


Characteristics of Developmental and Epileptic Encephalopathy Associated with PACS2 p.Glu209Lys Pathogenic Variant—Our Experience and Systematic Review of the Literature
 , ,
, ,  , and
, and
Abstract
1. Introduction
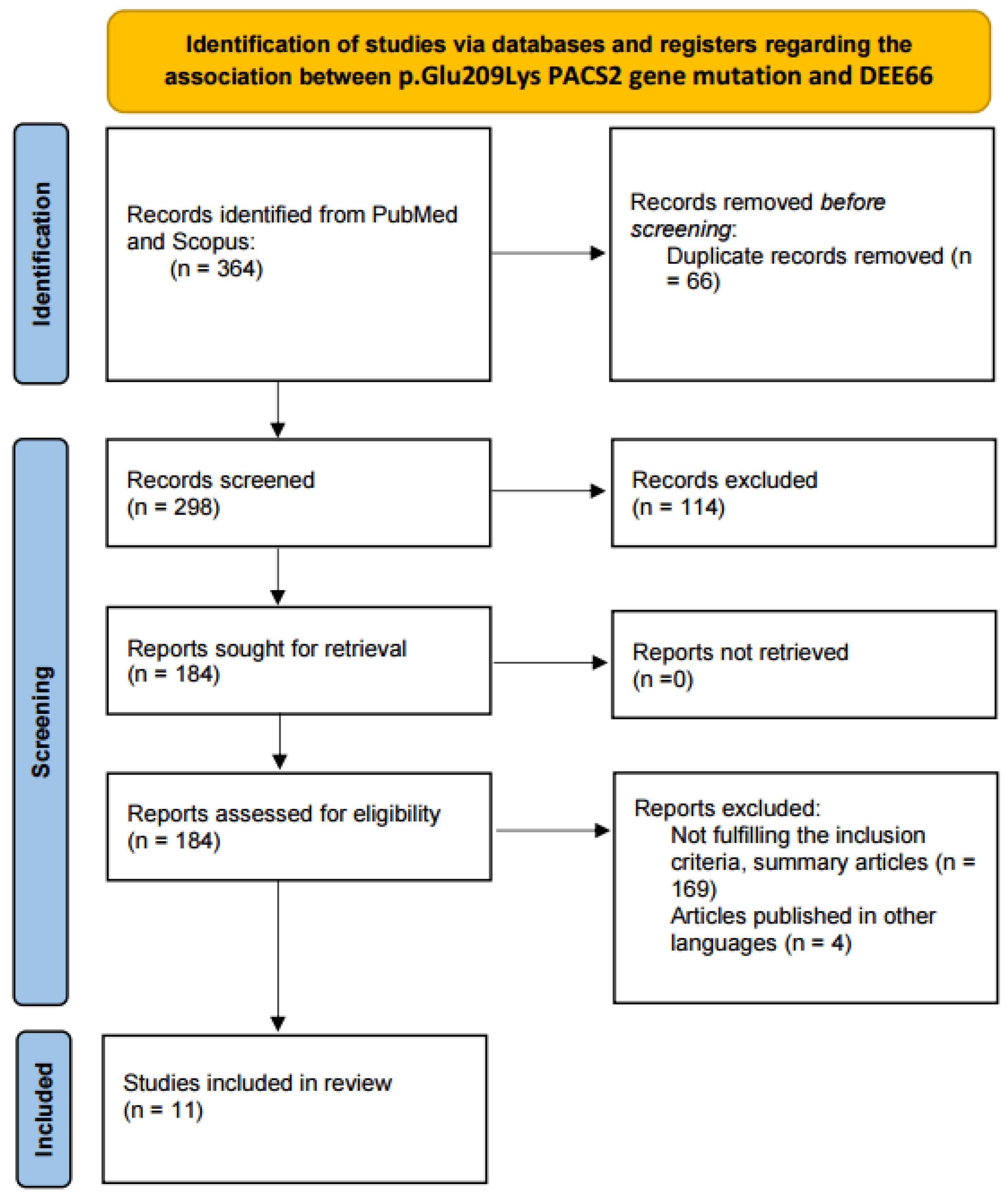
2. Material and Methods
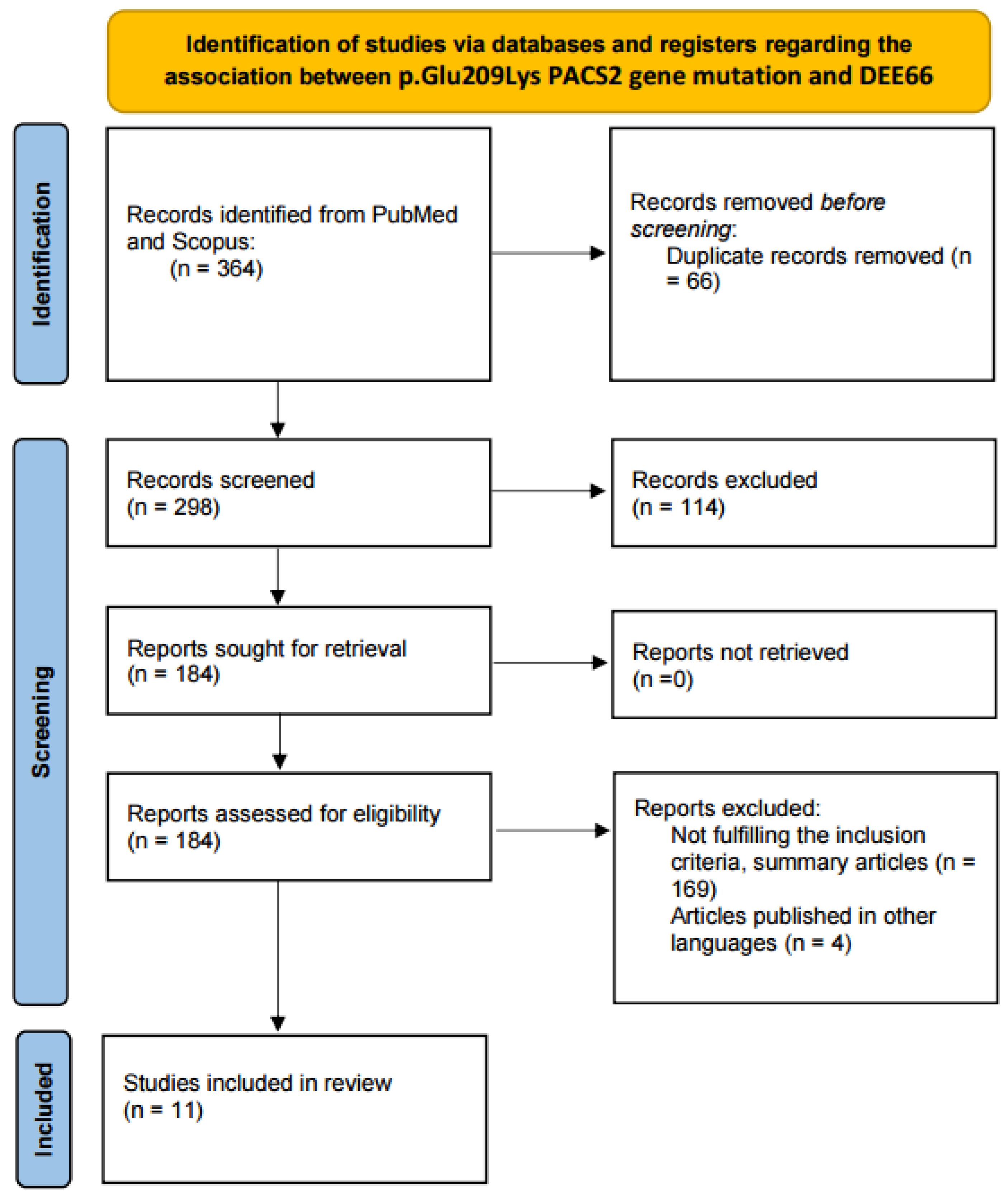
2.1. Literature Review
2.1.1. Information Sources and Search Strategy
2.1.2. Eligibility Criteria
2.2. The Methodology Used to Examine Our Own Patient
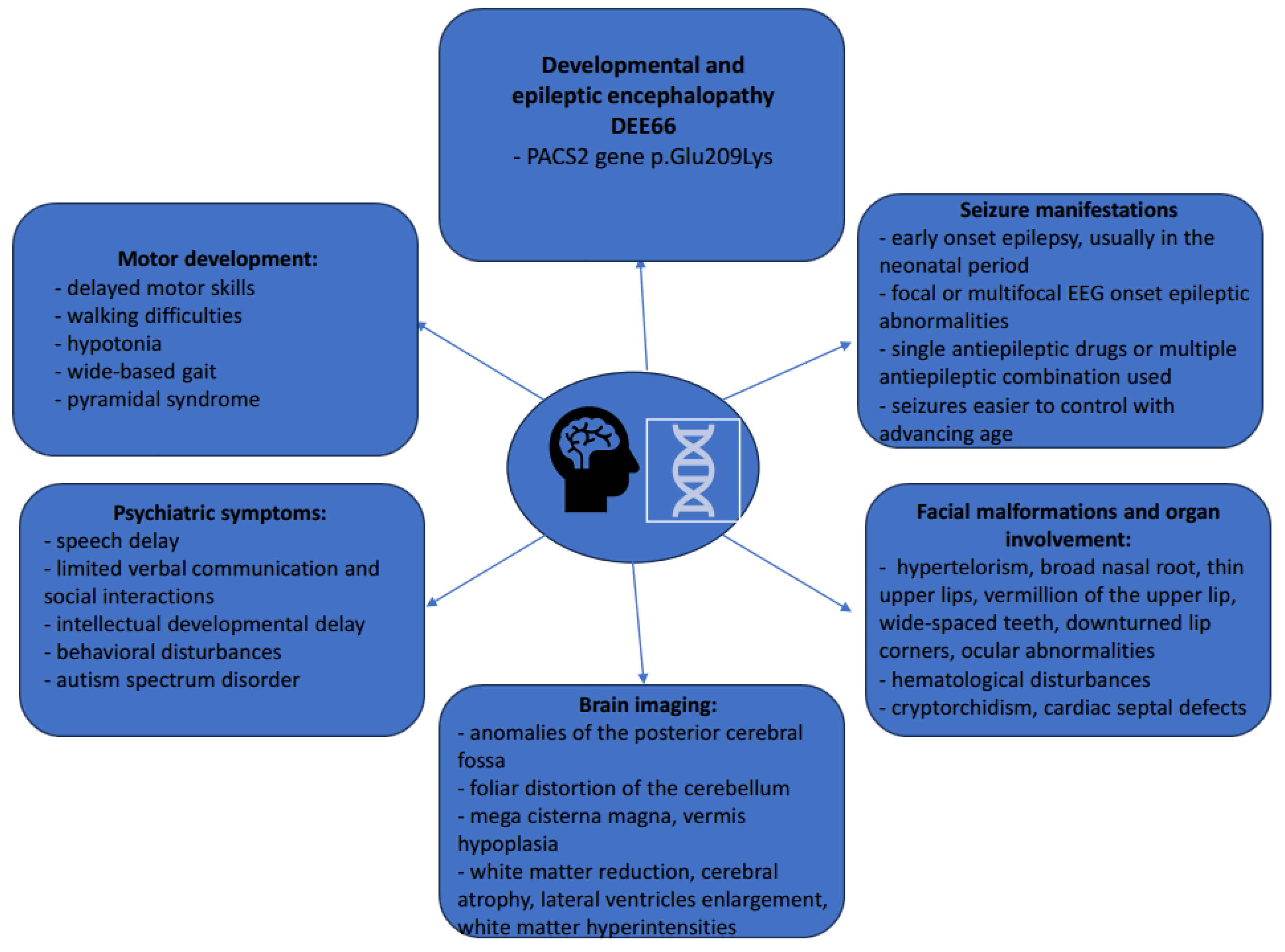
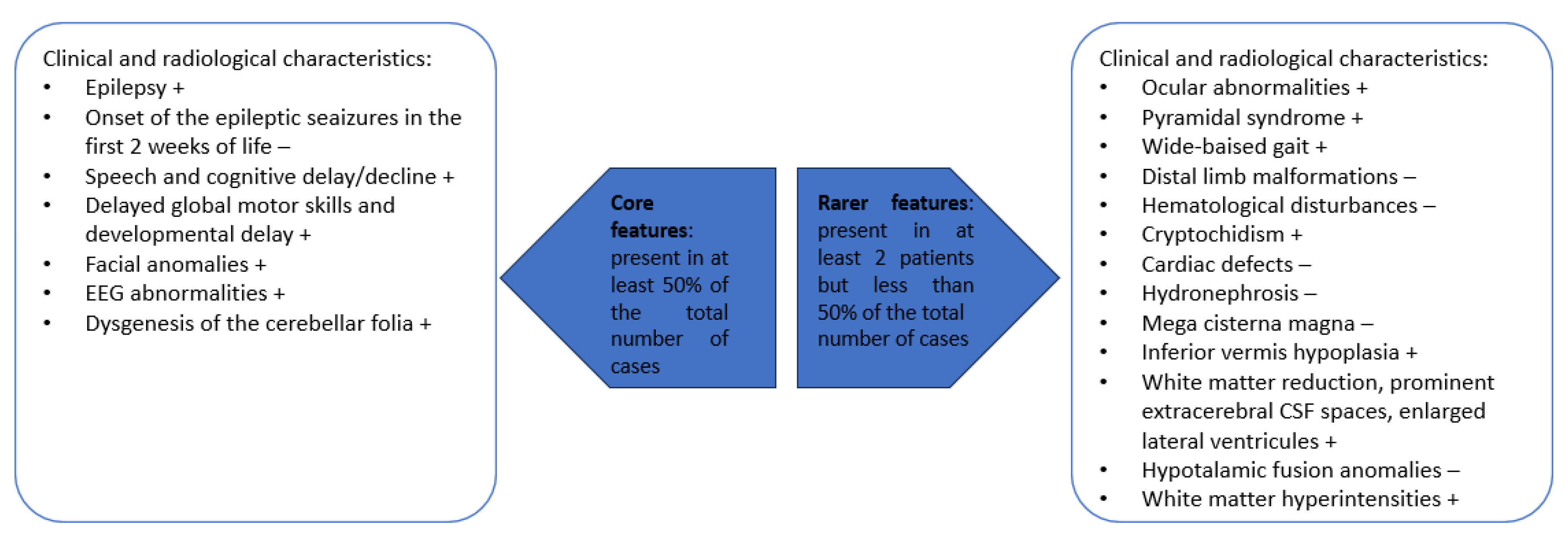
3. Results
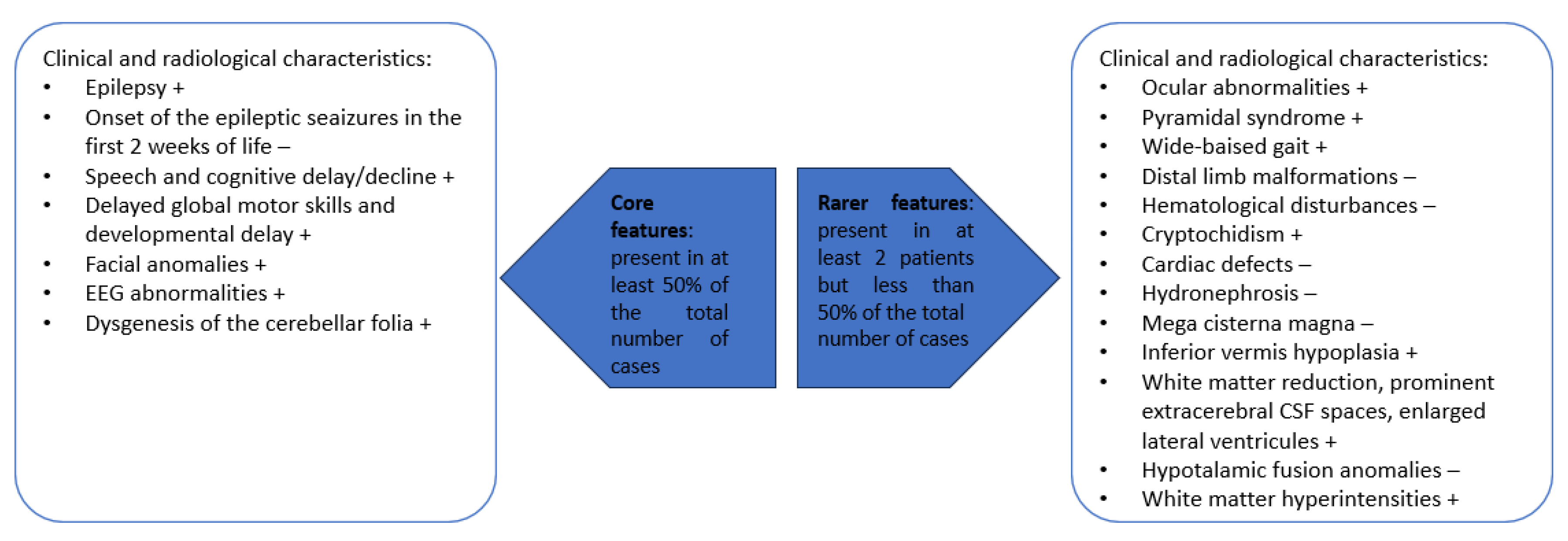
