
α-Methylacyl-CoA Racemase from Mycobacterium tuberculosis—Detailed Kinetic and Structural Characterization of the Active Site


Abstract
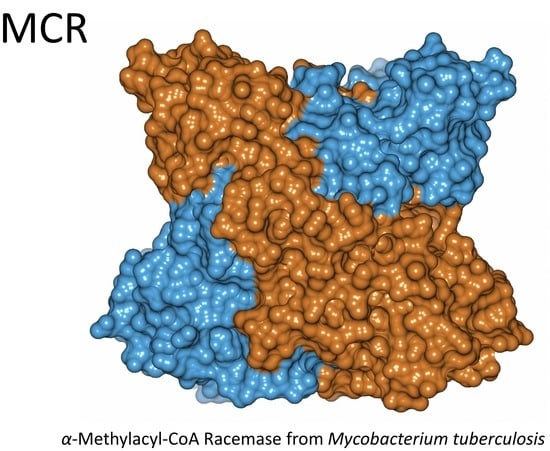
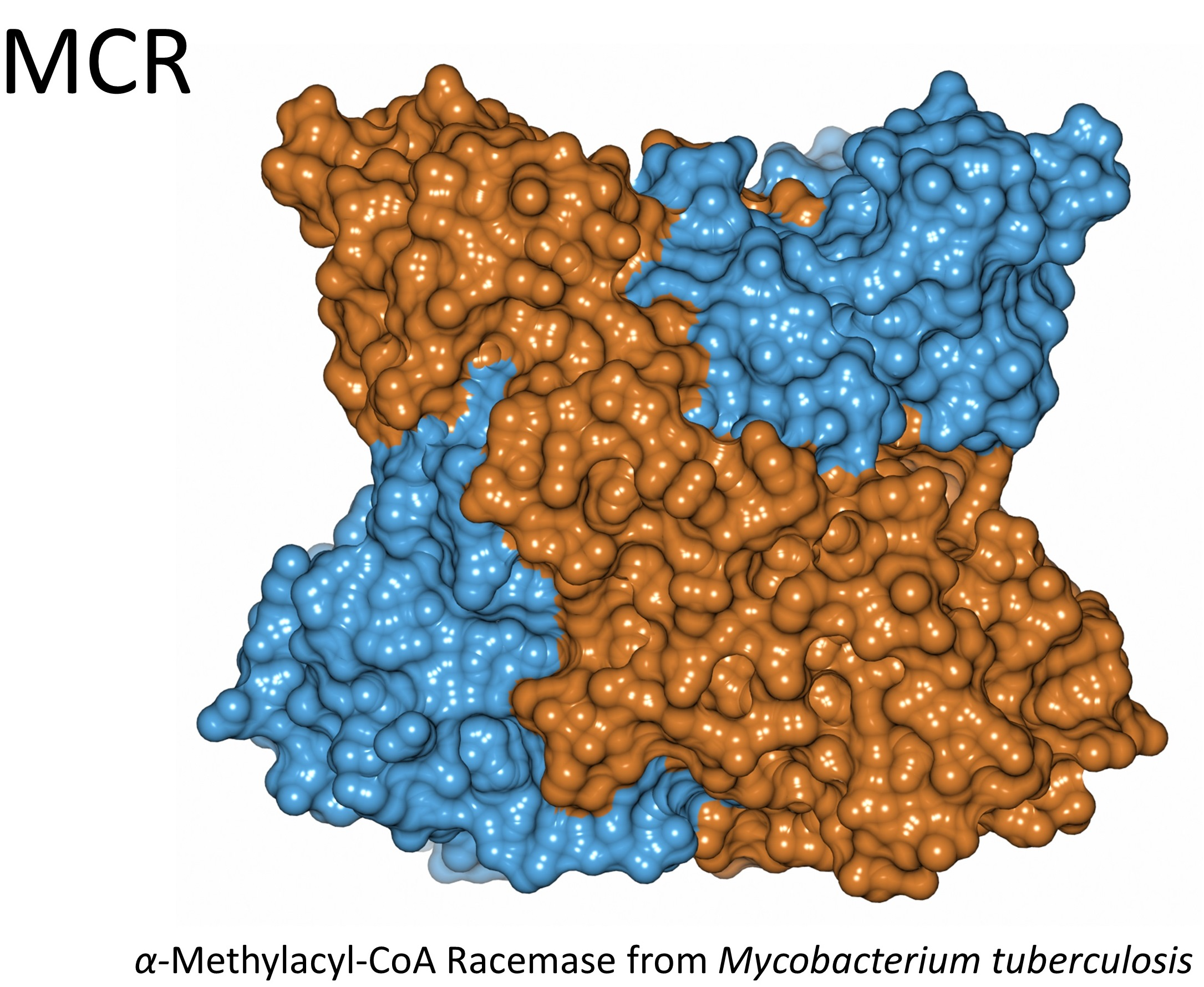
1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. 1H NMR Analysis
2.3. Colorimetric Assay
2.4. X-ray Crystallography
3. Results and Discussion
3.1. Biophysical Characterization of the Purified Wild-Type MCR and Three Active-Site Mutants
3.2. Enzymatic Activity Assays
3.3. Kinetic Assay
3.4. Crystal Structure of Wild-Type MCR in a New Form
3.5. Crystal Structures and Hydrogen Bond Interactions of Three MCR Mutants at the Active Site
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lloyd, M.D.; Darley, D.J.; Wierzbicki, A.S.; Threadgill, M.D. α-Methylacyl-CoA racemase: An ‘obscure’ metabolic enzyme takes centre stage. FEBS J. 2008, 275, 1089–1102. [Google Scholar] [CrossRef]
- Lloyd, M.D.; Yevglevskis, M.; Lee, G.L.; Wood, P.J.; Threadgill, M.D.; Woodman, T.J. α-Methylacyl-CoA racemase (AMACR): Metabolic enzyme, drug metabolizer and cancer marker P504S. Prog. Lipid Res. 2013, 52, 220–230. [Google Scholar] [CrossRef]
- Sakai, Y.; Takahashi, H.; Wakasa, Y.; Kotani, T.; Yurimoto, H.; Miyachi, N.; Van Veldhoven, P.P.; Kato, N. Role of α-methylacyl coenzyme A racemase in the degradation of methyl-branched alkanes by Mycobacterium sp strain P101. J. Bacteriol. 2004, 186, 7214–7220. [Google Scholar] [CrossRef]
- Carnell, A.J.; Hale, I.; Denis, S.; Wanders, R.J.A.; Isaacs, W.B.; Wilson, B.; Ferdinandusse, S. Design, synthesis, and in vitro testing of α-methylacyl-CoA racemase inhibitors. J. Med. Chem. 2007, 50, 2700–2707. [Google Scholar] [CrossRef]
- Carnell, A.J.; Kirk, R.; Smith, M.; McKenna, S.; Lian, L.-Y.; Gibson, R. Inhibition of human α-methylacyl-CoA racemase (AMACR): A target for prostate cancer. ChemMedChem 2013, 8, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.A.P.; Wang, H.; Nacev, B.A.; Mease, R.C.; Liu, J.O.; Pomper, M.G.; Isaacs, W.B. High-throughput screen identifies novel inhibitors of cancer biomarker α-methylacyl coenzyme A racemase (AMACR/P504S). Mol. Cancer Ther. Mol. Cancer Ther. 2011, 10, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Festuccia, C.; Gravina, G.L.; Mancini, A.; Muzi, P.; Di Cesare, E.; Kirk, R.; Smith, M.; Hughes, S.; Gibson, R.; Lian, L.