
In-Depth Phenotyping of PIGW-Related Disease and Its Role in 17q12 Genomic Disorder

 , , , , , , ,
, , , , , , ,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. WES Analysis, Interpretation and Validation
2.3. Immune Cytometry
2.4. Graphs
2.5. Search Method for Literature Review
3. Results
3.1. Clinical Reports
3.2. Molecular Findings
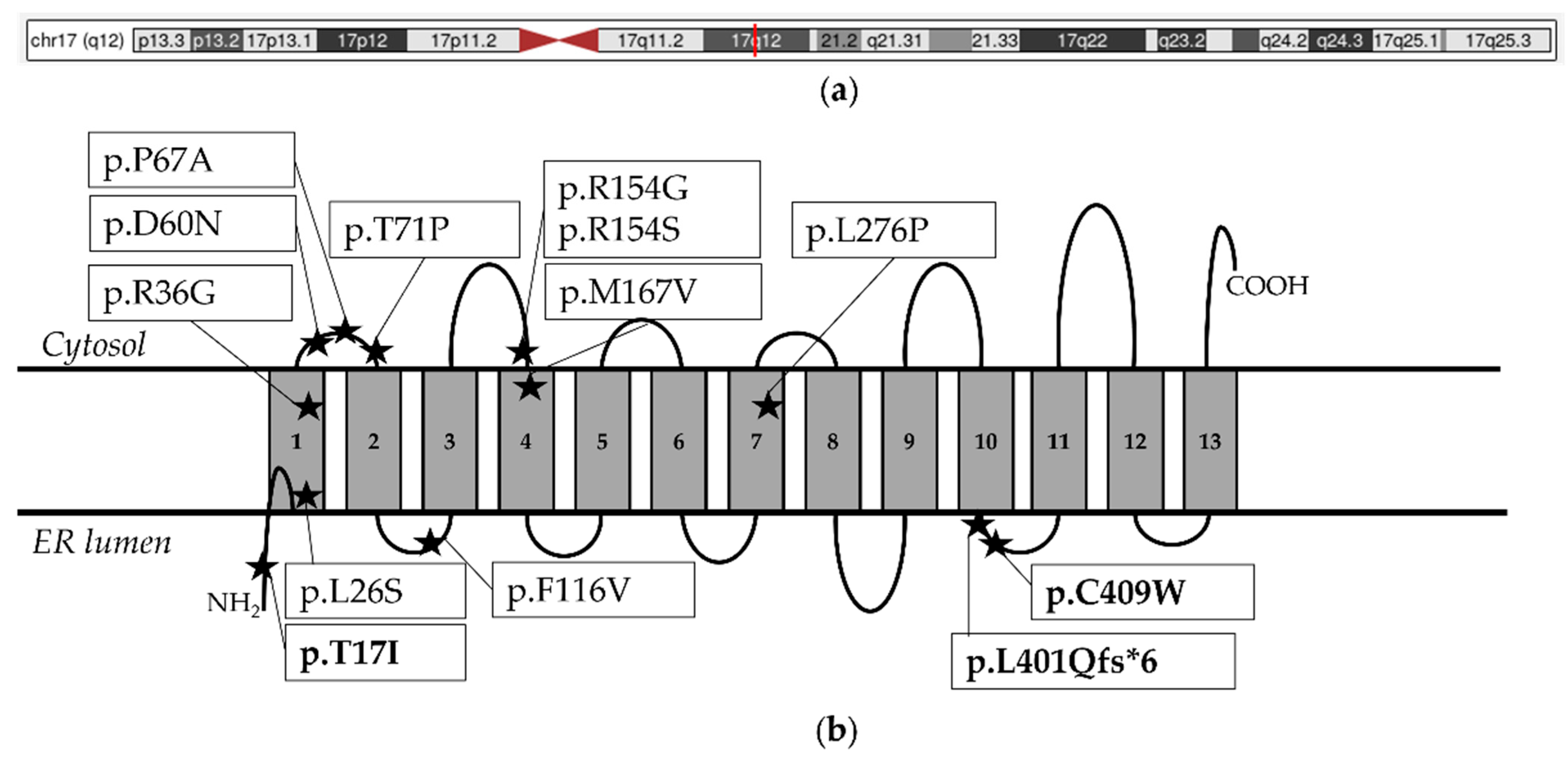
3.3. Literature Review
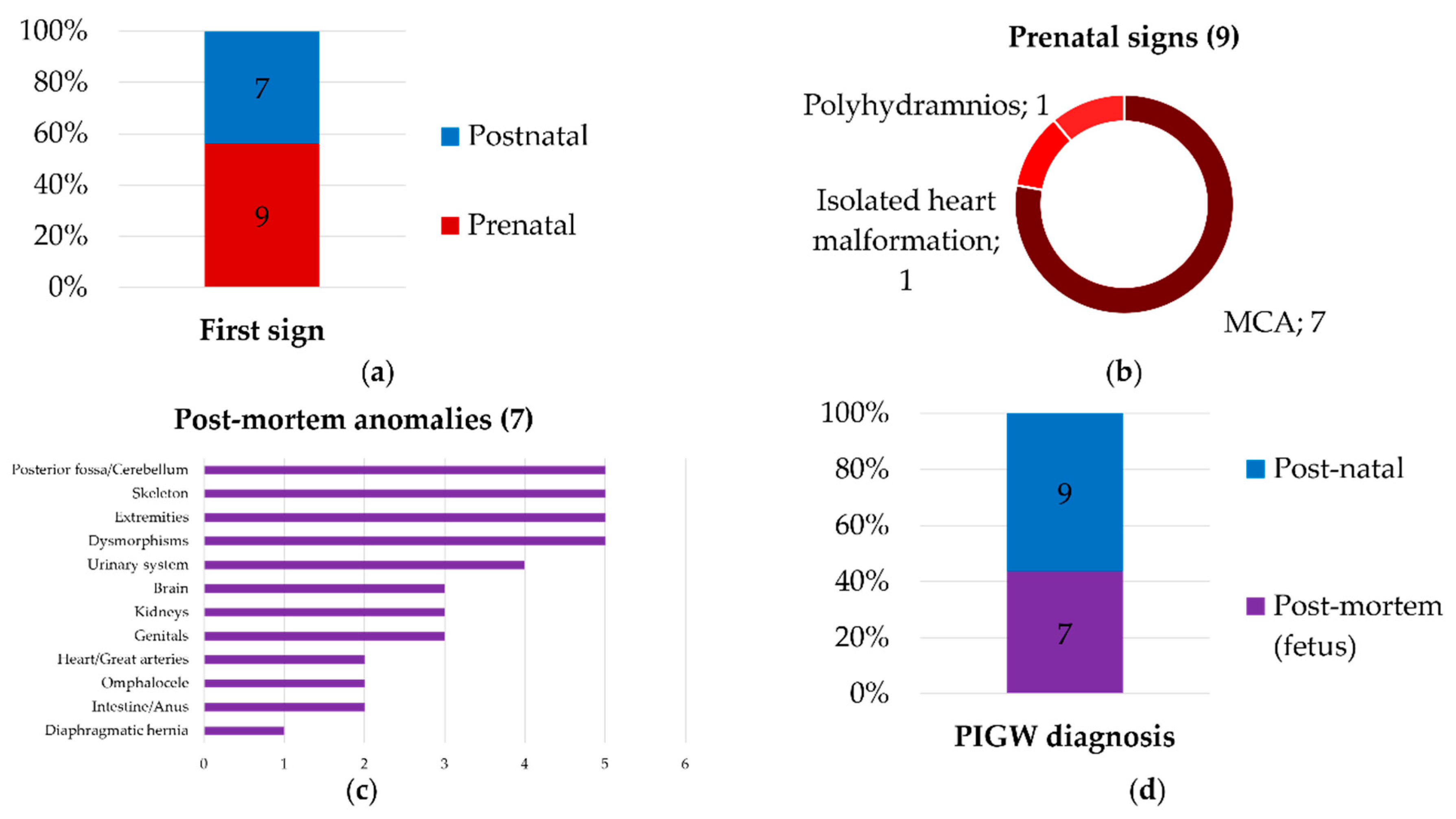
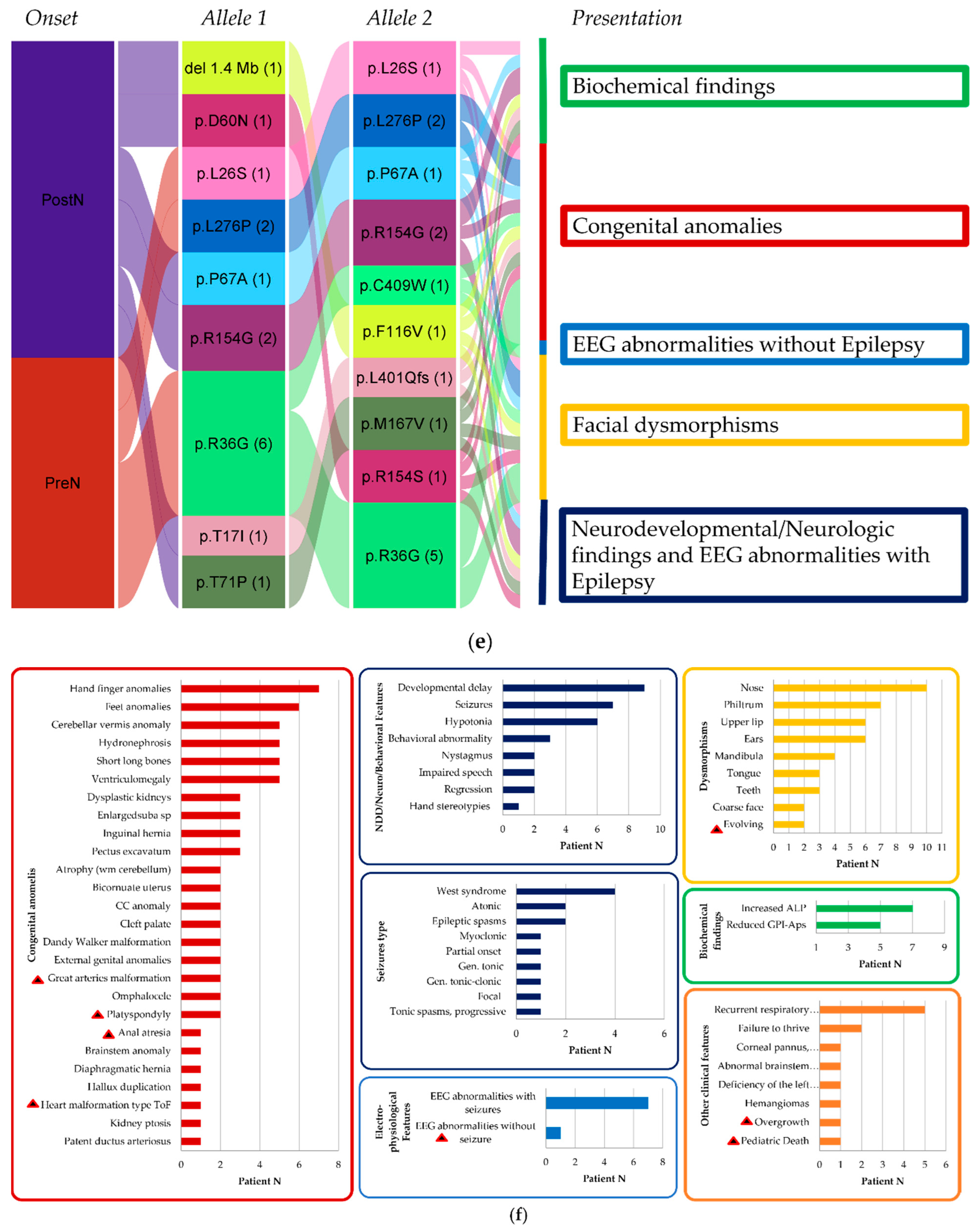
4. Discussion
4.1. New Clinical Findings
4.2. A Continuous and Broad Phenotypic Spectrum
4.3. New Molecular Findings
4.4. Considerations on PIGW Variants
4.5. Challenging Genotype-Driven Follow-Up
4.6. Perspectives
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ohishi, K.; Inoue, N.; Kinoshita, T. PIG-S and PIG-T, Essential for GPI Anchor Attachment to Proteins, Form a Complex with GAA1 and GPI8. EMBO J. 2001, 20, 4088–4098. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Fujita, M. Biosynthesis of GPI-Anchored Proteins: Special Emphasis on GPI Lipid Remodeling. J. Lipid Res. 2016, 57, 6–24. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J.; Péanne, R. What Is New in CDG? J. Inherit. Metab. Dis. 2017, 40, 569–586. [Google Scholar] [CrossRef] [PubMed]
- Foskett, G.K.; Engleman, E.; Klotz, J.; Choi, O.; Tolentino, L.; Kochhar, A.; Yang, Q.Z.; Stevenson, D.A. Use of Flow Cytometry for Diagnosis of Epilepsy Associated With Homozygous PIGW Variants. Pediatr. Neurol. 2018, 85, 67–70. [Google Scholar] [CrossRef]
- Wu, T.; Yin, F.; Guang, S.; He, F.; Yang, L.; Peng, J. The Glycosylphosphatidylinositol Biosynthesis Pathway in Human Diseases. Orphanet J. Rare Dis. 2020, 15, 129. [Google Scholar] [CrossRef]
- Nguyen, T.T.M.; Murakami, Y.; Wigby, K.M.; Baratang, N.V.; Rousseau, J.; St-Denis, A.; Rosenfeld, J.A.; Laniewski, S.C.; Jones, J.; Iglesias, A.D.; et al. Mutations in PIGS, Encoding a GPI Transamidase, Cause a Neurological Syndrome Ranging from Fetal Akinesia to Epileptic Encephalopathy. Am. J. Hum. Genet. 2018, 103, 602–611. [Google Scholar] [CrossRef]