3.1. Literature Review
3.2. Our Clinical Experience
4. Discussion
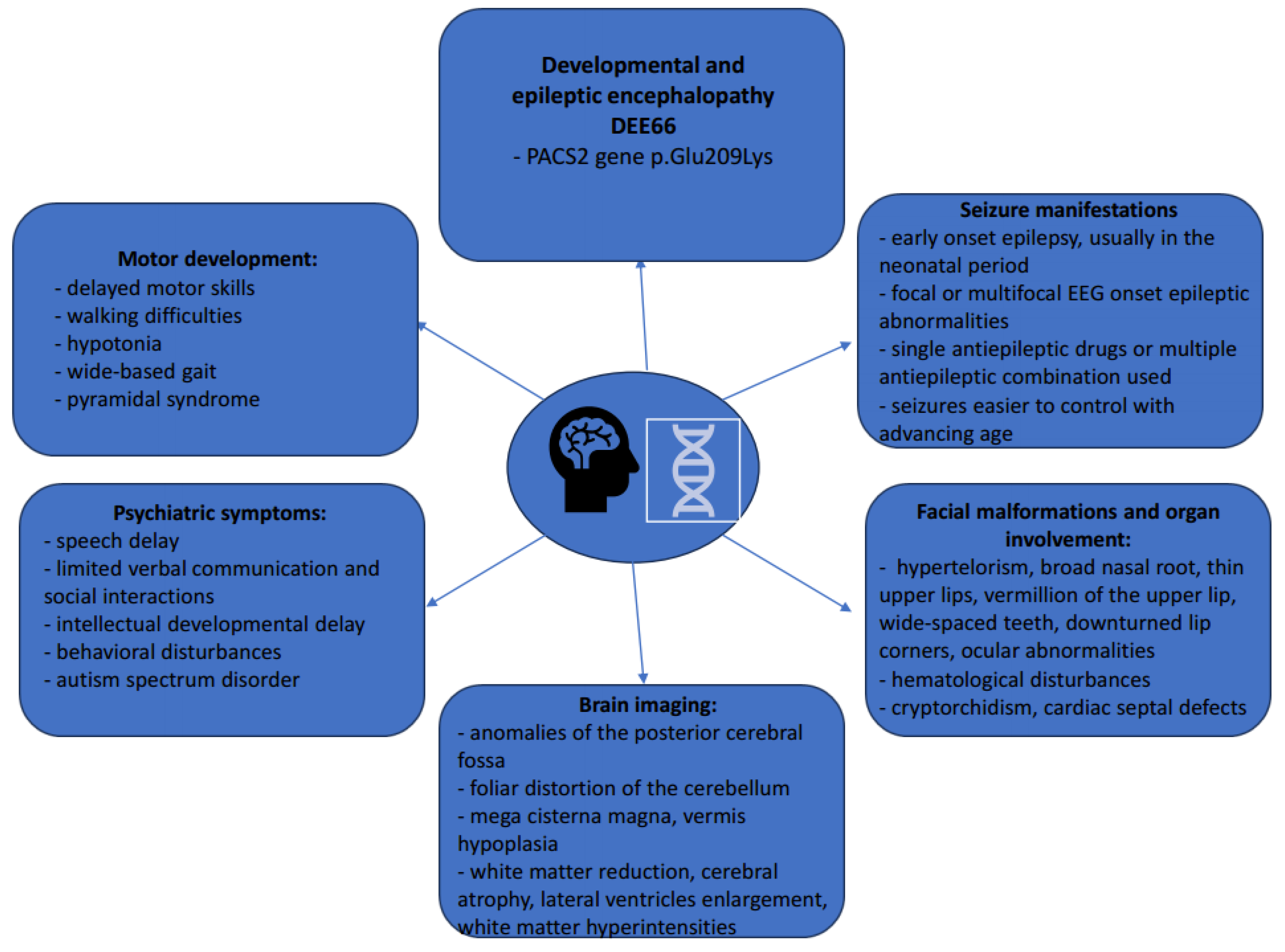
4.1. DEE66 General Considerations
4.2. DEE66 Molecular and Pathophysiologic Considerations
- (1)
- The N-terminal furin binding region (FBR) can recognize and bind with various cargo proteins. It plays a role in intracellular protein trafficking by recognizing acidic amino acid residues on cargo proteins [60]. The PACS2 p.Glu209Lys mutation leads to reduction/loss-of-function-like of PACS2 and alters the capacity of this region to form reversible bindings, disturbing cellular homeostasis [13].