-Y.; et al. Trifluoroibuprofen inhibits α-methylacyl coenzyme A racemase (AMACR/P504S), reduces cancer cell proliferation and inhibits in vivo tumor growth in aggressive prostate cancer models. Anti-Cancer Agents Med. Chem. 2014, 14, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, M.D.; Yevglevskis, M.; Nathubhai, A.; James, T.D.; Threadgill, M.D.; Woodman, T.J. Racemases and epimerases operating through a 1,1-proton transfer mechanism: Reactivity, mechanism and inhibition. Chem. Soc. Rev. 2021, 50, 5952–5984. [Google Scholar] [CrossRef] [PubMed]
- Pal, M.; Khanal, M.; Marko, R.; Thirumalairajan, S.; Bearne, S.L. Rational design and synthesis of substrate-product analogue inhibitors of α-methylacyl-coenzyme A racemase from Mycobacterium tuberculosis. Chem. Commun. (Camb.) 2016, 52, 2740–2743. [Google Scholar] [CrossRef] [PubMed]
- Pal, M.; Easton, N.M.; Yaphe, H.; Bearne, S.L. Potent dialkyl substrate-product analogue inhibitors and inactivators of α-methylacyl-coenzyme A racemase from Mycobacterium tuberculosis by rational design. Bioorg. Chem. 2018, 77, 640–650. [Google Scholar] [CrossRef]
- Savolainen, K.; Bhaumik, P.; Schmitz, W.; Kotti, T.J.; Conzelmann, E.; Wierenga, R.K.; Hiltunen, J.K. α-Methylacyl-CoA racemase from Mycobacterium tuberculosis: Mutational and structural characterization of the active site and the fold. J. Biol. Chem. 2005, 280, 12611–12620. [Google Scholar] [CrossRef]
- Sharma, S.; Bhaumik, P.; Schmitz, W.; Venkatesan, R.; Hiltunen, J.K.; Conzelmann, E.; Juffer, A.H.; Wierenga, R.K. The enolization chemistry of a thioester-dependent racemase: The 1.4 Å crystal structure of a reaction intermediate complex characterized by detailed QM/MM calculations. J. Phys. Chem. B 2012, 116, 3619–3629. [Google Scholar] [CrossRef]
- Bhaumik, P.; Schmitz, W.; Hassinen, A.; Hiltunen, J.K.; Conzelmann, E.; Wierenga, R.K. The catalysis of the 1,1-proton transfer by α-methyl-acyl-CoA racemase is coupled to a movement of the fatty acyl moiety over a hydrophobic, methionine-rich surface. J. Mol. Biol. 2007, 367, 1145–1161. [Google Scholar] [CrossRef]