- Bayat, A.; Knaus, A.; Pendziwiat, M.; Afenjar, A.; Barakat, T.S.; Bosch, F.; Callewaert, B.; Calvas, P.; Ceulemans, B.; Chassaing, N.; et al. Lessons Learned from 40 Novel PIGA Patients and a Review of the Literature. Epilepsia 2020, 61, 1142–1155. [Google Scholar] [CrossRef]
- Loong, L.; Tardivo, A.; Knaus, A.; Hashim, M.; Pagnamenta, A.T.; Alt, K.; Böhrer-Rabel, H.; Caro-Llopis, A.; Cole, T.; Distelmaier, F.; et al. Biallelic Variants in PIGN Cause Fryns Syndrome, Multiple Congenital Anomalies-Hypotonia-Seizures Syndrome, and Neurologic Phenotypes: A Genotype-Phenotype Correlation Study. Genet. Med. 2023, 25, 37–48. [Google Scholar] [CrossRef]
- Chiyonobu, T.; Inoue, N.; Morimoto, M.; Kinoshita, T.; Murakami, Y. Glycosylphosphatidylinositol (GPI) Anchor Deficiency Caused by Mutations in PIGW Is Associated with West Syndrome and Hyperphosphatasia with Mental Retardation Syndrome. J. Med. Genet. 2014, 51, 203–207. [Google Scholar] [CrossRef]
- Fang, Z.; Hu, C.; Zhou, S.; Yu, L. PIGW-Related Glycosylphosphatidylinositol Deficiency: A Case Report and Literature Review. Neurol. Sci. 2024, 45, 2253–2260. [Google Scholar] [CrossRef]
- Fu, L.; Liu, Y.; Chen, Y.; Yuan, Y.; Wei, W. Mutations in the PIGW Gene Associated with Hyperphosphatasia and Mental Retardation Syndrome: A Case Report. BMC Pediatr. 2019, 19, 68. [Google Scholar] [CrossRef] [PubMed]
- Hogrebe, M.; Murakami, Y.; Wild, M.; Ahlmann, M.; Biskup, S.; Hörtnagel, K.; Grüneberg, M.; Reunert, J.; Linden, T.; Kinoshita, T.; et al. A Novel Mutation in PIGW Causes Glycosylphosphatidylinositol Deficiency without Hyperphosphatasia. Am. J. Med. Genet. Part A 2016, 170, 3319–3322. [Google Scholar] [CrossRef] [PubMed]
- Meier, N.; Bruder, E.; Lapaire, O.; Hoesli, I.; Kang, A.; Hench, J.; Hoeller, S.; De Geyter, J.; Miny, P.; Heinimann, K.; et al. Exome Sequencing of Fetal Anomaly Syndromes: Novel Phenotype–Genotype Discoveries. Eur. J. Hum. Genet. 2019, 27, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Peron, A.; Iascone, M.; Salvatici, E.; Cavirani, B.; Marchetti, D.; Corno, S.; Vignoli, A. PIGW-Related Glycosylphosphatidylinositol Deficiency: Description of a New Patient and Review of the Literature. Am. J. Med. Genet. A 2020, 182, 1477–1482. [Google Scholar] [CrossRef]
