

Druggable Sterol Metabolizing Enzymes in Infectious Diseases: Cell Targets to Therapeutic Leads




Abstract
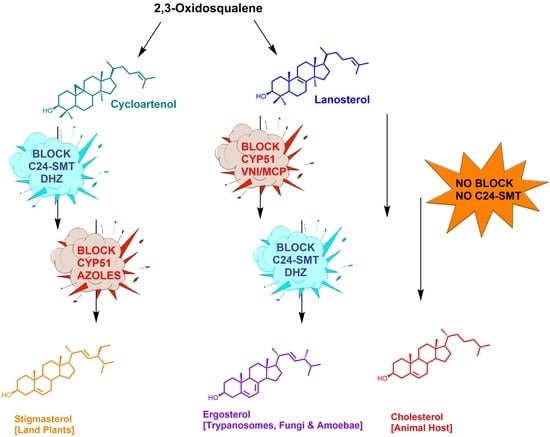
1. Introduction
2. For Context: Background for C24-SMT and C14-SDM Enzymes in Sterol Biosynthesis Pathways
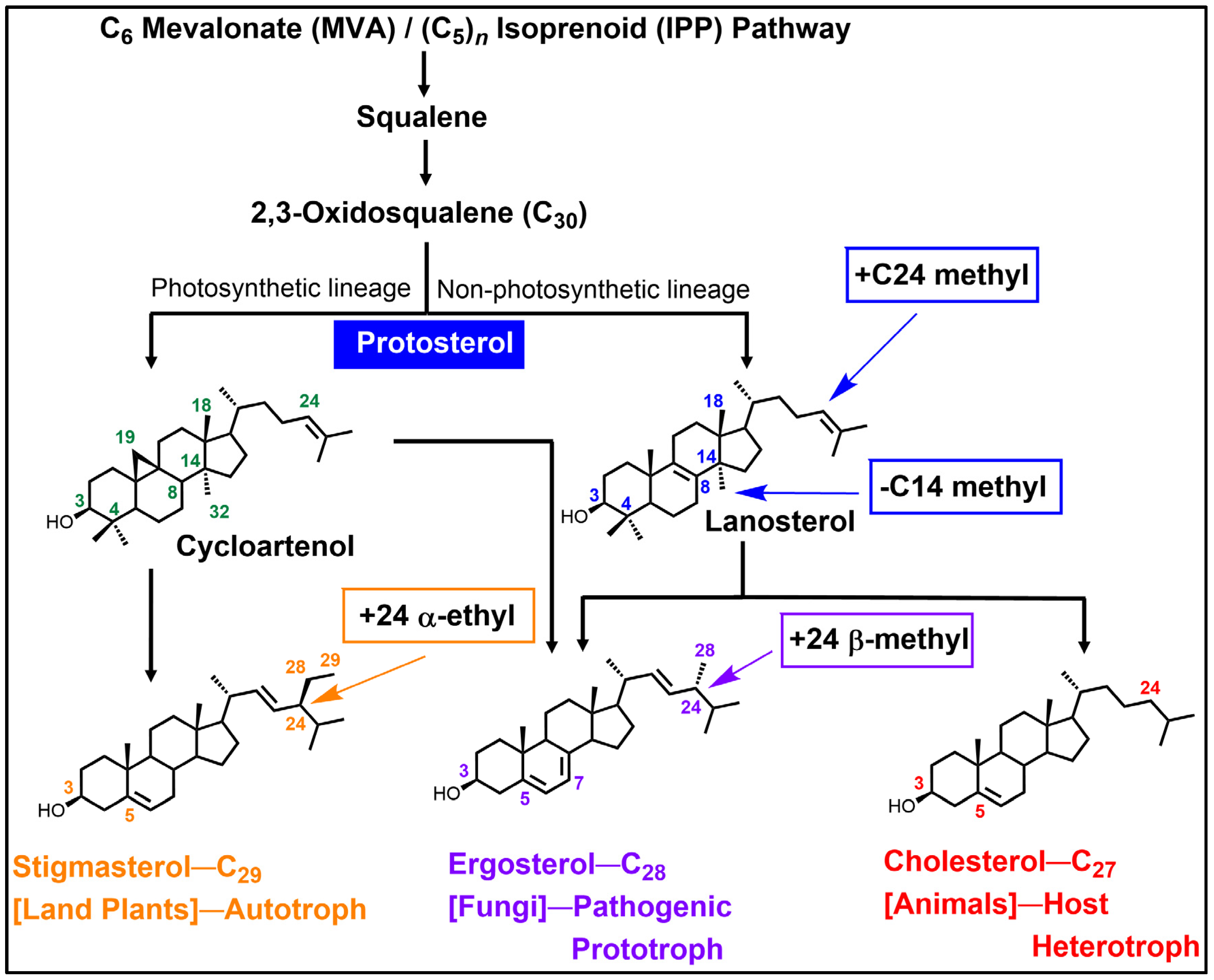
2.1. Substrates in Ergosterol Biosynthesis
2.2. The Evolution of Antifungal Drugs Targeting C24-SMT and C14-SDM in Ergosterol Biosynthesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanism of Action | Block C14-Demethylation | Block C24-Methylation | Complex with C28-Sterol | ||
|---|---|---|---|---|---|
| Biosynthesis Pathway | 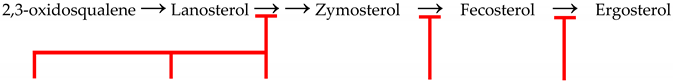 | ||||
| Time Introduced | Early 1980s | Early 1990s | 2022 | 2007 | Early 1960s |
| Drug Class | Imidazoles | Triazoles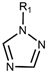 | Tetrazoles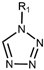 | Arylguanidines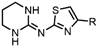 | Polyene Antibiotics |
| Drug Examples |
|
| Oteseconazole | Abafungin |
|
| Drug/Use/Examples |
|
| Vivjoa/vaginal Yeast Infections/ Candidiasis | Abasol/ Nail Fungus- Onychomycosis/Dermatomycoses |
|
2.3. C24-SMT: Differences in Catalytic Competence and Product Distributions in Non-Pathogenic and Pathogenic Organisms
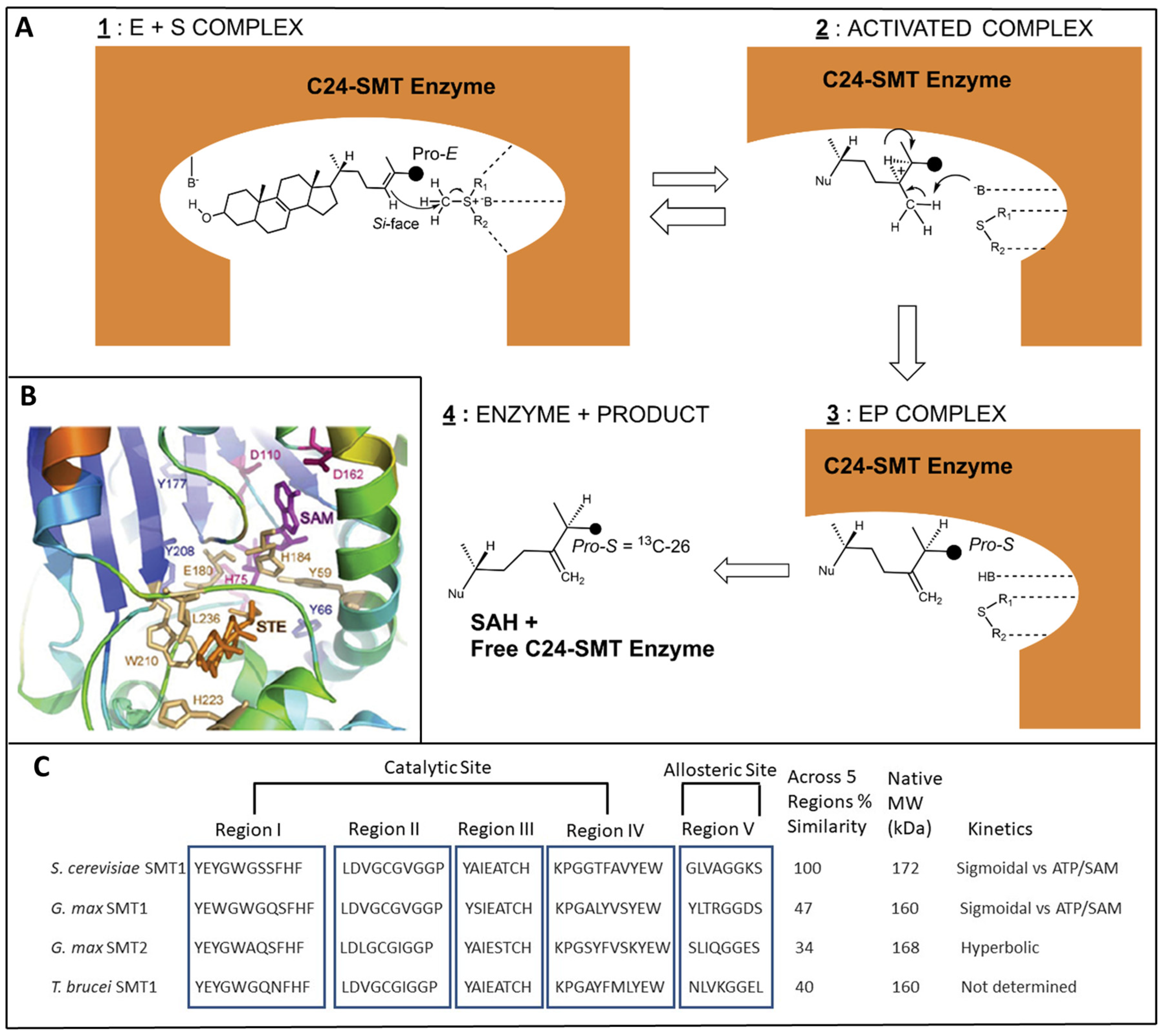
3. C24-SMT Sterol Biosynthesis Inhibitors
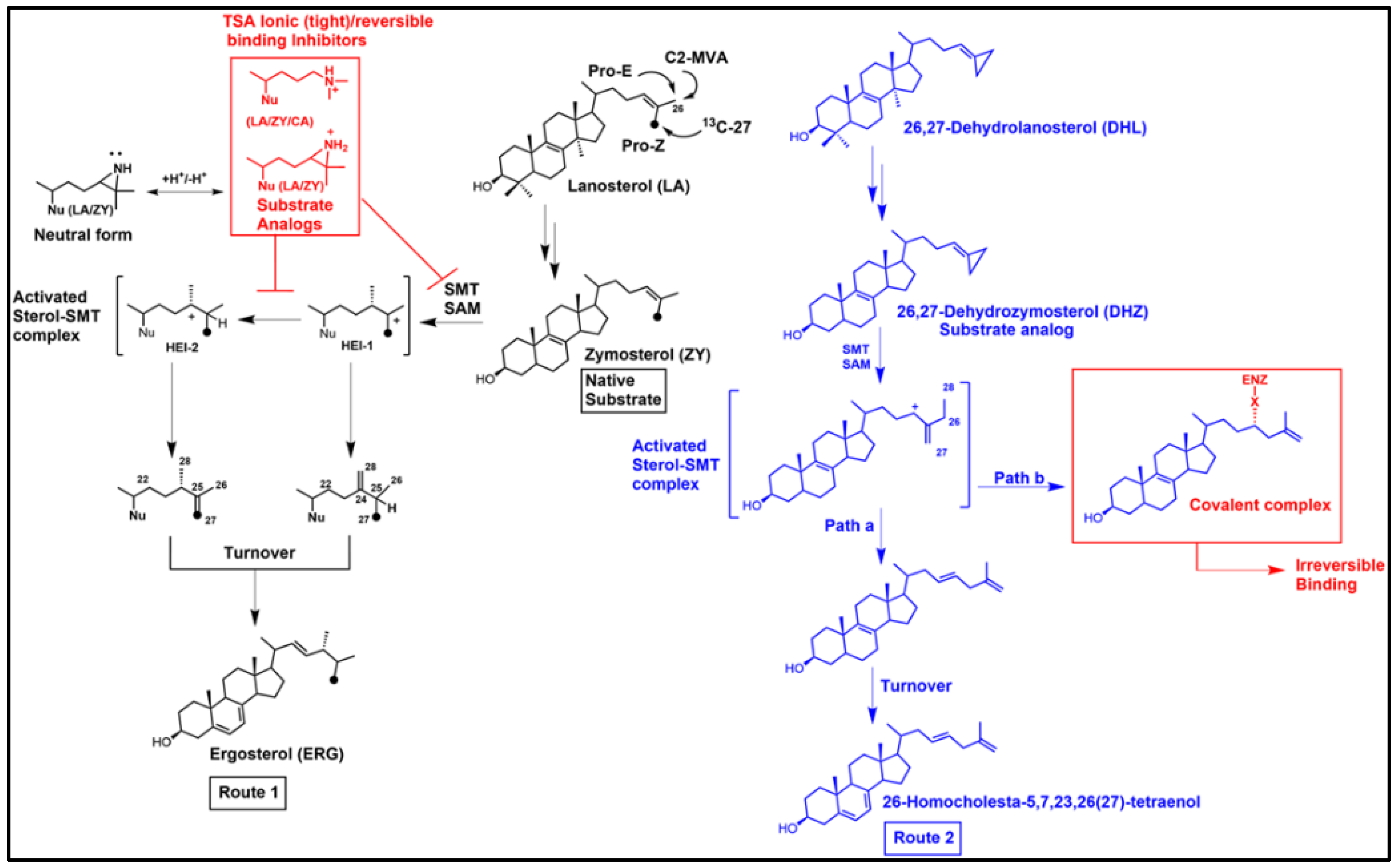
3.1. Inhibition of Ergosterol Biosynthesis Using Reversible-Type Inhibitors: The Transition State Analogs (TSA) Targeting C24-SMT
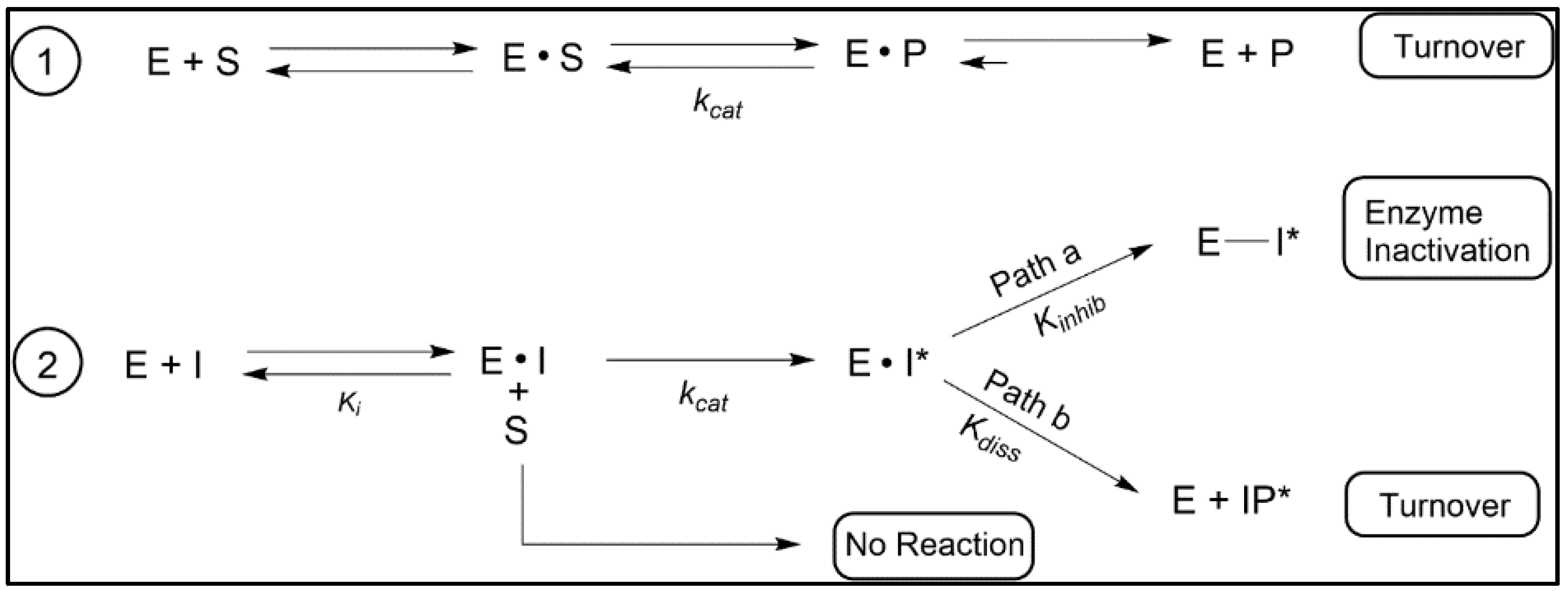
3.2. “Bait and Switch” to Irreversibly Inhibit Ergosterol Synthesis in Parasites, a New Concept
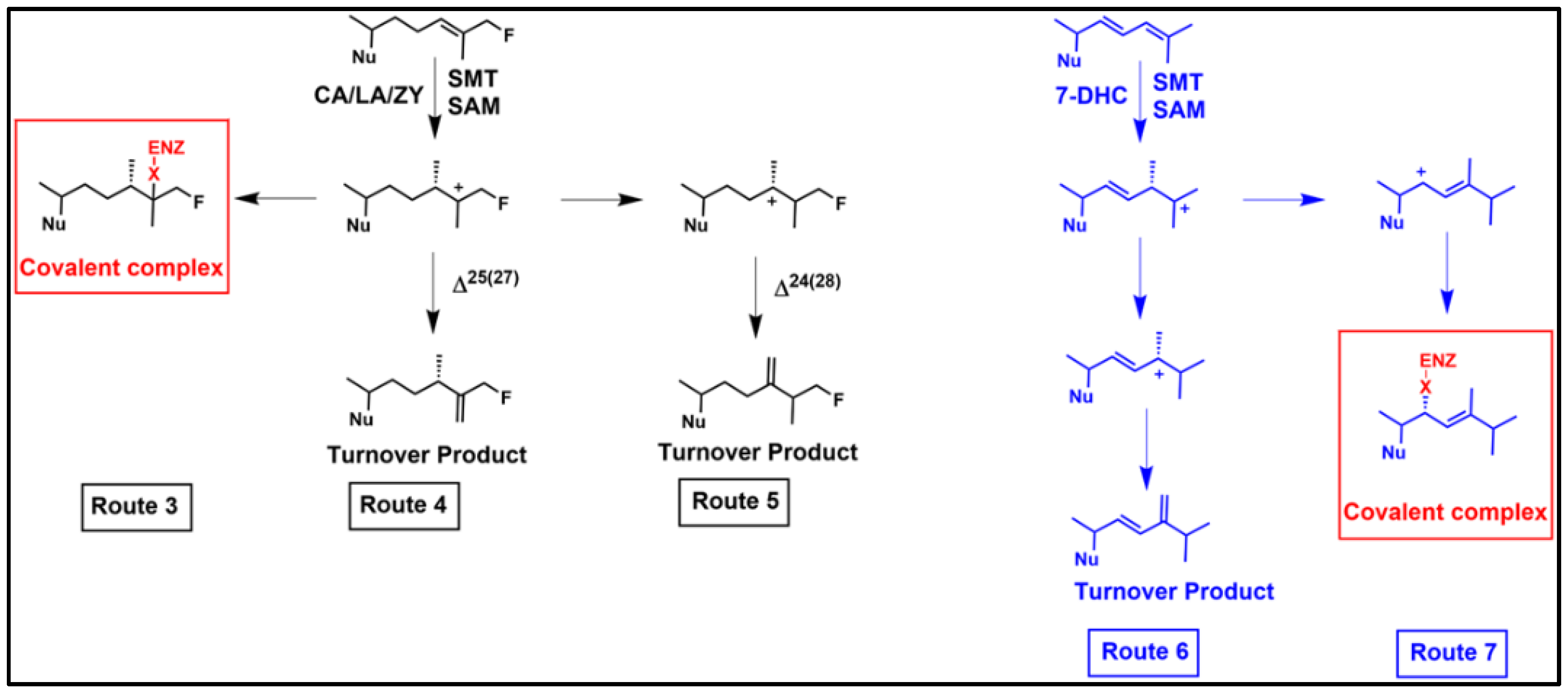
3.3. Aberrant Electrophiles and Strategies for Irreversible-Type C24-SMT Inhibitors as Therapeutic Leads
4. C14-SDM Sterol Biosynthesis Inhibitors
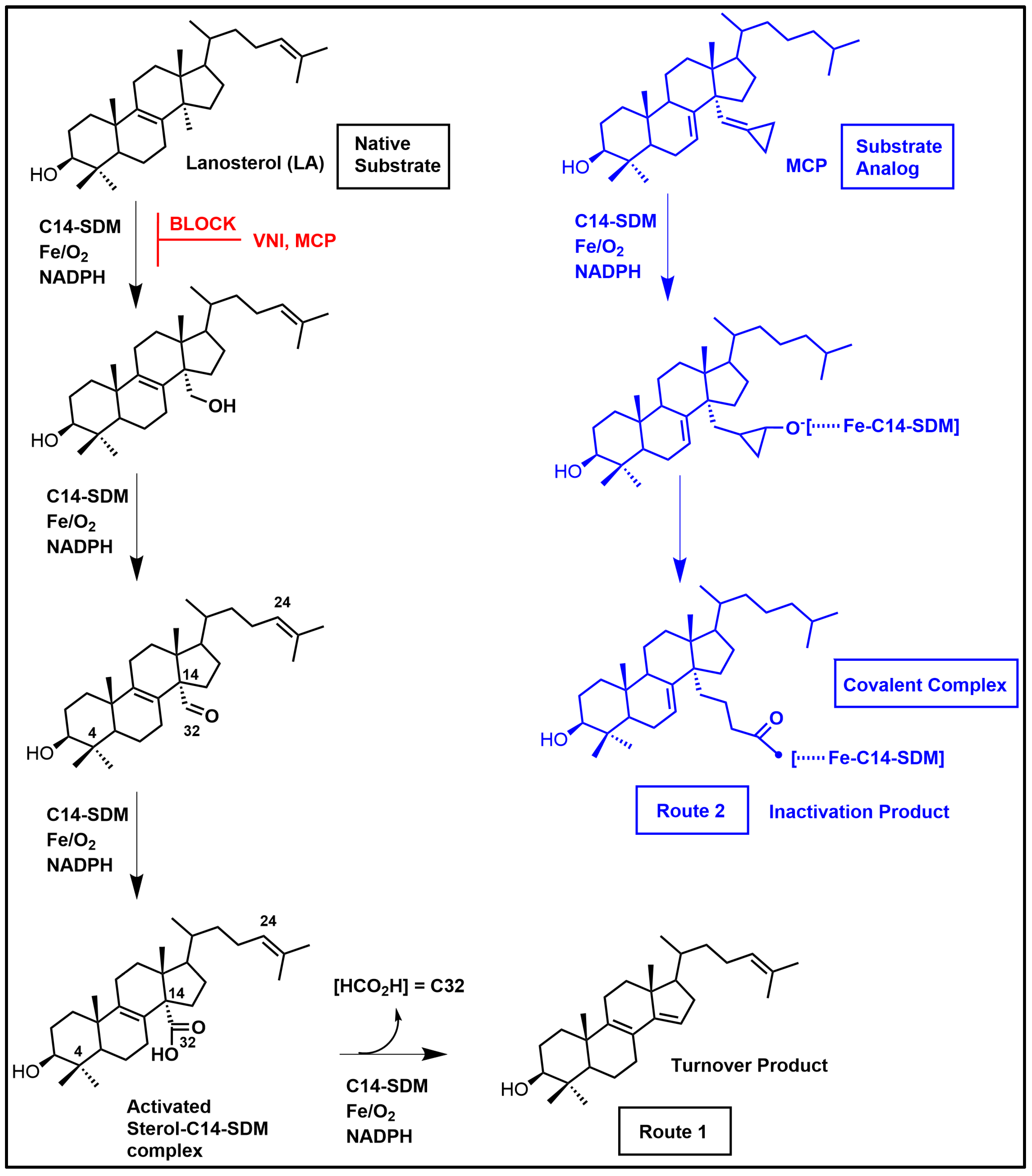
4.1. C14-SDM Catalytic Competence and Properties
4.2. Phenotypic Screening of Azoles—Basis for the Druggability of Protozoan CYP51 Enzymes
4.3. CYP51 Inhibitors: First Efforts for Sterol-Based Substrate Analogs in Treating Heart Disease
4.4. CYP51 Inhibitors: Sterol-Based Analogs for Treating Parasitic Disease
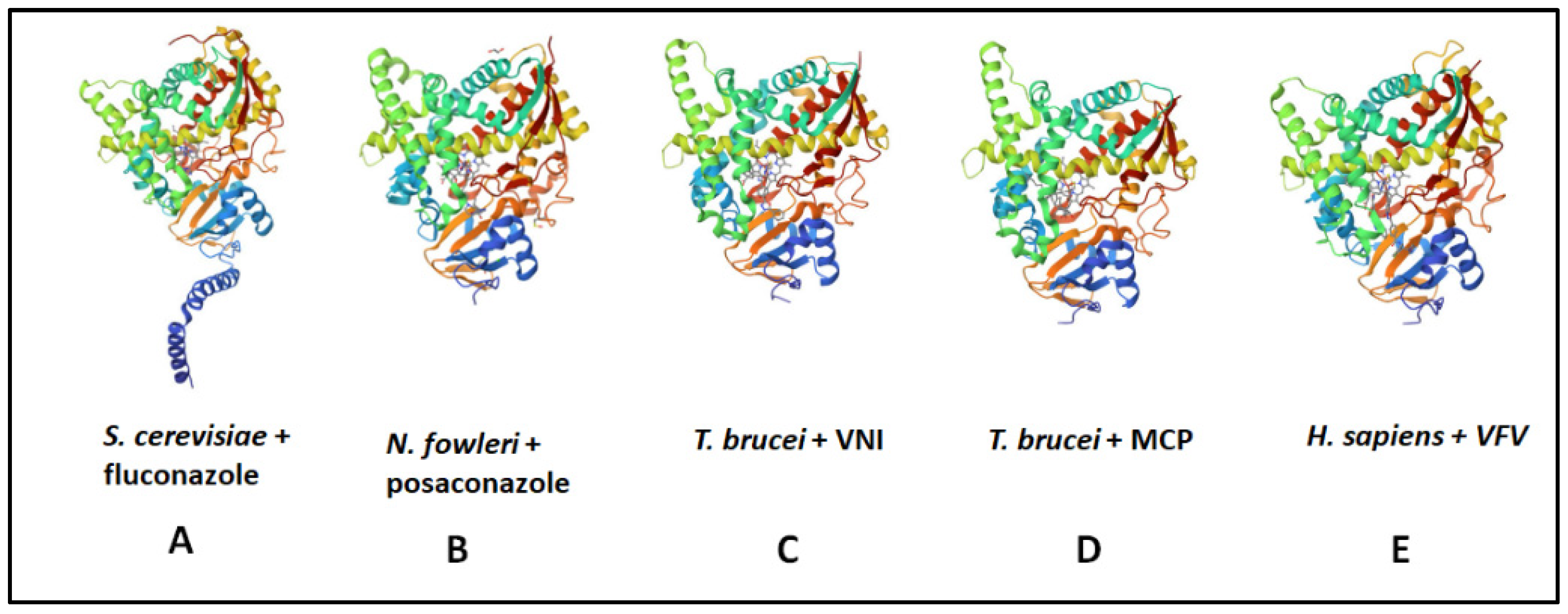
4.5. CYP51 Inhibitors:Azole-Based Time-Dependent Inhibitors for Treating Parasitic Diseases
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nes, W.D. Biosynthesis of Cholesterol and Other Sterols. Chem. Rev. 2011, 111, 6423–6451. [Google Scholar] [CrossRef]
- Madan, B.; Virshup, D.M.; Nes, W.D.; Leaver, D.J. Unearthing the Janus-faced Cholesterogenesis Pathways in Cancer. Biochem. Pharmacol. 2022, 196, 114611. [Google Scholar] [CrossRef]
- Bloch, K.E. Sterol Structure and Membrane Function. Crit. Rev. Biochem. 1983, 14, 47–92. [Google Scholar] [CrossRef] [PubMed]
- Nes, W.R.; Nes, W.D. Lipids in Evolution; Plenum Press: New York, NY, USA, 1980; 144p. [Google Scholar]
- Roberts, C.W.; McLeod, R.; Rice, D.W.; Ginger, M.; Chance, M.L.; Goad, L.J. Fatty and Sterol Metabolism: Potential antimicrobial targets of apicomplexan and trypanosomatid parasitic protozoa. Mol. Biochem. Parasitol. 2003, 126, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Nes, W.R.; McKean, M.L. Biochemistry of Steroids and Other Isopentenoids; University Park Press: Baltimore, MD, USA, 1977. [Google Scholar]
- Ganapathy, K.; Jones, C.W.; Stephens, C.M.; Vatsyayan, R.; Marshall, J.A.; Nes, W.D. Molecular Probing of the Saccharomyces cerevisiae Sterol 24-C methyltransferase Reveals Multiple Amino Acid Residues Involved with C2-transfer Activity. Biochim. Biophys. Acta 2008, 1781, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.M.; Carvalho, D.T.; Sousa, E.; Pinto, E. New Antifungal Agents with Azole Moieties. Pharmaceuticals 2022, 17, 1427. [Google Scholar] [CrossRef]
- de Macedo-Silva, S.T.; Visbal, G.; Urbina, J.A.; de Souza, W.; Rodriques, J.C.F. Potent In Vitro Antiproliferative Synergism of Combinations of Ergosterol Biosynthesis Inhibitors against Leishmania amazonensis. Antimicrob. Agents Chemother. 2015, 59, 6402–6418. [Google Scholar] [CrossRef] [PubMed]
- Shing, B.; Singh, S.; Podust, L.M.; McKerrow, J.H.; Debnath, A. The Antifungal Drug Isovuconazole is both Amoebicidal and Cystcidal against Acanthamoeba castellanii. Antimicrob. Agents Chemother. 2020, 64, e02223-19. [Google Scholar] [CrossRef]
- Moulin, P.C.; Vollrath, J.; Won, M.M.; Wang, J.X.; Burleigh, B.A. Endogenous Sterol Synthesis is Dispensable for Trypanosoma cruzi epimastigote Growth but not Stress Tolerance. Front. Microbiol. 2022, 13, 937910. [Google Scholar] [CrossRef]
- Lu, Z.; Li, Y.; Wei, D.; Wang, J.; Qiao, C.; Li, G.-y.; Zhang, G.; Luo, Y. Biosensor-Enabled Discovery of CaERG6 Inhibitors and Their Antifungal Mode of Action Against Candida albicans. ACS Infect. Dis. 2023, 9, 785–800. [Google Scholar] [CrossRef]
- Leaver, D.J. Synthesis and Biological Activity of Sterol 14α-Demethylase and Sterol C24-methyl transferase Inhibitors. Molecules 2018, 23, 1753. [Google Scholar] [CrossRef]
- Zhou, W.; Warrilow, A.G.S.; Thomas, C.D.; Ramos, E.; Parker, J.E.; Price, C.L.; Vanderloop, B.H.; Fisher, P.M.; Loftis, M.D.; Kelley, D.E.; et al. Functional Importance for Developmental Regulation of Sterol Biosynthesis in Acanthamoeba castellanii. BBA Mol. Cell Biol. Lipids 2018, 1863, 1164–1178. [Google Scholar] [CrossRef]