- (2)
- The middle region (MR) contains phosphorylation sites and plays a major regulatory role. It promotes interactions between proteins and regulates PACS2 functions during cellular growth, repair, and death [46]. This region contains a binding site that becomes active after phosphorylation for 14-3-3, forming together a complex that reduces tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) levels, inhibits apoptosis, and promotes lipid biogenesis [19,61].
- (3)
5. Study Limitations
6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviation | Meaning |
| CK2A1 | casein kinase 2 alpha 1 |
| CNS | central nervous system |
| COVID-19 | corona virus disease 2019 |
| CSF | cerebrospinal fluid |
| CT | computed tomography |
| CTR | C-terminal region |
| DEE | developmental and epileptic encephalopathies |
| DNA | deoxyribonucleic acid |
| EEG | electroencephalogram |
| EMG | electromyography |
| ER | endoplasmic reticulum |
| FLAIR | fluid attenuated inversion recovery |
| FBR | furin binding region |
| IQ | intelligence quotient |
| LP | lumbar puncture |
| MAMs | mitochondria-associated membranes |
| MMSE | mini-mental state examination |
| MR | middle region |
| MRC | Medical Research Council scale |
| MRI | magnetic resonance imaging |
| NIH | National Institute of Health |
| PACS2 | phosphofurin acidic cluster sorting protein 2 |
| PRISMA | Preferred Reporting Items for Systematic Review and Meta-Analyses |
| TNF | tumor necrosis factor |
| TRAIL | TNF-related apoptosis-inducing ligand |
References
- Dentici, M.L.; Barresi, S.; Niceta, M.; Ciolfi, A.; Trivisano, M.; Bartuli, A.; Digilio, M.C.; Specchio, N.; Dallapiccola, B.; Tartaglia, M. Expanding the clinical spectrum associated with PACS2 mutations. Clin. Genet. 2019, 95, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Eltze, C.M.; Chong, W.K.; Cox, T.; Whitney, A.; Cortina-Borja, M.; Chin, R.F.; Scott, R.C.; Cross, J.H. A population-based study of newly diagnosed epilepsy in infants. Epilepsia 2013, 54, 437–445. [Google Scholar] [CrossRef] [PubMed]
- von Deimling, M.; Helbig, I.; Marsh, E.D. Epileptic Encephalopathies-Clinical Syndromes and Pathophysiological Concepts. Curr. Neurol. Neurosci. Rep. 2017, 17, 10. [Google Scholar] [CrossRef] [PubMed]
- Raga, S.; Specchio, N.; Rheims, S.; Wilmshurst, J.M. Developmental and epileptic encephalopathies: Recognition and approaches to care. Epileptic Disord. 2021, 23, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; French, J.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; Perucca, E.; Tomson, T.; Wiebe, S.; Zhang, Y.H.; et al. Classification of the epilepsies: New concepts for discussion and debate-Special report of the ILAE Classification Task Force of the Commission for Classification and Terminology. Epilepsia Open 2016, 1, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Olson, H.E.; Jean-Marçais, N.; Yang, E.; Heron, D.; Tatton-Brown, K.; van der Zwaag, P.A.; Bijlsma, E.K.; Krock, B.L.; Backer, E.; Kamsteeg, E.J.; et al. A Recurrent De Novo PACS2 Heterozygous Missense Variant Causes Neonatal-Onset Developmental Epileptic Encephalopathy, Facial Dysmorphism, and Cerebellar Dysgenesis. Am. J. Hum. Genet. 2018, 102, 995–1007, Erratum in Am. J. Hum. Genet. 2018, 103, 631. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Deng, J.; Chen, C.; Wang, X.; Fang, F.; Li, H.; Luo, J.; Wu, J. Developmental and Epileptic Encephalopathy 76: Case Report and Review of Literature. Children 2022, 9, 1967. [Google Scholar] [CrossRef]
- Simmen, T.; Aslan, J.E.; Blagoveshchenskaya, A.D.; Thomas, L.; Wan, L.; Xiang, Y.; Feliciangeli, S.F.; Hung, C.H.; Crump, C.M.; Thomas, G. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 2005, 24, 717–729, Erratum in EMBO J. 2005, 24, 1301. [Google Scholar] [CrossRef]
- Köttgen, M.; Benzing, T.; Simmen, T.; Tauber, R.; Buchholz, B.; Feliciangeli, S.; Huber, T.B.; Schermer, B.; Kramer-Zucker, A.; Höpker, K.; et al. Trafficking of TRPP2 by PACS proteins represents a novel mechanism of ion channel regulation. EMBO J. 2005, 24, 705–716. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.; Aslan, J.E.; Thomas, L.; Shinde, P.; Shinde, U.; Simmen, T. Caught in the act—Protein adaptation and the expanding roles of the PACS proteins in tissue homeostasis and disease. J. Cell Sci. 2017, 130, 1865–1876. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, K.; Wang, S.; Zhang, Y.H.; Yang, Z.X.; Wu, Y.; Jiang, Y.W. PACS gene family-related neurological diseases: Limited genotypes and diverse phenotypes. World J. Pediatr. 2023; Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Aslan, J.E.; You, H.; Williamson, D.M.; Endig, J.; Youker, R.T.; Thomas, L.; Shu, H.; Du, Y.; Milewski, R.L.; Brush, M.H.; et al. Akt and 14-3-3 control a PACS-2 homeostatic switch that integrates membrane traffic with TRAIL-induced apoptosis. Mol. Cell 2009, 34, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Terrone, G.; Marchese, F.; Vari, M.S.; Severino, M.; Madia, F.; Amadori, E.; Del Giudice, E.; Romano, A.; Gennaro, E.; Zara, F.; et al. A further contribution to the delineation of epileptic phenotype in PACS2-related syndrome. Seizure 2020, 79, 53–55. [Google Scholar] [CrossRef]
- Sakaguchi, Y.; Yoshihashi, H.; Uehara, T.; Miyama, S.; Kosaki, K.; Takenouchi, T. Coloboma may be a shared feature in a spectrum of disorders caused by mutations in the WDR37-PACS1-PACS2 axis. Am. J. Med. Genet. A 2021, 185, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, I.; Guillén Benítez, E.; Sanchez-Montanez, A.; Limeres, J.; López-Grondona, F.; Cuscó, I.; Tizzano, E.F. Vein of Galen aneurysm, dilated cardiomyopathy, and slender habitus in a patient with a recurrent pathogenic variant in PACS2. Am. J. Med. Genet. A 2022, 188, 991–995. [Google Scholar] [CrossRef]
- Mizuno, T.; Miyata, R.; Hojo, A.; Tamura, Y.; Nakashima, M.; Mizuguchi, T.; Matsumoto, N.; Kato, M. Clinical variations of epileptic syndrome associated with PACS2 variant. Brain Dev. 2021, 43, 343–347. [Google Scholar] [CrossRef]
- Cesaroni, E.; Matricardi, S.; Cappanera, S.; Marini, C. First reported case of an inherited PACS2 pathogenic variant with variable expression. Epileptic Disord. 2022, 24, 572–576. [Google Scholar] [CrossRef]
- Sánchez-Soler, M.J.; Serrano-Antón, A.T.; López-González, V.; Guillén-Navarro, E. New case with the recurrent c.635G>A pathogenic variant in the PACS2 gene: Expanding the phenotype. Neurologia 2021, 36, 716–719. [Google Scholar] [CrossRef]
- Perulli, M.; Picilli, M.; Contaldo, I.; Amenta, S.; Gambardella, M.L.; Quintiliani, M.; Musto, E.; Turrini, I.; Veredice, C.; Zollino, M.; et al. Pyridoxine supplementation in PACS2-related encephalopathy: A case report of possible precision therapy. Seizure 2023, 105, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Chou, I.J.; Hou, J.Y.; Fan, W.L.; Tsai, M.H.; Lin, K.L. Long-Term Outcome of Neonatal Seizure with PACS2 Mutation: Case Series and Literature Review. Children 2023, 10, 621. [Google Scholar] [CrossRef] [PubMed]
- Checri, R.; Dozières-Puyravel, B.; Elmaleh-Bergès, M.; Verloes, A.; Auvin, S. PACS2 pathogenic variant associated with malformation of cortical development and epilepsy. Epileptic Disord. 2023, 00, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Moulis, M.; Grousset, E.; Faccini, J.; Richetin, K.; Thomas, G.; Vindis, C. The Multifunctional Sorting Protein PACS-2 Controls Mitophagosome Formation in Human Vascular Smooth Muscle Cells through Mitochondria-ER Contact Sites. Cells 2019, 8, 638. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.ncbi.nlm.nih.gov/clinvar (accessed on 5 December 2023).
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.ensembl.org/Homo_sapiens/Tools/VEP (accessed on 5 December 2023).
- Available online: https://www.ncbi.nlm.nih.gov/medgen/1648486 (accessed on 5 December 2023).
