular disorders caused by hyperfibrinogenemia. J. Physiol. 2016, 594, 5941–5957. [Google Scholar] [CrossRef]
- Clark, V.D.; Layson, A.; Charkviani, M.; Muradashvili, N.; Lominadze, D. Hyperfibrinogenemia-mediated astrocyte activation. Brain Res. 2018, 1699, 158–165. [Google Scholar] [CrossRef]
- Sulimai, N.; Brown, J.; Lominadze, D. Fibrinogen Interaction with Astrocyte ICAM-1 and PrPC Results in the Generation of ROS and Neuronal Death. Int. J. Mol. Sci. 2021, 22, 2391. [Google Scholar] [CrossRef]
- Sulimai, N.; Brown, J.; Lominadze, D. The Role of Nuclear Factor-Kappa B in Fibrinogen-Induced Inflammatory Responses in Cultured Primary Neurons. Biomolecules 2022, 12, 1741. [Google Scholar] [CrossRef]
- Muradashvili, N.; Charkviani, M.; Sulimai, N.; Tyagi, N.; Crosby, J.; Lominadze, D. Effects of fibrinogen synthesis inhibition on vascular cognitive impairment during traumatic brain injury in mice. Brain Res. 2020, 1751, 147208. [Google Scholar] [CrossRef]
- Wilberding, J.A.; Ploplis, V.A.; McLennan, L.; Liang, Z.; Cornelissen, I.V.O.; Feldman, M.; Deford, M.E.; Rosen, E.D.; Castellino, F.J. Development of pulmonary fibrosis in fibrinogen-deficient mice. Ann. N. Y. Acad. Sci. 2001, 936, 542–548. [Google Scholar] [CrossRef]
- Ploplis, V.A.; Wilberding, J.; McLennan, L.; Liang, Z.; Cornelissen, I.; DeFord, M.E.; Rosen, E.D.; Castellino, F.J. A Total Fibrinogen Deficiency Is Compatible with the Development of Pulmonary Fibrosis in Mice. Am. J. Pathol. 2000, 157, 703–708. [Google Scholar] [CrossRef]
- Chuah, Y.M.L.; Maybery, M.T.; Fox, A.M. The long-term effects of mild head injury on short-term memory for visual form, spatial location, and their conjunction in well-functioning university students. Brain Cogn. 2004, 56, 304–312. [Google Scholar] [CrossRef]
- Muradashvili, N.; Benton, R.L.; Saatman, K.E.; Tyagi, S.C.; Lominadze, D. Ablation of matrix metalloproteinase-9 gene decreases cerebrovascular permeability and fibrinogen deposition post traumatic brain injury in mice. Metab. Brain Dis. 2015, 30, 411–426. [Google Scholar] [CrossRef]
- Chung, E.; Ji, Y.; Sun, Y.; Kascsak, R.; Kascsak, R.; Mehta, P.; Strittmatter, S.; Wisniewski, T. Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer’s disease model mouse. BMC Neurosci. 2010, 11, 130. [Google Scholar] [CrossRef]
- Gimbel, D.A.; Nygaard, H.B.; Coffey, E.E.; Gunther, E.C.; Laurén, J.; Gimbel, Z.A.; Strittmatter, S.M. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 2010, 30, 6367–6374. [Google Scholar] [CrossRef]
- Chhabra, G.; Rangarajan, K.; Subramanian, A.; Agrawal, D.; Sharma, S.; Mukhopadhayay, A. Hypofibrinogenemia in isolated traumatic brain injury in Indian patients. Neurol. India 2010, 58, 756–757. [Google Scholar]
- Meizoso, J.P.; Moore, E.E.; Pieracci, F.M.; Saberi, R.A.; Ghasabyan, A.; Chandler, J.; Namias, N.; Sauaia, A. Role of Fibrinogen in Trauma-Induced Coagulopathy. J. Am. Coll. Surg. 2022, 234, 465–473. [Google Scholar] [CrossRef]
- Davalos, D.; Akassoglou, K. Fibrinogen as a key regulator of inflammation in disease. Semin. Immunopathol. 2012, 34, 43–62. [Google Scholar] [CrossRef]