- Zhou, W.; Debnath, A.; Jennings, G.; Hahn, H.J.; Vanderloop, B.H.; Chaudhuri, M.; Nes, W.D.; Podust, L.M. Enzymatic chokepoints and synergistic drug targets in the sterol biosynthetic pathway of Nageleria fowleri. PLoS Pathog. 2018, 14, e1007245. [Google Scholar] [CrossRef] [PubMed]
- Haubrich, B.A.; Singha, U.K.; Miller, M.B.; Nes, C.R.; Anyatonwu, H.; Lecordier, L.; Patkar, P.; Leaver, D.J.; Villalta, F.; Vanhollebeke, B.; et al. Discovery of an Ergosterol-signaling Factor that Regulates Trypanosome brucei Growth. J. Lipid Res. 2015, 56, 331–341. [Google Scholar] [CrossRef]
- Lepesheva, G.I.; Ott, R.D.; Hargrove, T.Y.; Kleshchenko, Y.Y.; Schuster, I.; Nes, W.D.; Waterman, M.R. Sterol 14α-Demethylase as a Potential Target for Antitrypanosomal Therapy: Enzyme Inhibition and Parasite Cell Growth. Chem. Biol. 2007, 14, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Kanagasabai, R.; Zhou, W.; Nguyen, T.T.; Veeramachaneni, P.; Nes, W.D. Disruption of Ergosterol Biosynthesis, Growth, and the Morphological Transition in Candida albicans by Sterol Methyltransferase Inhibitors Containing Sulfur at C-25 in the Sterol Side Chain. Lipids 2004, 39, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Warrilow, A.G.S.; Martel, C.M.; Parker, J.E.; Melo, N.; Lamb, D.C.; Nes, W.D.; Kelly, D.E.; Kelly, S.L. Azole Binding Properties of Candida albicans Sterol 14α-Demethylase (CaCYP51). Antimicrob. Agents Chemother. 2010, 54, 4235–4245. [Google Scholar] [CrossRef] [PubMed]
- Warrilow, A.G.S.; Parker, J.E.; Price, C.L.; Nes, W.D.; Garvey, E.P.; Hoekstra, W.J.; Schotzinger, R.E.; Kelly, D.E.; Kelly, S.L. The investigational Drug VT-1129 is a Highly Potent Inhibitor of Cryptococcus species CYP51 but only weakly Inhibits the Human Enzyme. Antimicrob. Agents Chemother. 2016, 60, 4530–4538. [Google Scholar] [CrossRef] [PubMed]
- Nes, W.D.; Zhou, W.; Ganapathy, K.; Liu, J.; Vatasvan, R.; Chamala, S.; Hernandez, K.; Miranda, M. Sterol 24-C-Methyltransferase: An Enzymatic Target for the Disruption of Ergosterol Biosynthesis and Homeostasis in Cryptococcus neoformans. Arch. Biochem. Biophys. 2009, 481, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Warrilow, A.G.S.; Parker, J.E.; Price, C.L.; Rolley, N.J.; Nes, W.D.; Kelly, D.E.; Kelly, S.L. Isavucaonazole and Voriconazole Inhibition of Sterol 14α-Demethylases (CYP51) from Aspergillus fumigatus and Homo Sapiens. Int. J. Antimicob. Agents 2019, 54, 449–455. [Google Scholar] [CrossRef]
- Muller, C.; Binder, U.; Bracher, F.; Giera, M. Antifungal drug testing by combining minimal inhibitory concentration testing with target identification by gas-chromatography-mass spectroscopy. Nat. Protoc. 2017, 12, 947–963. [Google Scholar] [CrossRef]
- Zu, P.; Koch, H.; Schwery, O.; Pironon, S.; Phillips, C.; Ondo, I.; Farrell, I.W.; Nes, W.D.; Moore, E.; Wright, G.A.; et al. Pollen sterols are associated with phylogeny and environment but not with pollinator guilds. New Phytol. 2021, 230, 1169–1184. [Google Scholar] [CrossRef] [PubMed]
- Darnet, S.; Blary, A.; Chevalier, Q.; Schaller, H. Phytosterol Profiles, Genomes, and Enzymes. An Overview. Front. Plant Sci. 2021, 12, 665206. [Google Scholar] [CrossRef] [PubMed]
- Nes, W.D.; Norton, R.A.; Crumley, F.G.; Madigan, S.J.; Katz, E.R. Sterol phylogenesis and algal evolution. Proc. Natl. Acad. Sci. USA 1990, 87, 7565–7569. [Google Scholar] [CrossRef]
- Nes, W.D.; Xu, S.; Haddon, W.F. Evidence for Similarities and Differences in the Biosynthesis of Fungal Sterols. Steroids 1989, 53, 533–558. [Google Scholar] [CrossRef]
- Nes, C.R.; Singha, U.K.; Liu, J.; Ganapathy, K.; Villalta, F.; Waterman, M.R.; Lepesheva, G.I.; Chaudhuri, M.; Nes, W.D. Novel sterol metabolic network of Trypanosoma brucei procyclic and blood stream forms. Biochem. J. 2012, 443, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Nes, W.R.; Sekula, B.C.; Nes, W.D.; Adler, J.H. The Functional Importance of Structural Features of Ergosterol in Yeast. J. Biol. Chem. 1978, 253, 6218–6225. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.R.; Nes, W.D. Regulation of Sterol Biosynthesis and Its Phylogenetic Implications. In Regulation of Isopentenoid Metabolism; ACS Symposium Series; Nes, W.D., Parish, E.J., Trazasko, J.M., Eds.; American Chemical Society: Washington, DC, USA, 1992; Volume 497, pp. 110–145. [Google Scholar]
- Bouvier-Nave, P.; Rahier, A.; Camara, B. Biogenesis, Molecular Recognition and Function of Plant Isoprenoids. Prog. Lipid Res. 2005, 44, 357–429. [Google Scholar] [CrossRef]
- Nes, W.D.; Janssen, G.G.; Bergenstrahle, A. Structural Requirements for Transformation of Substrates by the (S)-Adenosyl-L-methionine: Δ24(25)-Sterol Methyl Transferase. J. Biol. Chem. 1991, 266, 15202–15212. [Google Scholar] [CrossRef]
- Gaylor, J.L. Membrane-bound Enzymes of Cholesterol Synthesis from Lanosterol. Biochem. Biophys. Res. Commun. 2002, 292, 1139–1146. [Google Scholar] [CrossRef]
- Nes, W.D.; Venkatramesh, M. Enzymology of Phytosterol Transformations. In Biochemistry and Function of Sterols; Parish, E.J., Nes, W.D., Eds.; CRC Press: Boca Raton, FL, USA, 1997; pp. 111–122. [Google Scholar]