- Stoian, A.; Bajko, Z.; Maier, S.; Cioflinc, R.A.; Grigorescu, B.L.; Moțățăianu, A.; Bărcuțean, L.; Balașa, R.; Stoian, M. High-dose intravenous immunoglobulins as a therapeutic option in critical illness polyneuropathy accompanying SARS-CoV-2 infection: A case-based review of the literature (Review). Exp. Ther. Med. 2021, 22, 1182. [Google Scholar] [CrossRef]
- Amore, G.; Spoto, G.; Ieni, A.; Vetri, L.; Quatrosi, G.; Di Rosa, G.; Nicotera, A.G. A Focus on the Cerebellum: From Embryogenesis to an Age-Related Clinical Perspective. Front. Syst. Neurosci. 2021, 15, 646052. [Google Scholar] [CrossRef]
- Divya, K.P.; Kishore, A. Treatable cerebellar ataxias. Clin. Park. Relat. Disord. 2020, 3, 100053. [Google Scholar] [CrossRef]
- Maier, S.; Moțățăianu, A.; Bărcuțean, L.; Balint, A.; Huțanu, A.; Bajko, Z.; Stoian, A.; Andone, S.; Bălașa, R. Interferon β 1A, an immunomodulator in relapsing remitting multiple sclerosis patients: The effect on pro inflammatory cytokines. Farmacia 2020, 68, 65–75. [Google Scholar] [CrossRef]
- Stoian, A.; Bajko, Z.; Stoian, M.; Cioflinc, R.A.; Niculescu, R.; Arbănași, E.M.; Russu, E.; Botoncea, M.; Bălașa, R. The Occurrence of Acute Disseminated Encephalomyelitis in SARS-CoV-2 Infection/Vaccination: Our Experience and a Systematic Review of the Literature. Vaccines 2023, 11, 1225. [Google Scholar] [CrossRef] [PubMed]
- Stoian, A.; Stoian, M.; Bajko, Z.; Maier, S.; Andone, S.; Cioflinc, R.A.; Moțățăianu, A.; Barcuțean, L.; Bălașa, R. Autoimmune Encephalitis in COVID-19 Infection: Our Experience and Systematic Review of the Literature. Biomedicines 2022, 10, 774. [Google Scholar] [CrossRef] [PubMed]
- Balasa, R.; Maier, S.; Barcutean, L.; Stoian, A.; Motataianu, A. The direct deleterious effect of Th17 cells in the nervous system compartment in multiple sclerosis and experimental autoimmune encephalomyelitis: One possible link between neuroinflammation and neurodegeneration. Rev. Română Med. Lab. 2020, 28, 9–17. [Google Scholar] [CrossRef]
- Gorodetsky, C.; Fasano, A. Developmental and Epileptic Encephalopathies in Adults: An Evolving Field. Neurology 2022, 99, 89–91. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef]
- Youker, R.T.; Shinde, U.; Day, R.; Thomas, G. At the crossroads of homoeostasis and disease: Roles of the PACS proteins in membrane traffic and apoptosis. Biochem. J. 2009, 421, 1–15. [Google Scholar] [CrossRef]
- Schuurs-Hoeijmakers, J.H.; Oh, E.C.; Vissers, L.E.; Swinkels, M.E.; Gilissen, C.; Willemsen, M.A.; Holvoet, M.; Steehouwer, M.; Veltman, J.A.; De Vries, B.B.; et al. Recurrent de novo mutations in PACS1 cause defective cranial-neural-crest migration and define a recognizable intellectual-disability syndrome. Am. J. Hum. Genet. 2012, 91, 1122–1127. [Google Scholar] [CrossRef]
- Neagu, A.C.; Budișteanu, M.; Gheorghe, D.C.; Mocanu, A.I.; Mocanu, H. Rare Gene Mutations in Romanian Hypoacusis Patients: Case Series and a Review of the Literature. Medicina 2022, 58, 1252. [Google Scholar] [CrossRef]
- Wan, L.; Molloy, S.S.; Thomas, L.; Liu, G.; Xiang, Y.; Rybak, S.L.; Thomas, G. PACS-1 defines a novel gene family of cytosolic sorting proteins required for trans-Golgi network localization. Cell 1998, 94, 205–216. [Google Scholar] [CrossRef]
- Li, C.; Li, L.; Yang, M.; Zeng, L.; Sun, L. PACS-2: A key regulator of mitochondria-associated membranes (MAMs). Pharmacol. Res. 2020, 160, 105080. [Google Scholar] [CrossRef]
- Pinton, P. Mitochondria-associated membranes (MAMs) and pathologies. Cell Death Dis. 2018, 9, 413. [Google Scholar] [CrossRef]
- Area-Gomez, E.; de Groof, A.; Bonilla, E.; Montesinos, J.; Tanji, K.; Boldogh, I.; Pon, L.; Schon, E.A. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 2018, 9, 335. [Google Scholar] [CrossRef]
- Zang, R.X.; Mumby, M.J.; Dikeakos, J.D. The Phosphofurin Acidic Cluster Sorting Protein 2 (PACS-2) E209K Mutation Responsible for PACS-2 Syndrome Increases Susceptibility to Apoptosis. ACS Omega 2022, 7, 34378–34388. [Google Scholar] [CrossRef] [PubMed]
- Wieckowski, M.R.; Giorgi, C.; Lebiedzinska, M.; Duszynski, J.; Pinton, P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 2009, 4, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- Perrone, M.; Caroccia, N.; Genovese, I.; Missiroli, S.; Modesti, L.; Pedriali, G.; Vezzani, B.; Vitto, V.A.M.; Antenori, M.; Lebiedzinska-Arciszewska, M.; et al. The role of mitochondria-associated membranes in cellular homeostasis and diseases. Int. Rev. Cell Mol. Biol. 2020, 350, 119–196. [Google Scholar] [CrossRef] [PubMed]
- Thi My Nhung, T.; Phuoc Long, N.; Diem Nghi, T.; Suh, Y.; Hoang Anh, N.; Jung, C.W.; Minh Triet, H.; Jung, M.; Woo, Y.; Yoo, J.; et al. Genome-wide kinase-MAM interactome screening reveals the role of CK2A1 in MAM Ca2+ dynamics linked to DEE66. Proc. Natl. Acad. Sci. USA 2023, 120, e2303402120. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Pellegrini, L. The coming of age of the mitochondria-ER contact: A matter of thickness. Cell Death Differ. 2016, 23, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Kravic, B.; Harbauer, A.B.; Romanello, V.; Simeone, L.; Vögtle, F.N.; Kaiser, T.; Straubinger, M.; Huraskin, D.; Böttcher, M.; Cerqua, C.; et al. In mammalian skeletal muscle, phosphorylation of TOMM22 by protein kinase CSNK2/CK2 controls mitophagy. Autophagy 2018, 14, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Afzal, M.; Kren, B.T.; Naveed, A.K.; Trembley, J.H.; Ahmed, K. Protein kinase CK2 impact on intracellular calcium homeostasis in prostate cancer. Mol. Cell. Biochem. 2020, 470, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Zeng, K.W.; Wang, J.K.; Wang, L.C.; Guo, Q.; Liu, T.T.; Wang, F.J.; Feng, N.; Zhang, X.W.; Liao, L.X.; Zhao, M.M.; et al. Small molecule induces mitochondrial fusion for neuroprotection via targeting CK2 without affecting its conventional kinase activity. Signal Transduct. Target. Ther. 2021, 6, 71, Erratum in Signal Transduct. Target. Ther. 2021, 6, 120. [Google Scholar] [CrossRef]
- Lynes, E.M.; Raturi, A.; Shenkman, M.; Ortiz Sandoval, C.; Yap, M.C.; Wu, J.; Janowicz, A.; Myhill, N.; Benson, M.D.; Campbell, R.E.; et al. Palmitoylation is the switch that assigns calnexin to quality control or ER Ca2+ signaling. J. Cell Sci. 2013, 126 Pt 17, 3893–3903. [Google Scholar] [CrossRef]
- Bravo-Sagua, R.; Parra, V.; López-Crisosto, C.; Díaz, P.; Quest, A.F.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. Compr. Physiol. 2017, 7, 623–634. [Google Scholar] [CrossRef]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef]
- Porter, R.J.; Dhir, A.; Macdonald, R.L.; Rogawski, M.A. Mechanisms of action of antiseizure drugs. Handb. Clin. Neurol. 2012, 108, 663–681. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Frew, A.J.; Smyth, M.J. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat. Rev. Cancer 2008, 8, 782–798. [Google Scholar] [CrossRef] [PubMed]
- Aslan, J.E.; Thomas, G. Death by committee: Organellar trafficking and communication in apoptosis. Traffic 2009, 10, 1390–1404. [Google Scholar] [CrossRef] [PubMed]