- Rhee, K.H.; Lee, K.S.; Priyadarshi, A.; Kim, E.E.; Hwang, K.Y. Crystallization and preliminary X-ray crystallographic studies of fatty acid-CoA racemase from Mycobacterium tuberculosis H37Rv. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2005, 61, 1017–1019. [Google Scholar] [CrossRef]
- Lee, K.S.; Park, S.M.; Rhee, K.H.; Bang, W.G.; Hwang, K.; Chi, Y.M. Crystal structure of fatty acid-CoA racemase from Mycobacterium tuberculosis H37Rv. Prot. Struct. Funct. Bioinform. 2006, 64, 817–822. [Google Scholar] [CrossRef]
- Mojanaga, O.O.; Acharya, K.R.; Lloyd, M.D. Recombinant protein production for structural and kinetic studies: A case study using M. tuberculosis α-methylacyl-CoA racemase (MCR). Methods Enzymol. 2023, 690, 1–37. [Google Scholar] [CrossRef]
- Yevglevskis, M.; Lee, G.L.; Nathubhai, A.; Petrova, Y.D.; James, T.D.; Threadgill, M.D.; Woodman, T.J.; Lloyd, M.D. A novel colorimetric assay for α-methylacyl-CoA racemase 1A (AMACR; P504S) utilizing the elimination of 2,4-dinitrophenolate. Chem. Commun. (Camb.) 2017, 53, 5087–5090. [Google Scholar] [CrossRef] [PubMed]
- Woodman, T.J.; Lloyd, M.D. Analysis of enzyme reactions using NMR techniques: A case study with α-methylacyl-CoA racemase (AMACR). Methods Enzymol. 2023, 690, 159–209. [Google Scholar] [CrossRef] [PubMed]
- Olp, M.D.; Kalous, K.S.; Smith, B.C. ICEKAT: An interactive online tool for calculating initial rates from continuous enzyme kinetic traces. BMC Bioinf. 2020, 21, 186. [Google Scholar] [CrossRef]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Agirre, J.; Atanasova, M.; Bagdonas, H.; Ballard, C.B.; Basle, A.; Beilsten-Edmands, J.; Borges, R.J.; Brown, D.G.; Burgos-Marmol, J.J.; Berrisford, J.M.; et al. The CCP4 suite: Integrative software for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2023, 79, 449–461. [Google Scholar] [CrossRef]
- Waterman, D.G.; Winter, G.; Gildea, R.J.; Parkhurst, J.M.; Brewster, A.S.; Sauter, N.K.; Evans, G. Diffraction-geometry refinement in the DIALS framework. Acta Crystallogr. D Struct. Biol. 2016, 72, 558–575. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2009, 66, 12–21. [Google Scholar] [CrossRef]