- Ronzoni, L.; Boito, S.; Meossi, C.; Cesaretti, C.; Rinaldi, B.; Agolini, E.; Rizzuti, T.; Pezzoli, L.; Silipigni, R.; Novelli, A.; et al. Prenatal Ultrasound Findings Associated with PIGW Variants: One More Piece in the FRYNS Syndrome Puzzle? PIGW-related Prenatal Findings. Prenat. Diagn. 2022, 42, 1493–1502. [Google Scholar] [CrossRef]
- Musante, L.; Costa, P.; Zanus, C.; Faletra, F.; Murru, F.M.; Bianco, A.M.; La Bianca, M.; Ragusa, G.; Athanasakis, E.; d’Adamo, A.P.; et al. The Genetic Diagnosis of Ultrarare DEEs: An Ongoing Challenge. Genes 2022, 13, 500. [Google Scholar] [CrossRef]
- Sobreira, N.; Schiettecatte, F.; Valle, D.; Hamosh, A. GeneMatcher: A Matching Tool for Connecting Investigators with an Interest in the Same Gene. Hum. Mutat. 2015, 36, 928–930. [Google Scholar] [CrossRef]
- SoFFœt—Società Francese Di Fetopatologia. Available online: https://soffoet.fr/ (accessed on 12 December 2024).
- Rolland, M.; Dubourg, C.; Cospain, A.; Droitcourt, C.; Pasquier, L. Child with a Mild CHIME Syndrome Phenotype and Carrying a Novel p.(Asp52Asn) PIGL Pathogenic Variant in Association with the Previously Reported p.(Leu167Pro) Variant: A Case Report. Pediatr. Dermatol. 2022, 39, 434–437. [Google Scholar] [CrossRef]
- Winter-Paquette, L.M.; Al Suwaidi, H.H.; Sajjad, Y.; Bricker, L. Congenital Diaphragmatic Hernia and Early Lethality in PIGL-Related Disorder. Eur. J. Med. Genet. 2022, 65, 104501. [Google Scholar] [CrossRef]
- van der Crabben, S.N.; Harakalova, M.; Brilstra, E.H.; van Berkestijn, F.M.C.; Hofstede, F.C.; van Vught, A.J.; Cuppen, E.; Kloosterman, W.; Ploos van Amstel, H.K.; van Haaften, G.; et al. Expanding the Spectrum of Phenotypes Associated with Germline PIGA Mutations: A Child with Developmental Delay, Accelerated Linear Growth, Facial Dysmorphisms, Elevated Alkaline Phosphatase, and Progressive CNS Abnormalities. Am. J. Med. Genet. A 2014, 164A, 29–35. [Google Scholar] [CrossRef]
- The Phenotype of a Germline Mutation in PIGA: The Gene Somatically Mutated in Paroxysmal Nocturnal Hemoglobinuria—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/22305531/ (accessed on 24 July 2024).