- Guo, D.; Venkatramesh, M.; Nes, W.D. Developmental Regulation of Sterol Biosynthesis in Zea mays. Lipids 1995, 30, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Oehlschlager, A.C.; Angus, R.H.; Pierce, A.M.; Pierce, H.D.; Srinivasan, R. Azasterol inhibition of Δ24-sterol methyltransferase in Saccharomyces cerevisiae. Biochemistry 1984, 23, 3582–3589. [Google Scholar] [CrossRef] [PubMed]
- Nes, W.D. Sterol methytransferase: Enzymology and Inhibition. Biochim. Biophys. Acta 2000, 1529, 63–88. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, T.Y.; Wawrzak, Z.; Liu, J.; Nes, W.D.; Waterman, M.R. Substrate Preferences and Catalytic Parmeters Determined by Structural Characteristics of Sterol 14α-Demethylase from Leishmania infatum. J. Biol. Chem. 2011, 286, 26838–26848. [Google Scholar] [CrossRef]
- Burden, R.S.; Cooke, D.T.; Carter, G.A. Inhibitors of Sterol biosynthesis and Growth in Plants and Fungi. Phytochemistry 1989, 28, 1791–1804. [Google Scholar] [CrossRef]
- Mercer, E.I. Inhibitors of Sterol Biosynthesis, and their Applications. Prog. Lipid Res. 1993, 32, 357–416. [Google Scholar] [CrossRef]
- Nes, W.D.; McCourt, B.S.; Zhou, W.; Ma, J.; Marshall, J.A.; Peek, L.-A.; Brenan, M. Overexpression, Purification, and Steroechemical Studies of the Recombinant (S)-Adenosyl-L-methionine: Δ24(25)- to Δ24(28)-Sterol Methyl Transferase Enzyme from Saccharomyces cerevisiae. Arch. Biochem. Biophys. 1998, 353, 297–311. [Google Scholar] [CrossRef]
- Bellamine, A.; Mangla, A.T.; Nes, W.D.; Waterman, M.R. Characterization and Catalytic Properties of the sterol 14α-Demethylase from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 1999, 96, 8937–8942. [Google Scholar] [CrossRef]
- Fromtling, R.A. Overview of Medically Important Antifungal Azole Derivatives. Clin. Microbiol. Rev. 1988, 1, 187–217. [Google Scholar] [CrossRef]
- Palacias, D.S.; Paizley, I.; Burke, M.D. Amphotericin B Kills Yeast By Simply Binding Ergosterol. Proc. Natl. Acad. Sci. USA 2012, 109, 2234–2239. [Google Scholar] [CrossRef]
- Yocum, R.R. The Mechanism of Action of Penicillin. Penicillin Acylated the Active Site of Bacillus stearothermophilus D-Alanine Carboxypeptidase. J. Biol. Chem. 1980, 10, 3977–3986. [Google Scholar] [CrossRef]
- Vanden Bossche, H.; Marichal, P.; Coene, M.-C.; Willemsens, G.; Jeune, L.L.; Cools, W.; Verhoeven, H. Cytochrome-P450-Dependent 14α-Demethylase: Target for Antifungal Agents and Herbicides. In Regulation of Isopentenoid Metabolism; ACS Symposium Series; Nes, W.D., Parish, E.J., Trzaskos, J.M., Eds.; American Chemical Society: Washington, DC, USA, 1992; Volume 497, pp. 219–230. [Google Scholar]
- Musiol, R.; Kowalczyk, W. Azole Antibiotics—A Highway to New Drugs or a Dead End? Curr. Med. Chem. 2012, 19, 1378–1388. [Google Scholar] [CrossRef] [PubMed]
- Trazaskos, J.M.; Fisher, R.T.; Favata, M.F. Mechanistic Studies of C-32 Demethylation. Conditions which Promote Oxysterol Intermediate Accumulation During the Demethylation Process. J. Biol. Chem. 1986, 36, 16397–16942. [Google Scholar] [CrossRef]
- Favata, M.F.; Trazaskos, J.M.; Chen, H.W.; Fisher, R.T.; Greenberg, R.S. Modulation of 3-Hydroxy-3- Methylglutarylcoenzyme A Reductase by Azole Antimycotics Requires Lanosterol Demethylation, but not 24,25-Epoxylanosterol Formation. J. Biol. Chem. 1987, 262, 12254–12260. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.I.; Hargrove, T.Y.; Anderson, S.; Kleshchenko, Y.; Futak, V.; Wawrzak, Z.; Villalta, F.; Waterman, M.R. Structural Insights into Inhibition of Sterol 14α-demethylase in the Human Pathogen Trypanosoma cruzi. J. Biol. Chem. 2010, 285, 25582–25590. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, W.J.; Garvey, E.P.; Moore, W.R.; Rafferty, S.W.; Yates, C.M.; Schotzinger, R.J. Design and Optimization of Highly-selective Fungal CYP51 Inhibitors. Bioorgan. Med. Chem. Lett. 2014, 24, 3455–3458. [Google Scholar] [CrossRef]
- Worthington, P. Sterol Biosynthesis Inhibiting Triazole Fungicides. In Bioactive Heterocyclic Compound Classes: Agrochemicals; Lamberth, C., Dinges, J., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp. 129–204. [Google Scholar]
- Zhang, J.; Li, L.; Lv, Q.; Yan, L.; Wang, Y.; Jiang, Y. The Fungal CYP51s: Their Functions, Structures, Related Drug Resistance, and Inhibitors. Front. Microbiol. 2019, 10, 691. [Google Scholar] [CrossRef]
- Borelli, C.; Schaller, M.; Niewerth, M.; Nocker, K.; Basner, B.; Berg, D.; Tieman, R.; Tietjen, K.; Fugmann, B.; Lang-Fugmann, S.; et al. Modes of Action of the Arylquanidine Abufungin beyond interference with Ergosterol Biosynthesis and in vitro Activity Against Medically Important Fungi. Chemotherapy 2008, 54, 245–259. [Google Scholar] [CrossRef]
- Kontoyiannis, D.P.; Lewis, R.E. Antifungal Drug Resistance of Pathogenic Fungi. Lancet 2022, 359, 1135–1144. [Google Scholar] [CrossRef]
- Alpizar-Sosa, E.A.; Ithnin, N.R.B.; Wei, W.; Poutain, A.W.; Weidt, S.K.; Donachie, A.M.; Ritchie, R.; Dickie, E.A.; Burchmore, R.J.S.; Denny, P.W.; et al. Amphotericin B Resistance in Leishmania mexicana: Alterations to Sterol Metabolism and Oxidative Stress Repsonse. PLoS Negl. Trop. Dis. 2022, 16, e0010779. [Google Scholar] [CrossRef]
- Malhotra, H.C.; Nes, W.R. The Mechanism of Introduction of Alkyl Groups at C-24 of Sterols IV: Inhibition by Triparinol. J. Biol. Chem. 1971, 246, 4934–4937. [Google Scholar] [CrossRef] [PubMed]