- McNicholas, S.; Potterton, E.; Wilson, K.S.; Noble, M.E.M. Presenting your structures: The CCP4mg molecular-graphics software. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 386–394. [Google Scholar] [CrossRef]
- Zarycz, M.N.C.; Fonseca Guerra, C. NMR 1H-shielding constants of hydrogen-bond donor reflect manifestation of the Pauli principle. J. Phys. Chem. Lett. 2018, 9, 3720–3724. [Google Scholar] [CrossRef]
- Yevglevskis, M.; Bowskill, C.R.; Chan, C.C.Y.; Heng, J.H.-J.; Threadgill, M.D.; Woodman, T.J.; Lloyd, M.D. A study on the chiral inversion of mandelic acid in humans. Org. Biomol. Chem. 2014, 12, 6737–6744. [Google Scholar] [CrossRef]
- Woodman, T.J.; Wood, P.J.; Thompson, A.S.; Hutchings, T.J.; Steel, G.R.; Jiao, P.; Threadgill, M.D.; Lloyd, M.D. Chiral inversion of 2-arylpropionyl-CoA esters by α-methylacyl-CoA racemase 1A (AMACR, P504S). Chem. Commun. (Camb.). 2011, 47, 7332–7334. [Google Scholar] [CrossRef]
- Yevglevskis, M.; Lee, G.L.; Nathubhai, A.; Petrova, Y.D.; James, T.D.; Threadgill, M.D.; Woodman, T.J.; Lloyd, M.D. Structure-activity relationships of rationally designed AMACR 1A inhibitors. Bioorg. Chem. 2018, 79, 145–154. [Google Scholar] [CrossRef]
- Ouazia, D.; Bearne, S.L. A continuous assay for α-methylacyl-coenzyme A racemase using circular dichroism. Anal. Biochem. 2010, 398, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Shieh, W.R.; Chen, C.S. Purification and characterization of novel 2-arylpropionyl-CoA epimerases from rat-liver cytosol and mitochondria. J. Biol. Chem. 1993, 268, 3487–3493. [Google Scholar] [CrossRef] [PubMed]
- Darley, D.J.; Butler, D.S.; Prideaux, S.J.; Thornton, T.W.; Wilson, A.D.; Woodman, T.J.; Threadgill, M.D.; Lloyd, M.D. Synthesis and use of isotope-labelled substrates for a mechanistic study on human α-methylacyl-CoA racemase 1A (AMACR, P504S). Org. Biomol. Chem. 2009, 7, 543–552. [Google Scholar] [CrossRef]
- Lloyd, M.D. High-throughput screening for the discovery of enzyme inhibitors. J. Med. Chem. 2020, 63, 10742–10772. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Km (µM) | Vmax (µmol.min−1.mg−1) | kcat (s−1) | kcat/Km (M−1.s−1) |
|---|---|---|---|---|
| MCR | 96 ± 14 | 157 ± 13 | 102 ± 8.3 | 1.07 × 106 |
| MCR H126A | 63.0 ± 5.5 | 11 ± 0.4 | 7.2 ± 0.3 | 0.11 × 106 |