- Bayat, A.; Lindau, T.; Aledo-Serrano, A.; Gil-Nagel, A.; Barić, I.; Bartoniček, D.; Mateševac, J.; Ramadža, D.P.; Žigman, T.; Pušeljić, S.; et al. GPI-Anchoring Disorders and the Heart: Is Cardiomyopathy an Overlooked Feature? Clin. Genet. 2023, 104, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Bayat, A.; Kløvgaard, M.; Johannesen, K.M.; Barakat, T.S.; Kievit, A.; Montomoli, M.; Parrini, E.; Pietrafusa, N.; Schelhaas, J.; van Slegtenhorst, M.; et al. Deciphering the Premature Mortality in PIGA-CDG—An Untold Story. Epilepsy Res. 2021, 170, 106530. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Kanzawa, N.; Saito, K.; Krawitz, P.M.; Mundlos, S.; Robinson, P.N.; Karadimitris, A.; Maeda, Y.; Kinoshita, T. Mechanism for Release of Alkaline Phosphatase Caused by Glycosylphosphatidylinositol Deficiency in Patients with Hyperphosphatasia Mental Retardation Syndrome. J. Biol. Chem. 2012, 287, 6318–6325. [Google Scholar] [CrossRef] [PubMed]
- Clinical Variability in Inherited Glycosylphosphatidylinositol Deficiency Disorders—Bellai-Dussault—2019—Clinical Genetics—Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1111/cge.13425 (accessed on 1 May 2024).
- Large Genomic Rearrangements in the Hepatocyte Nuclear Factor-1β (TCF2) Gene Are the Most Frequent Cause of Maturity-Onset Diabetes of the Young Type 5|Diabetes|American Diabetes Association. Available online: https://diabetesjournals.org/diabetes/article/54/11/3126/25457/Large-Genomic-Rearrangements-in-the-Hepatocyte (accessed on 5 May 2024).
- Mefford, H.C.; Clauin, S.; Sharp, A.J.; Moller, R.S.; Ullmann, R.; Kapur, R.; Pinkel, D.; Cooper, G.M.; Ventura, M.; Ropers, H.H.; et al. Recurrent Reciprocal Genomic Rearrangements of 17q12 Are Associated with Renal Disease, Diabetes, and Epilepsy. Am. J. Hum. Genet. 2007, 81, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Moreno-De-Luca, D.; Mulle, J.G.; Kaminsky, E.B.; Sanders, S.J.; Myers, S.M.; Adam, M.P.; Pakula, A.T.; Eisenhauer, N.J.; Uhas, K.; Weik, L.; et al. Deletion 17q12 Is a Recurrent Copy Number Variant That Confers High Risk of Autism and Schizophrenia. Am. J. Hum. Genet. 2010, 87, 618–630. [Google Scholar] [CrossRef]
- Nagamani, S.C.S.; Erez, A.; Shen, J.; Li, C.; Roeder, E.; Cox, S.; Karaviti, L.; Pearson, M.; Kang, S.-H.L.; Sahoo, T.; et al. Clinical Spectrum Associated with Recurrent Genomic Rearrangements in Chromosome 17q12. Eur. J. Hum. Genet. 2010, 18, 278–284. [Google Scholar] [CrossRef]
- Loirat, C.; Bellanné-Chantelot, C.; Husson, I.; Deschênes, G.; Guigonis, V.; Chabane, N. Autism in Three Patients with Cystic or Hyperechogenic Kidneys and Chromosome 17q12 Deletion. Nephrol. Dial. Transplant. 2010, 25, 3430–3433. [Google Scholar] [CrossRef]
- Mefford, H. 17q12 Recurrent Duplication. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Jing, X.-Y.; Huang, L.-Y.; Zhen, L.; Han, J.; Li, D.-Z. Prenatal Diagnosis of 17q12 Deletion Syndrome: A Retrospective Case Series. J. Obstet. Gynaecol. 2019, 39, 323–327. [Google Scholar] [CrossRef]
- Verscaj, C.P.; Velez-Bartolomei, F.; Bodle, E.; Chan, K.; Lyons, M.J.; Thorson, W.; Tan, W.-H.; Rodig, N.; Graham, J.M.; Peron, A.; et al. Characterization of the Prenatal Renal Phenotype Associated with 17q12, HNF1B, Microdeletions. Prenat. Diagn. 2024, 44, 237–246. [Google Scholar] [CrossRef]
- Zhou, C.-X.; Zhu, X.-Y.; Zhu, Y.-J.; Gu, L.-L.; He, L.-L.; Liu, W.; Yang, Y.; Wu, X.; Duan, H.-L.; Ru, T.; et al. Prenatal Features of 17q12 Microdeletion and Microduplication Syndromes: A Retrospective Case Series. Taiwan. J. Obstet. Gynecol. 2021, 60, 232–237. [Google Scholar] [CrossRef]
- Musante, L.; Faletra, F.; Meier, K.; Tomoum, H.; Najarzadeh Torbati, P.; Blair, E.; North, S.; Gärtner, J.; Diegmann, S.; Beiraghi Toosi, M.; et al. TTC5 Syndrome: Clinical and Molecular Spectrum of a Severe and Recognizable Condition. Am. J. Med. Genet. A 2022, 188, 2652–2665. [Google Scholar] [CrossRef]
- Highnam, G.; Wang, J.J.; Kusler, D.; Zook, J.; Vijayan, V.; Leibovich, N.; Mittelman, D. An Analytical Framework for Optimizing Variant Discovery from Personal Genomes. Nat. Commun. 2015, 6, 6275. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Limongelli, I.; Marini, S.; Bellazzi, R. PaPI: Pseudo Amino Acid Composition to Score Human Protein-Coding Variants. BMC Bioinform. 2015, 16, 123. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 7, Unit7.20. [Google Scholar] [CrossRef]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT Web Server: Predicting Effects of Amino Acid Substitutions on Proteins. Nucleic. Acids Res. 2012, 40, W452–W457. [Google Scholar]