- Ator, M.A.; Schmidt, S.A.; Adams, J.L.; Dolle, R.E. Mechanism and Inhibition of Δ24-Sterol Methyltransferase from Candida albicans and Candida tropicalis. Biochemistry 1989, 28, 9633–9640. [Google Scholar] [CrossRef] [PubMed]
- Ator, M.A.; Schmidt, S.J.; Adams, J.L.; Dolle, R.E.; Kruse, L.I.; Frey, C.L.; Barone, J.M. Synthesis, specificity, and antifungal activity of inhibitors of the Candida albicans Δ24-sterol methyltransferase. J. Med. Chem. 1992, 35, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Arigoni, D. Stereochemical Studies of Enzymic C-Methylations. CIBA Found. Symp. 1978, 60, 243–260. [Google Scholar]
- Nes, W.D. Enzyme Mechanisms for Sterol C-Methylations. Phytochemistry 2003, 64, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Nes, W.D.; Norton, R.A.; Benson, M. Carbon-13 NMR Studies on Sitosterol Biosynthesized from [13C]Mevalonates. Phytochemistry 1992, 31, 805–811. [Google Scholar] [CrossRef]
- Zhou, W.; Nes, W.D. Stereochemistry of Hydrogen Introduction at C-25 in Ergosterol Synthesized by the Mevalonate-independent Pathway. Tetrahedron Lett. 2000, 41, 2791–2795. [Google Scholar] [CrossRef]
- Opitz, S.; Nes, W.D.; Gershenzon, J. Both Methylerythritol Phosphate and Mevalonate Pathways Contribute to Biosynthesis of Each of the Major Isoprenoid Classes in Young Cotton Seedlings. Phytochemistry 2014, 98, 110–119. [Google Scholar] [CrossRef]
- Guo, D.; Jia, Z.; Nes, W.D. Stereochemistry of Hydrogen Migration from C-24 to C-25 During Phytosterol Biomethylation. J. Am. Chem. Soc. 1996, 118, 8507–8508. [Google Scholar] [CrossRef]
- Tong, Y.; McCourt, B.S.; Guo, D.; Mangla, A.T.; Zhou, W.-X.; Jenkins, M.D.; Zhou, W.; Lopez, M.; Nes, W.D. Stereochemical Features of C-Methylations on the Path to Δ24(28)-methylene and Δ24(28)-Ethylidene Sterols: Studies on the Recombinant Phytosterol Methyltransferase from Arabidopsis thaliana. Tetrahedron Lett. 1997, 38, 6115–6118. [Google Scholar] [CrossRef]
- Nes, W.D.; Song, Z.; Dennis, A.L.; Zhou, W.; Nam, J.; Miller, M.B. Biosynthesis of Phytosterols: Kinetic Mechanism for the Enzymatic C-Methylation of Sterols. J. Biol. Chem. 2003, 278, 34505–34516. [Google Scholar] [CrossRef]
- Haubrich, B.A.; Collins, E.K.; Howard, A.L.; Wang, Q.; Snell, W.J.; Miller, M.B.; Thomas, C.D.; Pleasant, S.K.; Nes, W.D. Characterization, Mutagenesis, and Mechanistic Analysis of an Ancient Algal Sterol C24-Methyltransferase: Implications for Understanding Sterol Evolution in the Green Lineage. Phytochemistry 2015, 113, 64–72. [Google Scholar] [CrossRef]
- Mangla, A.T.; Nes, W.D. Sterol C-24 Methyltransferase from Prototheca wickerhamii: Mechanism, Specificity, and Inhibition. Bioorg. Chem. 2000, 8, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Venkatramesh, R.; Guo, D.; Jia, Z.; Nes, W.D. Mechanism and Structural Requirements for Transformation of Substates by the (S)-Adenosyl-L-methionine; Δ24(25)-Sterol Methyltransferase from Saccharomyces cerevisiae. Biochim. Biophys. Acta 1996, 1299, 313–324. [Google Scholar] [CrossRef]
- Howard, A.L.; Liu, J.; Elmegeed, G.A.; Collins, E.K.; Ganatra, K.S.; Nwogwugwu, C.A.; Nes, W.D. Sterol C24-Methyltransferase: Physio-and Stereo-chemical Features of the Sterol C3-Group Required for Catalytic Competence. Arch. Biochem. Biophys. 2012, 521, 43–50. [Google Scholar] [CrossRef]
- Kidane, M.E.; Vanderloop, B.H.; Zhou, W.; Thomas, C.D.; Ramos, E.; Singha, U.; Chaudhuri, C.; Nes, W.D. Sterol Methyltransferase a Target for Anti-amoeba Therapy: Towards Transition State Analog and Suicide Substrate Drug Design. J. Lipid Res. 2017, 58, 2310–2323. [Google Scholar] [CrossRef]
- Zhou, W.; Lepesheva, G.I.; Waterman, M.R.; Nes, W.D. Mechanistic Analysis of a Multiple Product Sterol Methyltransferase Implicated in Ergosterol Biosynthesis in Trypanosoma brucei. J. Biol. Chem. 2006, 281, 6290–6296. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ganapathy, K.; Wywial, E.; Bujnicki, J.M.; Nwogwugwu, C.A.; Nes, W.D. Effect of Substrate Features and Mutagenesis of Active Site Tyrosine Residues on the Reaction Course Catalyzed by Trypanosoma brucei Sterol C24-Methyltransferase. Biochem. J. 2011, 439, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Nes, W.D.; Jayasimha, P.; Zhou, W.; Kanagasbai, R.; Lin, C.; Jaradat, T.T.; Shaw, R.W.; Bujnicki, J.M. Sterol Methyltransferase. Functional Analysis of Highly Conserved Residues by Site-Directed Mutagenesis. Biochemistry 2004, 43, 569–576. [Google Scholar] [CrossRef]
- Marshall, J.A. Studies on the Enzymology of Sterol Methyltransferase from Saccharomyces cerevisiae. Ph.D. Dissertation, Texas Tech University, Lubbock, TX, USA, 2001. [Google Scholar]
- Patkar, P. Sterol Methyltransferase: Probing Its Drug Target and Allosteric Properties. Ph.D. Dissertation, Texas Tech University, Lubbock, TX, USA, 2016. [Google Scholar]
- Jayasimha, P. Sterol Methyltransferase: Protein Engineering, Molecular Mapping of AdoMet Binding Site, Thermodynamic Analysis, and Its Phylogenetic Implications. Ph.D. Dissertation, Texas Tech University, Lubbock, TX, USA, 2006. [Google Scholar]
- Neelakandan, A.K.; Song, Z.; Wang, J.; Richards, M.H.; Wu, X.; Valliyodan, B.; Nguyen, H.G.; Nes, W.D. Cloning, Functional Expression and Phylogenetic Analysis of Plant Sterol C24-Methyltransferases Involved in Sitosterol Biosynthesis. Phytochemistry 2009, 70, 1982–1988. [Google Scholar] [CrossRef]