| MCR D156A | 70.0 ± 9.7 | 48 ± 3 | 31 ± 2.1 | 0.44 × 106 |
| MCR E241A | 24.0 ± 3.1 | 23 ± 1 | 15 ± 0.7 | 0.63 × 106 |
| AMACR 1A [17] | 58.0 | 0.112 | 0.088 | 1517 |
| Wild-Type MCR | MCR H126A | MCR D156A | MCR E241A | |
|---|---|---|---|---|
| Beamline | I04 | I03 | I04 | I03 |
| Wavelength used (Å) | 0.9537 | 0.9762 | 0.9537 | 0.9762 |
| Crystallographic statistics | ||||
| Space group | C2 | C2 | C2 | C2 |
| Unit-cell dimensions | ||||
| a, b, c (Å) | 181.05, 79.09, 59.19 | 181.11, 79.15, 59.10 | 180.47, 78.75, 58.94 | 180.62, 78.99, 58.923 |
| α, β, ϒ (°) | 90.00, 92.01, 90.00 | 90.00, 91.66, 90.00 | 90.00, 92.151, 90.00 | 90.00, 92.19, 90.00 |
| Resolution-range (Å) | 90.47–1.65 (1.68–1.65) | 90.52–2.45 (2.55–2.45) | 90.17–1.64 (1.67–1.64) | 90.24–1.85 (1.89–1.85) |
| Rmerge (%) | 0.098 (2.622) | 0.422 (3.294) | 0.100 (2.265) | 0.139 (1.987) |
| Rpim (%) | 0.040 (1.065) | 0.159 (1.442) | 0.041 (0.908) | 0.057 (0.813) |
| CC1/2 (%) | 0.999 (0.320) | 0.982 (0.278) | 0.998 (0.372) | 0.998 (0.500) |
| Mean < I/σ(I) > | 8.9 (0.7) | 4.9 (0.8) | 9.9 (0.9) | 8.2 (0.9) |
| Completeness (%) | 100 (100) | 100 (100) | 99.0 (98.2) | 100 (100) |
| No. of observed reflections | 679637 (33777) | 214015 (20978) | 688384 (34415) | 478293 (30079) |
| No. of unique reflections | 100251 (4908) | 30851 (3454) | 99692 (4868) | 70688 (4349) |
| Multiplicity | 6.8 (6.9) | 6.9 (6.1) | 6.9 (7.1) | 6.8 (6.9) |
| Refinement statistics | ||||
| Rwork/Rfree | 0.19/0.23 | 0.19/0.26 | 0.18/0.21 | 0.19/0.24 |
| RMSD in bond lengths (Å) | 0.011 | 0.016 | 0.016 | 0.016 |
| RMSD in bond angles (°) | 1.77 | 2.58 | 2.14 | 2.38 |
| Ramachandran plot statistics (%) | ||||
| Favoured | 96.29 | 94.00 | 96.86 | 94.29 |
| Allowed | 2.79 | 5.71 | 3.14 | 5.28 |
| Outliers | 0.92 | 0.29 | 0.0 | 0.43 |
| Average B-Factors (Å2) | ||||
| Protein | 33.31 | 48.5 | 29.84 | 33.59 |
| Water | 37.16 | 33.02 | 34.48 | 37.34 |
| No. Atoms | ||||
| Protein | 5387 | 5373 | 5380 | 5371 |
| Water | 421 | 61 | 429 | 413 |
| Subunit 1 | |||||
|---|---|---|---|---|---|
| Donor | Acceptor | MCR Wild-Type | MCR H126A | MCR D156A | MCR E241A |
| His126 Nδ1 | PEG | 3.18 | - | 2.96 | 3.08 |
| PEG | 3.16 | - | - | 3.09 | |
| Water | 3.10 | - | - | - | |