- Jian, X.; Boerwinkle, E.; Liu, X. In Silico Prediction of Splice-Altering Single Nucleotide Variants in the Human Genome. Nucleic. Acids Res. 2014, 42, 13534–13544. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A General Framework for Estimating the Relative Pathogenicity of Human Genetic Variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster Evaluates Disease-Causing Potential of Sequence Alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef]
- Bayat, A.; Fenger, C.D.; Techlo, T.R.; Højte, A.F.; Nørgaard, I.; Hansen, T.F.; Rubboli, G.; Møller, R.S. Impact of Genetic Testing on Therapeutic Decision-Making in Childhood-Onset Epilepsies—A Study in a Tertiary Epilepsy Center. Neurotherapeutics 2022, 19, 1353–1367. [Google Scholar] [CrossRef]
- Lefebvre, M.; Bruel, A.-L.; Tisserant, E.; Bourgon, N.; Duffourd, Y.; Collardeau-Frachon, S.; Attie-Bitach, T.; Kuentz, P.; Assoum, M.; Schaefer, E.; et al. Genotype-First in a Cohort of 95 Fetuses with Multiple Congenital Abnormalities: When Exome Sequencing Reveals Unexpected Fetal Phenotype-Genotype Correlations. J. Med. Genet. 2021, 58, 400–413. [Google Scholar] [CrossRef]
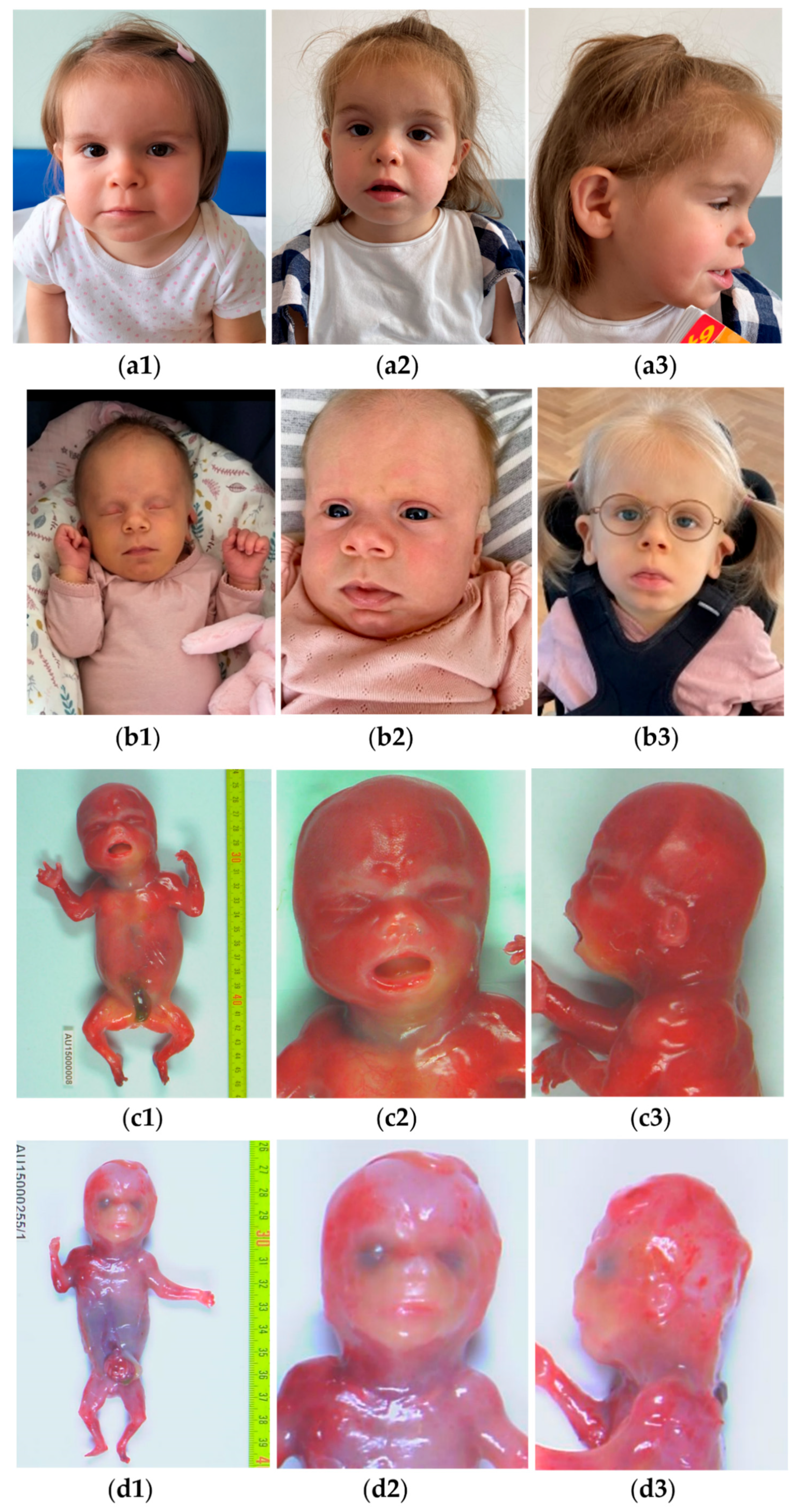

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Sex | Ancestry, Family History | PIGW (NM_178517.5) Variants, Status, and Inheritance | Presentation | Prenatal Findings | Birth Presentation | |
|---|---|---|---|---|---|---|---|
| P2 | F | Danish, nc | c.50C>T, p.(Thr17Ile)/c.1202del, p.(Leu401GlnfsTer6); ht parents | PostN | Normal | 37 + 5. Birth W 4040 g. | |
| [14] | F | Egyptian, 1st cousins | c.77C>T, p.(Leu26Ser), hz | PreN/PostN | Polyhydramnios, maternal diabetes | Sp delivery at term. W 3700 g (+0.98 SD), L 50 cm (+0.46 SD); APGAR 9–10 | |
| P1 | F | Italian, nc; previous miscarriage | c.106A>G, p.(Arg36Gly)pat/c.1227T>G, p.(Cys409Trp) mat | PreN/PostN | T1 increased risk, combined test for trisomy 21 (1/94); T2 (20 wgs): overriding aorta | Sp delivery 37 + 6 wgs. W 2.7 kg (90°pc); APGAR 9/9 | |
| P3 | F | France, previous miscarriage | c.106A>G, p.(Arg36Gly), h | PreN | Short long-bones with the femoral bowing, short hands and feet, cardiopathy and a Blake’s pouch cyst | TOP, 19 + 5 wgs, OFD < 3rd pc, W 50°pc, L 25°pc. Nuchal oedema | |
| P4 | M | France, sib of P3 | c.106A>G, p.(Arg36Gly), hz | PreN | Short long-bones, omphalocele, polydactyly | TOP 14 + 3 wgs | |
| [13] Family 9, II 1 | M | NR | c.106A>G, p.(Arg36Gly), hz | PreN (fetus) | T2 (18 wgs): MCA, posterior fossa anomaly; TOP | NA | |
| [13] Family 9, II 2 | F | NR | c.106A>G, p.(Arg36Gly), hz | PreN (fetus) | T1 (12 wgs): posterior fossa malformation, MCA; TOP | NA | |
| [15] fetus 3 | F | Italian, nc | c.106A>G, p.(Arg36Gly), hz | PreN (fetus) | T1: increased NT (3.8 mm), SUA and omphalocele; T2 (15 wgs): brain malformations, dysmorphisms; short limbs; TOP at 17 wgs | NA | |
| [11] | M | Nc; elder brother: DD, death at 7 mth, RRI | c.178G>A,p.(Asp60Asn)pat/c.462A>T, p.(Arg154Ser) mat | PostN | NR | Sp delivery after 39 wgs. Pneumonia at 15 days of life | |
| [4] | F | Mexico, likely c (runs of hz at SNP-array) | c.199C>G, p.(Pro67Ala), hz | PostN | NR | NR | |
| [9] | NR | Japanese, nc | c.211A>C, p.(Thr71Pro)/c.499A>G, p.(Met167Val), comp htz | PostN | NR | NR | |
| [12] patient 1 | M | 1st cousins | c.460A>G, p.(Arg154Gly), hz | PostN | No | W 3.8 kg, L 52 cm, OFC 36 cm. Bradycardia, muscle hypotonia; APGAR 2/6/9 | |
| [12] patient 2 | F | 2nd cousin of [9] patient 1 | c.460A>G, p.(Arg154Gly), hz | PostN | No | W 3.5 kg, L 51 cm, OFC 35 cm. Hypotonia and a large tongue; APGAR 9/10/10 | |
| [15] fetus 1 | M | Italian, the couple’s grandparents were 1st cousins | c.827T>C, p.(Leu276Pro), hz | PreN (fetus) | T1: increased NT (4.10 mm, >95th pct); T2: pernasal thickness, polyhydramnios, dysmorphisms and MCA; TOP at 21 wgs | NA | |
| [15] fetus 2 | F | Italian, the couple’s grandparents were 1st cousins, antecedent: [13] fetus 1 | c.827T>C, p.(Leu276Pro), hz | PreN (fetus) | T1: low-risk combined test, normal NT; T2 (16 wgs): MCA; 18 wgs: increased pernasal thickness, dysmorphisms, and MCA; 20 wgs: brain anomalies; TOP at 21 wgs | NA | |