- Feliciangeli, S.F.; Thomas, L.; Scott, G.K.; Subbian, E.; Hung, C.H.; Molloy, S.S.; Jean, F.; Shinde, U.; Thomas, G. Identification of a pH sensor in the furin propeptide that regulates enzyme activation. J. Biol. Chem. 2006, 281, 16108–16116. [Google Scholar] [CrossRef] [PubMed]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- Werneburg, N.W.; Bronk, S.F.; Guicciardi, M.E.; Thomas, L.; Dikeakos, J.D.; Thomas, G.; Gores, G.J. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) protein-induced lysosomal translocation of proapoptotic effectors is mediated by phosphofurin acidic cluster sorting protein-2 (PACS-2). J. Biol. Chem. 2012, 287, 24427–24437. [Google Scholar] [CrossRef] [PubMed]
- Guicciardi, M.E.; Werneburg, N.W.; Bronk, S.F.; Franke, A.; Yagita, H.; Thomas, G.; Gores, G.J. Cellular inhibitor of apoptosis (cIAP)-mediated ubiquitination of phosphofurin acidic cluster sorting protein 2 (PACS-2) negatively regulates tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) cytotoxicity. PLoS ONE 2014, 9, e92124. [Google Scholar] [CrossRef] [PubMed]
- Atkins, K.M.; Thomas, L.L.; Barroso-González, J.; Thomas, L.; Auclair, S.; Yin, J.; Kang, H.; Chung, J.H.; Dikeakos, J.D.; Thomas, G. The multifunctional sorting protein PACS-2 regulates SIRT1-mediated deacetylation of p53 to modulate p21-dependent cell-cycle arrest. Cell Rep. 2014, 8, 1545–1557. [Google Scholar] [CrossRef]
- Urbanska, M.; Gozdz, A.; Swiech, L.J.; Jaworski, J. Mammalian target of rapamycin complex 1 (mTORC1) and 2 (mTORC2) control the dendritic arbor morphology of hippocampal neurons. J. Biol. Chem. 2012, 287, 30240–30256. [Google Scholar] [CrossRef] [PubMed]
- Toyo-oka, K.; Wachi, T.; Hunt, R.F.; Baraban, S.C.; Taya, S.; Ramshaw, H.; Kaibuchi, K.; Schwarz, Q.P.; Lopez, A.F.; Wynshaw-Boris, A. 14-3-3ε and ζ regulate neurogenesis and differentiation of neuronal progenitor cells in the developing brain. J. Neurosci. 2014, 34, 12168–12181. [Google Scholar] [CrossRef] [PubMed]
- Pisano, T.; Numis, A.L.; Heavin, S.B.; Weckhuysen, S.; Angriman, M.; Suls, A.; Podesta, B.; Thibert, R.L.; Shapiro, K.A.; Guerrini, R.; et al. Early and effective treatment of KCNQ2 encephalopathy. Epilepsia 2015, 56, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Dilena, R.; Striano, P.; Gennaro, E.; Bassi, L.; Olivotto, S.; Tadini, L.; Mosca, F.; Barbieri, S.; Zara, F.; Fumagalli, M. Efficacy of sodium channel blockers in SCN2A early infantile epileptic encephalopathy. Brain Dev. 2017, 39, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Mallik, S.; Kundu, S. Topology and Oligomerization of Mono- and Oligomeric Proteins Regulate Their Half-Lives in the Cell. Structure 2018, 26, 869–878.e3. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Miura, M. Programmed cell death in neurodevelopment. Dev. Cell 2015, 32, 478–490. [Google Scholar] [CrossRef]
- Blanquie, O.; Kilb, W.; Sinning, A.; Luhmann, H.J. Homeostatic interplay between electrical activity and neuronal apoptosis in the developing neocortex. Neuroscience 2017, 358, 190–200. [Google Scholar] [CrossRef]
- Cocito, C.; Merighi, A.; Giacobini, M.; Lossi, L. Alterations of Cell Proliferation and Apoptosis in the Hypoplastic Reeler Cerebellum. Front. Cell. Neurosci. 2016, 10, 141. [Google Scholar] [CrossRef]
- Holder, J.L., Jr.; Lotze, T.E.; Bacino, C.; Cheung, S.W. A child with an inherited 0.31 Mb microdeletion of chromosome 14q32.33: Further delineation of a critical region for the 14q32 deletion syndrome. Am. J. Med. Genet. A 2012, 158, 1962–1966. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authors | Study Design | Sex/Mean Age at Seizures Onset | Seizures Characteristics | Age at Genetic Diagnosis | Neurological Symptoms | Eye Features and Facial Abnormalities | Psychiatric/Behavioral Features | Other Symptoms and Organ Involvement | EEG | Imagistic Investigations and CNS Alterations | Treatment | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Olson et al. [7], 2018 | Case series (Case 1) | Female/6 days | Focal seizures | 16 years | Moderate myopia, delayed motor skills, hypotonia, increased tendon reflexes | Synophris, hypertelorism, down-slanting palpebral fissures, thin upper lip, down-turned corners of the mouth, prominent incisors and widely-spaced teeth, everted vermillion of lower lip | Speech delay | Neutropenia, metatarsus varus | Not described | Brain (magnetic resonance imaging) MRI: mega cisterna magna, foliar distortion of the left cerebellar hemisphere | Carbamazepine | Not described |
| Olson et al. [7], 2018 | Case series (Case 2) | Female/4 days | Generalized tonic-clonic seizures | 4 years | Delayed motor skills, nystagmus, wide-based gait, | Synophris, hypertelorism, down-slanting palpebral fissures, broad nasal root, thin upper lip, down-turned corners of the mouth | Stereotypies, sleeping and behavioral disturbances | Anemia, dextrocardia, | EEG until 3.5 months: focal spikes on a normal background; normal aspect at 1-year-old | Brain MRI: inferior vermis hypoplasia, cisterna magna, foliar distortion of the cerebellum, hypothalamic fusion anomaly | Phenobarbital, valproate | Longest seizure-free interval: 6 months |
| Olson et al. [7], 2018 | Case series (Case 3) | Male/4 days | Not described | 15 years | Delayed motor acquisition | Synophris, hypertelorism, down-slanting palpebral fissures, thin upper lip, down-turned corners of the mouth, prominent incisors and widely-spaced teeth, everted vermillion of lower lip | Speech delay | Syndactyly of toes, cryptorchidism | EEG: not available | Brain MRI: increased subarachnoid spaces | Carbamazepine | Not described |
| Olson et al. [7], 2018 | Case series (Case 4) | Female/7 days | Not described | 8 years | Strabismus, delayed motor acquisition | Synophris, hypertelorism, down-slanting palpebral fissures, broad nasal root, thin upper lip, down-turned corners of the mouth, prominent incisors | Speech delay, mild autistic disorder | Anemia | EEG in neonatal period: multifocal sharp waves, intermittent generalized burst of epileptic activity | Brain MRI: inferior vermis hypoplasia, mega cisterna magna, mild distortion of the cerebellar folia | Phenobarbital, pyridoxal-5-phosphate, pyridoxine, valproate | 2 years seizure-free interval on antiepileptic treatment, reoccurrence of seizures at tempted withdrawal of valproate |
| Olson et al. [7], 2018 | Case series (Case 5) | Male/2 days | Clonic and generalized tonic-clonic seizures | 19 months | Strabismus, visual problems, delayed motor skills, | Broad nasal root, thin upper lip, down-turned corners of the lips | Not described | Anemia, V finger clinodactyly, variant transverse palmar crease | EEG in the neonatal period: normal; 4 months: generalized slowing and multifocal sharp waves; frequent focal seizures | Brain MRI: inferior vermis hypoplasia, mega cisterna magna, foliar distortion of the left cerebellar hemisphere, hypothalamic fusion anomaly | Levetiracetam, phenobarbital, carbamazepine | The longest seizure-free interval: 9 months |
| Olson et al. [7], 2018 | Case series (Case 6) | Male/2 days | Not described | 8 years | Strabismus, astigmatism, myopia, anisocoria, delayed motor acquisitions, | Thin upper lip, down-slanting palpebral fissures, everted vermillion of lower lip | Obsessive compulsive disorder | Finger pads, small ventricular septal defects, testis ectopia | EEG in the neonatal period: left temporal spikes; | Brain MRI performed at 10 days and 4 months: no pathological findings | Topiramate | The longest seizure-free interval: 3.5 year |