- Danesh, J.; Lewington, S.; Thompson, S.G.; Lowe, G.D.; Collins, R.; Kostis, J.B.; Wilson, A.C.; Folsom, A.R.; Wu, K.; Benderly, M.; et al. Plasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality: An individual participant meta-analysis. JAMA 2005, 294, 1799–1809. [Google Scholar]
- Sulimai, N.; Brown, J.; Lominadze, D. The Effects of Fibrinogen’s Interactions with Its Neuronal Receptors, Intercellular Adhesion Molecule-1 and Cellular Prion Protein. Biomolecules 2021, 11, 1381. [Google Scholar] [CrossRef]
- Hsiao, T.W.; Swarup, V.P.; Kuberan, B.; Tresco, P.A.; Hlady, V. Astrocytes specifically remove surface-adsorbed fibrinogen and locally express chondroitin sulfate proteoglycans. Acta Biomater. 2013, 9, 7200–7208. [Google Scholar] [CrossRef]
- Kwon, J.W.; Kwon, H.K.; Shin, H.J.; Choi, Y.M.; Anwar, M.A.; Choi, S. Activating transcription factor 3 represses inflammatory responses by binding to the p65 subunit of NF-κB. Sci. Rep. 2015, 5, 14470. [Google Scholar] [CrossRef]
- Shih, R.-H.; Wang, C.-Y.; Yang, C.-M. NF-kappaB signaling pathways in neurological inflammation: A mini review. Front. Mol. Neurosci. 2015, 8, 77. [Google Scholar] [CrossRef]
- Xu, G.; Zhang, H.; Zhang, S.; Fan, X.; Liu, X. Plasma fibrinogen is associated with cognitive decline and risk for dementia in patients with mild cognitive impairment. Int. J. Clin. Pract. 2008, 62, 1070–1075. [Google Scholar] [CrossRef]
- Horsburgh, K.; Cole, G.M.; Yang, F.; Savage, M.J.; Greenberg, B.D.; Gentleman, S.M.; Graham, D.I.; Nicoll, J.A. beta-amyloid (Abeta)42(43), abeta42, abeta40 and apoE immunostaining of plaques in fatal head injury. Neuropathol. Appl. Neurobiol. 2000, 26, 124–132. [Google Scholar] [CrossRef]
- Ahn, H.; Glickman, J.; Poon, K.; Zamolodchikov, D.; Jno-Charles, O.; Norris, E.; Strickland, S. A novel A[beta]-fibrinogen interaction inhibitor rescues altered thrombosis and cognitive decline in Alzheimer’s disease mice. J. Exp. Med. 2014, 211, 1049–1062. [Google Scholar] [CrossRef]
- Ahn, H.J.; Zamolodchikov, D.; Cortes-Canteli, M.; Norris, E.H.; Glickman, J.F.; Strickland, S. Alzheimer’s disease peptide β-amyloid interacts with fibrinogen and induces its oligomerization. Proc. Natl. Acad. Sci. USA 2010, 107, 21812–21817. [Google Scholar] [CrossRef]
- Rubenstein, R.; Chang, B.; Grinkina, N.; Drummond, E.; Davies, P.; Ruditzky, M.; Sharma, D.; Wang, K.; Wisniewski, T. Tau phosphorylation induced by severe closed head traumatic brain injury is linked to the cellular prion protein. Acta Neuropathol. Commun. 2017, 5, 30. [Google Scholar] [CrossRef]
- Pekny, M.; Pekna, M. Astrocyte reactivity and reactive astrogliosis: Costs and benefits. Physiol. Rev. 2014, 94, 1077–1098. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef]
- Ota, Y.; Zanetti, A.T.; Hallock, R.M. The Role of Astrocytes in the Regulation of Synaptic Plasticity and Memory Formation. Neural Plast. 2013, 2013, 11. [Google Scholar] [CrossRef]
- Ryu, J.K.; Petersen, M.A.; Murray, S.G.; Baeten, K.M.; Meyer-Franke, A.; Chan, J.P.; Vagena, E.; Bedard, C.; Machado, M.R.; Rios Coronado, P.E.; et al. Blood coagulation protein fibrinogen promotes autoimmunity and demyelination via chemokine release and antigen presentation. Nat. Commun. 2015, 6, 8164. [Google Scholar] [CrossRef]