| [10] | F | Chinese, nc | 17q12 deletion ~1.4 Mb pat/c.346 T>G, p.(Phe116Val) mat | PostN | NR | Sp delivery at term. W 3200 g; APGAR after resuscitation 7–8–10 | |
| Statistics (%) | F 10/15 (66.7) | C 4/16 fam (25.0)|Dead sibling/miscarriage 3/16 (18.7) | Missense (12/14); frameshift (1/14); deletion (1/14) | PreN 9/16 (56.2); fetus 7/16 (43.7); PostN (43.7) | Fetal signs 9/16 (56.2)|T1 7/9 (77.7) | NA | |
| Patient | Heart Anomalies | Brain Anomalies | Other Congenital Anomalies | Skeletal Anomalies | Extremities Anomalies | ||
| P2 | Normal US at 1 and 2 yrs | Normal MRI | Bil hydronephrosis | NA | Normal | ||
| [14] | Normal US | MRI: Bil enlargement of the cortical fronto-insular subarachnoid space and of the ventricles, thin CC and brainstem, white matter atrophy, moderate cerebellar atrophy | NR | Pectus excavatum, scoliosis | Long fingers with right fifth finger clinodactyly, and rocker bottom feet | ||
| P1 | US: vsd type ToF, altered pressures in the pulmonary artery, asd, ostium secundum type | Normal MRI | Right inguinal hernia | NR | Low-set left toe, overcrowding of toes | ||
| P3 | Overriding aorta | Microcephaly, enlarged ventricles and cerebellum hypoplasia. | Bil renal dysplasia | PME XR: shortened long bones, enlarged metaphysis, short ribs, and platyspondyly | Brachypahalangy | ||
| P4 | Asymmetry of the great arteries | NR | Bil renal dysplasia, omphalocele, anal atresia | PME XR: shortened long bones, platyspondyly | Right duplicated hallux | ||
| [13] Family 9, II 1 | NR | US (18 wgs): Dandy Walker malformation | US: hydronephrosis, genital hypoplasia | NR | NR | ||
| [13] Family 9, II 2 | NR | US (12 wgs): Dandy Walker malformation | US: diaphragmatic hernia, dysplastic kidneys, hydronephrosis | NR | NR | ||
| [15] fetus 3 | NR | US (15 wgs): third ventricle enlargement and cyst in the posterior fossa; MRI (16 wgs) dysmorphic and extra-rotated cerebellar vermis | US: SUA and a bowel-containing omphalocele; PME: bil renal ptosis and bicornuate uterus | US/PME: short limbs | PME: short halluces | ||
| [11] | Normal US | MRI: widening of the subarachnoid space in both frontotemporal regions | Umbilical hernia and bilateral indirect inguinal hernias | Pectus excavatum | Bil mild flexion contractures of all fingers | ||
| [4] | NR | NR | NR | Pectus excavatum | Bil mild flexion contractures of all fingers | ||
| [9] | NR | NR | Inguinal hernia | NR | Normal | ||
| [12] patient 1 | NR | Normal MRI | Ankiloglossia | NR | Thumb-in-fist posture | ||
| [12] patient 2 | NR | Normal MRI | NR | NR | NR | ||
| [15] fetus 1 | NR | NR | PME: bil hydronephrosis | US/PME: short limbs | PME: bil brachydactyly with clinodactyly of the 5th finger, clubfeet | ||
| [15] fetus 2 | NR | US/MRI (20 wgs): enlargement of sub-arachnoid spaces, mild ventriculomegaly, cerebellar vermis hypoplasia, CC hypoplasia and dysmorphic features of brainstem with a suspected diencephalic–mesencephalic junction dysplasia | US/PME: hepatomegaly, bil hydronephrosis; PME: incomplete lung lobulation, intestinal malrotation, bicornuate uterus, and clitoris hypertrophy | US/PME: short limbs | US/PME: bil brachydactyly with clinodactyly of the 5th finger, clubfeet, short halluces | ||
| [10] | Patent ductus arteriosus | MRI: cerebellar atrophy and bil enlargement of the cortical subarachnoid space and ventricles | NR | NR | NR | ||
| Statistics (%) | 4/16 (25.0) | 8/16 (50.0) | 12/16 (75.0) | 8/16 (50.0) | 10/16 (62.5) | ||
| Patient | Facial Dysmorphisms | Neurodevelopment | Seizure Onset | Seizure Semiology | EEG Abnormalities | Seizure Management | |
| P2 | (photo) Brachycephaly, dolichocephaly, thin feathery hair, hypotelorism, deep-set eyes, large cup-formed ears, and flat nasal bridge | Global DD with delayed milestones, limited head control. Developmental regression (10 mth). Profound, global DD (no eye contact, no speech, 3 yrs) | 10 mth | Tonic-spams, progressive epilepsy | EEG at 1 yr: overall hypsarrhythmia, characterized by cyclical variations during wakefulness and sleep. EEG during wakefulness showed periods of well-organized activity beyond 12 Hz, diffusely mixed with periods of theta activity without lateralization, alternating with periods of rhythmic delta activity mixed with sharp-waves, localized in multiple foci in both hemispheres. EEG during sleep: The lack of definitive sleep spindles and K-complexes, instead is seen a more persistent slow-wave pattern with 1.5–2 Hz activity, intermixed with sharp-waves as periodic discharges, where there may be asynchronous or synchronous activity in the two hemispheres. Ictal EEG at 3 yrs showed multifocal epileptiform changes. | Difficult to control: tried topiramat, levetiracetam, vigabatrin, pyrodoxin, clobazam, initiation of a ketogenic diet | |