| Olson et al. [7], 2018 | Case series (Case 7) | Male/2 days | Focal tonic stiffening, later clonic seizures, autonomic features | 16 months | Mild conductive hearing loss, poor feeding, axial hypotonia, unable to walk, slightly increased tone in hands | Broad nasal root, thin upper lip | Speech delay | Mild anemia, café au lait birthmark, frequent ear and respiratory infections, cryptorchidism | EEG at 6–7 weeks: epileptiform activity; 9 months: normal aspect | Brain MRI: inferior vermis hypoplasia, left retro cerebellar cysts, distortion of the left cerebellar hemisphere | Phenobarbital | Not described |
| Olson et al. [7], 2018 | Case series (Case 8) | Female/2 weeks | Focal, latter tonic seizures | 5 years | Hypermetropic astigmatism, unable to walk | Hypertelorism, broad nasal root, thin upper lip, down-slanting palpebral fissures | Atypical social and behavioral features | Not described | EEG at 6 weeks: right frontocentral and left temporal subtle aberration | Brain MRI in the neonatal period: normal aspect | Phenobarbital, sodium valproate | Seizure-free for 3.5 year |
| Olson et al. [7], 2018 | Case series (Case 9) | Female/2 days of life | Focal tonic, tonic-clonic, myoclonic; in evolution generalized tonic-clonic and tonic seizures | 3 years | Myopia, astigmatism, cortical visual impairment, delayed motor skills, diffuse hypotonia | Synophris, broad nasal root, thin upper lip | Atypical social and behavioral features | Accessory caudally placed nipples | EEG in the neonatal period: excess discontinuity and multifocal sharp waves; EEG at the age of 2: intermittent generalized slowing, intermittent left temporal slowing | Brain MRI: mega cisterna magna, cerebellar foliar distortion, hypothalamic fusion anomaly | Levetiracetam, phenobarbital | The longest seizure-free interval under treatment: 2 years |
| Olson et al. [7], 2018 | Case series (Case 10) | Male/1–2 months | Eye deviation and clonic seizure, later in evolution generalized tonic-clonic seizure | 7 years | Delayed motor acquisitions, diffuse hypotonia | Mildly dysmorphic | Autism spectrum disorder | Bilateral palmar crease | EEG at 4 months: normal; EEG at 17 months: rare generalized spikes | Brain MRI: mega cisterna magna | Levetiracetam | The longest seizure-free interval: 2 years |
| Olson et al. [7], 2018 | Case series (Case 11) | Male/1 day | Focal seizures until 2 months of age; generalized tonic-clonic seizures latter in evolution | 12.5 years | Hypermetropic astigmatism, transient nystagmus, delay motor skills, speech delay | Broad nasal root, thin upper lip, down-turned corners of the lips, widely-spaced teeth | Autism spectrum disorder | V finger brachyclino-dactyly, broad and tapering short fingers; frequent otitis; micro penis, unilateral cryptorchidism, velo pharyngeal hypotonia, central precocious puberty, infantile hypertrophic pyloric stenosis | EEG in the neonatal period: epileptic discharges in the left Rolandic region | Brain MRI: thick corpus callosum, inferior vermis hypoplasia | Valproate | Not described |
| Olson et al. [7], 2018 | Case series (Case 12) | Female/3 days | Tonic and focal tonic-clonic; status epilepticus | 9 months | Hypotonia | Hypertelorism, thin upper lip, down-turned corners of the lips, prominent incisors | Transient stereotypes | V finger clinodactyly, brachycephaly, inverted nipples | EEG in the neonatal period: sharp waves and excess multifocal spikes more evident in the bilateral temporal regions; 2 months: high amplitude background, multifocal spikes | Brain MRI: mega cisterna magna, severe foliar distortion of the cerebellum | Levetiracetam phenobarbital oxcarbazepine | Not described |
| Olson et al. [7], 2018 | Case series (Case 13) | Female/2 weeks | Focal tonic-clonic seizures; latter in evolution: focal or generalized seizures | 3.5 years | Delayed motor acquisition, axial hypotonia, increased tendon reflexes | Hypertelorism, thin upper lip, down-turned corners of the lips, down-turned corners of the lips, everted vermillion of lower lip | Stereotypes, severe speech delay | Atrial septum defect, sacral pit | EEG: normal at six days; EEG in the neonatal period: multifocal epileptic activity, slow spikes with high amplitude; EEG at 17 months: spikes at vertex, enhanced in sleep | Brain MRI: cerebellar foliar distortion | Vigabatrin, levetiracetam, pyridoxal phosphate, pyridoxine, lamotrigine, valproate, clobazam | Not described |
| Olson et al. [7], 2018 | Case series (Case 14) | Female/3 days | Tonic and later in evolution also generalized tonic-clonic seizures | 8 years | Delayed motor acquisition, diffuse hypotonia, wide-based gait, | Broad nasal root, thin upper lip, everted vermillion of lower lips | Speech delay, selective mutism | V finger clinodactyly, right transverse palmar crease | EEG in the neonatal period: bilateral central and temporal sharp waves; EEG at 10 months: left frontocentral spikes; EEG at 22 months: spike or poly spike waves with high amplitude; EEG at 3 years: generalized slowing, left temporal epileptic discharges | Brain MRI: mild subarachnoid hemorrhage; prominent cisterna magna; slight foliar distortion of the left cerebellar hemisphere | Phenobarbital, pyridoxine, levetiracetam lacosamide | Not described |
| Dentici et al. [1], 2019 | Case report | Male/3 days of life | Impairment of consciousness, upward rolling of the eyes | 7 years | Delayed global motor skills during childhood; dysphagia, difficult chewing | Relative macrocephaly, broad and highly arched eyebrows, long eyelashes, broad nasal tip, rotated years, thin upper lip, spaced teeth, down-turned corners of the lips | Limited verbal speech; reduced intelligence quotient (IQ) (47) | Reduced growth velocity (required GH treatment) | Epileptic abnormalities in central region, bilaterally; Normal EEG at 7 years | Brain MRI: abnormal cerebellar foliation pattern in the posterior and basal portions of the right hemisphere; white matter reduction in the parietal regions and in the periventricular posterior regions, lateral ventricles asymmetry, centrum semiovale hyperintensities in the frontal regions | Phenobarbital; Vigabatrin and valproic acid | with seizures remission for one month; recurrence with blank staring, limb hypertonia, incontrollable crying; good control of seizures; antiepileptic drug discontinuation at 6 years |
| Terrone et al. [15], 2020 | Case report | Female/48 h of life | Brief, tonic seizures of the upper limbs accompanied by autonomic features (facial flushing, perioral cyanosis); at 4 months: oro masticatory automatism, eye staring, dystonic postures of limb and trunk | 4 months | Truncal hypotonia | Hypertelorism, broad nasal root, down-turned corners of the mouth, down-slanting palpebral fissures | Developmental delay in language and social communication | Monoarticular arthritis of the left knee | Interictal EEG: Multifocal epileptic abnormalities on the anterior and central areas of the right hemisphere; ictal EEG: right hemisphere focal seizures with rapid generalization | Brain MRI: enlargement of the left posterior horn of the lateral ventricle, irregular hemispheric cerebellar foliation | Phenobarbital, levetiracetam; Carbamazepine was added with gradual withdrawn of phenobarbital | 3 months seizures free; Recurrence of seizures at 4 months; At 20 months of life: seizures free with carbamazepine |