- Song, Z.; Nes, W.D. Sterol Biosynthesis Inhibitors: Potential for Transition State Analogs and Mechanism-based Inactivators Targeted at Sterol Methyltransferase. Lipids 2007, 42, 15–33. [Google Scholar] [CrossRef]
- Ganapathy, K.R.; Kanagasabai, R.; Nguyen, T.T.M.; Nes, W.D. Purification, Characterization, and Inhibition, of Sterol C24-Methyltransferase from Candida albicans. Arch. Biochem. Biophys. 2011, 505, 194–201. [Google Scholar] [CrossRef]
- Soape, M.P. Protein Chemistry, Peptide Mapping, and Preliminary Structural Characterization of Saccharomyces cerevisiae Sterol C24-Methyltransferase Expressed in Escherichia coli. Master’s Thesis, Texas Tech University, Lubbock, TX, USA, 2007. [Google Scholar]
- Nes, W.D.; Sinha, A.; Jayasimha, P.; Zhou, W.; Song, Z.; Dennis, A.L. Probing the Sterol Binding Site of Soybean Sterol Methyltransferase by Site-Directed Mutagenesis: Functional Analysis of Conserved Amino Acids in Region I. Arch. Biochem. Biophys. 2006, 448, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Nes, W.D.; NcCourt, B.S.; Marshall, J.A.; Dennis, A.L.; Lopez, M.; Le, H. Site-directed Mutagenesis of the Sterol Methyltransferase Active Site from Saccharomyces cerevisiae Results in Novel 24-Ethyl Sterols. J. Org. Chem. 1999, 64, 1535–1542. [Google Scholar] [CrossRef]
- Jayasimha, P.J.; Nes, W.D. Photoaffinity Labeling and Mutational Analysis of the 24C- Methyltransferase Defines the AdoMet Binding Site. Lipids 2008, 43, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Nes, W.D.; Marshall, J.A.; Jia, Y.; Jaradat, T.T.; Song, Z. Active Site Mapping and Substrate Channeling in the Sterol Methyltransferase Pathway. J. Biol. Chem. 2002, 277, 42549–42556. [Google Scholar] [CrossRef] [PubMed]
- Nes, W.D.; Jaysimha, P.; Song, Z. Yeast Sterol C24-methyltransferase: Role of Highly Conserved Tyrosine-81 in Catalytic Competence Studied by Site-Directed Mutagenesis and Thermodynamic Analysis. Arch. Biochem. Biophys. 2008, 477, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Nes, W.D.; Hanners, P.K.; Parish, E.J. Control of Fungal Sterol C-24 Transalkylation: Importance to Developmental Regulation. Biochem. Biophys. Res. Commun. 1986, 139, 410–415. [Google Scholar] [CrossRef]
- Nes, W.D.; Janssen, G.G.; Norton, R.A.; Kalinowska, M.; Crumley, F.G.; Tal, B. Regulation of Sterol Biosynthesis in Sunflower by 24(R,S)-Epiminolanosterol, a Novel C24-Methyltransferase Inhibitor. Biochem. Biophys. Res. Commun. 1991, 177, 566–574. [Google Scholar] [CrossRef]
- Urbina, J.A.; Vivas, J.; Lazardi, J.; Molina, J.; Payares, G.; Piras, M.M.; Piras, R. Antiproliferative Effects of Δ24(25)-Sterol Methyltransferase Inhibitors on Trypanosoma (schizotrypanum) cruzi: In vitro and in vivo Studies. Chemotherapy 1996, 42, 294–307. [Google Scholar] [CrossRef]
- Sowa, M.A. Characterization, and Inhibition of C24-Methyltransferase of Trypanosoma cruzi. Master’s Thesis, Texas Tech University, Lubbock, TX, USA, 2016. [Google Scholar]
- Mohr, G. Sterol Methyltransferase Enzyme, and Its Chemotherapeutic Implications for Chagas Disease. Ph.D. Dissertation, Texas Tech University, Lubbock, TX, USA, 2014. [Google Scholar]
- Popjak, G.; Meenan, A.; Parish, E.J.; Nes, W.D. Inhibition of Cholesterol Synthesis and Cell Growth by 24(R,S), 25-Epiminolanosterol and Triparinol in Cultured Rat Hepatoma Cells. J. Biol. Chem. 1989, 264, 6230–6238. [Google Scholar] [CrossRef]
- Pereira, M.Z.; Song, Z.; Santos-Silva, L.K.; Richards, M.R.; Nguyen, T.T.; Liu, J.; Ganapathy, K.; Nes, W.D. Cloning, Mechanistic and Functional Analysis of a Fungal Sterol C-24 Methyltransferase Implicated in Brassicasterol Biosynthesis. Biochim. Biophys. Acta 2012, 1801, 1163–1174. [Google Scholar] [CrossRef]
- Zhou, W.; Cross, G.A.M.; Nes, W.D. Cholesterol Import Fails to Prevent Catalyst-based Inhibition of Ergosterol Synthesis and Cell Proliferation of Trypanosoma brucei. J. Lipid Res. 2007, 48, 655–673. [Google Scholar] [CrossRef]
- Nes, W.D.; Guo, D.; Zhou, W. Substrate-based Inhibitors of the (S)-Adenosyl-L-methionine: Δ24(25)-to Δ24(28)-Sterol Methyltransferase from Saccharomyces cerevisiae. Arch. Biochem. Biophys. 1997, 342, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Keeler, R.F. Mammalian Teratogenicity of Steroidal Alkaloids. In Isopentenoids in Plants; Nes, W.D., Fuller, G., Tsai, L.-S., Eds.; Marcel Dekker: New York, NY, USA, 1984; pp. 531–562. [Google Scholar]
- Sakyi, P.O.; Broni, E.; Amewu, R.K.; Miller, W.A., III.; Wilson, M.D.; Kwofie, S.K. Homology Modeling, de Novo Design of Ligands, and Molecular Docking Identity Potential Inhibitors of Leishmania donovani 24-Sterol Methyltransferase. Front. Cell. Infect. Microbiol. 2022, 12, 859981. [Google Scholar] [CrossRef] [PubMed]
- Leaver, D.J.; Patkar, P.; Singha, M.; Miller, M.B.; Haubrich, B.A.; Chaudhuri, M.; Nes, W.D. Fluorinated Sterols are Suicide Inhibitors of Ergosterol Biosynthesis that Prevents Growth of Trypanosoma brucei. Chem. Biol. 2015, 22, 1374–1383. [Google Scholar] [CrossRef]