| [14] | (photo) low frontal hairline, the wide nasal bridge with a bulbous nasal tip and protruding columella, short philtrum, wide-spaced teeth, and the stem of the antihelix was fused to the helix | Global DD (sat unsupported at 12 mth; control of the trunk at 36 mth; unable to stand only with support from age 10 years), severe ID with absent speech | 10 mth | Daily generalized tonic seizures with lip cyanosis since 10 mth; epileptic spasms/West syndrome since 13 mth; bilateral tonic seizure since 16 mth and focal-onset seizures; less than a seizure per year from 22 mth to 8 yrs; seizure free from 8 yrs to follow-up at 12 yrs | High-amplitude hypsarrhythmic pattern (13 mth); slowing of the background activity with single sharp-waves over the fronto-temporal bil regions during drowsiness (7 yrs) | Controlled with phenobarbital up to 13 mth; then treated with adenocorticotropic hormone (ACTH) with remission since 16 mth; then started Valproid acid without remission, Vigabatrin added at 20 mth | |
| P1 | (photo) squared/coarse face with large/prominent forehead. Anteverted, slightly low-set ears. Depressed nasal root, short nose, marked and protruding philtrum, slightly widened mouth with thin upper lip. Diastema. Protruding tongue. Micrognathia | Psychomotor DD, absent speech, immature gross and fine motor skills | No clinically recognizable epileptic episodes | NR | Atypical/disorganized, with unusually large amplitude, presence of continuous alpha band activity on the bilateral anterior areas and multifocal paroxysmal abnormalities (2 yrs 11 mth); EEG during sleep showed (a) the absence of the typical elements of physiological organization, while differentiating phases of drowsiness/sleepiness from phases of deeper sleep; (b) the presence of diffuse rapid activity that with falling asleep also increases in amplitude becoming dominant; (c) the presence of multifocal paroxysmal abnormalities (45 mth) | NR | |
| P3 | (photo) Brachycephaly, hypertelorism, low set ears | NA | NA | NA | NA | NA | |
| P4 | (photo) Cleft palate, turricephaly, long philtrum | NA | NA | NA | NA | NA | |
| [13] Family 9, II 1 | NR | NA | NA | NA | NA | NA | |
| [13] Family 9, II 2 | NR | NA | NA | NA | NA | NA | |
| [15] fetus 3 | (photo) US (15 wg): nasal bone hypoplasia, micrognathia; PME: brachycephaly, hypertelorism, anteverted nares, micrognathia, low-set ears | NA | NA | NA | NA | NA | |
| Patient | Facial Dysmorphisms | Neurodevelopment | Seizure Onset | Seizure Semiology | EEG Abnormalities | Seizure Management | |
| [11] | (reported) coarse facial features, wide nasal bridge, tent-shaped lips, high, narrow palatine arches | Severe psychomotor DD | 70 dys | Partial-onset epileptic seizures | Sharp waves, sharp, slow discharges in the temporal and frontal regions | Treated by oxcarbazepine and anti-epileptic drugs but not be completely controlled even after temperature fall | |
| [4] | (reported) anteverted nares, tented upper lip | Profound, global DD: inability to roll, poor head control, absence of tracking | 7 mth | Infantile spasms | NR | NR | |
| [9] | (reported) broad nasal bridge and tented upper lip | Profound DD | NR | Clusters of tonic spasms, West syndrome | High-amplitude hypsarrhythmic pattern | NR | |
| [12] patient 1 | NR | Psychomotor DD (sit at 11 mth, crawl at 13 mth, walk at 17 mth) | 5 yrs | Seizure with atonia of the right leg and tendency to fall backwards | Multifocal spike-wave complexes especially while asleep | Improvement with sulthiame and clobazam therapy | |
| [12] patient 2 | (photo, partial; reported) macroglossia | Inactivity; Psychomotor DD (sit at 12 mth, crawl at 30 mth, not walking at 4 yrs) | 2 yrs | First episode of atonic seizure followed by frequent seizures with myoclonic tongue movements, profuse salivation, fecal incontinence, and increased arm stiffness | Multifocal spike-wave complexes | Anti-convulsive treatment was only partially effective | |
| [15] fetus 1 | (photo) US: long philtrum; PME: dysmorphic ears, flat nose with anteverted nares, long philtrum, microretrognathia | NA | NA | NA | NA | NA | |