| Sakaguchi Y et al. [16], 2020 | Case report | Male/2 months | Tonic seizures | 23-year-old | Hypotonia during neonatal period, delayed head control, delayed motor skills; upper visual field defect in his left eye; left ear sensorineural deafness; adult life: diffuse hypotonia, wide-based gait | Irideal and choroidal coloboma in the inferior quadrant of the left eye, astigmatism; highly arched eyebrows, down-slanting palpebral fissures, bilateral mild ptosis, broad nasal root, low-set ears | Delayed speaking and language comprehension | Pectus excavates, slender fingers, talipes plantaris | EEG: epileptic discharges in the right occipital region; | Brain MRI: dysgenesis and progressive atrophy of the cerebellum, foliar distortion of the inferior hemisphere more evident on the left side; hyperplasia of the anterior pituitary gland | Diazepam (ineffective), phenobarbital | Controlled seizures at 6 months; medication free since the age of 9; able to attend college |
| Valenzuela I et al. [17], 2021 | Case report | Male/3 days after birth | Hypertonic seizures with generalized cyanosis | 25-year-old | Hypotonia and hypoactivity evident since birth; mild global developmental delay with delayed motor skills; oropharyngeal dysphagia diagnosed at 20 years | Widely-spaced eyes, down slanted palpebral fissure, strabismus, laterally extended thick eyebrows, retrognathia, thick upper lip, high palate, low-set ears | Mild global developmental delay, reduced cognitive performance | Left cryptorchidism, pectus carinatum, distal joint laxity, slender fingers; progressive congestive heart failure with idiopathic dilated cardiomyopathy diagnosed at 14-years old | Not available/not described by the authors | Brain MRI: severe dysmorphic colpocephalic lateral ventriculomegaly with chronic hydrocephalus considered to be secondary to the vascular malformation; vein of Galen aneurysm present | Levetiracetam | Seizure-free under treatment with levetiracetam; stable under treatment for cardiac failure |
| Mizuno T et al. [18], 2021 | Case series (Case 1) | Female/2 weeks after birth | Frequent tonic convulsions with head turning | 2-year-old | Hypotonia, mild psychomotor developmental delay | Hypertelorism, downward-slanting palpebral fissures, thin upper lip, down-turned corners of the mouth, broad nasal root | Mild psychomotor developmental delay | Not described | High-voltage slow waves in the left posterior-temporal areas | Brain MRI: decreased posterior periventricular white matter volume, old subependymal hemorrhage (perinatal injury) | Carbamazepine, clobazam | Almost controlled at 2 years of age |
| Mizuno T et al. [18], 2021 | Case series (Case 2) | Female/3 days after birth | Focal or generalized tonic convulsions | 12-years old | Hypotonia | Hypertelorism, downward-slanting palpebral fissures, thin upper lip, down-turned corners of the mouth, broad nasal root | Autism spectrum disorder, severe intellectual disability | Not described | Spikes over the right frontal areas | Brain MRI: normal findings | Topiramate, zonisamide, clonazepam; latter: lamotrigine, valproic acid, clonazepam | Controlled between 4-year-old and 9 years old; controlled latter again after medication changes |
| Mizuno T et al. [18], 2021 | Case series (Case 3) | Female/3 days after birth | Tonic convulsions | 3-year-old | Normal psychomotor development | Hypertelorism, downward-slanting palpebral fissures, everted vermillion of lower lip, broad nasal root | Normal psychomotor development | Not described | Ictal EEG (6 months): epileptic discharges in the left hemisphere | Brain MRI: right venous sinus thrombosis (at 3 days of life) | Carbamazepine | Controlled at 9 months; latter seizure-free without medication |
| Cesaroni E et al. [19], 2022 | Case report (Case 1) | Male/7 days after birth | Focal seizures with loss of contact, staring, crying, apnea with severe cyanosis, followed by tonic and vibratory prolonged phase | 11-month-old | Hypotonia, delayed motor skills | Long eyelashes, highly arched and sparse broad eyebrows, broad nasal tip, smooth philtrum, down-turned corners of the mouth, thin and everted upper lip vermillion | Delayed personal/social skills | Not described | Generalized slowing and with sporadic multifocal sharp waves Ictal EEG: focal rhythmic sharp waves, polyspike activity | Brain MRI: prominence of cisterna magna, mild inferior vermis and cerebellar hypoplasia | Phenobarbital; adjustments of the dose at 3 months; substitution with VPA and nitrazepam at 9 months; 12 moths: substitution with carbamazepine | Seizure remission with reoccurrence after 3 months, 9 months, 12 months; seizure-free at 19 months |
| Cesaroni E et al. [19], 2022 | Case report (Case 2) | Female/a few weeks after birth | Three brief febrile tonic seizures | 37-year-old | Not described | Synophyris, highly arched and sparse broad eyebrows, long eyelashes, broad nasal tip, thin everted upper lip, down-turned corners of the mouth | Learning disabilities during childhood | Not described | EEG (during childhood): reported to be normal | Brain computed tomography (CT) scan (at 3 months): no alterations | Phenobarbital | Seizure-free from 3 months of age; without medication since childhood |
| Sánchez-Soler et al. [20], 2021 | Case report (Case 3) | Female/3 months of age | Right-sided deviation of the eyes and head, dystonic posturing of the limbs, generalized hypertonia | 3-year-old | Psychomotor retardation; developmental delay; expressive language was affected | Facial dysmorphism | Occasional episodes of self and hetero-aggression, psychomotor retardation | Polydactyly in both feet | EEG at admission: diffuse background slowing, without focal abnormalities; interictal EEG at 24 and 30 moths: normal results | Brain CT scan: no alterations | Midazolam and phenytoin in status epilepticus; Valproic acid; zonisamide | No response at the initial treatment with valproic acid; exacerbated the symptoms; Presented other 3 seizures (5 months of life, respectively 24 and 30 months); seizure-free until the age of 3 when the case was reported |
| Perulli M et al. [21], 2023 | Case report | Male/8 days of life | 2 months: repetitive focal to bilateral motor seizures, severe desaturation; frequent episodes of status epilepticus | 12-year-old | Developmental delay | Facial dysmorphism present | Autism spectrum disorder, severe speech delay | Hand dysmorphism present | EEG (8 days): sporadic sharp waves in the anterior regions EEG (4.5 years): multifocal EEG pattern, overactivation in sleep EEG (11 years): theta background activity in awake state, no epileptiform activity | Brain MRI in the first month of life: hyperintensity in the periventricular white matter Brain MRI at 2.5 months: other changes occurred with global thickening of the corpus callosum, mega cisterna permagna | Diazepam, phenobarbital, phenytoin, levetiracetam, midazolam to whom the patient was refractory; improvement with pyridoxine, folinic acid, pyridoxal phosphate | Seizure-free since the age of 6 years, depends on his parents |