| His126 Nε2 | Glu241 Oε1/B | 2.70 | - | 2.65 | - |
| Glu241 Oε2/B | 3.24 | - | 3.23 | - | |
| Water | - | - | - | 2.84 | |
| Asp156 Oδ1 | Water | 2.64 | 2.70 | - | 2.55 |
| Asp156 Oδ2 | Asn152 Nδ2 | 2.82 | 2.99 | - | 2.83 |
| PEG | 2.78 | 2.51 | - | 2.77 | |
| Ala241 N | Water | - | - | - | 3.12 |
| Glu241 Oε1 | His126 Nε2/B | 2.65 | - | 2.61 | - |
| Glu241 Oε2 | His126 Nε2/B | 3.27 | - | 3.25 | - |
| Water | 3.04 | - | 3.17 | - | |
| Subunit 2 | |||||
| Donor | Acceptor | MCR Wild-Type | MCR H126A | MCR D156A | MCR E241A |
| His126 Nδ1 | PEG | 3.19 | - | 3.25 | 3.00 |
| PEG | - | - | - | 3.30 | |
| Water | 3.14 | - | - | - | |
| His126 Nε2 | Glu241 Oε1/A | 2.65 | - | 2.61 | - |
| Glu241 Oε2/A | 3.27 | - | 3.25 | - | |
| Water | - | - | - | 2.80 | |
| Asp156 Oδ1 | Water | 2.72 | - | - | 2.57 |
| Asp156 Oδ2 | Asn152 Nδ2 | 2.87 | 2.99 | - | 2.98 |
| PEG | 2.67 | 2.69 | - | 2.65 | |
| Water | - | 2.85 | - | 2.67 | |
| Ala241 N | Water | 3.00 | |||
| Glu241 Oε1 | His126 Nε2/A | 2.70 | - | 2.65 | - |
| Glu241 Oε2 | His126 Nε2/A | 3.24 | - | 3.23 | - |
| Water | - | - | - | ||
| Gln243 Nε2/B | - | 3.07 | - | - | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mojanaga, O.O.; Woodman, T.J.; Lloyd, M.D.; Acharya, K.R. α-Methylacyl-CoA Racemase from Mycobacterium tuberculosis—Detailed Kinetic and Structural Characterization of the Active Site. Biomolecules 2024, 14, 299. https://doi.org/10.3390/biom14030299
Mojanaga OO, Woodman TJ, Lloyd MD, Acharya KR. α-Methylacyl-CoA Racemase from Mycobacterium tuberculosis—Detailed Kinetic and Structural Characterization of the Active Site. Biomolecules. 2024; 14(3):299. https://doi.org/10.3390/biom14030299
Chicago/Turabian StyleMojanaga, Otsile O., Timothy J. Woodman, Matthew D. Lloyd, and K. Ravi Acharya. 2024. "α-Methylacyl-CoA Racemase from Mycobacterium tuberculosis—Detailed Kinetic and Structural Characterization of the Active Site" Biomolecules 14, no. 3: 299. https://doi.org/10.3390/biom14030299
APA StyleMojanaga, O. O., Woodman, T. J., Lloyd, M. D., & Acharya, K. R. (2024). α-Methylacyl-CoA Racemase from Mycobacterium tuberculosis—Detailed Kinetic and Structural Characterization of the Active Site. Biomolecules, 14(3), 299. https://doi.org/10.3390/biom14030299