- Suh, T.T.; Holmback, K.; Jensen, N.J.; Daugherty, C.C.; Small, K.; Simon, D.I.; Potter, S.; Degen, J.L. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev. 1995, 9, 2020–2033. [Google Scholar] [CrossRef]
- Huang, S.; Mulvihill, E.R.; Farrell, D.H.; Chung, D.W.; Davie, E.W. Biosynthesis of human fibrinogen. Subunit interactions and potential intermediates in the assembly. J. Biol. Chem. 1993, 268, 8919–8926. [Google Scholar] [CrossRef]
- Altieri, D.C.; Duperray, A.; Plescia, J.; Thornton, G.B.; Languino, L.R. Structural recognition of a novel fibrinogen gamma chain sequence (117–133) by intercellular adhesion molecule-1 mediates leukocyte-endothelium interaction. J. Biol. Chem. 1995, 270, 696–699. [Google Scholar] [CrossRef]
- Plow, E.F.; Haas, T.A.; Zhang, L.; Loftus, J.; Smith, J.W. Ligand binding to integrins. J. Biol. Chem. 2000, 275, 21785–21788. [Google Scholar] [CrossRef]
- Pluskota, E.; D’Souza, S.E. Fibrinogen interactions with ICAM-1 (CD54) regulate endothelial cell survival. Eur. J. Biochem. 2000, 267, 4693–4704. [Google Scholar] [CrossRef]
- Fazio, S. Fibrates—The Other Life-saving Lipid Drugs. US Cardiol. 2004, 1, 1–6. [Google Scholar] [CrossRef]
- Mazoyer, E.; Ripoll, L.; Boisseau, M.R.; Drouet, L. How does ticlopidine treatment lower plasma fibrinogen? Thromb. Res. 1994, 75, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Wager-Miller, J.; Murphy Green, M.; Shafique, H.; Mackie, K. Collection of Frozen Rodent Brain Regions for Downstream Analyses. J. Vis. Exp. 2020, 158, e60474. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment Groups | WT-Sham | WT-CCI | Fg γ+/−-Sham | Fg γ+/−-CCI |
|---|---|---|---|---|
| Total area, μm2 | 9.6 ± 3.7 | 63.7 ± 2.5 | 0.2 ± 0.2 | 23.0 ± 4.8 |
| Area Fraction, % | 0.013 ± 0.008 | 0.11 0 ± 0.004 | 0.033 ± 0.020 | 0.030 ± 0.012 |
| R(r) | 0.20 ± 0.05 | 0.44 ± 0.05 | 0.135 ± 0.141 | 0.103 ± 0.072 |
| M1 | 0 | 0.008 ± 0.003 | 0.003 ± 0.003 | 0 |
| M1 | 0.018 ± 0.005 | 0.105 ± 0.023 | 0.030 ± 0.015 | 0.043 ± 0.012 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sulimai, N.; Brown, J.; Lominadze, D. The Effect of Reduced Fibrinogen on Cerebrovascular Permeability during Traumatic Brain Injury in Fibrinogen Gene Heterozygous Knockout Mice. Biomolecules 2024, 14, 385. https://doi.org/10.3390/biom14040385
Sulimai N, Brown J, Lominadze D. The Effect of Reduced Fibrinogen on Cerebrovascular Permeability during Traumatic Brain Injury in Fibrinogen Gene Heterozygous Knockout Mice. Biomolecules. 2024; 14(4):385. https://doi.org/10.3390/biom14040385
Chicago/Turabian StyleSulimai, Nurul, Jason Brown, and David Lominadze. 2024. "The Effect of Reduced Fibrinogen on Cerebrovascular Permeability during Traumatic Brain Injury in Fibrinogen Gene Heterozygous Knockout Mice" Biomolecules 14, no. 4: 385. https://doi.org/10.3390/biom14040385
APA StyleSulimai, N., Brown, J., & Lominadze, D. (2024). The Effect of Reduced Fibrinogen on Cerebrovascular Permeability during Traumatic Brain Injury in Fibrinogen Gene Heterozygous Knockout Mice. Biomolecules, 14(4), 385. https://doi.org/10.3390/biom14040385