- Miller, M.B.; Patkar, P.; Singha, U.K.; Chaudhuri, M.; Nes, W.D. 24-Methylenecyclopropane Steroidal Inhibitors: A Trojan Horse in Ergosterol Biosynthesis that Prevents Growth of Trypanosoma brucei. Biochim. Biophys. Acta 2017, 1862, 305–317. [Google Scholar] [CrossRef]
- Patkar, P.; Haubrich, B.A.; Qi, M.; Nguyen, T.M.; Thomas, C.D.; Nes, W.D. C24-Sterol Methylation of 26-Fluorocycloartenols by Recombinant Sterol C24-Methyltransferase: Evidence for Channel Switching and Its Phylogenetic Implications. Biochem. J. 2015, 456, 253–262. [Google Scholar] [CrossRef]
- Ray, S.; Murkin, A.S. New Electrophiles and Strategies for Mechanism-based and Targeted Covalent Inhibitor Design. Biochemistry 2019, 58, 5234–5244. [Google Scholar] [CrossRef]
- Hargrove, T.Y.; Wawrzak, Z.; Liu, J.; Waterman, M.R.; Nes, W.D.; Lepesheva, G.I. Structural Complex of Sterol 14α-Deme-1447 thylase (CYP51) with 14α-Methylenecyclopropyl-7,24-dihydrolanosterol. J. Lipid Res. 2012, 53, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Heby, O.; Roberts, S.C.; Ullman, B. Polyamine Biosynthetic Enzymes as Drug Targets in Parasitic Protozoa. Biochem. Soc. Trans. 2003, 31, 7710–7726. [Google Scholar] [CrossRef]
- Venkatramesh, M.; Nes, W.D. Novel Sterol Transformations Promoted by Saccharomyces cerevisiae Strain GL7: Evidence for 9β,19-Cyclopropyl to 9(11)-Isomerizations and 14-Demethylation to 8(14)-Sterols. Arch. Biochem. Biophys. 1995, 324, 189–199. [Google Scholar] [CrossRef]
- De Souza, W.; Rodrigues, J.C.F. Sterol Biosynthesis Pathway as Target for Anti-trypansomal Drugs Interdisciplin. Perspect. Infect. Dis. 2009, 2009, 642502. [Google Scholar] [CrossRef]
- Gilbert, L.H. Drug Discovery for Neglected Diseases: Molecular Target-based and Phenotypic Approaches. J. Med. Chem. 2013, 56, 7719–7726. [Google Scholar] [CrossRef] [PubMed]
- Nes, W.D.; Stafford, A.E. Evidence for Metabolic and Functional Discrimination of Sterols by Phytophthora cactorum. Proc. Natl. Acad. Sci. USA 1983, 80, 3227–3231. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Fisher, P.M.; Vanderloop, B.H.; Shen, Y.; Shi, H.; Maldonado, A.J.; Leaver, D.J.; Nes, W.D. A Nematode Sterol 4-Methyltransferase Catalyzes a New Methylation Reaction Responsible for Sterol Diversity. J. Lipid Res. 2020, 61, 192–2020. [Google Scholar] [CrossRef] [PubMed]
- Zundel, M.; Nambiar, K.P.; Boswell, G.; Bloch, K. 6-Fluorocholesterol a Growth Factor for the Yeast Mutant GL7. Biochemistry 1989, 28, 5161–5164. [Google Scholar] [CrossRef] [PubMed]
- Janssen, G.G.; Nes, W.D. Structural Requirements for Transformation of Substrates by the S-Adenosyl-L-methionine: Δ24(25)-Sterol Methyltransferase. Inhibition by Analogs of the Transition State Coordinate. J. Biol. Chem. 1992, 267, 25856–25863. [Google Scholar] [CrossRef]
- Zhou, W.; Ramos, E.; Zhu, X.; Fisher, P.M.; Kidane, M.E.; Vanderloop, B.H.; Thomas, C.D.; Yan, Y.; Singha, U.; Chaudhuri, M.; et al. Steroidal Antibiotics Are Antimetabolites of Acanthamoeba Steroidogenesis with Phylogenetic Implications. J. Lipid Res. 2019, 60, 981–994. [Google Scholar] [CrossRef]
- Chaudhuri, M.; Singha, U.K.; Vanderloop, B.H.; Tripathi, A.; Nes, W.D. Steroidal Antimetabolites Protect Mice Against Trypanosoma brucei. Molecules 2022, 27, 4088. [Google Scholar] [CrossRef]
- Hargrove, T.Y.; Lamb, D.C.; Smith, J.A.; Wawrzak, Z.; Kelly, S.L.; Lepesheva, G.I. Unraveling the Role of Transient Redox Partner Complexes in P450 Electron Transfer Mechanics. Sci. Rep. 2022, 12, 16232. [Google Scholar] [CrossRef]
- Lamb, D.C.; Hargrove, T.Y.; Zhao, B.; Wawrzak, Z.; Goldstone, J.V.; Nes, W.D.; Kelly, S.L.; Waterman, M.R.; Stegeman, J.J.; Lepesheva, G.I. Concerning P450 Evolution: Structural Analyses Support Bacterial Origin of Sterol 14-Demethylases. Mol. Biol. Evol. 2021, 38, 952–967. [Google Scholar] [CrossRef]
- Nes, W.D.; Koike, K.; Jia, Z.; Sakamoto, Y.; Satou, T.; Nikaido, T.; Griffin, J.F. 9β,19-Cyclosterol Analysis by 1HNMR and 13CNMR, Crystallographic Observations, and Molecular Mechanics Calculations. J. Am. Chem. Soc. 1998, 120, 5970–5980. [Google Scholar] [CrossRef]
- Hargrove, T.Y.; Wawrzak, Z.; Guengerich, F.P.; Lepesheva, G.I. A Requirement for an Active Proton Delivery Network Supports a Compound-I-Mediated C-C Bond Cleavage in CYP51 Catalysis. J. Biol. Chem. 2020, 295, 9998–10007. [Google Scholar] [CrossRef]
- Hargrove, T.Y.; Friggeri, L.; Wawrzak, Z.; Sivakumaran, S.; Yazlovitskaya, E.M.; Hiebert, S.W.; Guengerich, F.P.; Waterman, M.R.; Lepesheva, G.I. Human sterol 14α-demethylase as a target for anticancer. J. Lipid Res. 2016, 57, 1552–1563. [Google Scholar] [CrossRef] [PubMed]
- Strushkevich, N.; Usanov, S.A.; Park, H.-W. Structural Basis of Human CYP51 Inhibition by Antifungal Azoles. J. Mol. Biol. 2010, 397, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, T.Y.; Wawrzak, Z.; Lamb, D.C.; Guengerich, F.P.; Lepesheva, G.I. Structure-function Characterization of Cytochrome P450 Sterol 14α-Demethylase (CYP51B) from Aspergillus fumigatus and Molecular Basis for the Development of Antifungal Drugs. J. Biol. Chem. 2015, 290, 23916–32934. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, N.J.; Cools, H.J.; Sierotzki, H.; Shaw, M.W.; Knogge, W.; Kelly, S.L.; Kelly, D.E.; Fraaije, B.A. Paralog Re-emergence: A Novel, Historically Contingent Mechanism in the Evolution of Antimicrobial Resistance. Mol. Biol. Evol. 2014, 31, 1793–1802. [Google Scholar] [CrossRef] [PubMed]