| Chou IJ et al. [22], 2023 | Case series and literature review (case 1) | Male/2 weeks of age | Generalized tonic-clonic seizures; repetitive spasms in the left face, left arm and hand and postural tonic spasms; | NA | Gross motor delay, speech delay, hypotonia | Synophrys, wide-spaced teeth | Speech delay | Right hydronephrosis, horseshoe kidney | Focal cortical dysfunction over the right frontal area; Video EEG performed at 6 months: interictal epileptic discharges located at Cz, C4, C3 and F3 | Brain ultrasonography and MRI: mega cisterna magna with prominent extracerebral cerebrospinal fluid spaces | Oral diazepam for the first seizures for 1 month; Phenobarbital and oxcarbazepine; Intravenous pyridoxal phosphate then oral inactive vitamin B6; ketogenic diet latter discontinued for dietary intolerance; levetiracetam was added | Seizure-free until 3 months of age; Seizures recurrence at 3 months in febrile context (pneumonia); Recurrence of seizure at the age of 6 months: effective control of seizures |
| Chou IJ et al. [22], 2023 | Case series and literature review (case 2) | Female/1 month of age | Grasping, tonic trunk posture; tonic movements in all upper and lower limbs, cyanosis of the lips, facial flushing, upward gaze, postictal lethargy; latter in evolution tonic-clonic seizures | NA | Gross motor and speech delay; | Facial dysmorphism, hypertelorism | Speech delay; attention deficit hyperactivity disorder | Repetitive urinary infections; right hydronephrosis, bilateral vesicoureteral reflux, atrial septal defect | Initial interictal EEG: slow waves in both hemispheres, focal spikes in the bilateral centrotemporal areas; After 3 years: generalized spikes; in evolution: interictal EEG: activity in the right centrotemporal area; normal EEG at 10 years of age under treatment | Brain MRI: vermis hypoplasia, mega cisterna magna; MRI at 7 years: mild prominence of the sulci and cisterns in both cerebral hemispheres, left mesial temporal sclerosis | Intravenous pyridoxal phosphate, shifted latter to oral vitamin B6; Oxcarbazepine was associated; Levetiracetam and valproic acid were added | 3 years seizure-free; Recurrence of tonic-clonic seizures |
| Chou IJ et al. [22], 2023 | Case series and literature review (case 3) | Female/2 weeks of age | Screaming, facial flush, general muscle tightening; focal seizure with: upward gaze, eye deviation to the left, dilated pupils without light reflex, head turning to the right, jerking of the left limbs, oxygen desaturation (increased frequency of the focal seizures that occurred between generalized seizures: up to 20/day); other seizures: yawning, hiccupping, bicycling movements of the legs, grasping, facial flushing | NA | Poor sucking and feeding; Global developmental delay; hypotonia, horizontal nystagmus, microcephaly; failure to thrive | Facial dysmorphism | Global developmental delay | Gastroesophageal reflux disease; atrial septal defect | Initial 24-h video EEG: diffuse cortical dysfunction, multifocal epileptiform discharges; Follow-up EEG: similar findings with the initial EEG | Brain MRI: mega cisterna magna | Phenobarbital; Levetiracetam and intravenous pyridoxal phosphate were added (shifted later on oral vitamin B6); Later in evolution a combination of vigabatrin, phenobarbital, clonazepam | Seizure persistence The seizure frequency was reduced to once or twice monthly |
| Checri R et al. [23], 2023 | Case report | Male/10 months | Asymmetrical epileptic spasms | 7-year-old child | Developmental delay with hypotonia, poor sucking, left hemiparesis | Round face with hypertelorism, oblique downward and outward palpebral clefts, wide nose, small teeth widely-spaced | Mild cognitive disabilities | Short extremities | Multifocal spikes associated with 1–2 Hz slow waves | Brain MRI: right insular polymicrogyria and pachygyria, cerebellar dysgenesis with disorganization of the cerebellar foliation mostly in the vermis | Resistant to prednisolone, vigabatrin, valproate, levetiracetam and lamotrigine; cessation of epileptic seizures after radiprodil (allosteric modulator of NMDA receptors) association | Seizure-free at 7 years on monotherapy (valproate) |
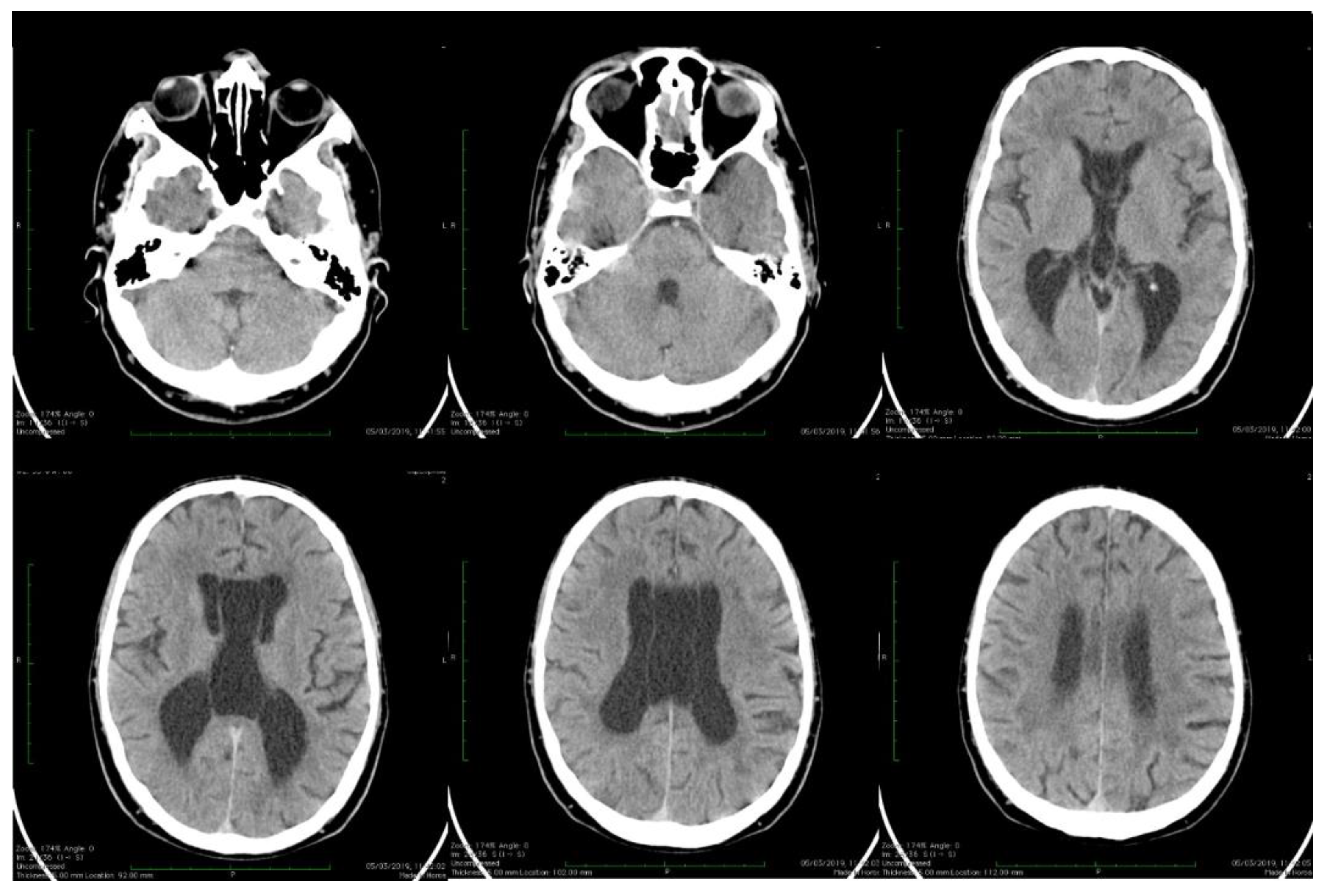
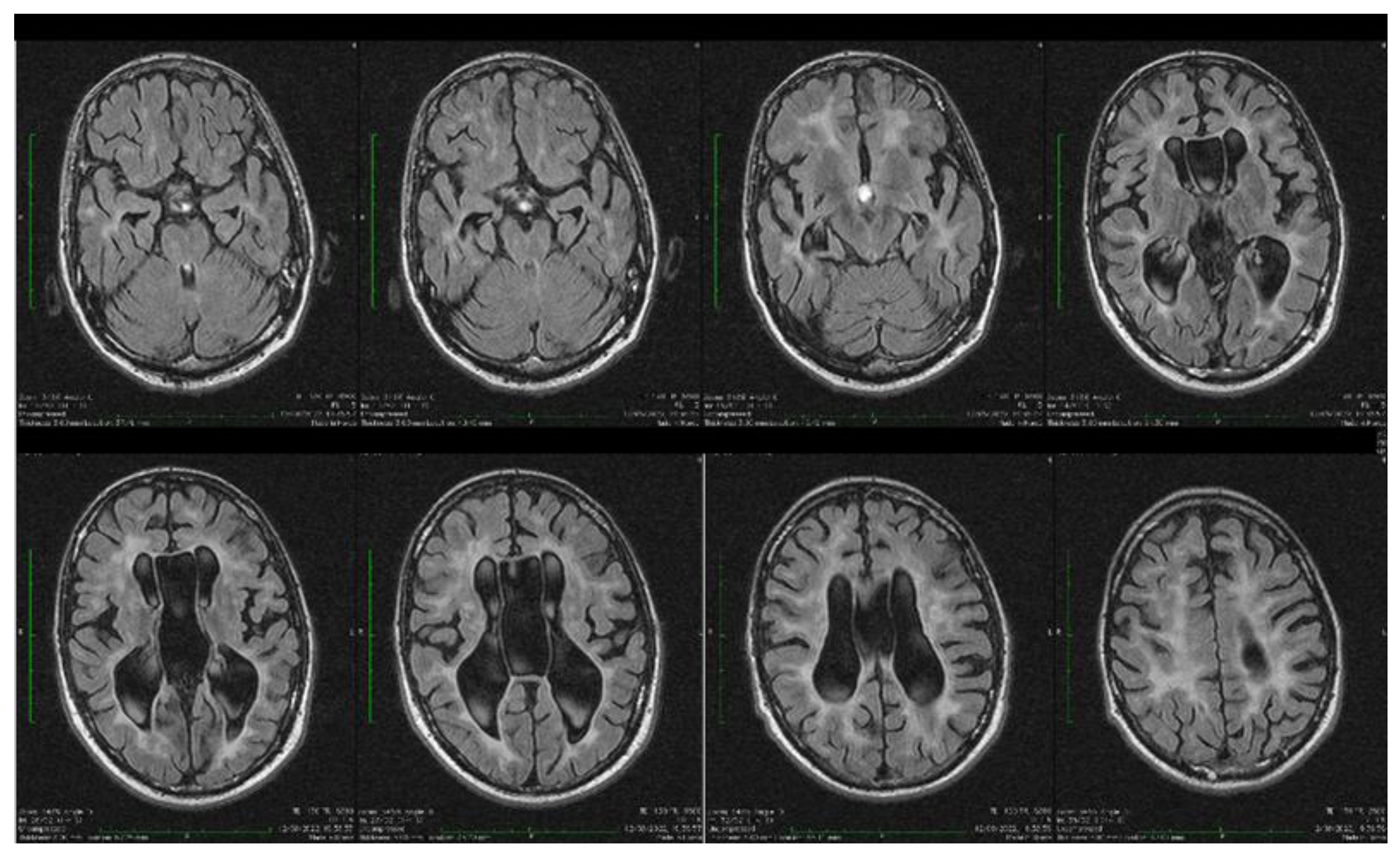
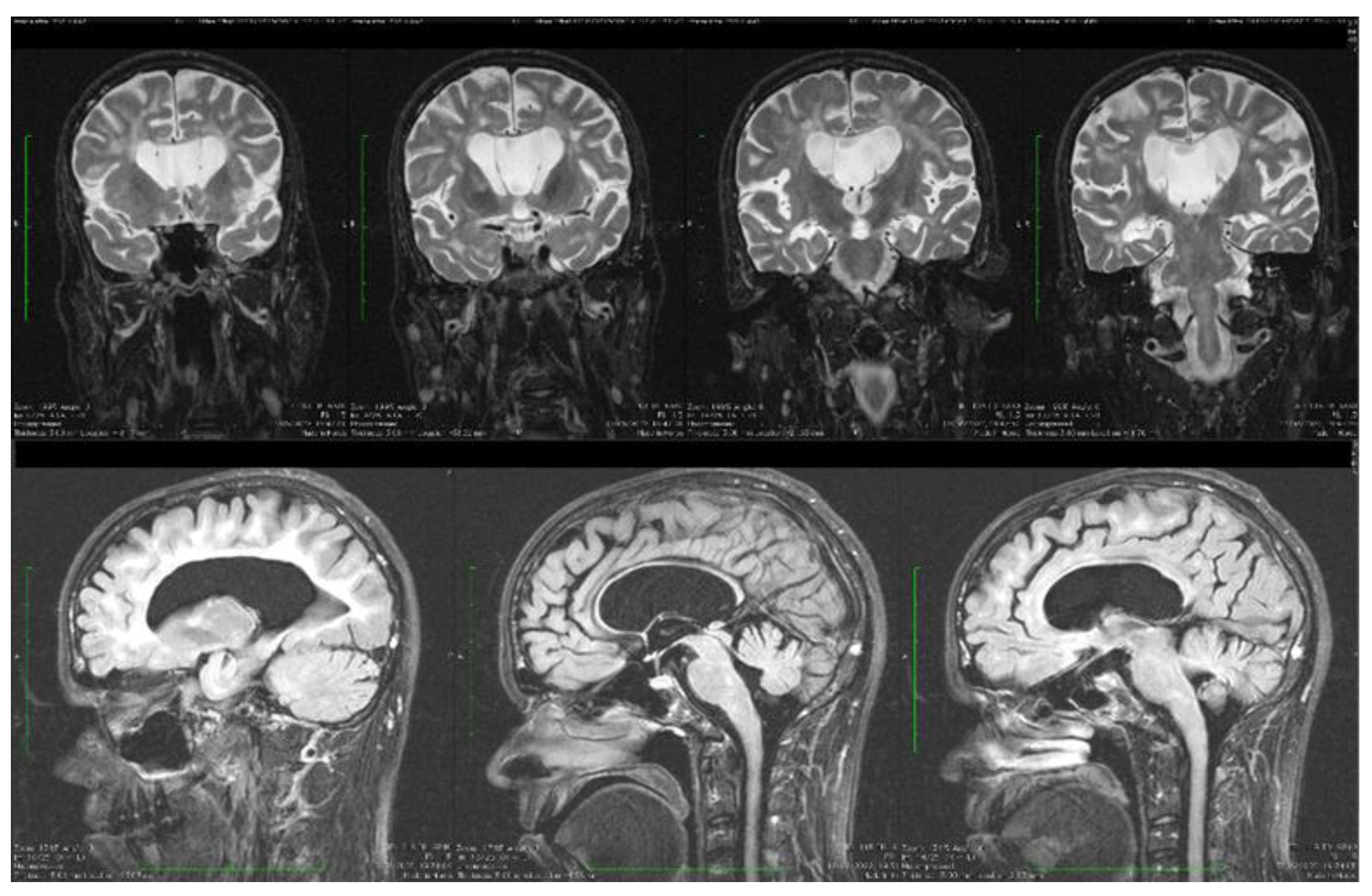
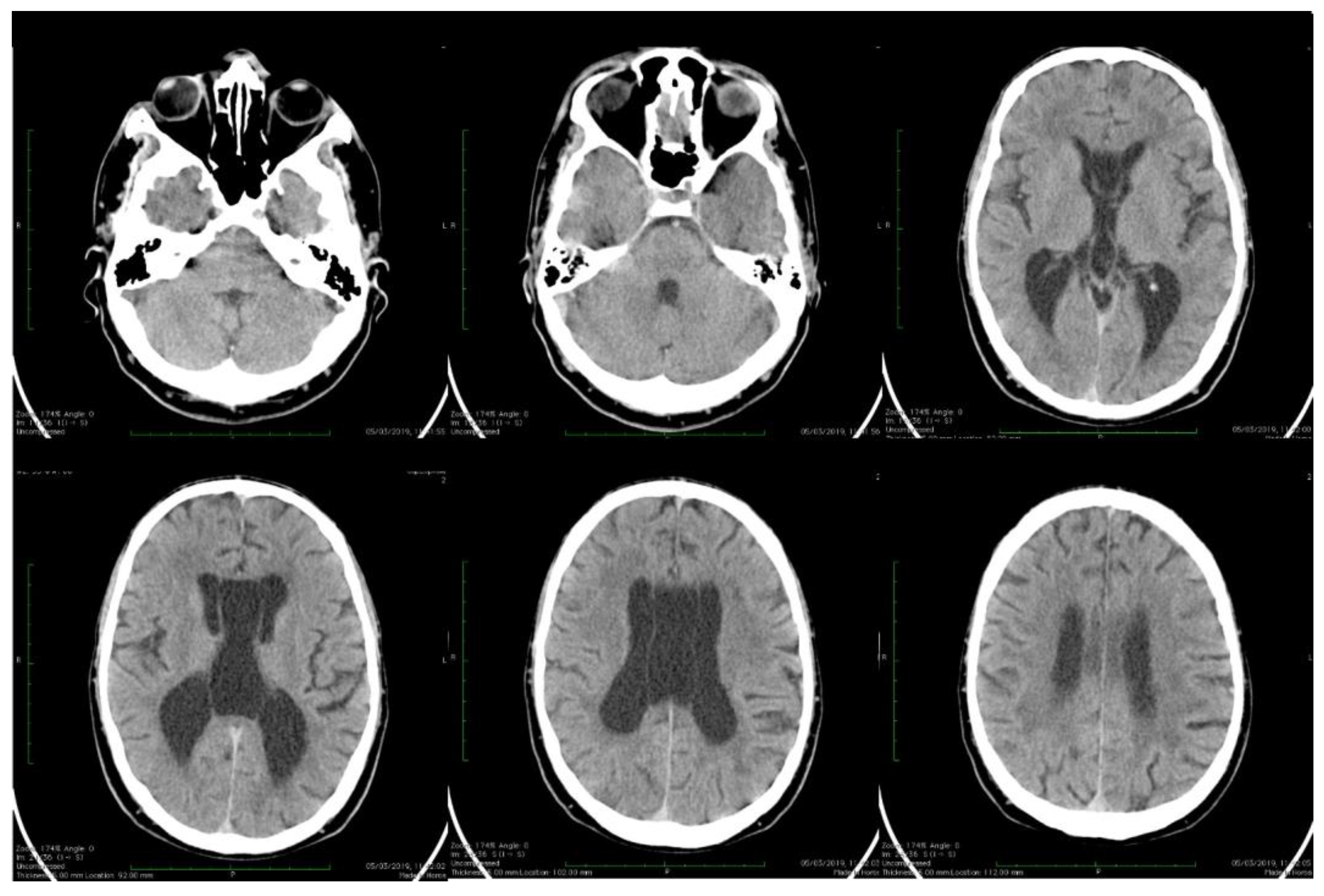
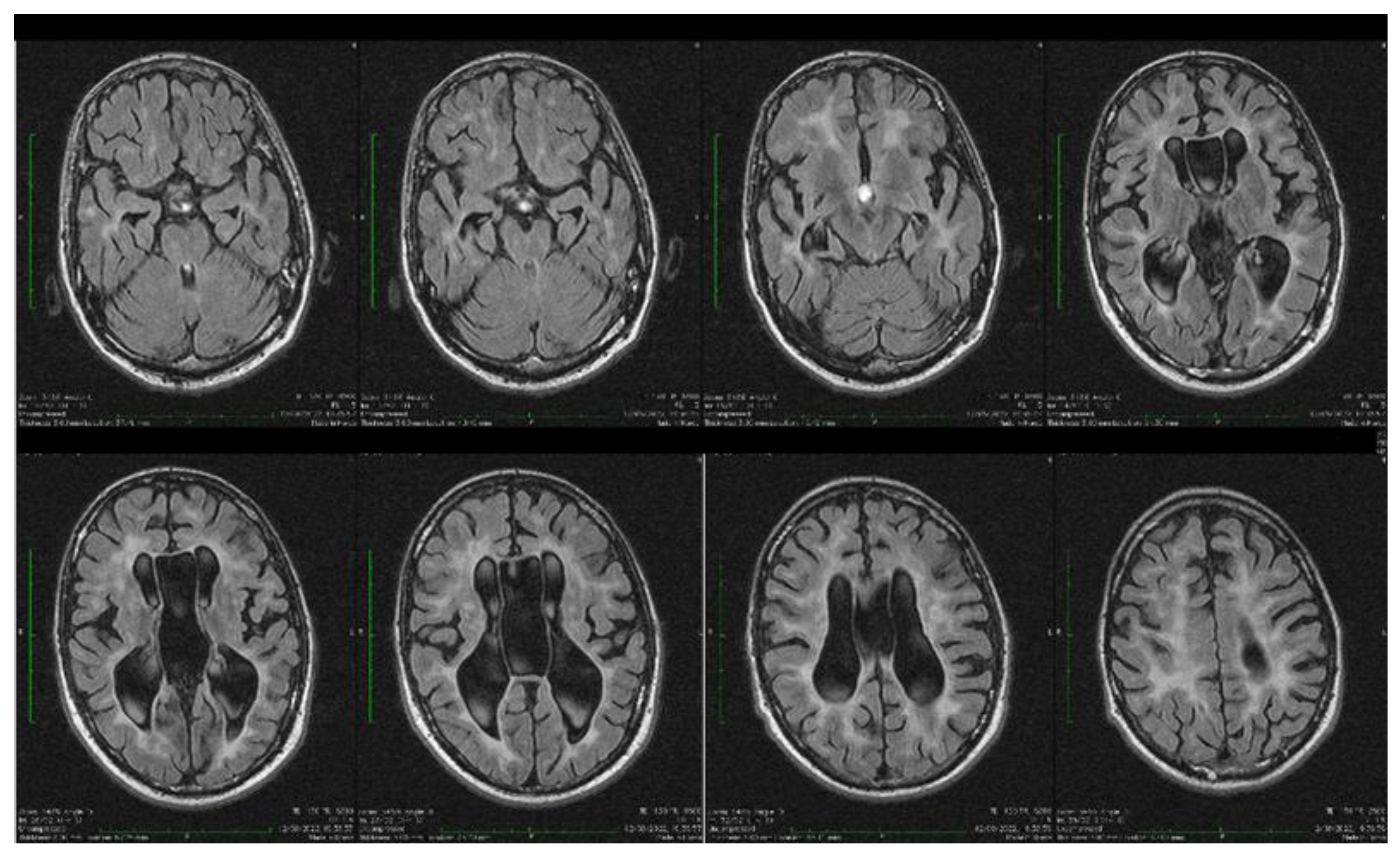
| Stoian et al. (our own case) | Case report | Male/4.5 months | Tonic seizures; later in evolution a left facial hemi spasm | 31 years | Reduced visual acuity (myopia), one and a half syndrome with left abduction nystagmus, left facial hemi spasms, divergent strabismus, flaccid tetra paresis, bilateral inconstant Babinski sign, gait instability, truncal ataxia, | Mild facial dysmorphism with thin upper lip and slightly spaced teeth | Social isolation, bradypsychia, bradylalia, echolalia, minimal linguistic baggage, behavioral stereotypes, MMSE17 points | Cryptorchidism | EEG: Poor unmodulated alpha activity, frontal intermittent theta activity; sharp waves and sharp peaks on the left central, parietal and temporal derivations | Brain CT: generalized sufferance pf white matter; cavum septum pellucidi, enlarged ventricular system, cerebral and cerebellar atrophy. Brain MRI performed before hospital admission: severe cerebral and cerebellar atrophy, vermis hypoplasia, supratentorial and infratentorial demyelinating lesions, cavum septum pellucidi, severe enlargement of the CSF spaces | Valproic acid, later replaced with levetiracetam and after a while with brivaracetam | The seizures disappeared under treatment being seizure-free since childhood; a left facial hemi spasm occurred lately |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stoian, A.; Bajko, Z.; Bălașa, R.; Andone, S.; Stoian, M.; Ormenișan, I.; Muntean, C.; Bănescu, C. Characteristics of Developmental and Epileptic Encephalopathy Associated with PACS2 p.Glu209Lys Pathogenic Variant—Our Experience and Systematic Review of the Literature. Biomolecules 2024, 14, 270. https://doi.org/10.3390/biom14030270
Stoian A, Bajko Z, Bălașa R, Andone S, Stoian M, Ormenișan I, Muntean C, Bănescu C. Characteristics of Developmental and Epileptic Encephalopathy Associated with PACS2 p.Glu209Lys Pathogenic Variant—Our Experience and Systematic Review of the Literature. Biomolecules. 2024; 14(3):270. https://doi.org/10.3390/biom14030270
Chicago/Turabian StyleStoian, Adina, Zoltan Bajko, Rodica Bălașa, Sebastian Andone, Mircea Stoian, Ioana Ormenișan, Carmen Muntean, and Claudia Bănescu. 2024. "Characteristics of Developmental and Epileptic Encephalopathy Associated with PACS2 p.Glu209Lys Pathogenic Variant—Our Experience and Systematic Review of the Literature" Biomolecules 14, no. 3: 270. https://doi.org/10.3390/biom14030270
APA StyleStoian, A., Bajko, Z., Bălașa, R., Andone, S., Stoian, M., Ormenișan, I., Muntean, C., & Bănescu, C. (2024). Characteristics of Developmental and Epileptic Encephalopathy Associated with PACS2 p.Glu209Lys Pathogenic Variant—Our Experience and Systematic Review of the Literature. Biomolecules, 14(3), 270. https://doi.org/10.3390/biom14030270