- Mellado, E.; Diaz-Guerra, T.M.; Cuenca-Estrella, M.; Rodriquez-Tudela, J.L. Identification of Two Different 14-Sterol demethylated genes (CYP51A and CYP51B) in Aspergillus fumigatus and other Aspergillus species. J. Clin. Microbiol. 2001, 39, 2431–2438. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.I.; Waterman, M.R. Sterol 14α-Demethylase Cytochrome P450 (CYP51), a P450 in all Biological Kingdoms. Biocim. Biophys. Acta 2007, 1770, 467–477. [Google Scholar] [CrossRef]
- Beach, D.H.; Goad, L.J.; Holz, G.G. Effects of Ketoconazole on Sterol Biosynthesis by Trypanosoma cruzi Epimastogotes. Biochem. Biophys. Res. Commun. 1986, 14, 851–856. [Google Scholar] [CrossRef]
- Hirst, L.W.; Green, W.R.; Merz, W.; Kaufman, C.; Visvesvara, G.S.; Jensen, A.; Howard, M. Management of Acanthamoeba keratitis. A Case Report and Review of Literature. Ophthalmology 1984, 91, 1105–1111. [Google Scholar] [CrossRef]
- Urbina, J.A. Lipid Biosynthesis Pathways as Chemotherapeutic Targets in Kinetoplastid Parasites. Parasitology 1997, 114, S91–S99. [Google Scholar] [CrossRef]
- Lepesheva, G.I.; Friggeri, L.; Waterman, M.R. CYP51 as Drug Targets for Fungi and Protozoan Parasites: Past, Present and Future. Parasitology 2018, 145, 1820–1836. [Google Scholar] [CrossRef]
- Villalta, F.; Dobish, M.C.; Nde, P.N.; Kleschenko, Y.Y.; Hargrove, T.Y.; Johnson, C.A.; Waterman, M.R.; Johnston, J.N.; Lepesheva, G.I. VNI Cures Acute and Chronic Experimental Chagas Disease. J. Infect. Dis. 2013, 208, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Hankins, E.G.; Gillespie, J.R.; Aikenhead, K.; Buckner, F.S. Upregulation of Sterol C14-Demethylase Expression in Trypanosoma cruzi Treated with Sterol Biosynthesis Inhibitors. Mol. Biochem. Parasitol. 2005, 144, 68–75. [Google Scholar] [CrossRef]
- Debnath, A.; Calvet, C.M.; Jennings, G.; Zhou, W.; Aksenov, A.; Luth, M.R.; Abagyan, R.; Nes, W.D.; McKerrow, J.H.; Podust, L.M. CYP51 is an Essential Drug Target for the Treatment of Primary Amoebic Meningoencephalitis (PAM). PLoS Negl. Trop. Dis. 2017, 11, e0006104. [Google Scholar] [CrossRef] [PubMed]
- Lamb, D.C.; Warrilow, A.G.S.; Rolley, N.J.; Parker, J.E.; Nes, W.D.; Smith, S.N.; Kelly, D.E.; Kelly, S.L. Azole Antifungal Agents to Treat the Human Pathogens Acanthamoeba castellanii and Acanthamoeba polyphoga Through Inhibition of Sterol 14α-Demethylase. Antimicrob. Agents Chemotherp. 2015, 59, 4707–4713. [Google Scholar] [CrossRef] [PubMed]
- Frye, L.L.; Leonard, D.A. Dual-Action Inhibitors of Cholesterol Biosynthesis: Lanosterol Analogs That Inhibit Lanosterol 14α-Demethylase and Suppress 3-Hydroxy-3-methylglutaryl-Coenzyme A Reductase Activity. In Regulation of Isopentenoid Metabolism; ACS Symposium Series; Nes, W.D., Parish, E.J., Trzaskos, J.M., Eds.; American Chemical Society: Washington, DC, USA, 1992; Volume 497, pp. 94–108. [Google Scholar]
- Frye, L.L.; Leonard, D.A. Lanosterol Analogs: Dual Action Inhibitors of Cholesterol Biosynthesis. In Biochemistry and Function of Sterols; Parish, E.J., Nes, W.D., Eds.; CRC Press: Boca Raton, FL, USA, 1997; pp. 151–168. [Google Scholar]
- Trzakos, J.M.; Magolda, R.L.; Favata, M.F.; Leonard, D.A.; Gaylor, J.L. Modulation of 3-Hydroxy-3-methylglutaryl CoA Reductase by 15-alpha-fluorolanost-7-en-3beta-ol. A Mechanism-based Inhibitor of Cholesterol Biosynthesis. J. Biol. Chem. 1993, 268, 22591–22599. [Google Scholar] [CrossRef]
- Rando, R.R. Chemistry and Enzymology of kcat inhibitors. Science 1974, 185, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Sutanto, F.; Konstantinidou, M.; Domling, A. Covalent Inhibitors: A Rational Approach to Drug Discovery. RSC Med. Chem. 2020, 11, 876–884. [Google Scholar] [CrossRef]
- Holgate, G.A.; Meek, T.D.; Grimley, R.L. Mechanistic enzymology in Drug Discovery: A Fresh Perspective. Nat. Rev. Drug Discov. 2018, 17, 115–132. [Google Scholar] [CrossRef]
- Lepesheva, G.I.; Park, H.-W.; Hargrove, T.Y.; Vanhollebeke, B.; Wawrzak, Z.; Harp, J.M.; Sundaramoorthy, M.; Nes, W.D.; Pays, E.; Chaudhuri, M.; et al. Crystal Structures of Trypanosoma brucei Sterol 14-Demethylase and Implications for Selective Treatment of Human Infections. J. Biol. Chem. 2010, 265, 1773–17804. [Google Scholar] [CrossRef]
- Friggeri, L.; Hargrove, T.Y.; Wawrzak, Z.; Guengerich, F.P.; Lepesheva, G.I. Validation of Human Sterol 14α-Demethylase (CYP51) Druggability: Structure-guided Design, Synthesis, and Evaluation of Stoichiometric, Functionally Irreversible Inhibitors. J. Med. Chem. 2019, 62, 10391–10401. [Google Scholar] [CrossRef]
- Hargrove, T.Y.; Wawezak, Z.; Rachokonda, G.; Nes, W.D.; Villalta, F.; Guengerich, F.P.; Lepesheva, G.L. Relaxed Substrate Requirements of Sterol 14α-Demethylase from Naegleria fowleri Are Accompanied by Resistance to Inhibition. J. Med. Chem. 2021, 64, 17511–17522. [Google Scholar] [CrossRef]
- Chen, C.-K.; Leung, S.S.F.; Guilbert, C.; Jacobson, M.P.; McKerrow, J.H.; Podust, L.M. Structural Characterization of CYP51 from Trypansoma cruzi and Trypanosoma brucei Bound to the Antifungal Drugs Posaconazole and Fluconazole. PLoS Negl. Trop. Dis. 2010, 4, e651. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.I.; Nes, W.D.; Zhou, W.; Hill, H.C.; Waterman, M.R. CYP51 from Trypanosoma brucei is Obtusifoliol-specific. Biochemistry 2004, 43, 10789–10799. [Google Scholar] [CrossRef] [PubMed]
- Sagatova, A.A.; Keniya, M.V.; Wilson, R.K.; Monk, B.C.; Tyndall, J.D.A. Structural Insights into Binding of the Antifungal Drug Fluconazole to Saaccharomyces cerevisiae Lanosterol 14α-Demethylase. Antimicrob. Agents Chemother. 2015, 59, 4982–4989. [Google Scholar] [CrossRef] [PubMed]
- D Bloch, K. Sterol Molecule: Structure, Function and Biosynthesis. Steroids 1992, 57, 378–383. [Google Scholar] [CrossRef]
- Gibbons, G.F. From Gallstones to Genes: Two Hundred Years of Sterol Research. A Tribute to George J. Schroepfer Jr. Lipids 2002, 37, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.I.; Villalta, F.; Waterman, M.R. Targeting Trypanosoma cruzi Sterol 14α-Demethylase (CYP51). Adv. Parasitol. 2011, 75, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Kane, A.; Carter, D.A. Augmenting Azoles with Drug Synergy to Expand the Antifungal Toolbox. Pharmaceuticals 2022, 15, 482. [Google Scholar] [CrossRef] [PubMed]
- Weiderhold, N.P. Pharmacodynamics, Mechanisms of Action, Resistance. J. Fungi 2022, 8, 857. [Google Scholar] [CrossRef] [PubMed]
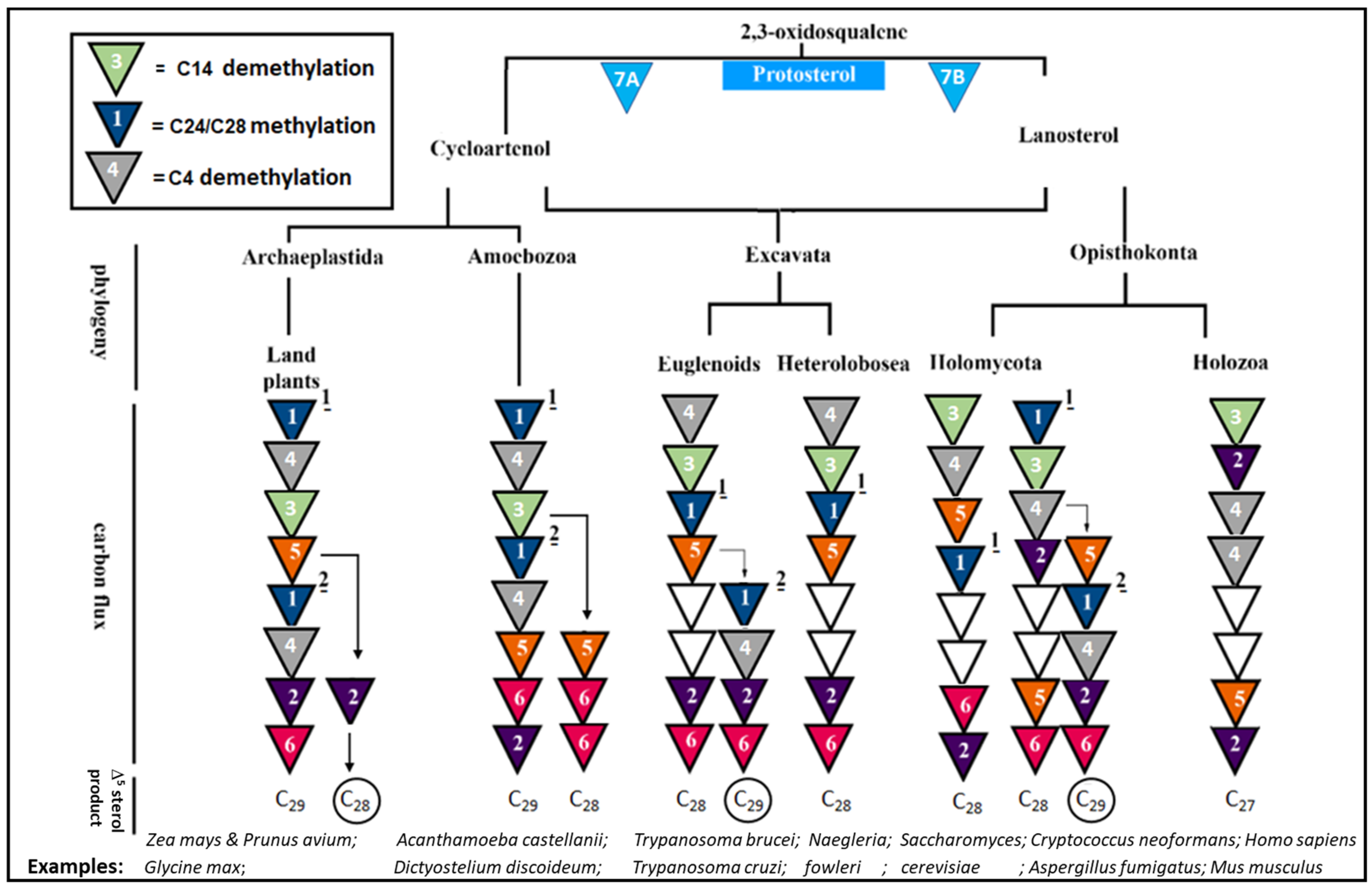



| Disease(s) | Sterol Biosynthetic Features 2 | Ergosterol Dependency | Infection Site | Representative Infectious Agent | Phylogenetic Lineage |
|---|---|---|---|---|---|
| Keratitis and granulomatous amoebic encephalitis | Prototroph: cycloartenol C28/C29 sterols | Yes | Blood and tissues | Acanthamoeba sp. | Amoebozoa |
| Naegleriasis and primary amoebic encephalitis | Prototroph: cycloartenol C28 sterol | Yes | Blood and tissues | Naegleria sp. | Heterolobosea |
| Sleeping sickness | Prototroph: lanosterol C28/C29 sterols | Yes | Blood and tissues | Trypanosoma brucei | Euglenoids |
| Chagas disease | Prototroph: lanosterol C28/C29 sterols | Yes | Blood and tissues | Trypanosoma cruzi | Euglenoids |
| Candidiasis | Prototroph: lanosterol C28 sterol | Yes | Skin/vagina/ mouth | Candida albicans | Holomycota |
| Cryptococcosis and meningitis | Prototroph: lanosterol C28 sterol | Yes | Lungs and brain | Cryptococcus sp. | Holomycota |
| Aspergillosis | Prototroph: lanosterol C28/C29 sterols | Yes | Lungs | Aspergillus sp. | Holomycota |
| None | Prototroph: lanosterol C27 sterol | No | NA | Homo sapiens | Holozoa |
| System(s) 1 | Function | Common Gene Name | Enzyme Nomenclature | Enzyme Commission (EC) Number | Cofactor | ||
|---|---|---|---|---|---|---|---|
| A. thaliana | S. cerevisiae | H. sapiens | |||||
 | 2,3- oxidosqualene cyclization | CAS (7A) | ERG7 (7B) | LSS (7B) | Lanosterol/ Cycloartenol synthase | 5.4.99.7/8 | NA 2 |
 | C14-demethylation | CYP51G1 | ERG11 | CYP51A1 | Sterol 14α-methyl demethylase | 1.14.13.70 | Heme NADPH |
 | C14-reduction | FK (TM7SF2) | ERG24 | DHCR14 | Sterol Δ14 -reductase | 1.3.1.70 | NADPH |
 | C4-methyl oxidation | SMO1 | ERG25 | SC4MOL | Sterol C4- methyloxidase | 1.14.13.72 | Oxo-diiron, NADPH |
 | C4-methyl acid elimination | AT3βHSD/ D1 | ERG26 | NSDHL | 3β-Hydroxy- Δ5-steroid dehydrogenase | 1.1.1.170 | NADH |
 | C3- ketoreduction | Predicted 3 | ERG27 | HSD17B7 | 3β-Keto-reductase | 1.1.1.270 | NADPH |
 | 9β, 19β- cyclopropane ring opening | CPI1 | NA 2 | NA 2 | Cycloeucalenol cycloisomerase | 5.5.1.9 | NA 2 |
 | Δ8-Δ7 isomerization | HYD1 | ERG2 | EBP | Sterol C8 isomerase | 5.3.3.5 | NA 2 |
 | Δ5-desaturation | DWF7 | ERG3 | SC5DL | Sterol C5-desaturase | 1.14.19.20 | Oxo-diiron, NADPH |
 | Δ7-reduction | DWF5 | NA 2 | DHCR7 | Sterol Δ7 reductase | 1.3.1.21 | NADPH |
 | Δ24-reduction | DWF1 | ERG4 | DHCR24 | Sterol Δ24(25)/Δ24(28) reductase | 1.3.1.72 | NADPH |
 | C22- desaturation | CYP710A | ERG5 | NA 2 | Sterol C22 desaturase | 1.14.19.41 | Heme NADPH |
 | C24/28-methylation | SMT1/2/3 | ERG6 | NA 2 | Sterol C24/28-methylase | 2.1.1.41/43 | SAM |
| Source | 24-SMT Substrate | Km | Kcat | Ea | Kinetic Mechanism | Reaction Pathway | Mr (kDa) |
|---|---|---|---|---|---|---|---|
| (μM) | (min−1) | (Kcat/mol) | |||||
| Fungi | S. cerevisiae SMT1 ZY | 17 | 0.6 | 13.7 | Random binding Concerted | SN2 Δ24(28) | 40.43 |
| Land Plant | G. max SMT1 CA | 30 | 1.3 | 15.2 | Ordered SAM Binds First | SN2 Δ24(28) | 40.40 |
| Land Plant | G. max SMT2 ML | 26 | 0.8 | 21.0 | ND 2 | SN2 Δ24(28) | 42.33 |
| Protozoa | T. brucei SMT1 ZY | 47 | 0.6 | 18.1 | ND 2 | SN2 Δ25(27) | 40.32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nes, W.D.; Chaudhuri, M.; Leaver, D.J. Druggable Sterol Metabolizing Enzymes in Infectious Diseases: Cell Targets to Therapeutic Leads. Biomolecules 2024, 14, 249. https://doi.org/10.3390/biom14030249
Nes WD, Chaudhuri M, Leaver DJ. Druggable Sterol Metabolizing Enzymes in Infectious Diseases: Cell Targets to Therapeutic Leads. Biomolecules. 2024; 14(3):249. https://doi.org/10.3390/biom14030249
Chicago/Turabian StyleNes, W. David, Minu Chaudhuri, and David J. Leaver. 2024. "Druggable Sterol Metabolizing Enzymes in Infectious Diseases: Cell Targets to Therapeutic Leads" Biomolecules 14, no. 3: 249. https://doi.org/10.3390/biom14030249
APA StyleNes, W. D., Chaudhuri, M., & Leaver, D. J. (2024). Druggable Sterol Metabolizing Enzymes in Infectious Diseases: Cell Targets to Therapeutic Leads. Biomolecules, 14(3), 249. https://doi.org/10.3390/biom14030249