| [15] fetus 2 | (photo) US: nasal bone hypoplasia, micrognathia; PME: brachycephaly, flat glabella, anteverted nares, long and deep philtrum, cleft palate, retrognathia | NA | NA | NA | NA | NA | |
| [10] | (reported) anteverted nares, tented upper lip, short philtrum, widely spaced teeth, and inner canthus | Psychomotor DD | 9 mth | Generalized tonic-clonic seizures (during pneumonia at 9 mth; without fever at 15 mth); clusters of epileptic spasms/West syndrome (in the course of pneumonia at 23 mth); refractory epilepsy; Recurrent epileptic spasms with muscular atrophy and hypotonia, asymmetric tonic seizures worsening during fever and infections | High-amplitude hypsarrhythmic pattern on an interictal electroencephalogram (23 mth) | Not controlled by poly-drug therapy (6 drugs) and vit B6 supplementation | |
| Statistics (%) | 12/16 (75.0) | 9/9 (100) | (70 dys–5 yrs); Me 44.1 mth | 8/9 (88.9) | 9/9 (100) | Partial control or uncontrolled epilepsy (5/8, 62.5) | |
| Patient | Other Neurologic Findings | Behavior | Infections | Other Clinical Findings | ALP Measurement | Other Biochemical Findings | |
| P2 | Mild axial hypotonia, difficulties swallowing | At 3 yrs, she had no eye contact and no speech | Frequent respiratory infections, annually 5–10 contacts to hospital. Pneumonia leading to lethal asystole at 3 yrs 10 mth | Height and weight > 3 SD. Hypermetropia, with +5.75 bilat | High (610–940 U/L), measured 12 times from 9 to 42 | NA | |
| [14] | Global hypotonia, intermittent hand stereotypies (hand washing and mouthing); limited, wide-based walking; ataxic gait | Limited social interaction, poor eye contact | RRI | Three congenital hemangiomas. Deficiency of the left central auditory pathway. Horizontal nystagmus | High at two measurements (425 and 380 U/L) | Normal metabolic tests and transferrin isoelectrofocusing | |
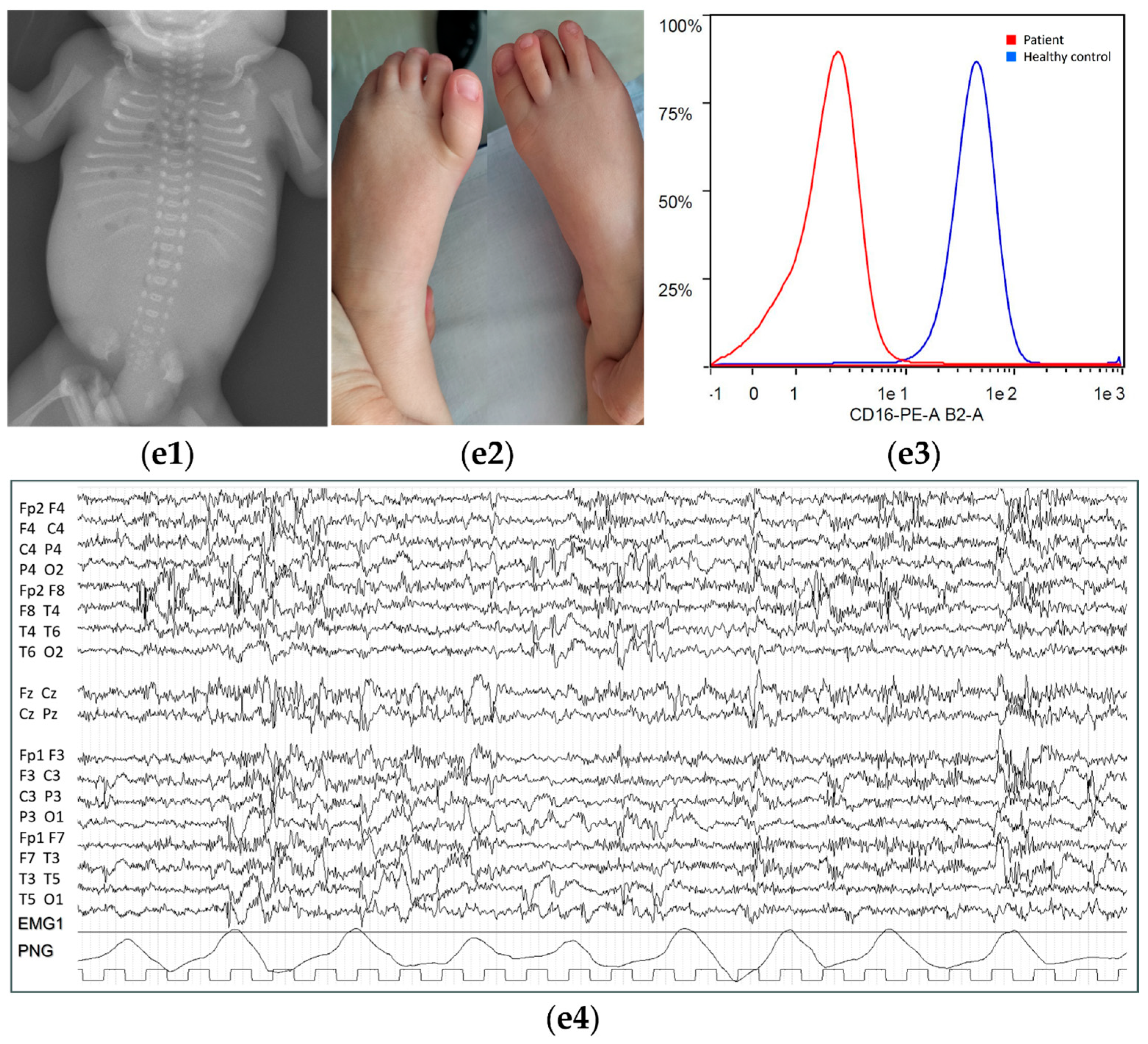
| P1 | NR | Absent speech, poor eye engagement, limited pointing | NR | Tooth agenesis. | High (519 U/L) at 3 yrs | Reduced CD16 at MFI from neutrophils | |
| P3 | NA | NA | NA | NR | NA | NA | |
| P4 | NA | NA | NA | NR | NA | NA | |
| [13] Family 9, II 1 | NA | NA | NA | NR | NA | NA | |
| [13] Family 9, II 2 | NA | NA | NA | NR | NA | NA | |
| [15] fetus 3 | NA | NA | NA | NR | NA | NA | |
| [11] | No | NR | RRI | Failure to thrive, poor weight gain. Bil lung pneumonia requiring intubation | High (414–798 U/L) | Normal levels of immunoglobulins and lymphocyte subsets; normal cerebrospinal fluid analysis | |
| [4] | NR | NR | Frequent aspiration pneumonia. | Failure to thrive. Poor tracking, vertical nystagmus | High (473 U/L and 495 U/L) pre-diagnosis, ranging from 235 to 575 U/L at 9–11 mth | FC: decreased anchor protein expression (82% granulocytes; 81% lymphocytes). Cluster differentiation: decreased CD16 and CD24 cell surface expression (>60% granulocytes), CD14 expression (88% on monocytes), and CD59 expression (42% lymphocytes) | |
| [9] | NR | NR | NR | No | High (2000 U/L) | FC: Reduced levels of the GPI-Aps. Reduced CD59 and FLAER on lymphocytes, reduced CD14 on monocytes. Normal routine laboratory investigations | |
| [12] patient 1 | Neonatal hypotonia | Absent speech (7 yrs), autistic traits | NR | Ankyloglossia | Up to 230 U/L (normal) | Low MFI for CD16 (2832); reduced CD16 (7214) | |
| [12] patient 2 | Neonatal hypotonia | NR | NR | Abnormal brainstem auditory evoked response | Up to 305 U/L (normal) | Low MFI for CD16 (3070); reduced CD16 (7439) | |
| [15] fetus 1 | NA | NA | NA | NR | NA | NA | |
| [15] fetus 2 | NA | NA | NA | NR | NA | NA | |
| [10] | Severe ID. Hypotonia. Muscular atrophy | NR | RRI | Corneal pannus, keratitis, and conjunctivitis | High (552–995 U/L) | NR | |
| Total (%) | 6/9 (88.9) | 4/9 (44.4) | 5/9 (55.6) | 8/16 (50.0) | High 7/9 (77.8) | FC: 5/6 (83.3) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feresin, A.; Lefebvre, M.; Sjøstrøm, E.; Zanus, C.; Paccagnella, E.; Bruno, I.; Valencic, E.; Morgan, A.; Tommasini, A.; Thauvin, C.; et al. In-Depth Phenotyping of PIGW-Related Disease and Its Role in 17q12 Genomic Disorder. Biomolecules 2024, 14, 1626. https://doi.org/10.3390/biom14121626
Feresin A, Lefebvre M, Sjøstrøm E, Zanus C, Paccagnella E, Bruno I, Valencic E, Morgan A, Tommasini A, Thauvin C, et al. In-Depth Phenotyping of PIGW-Related Disease and Its Role in 17q12 Genomic Disorder. Biomolecules. 2024; 14(12):1626. https://doi.org/10.3390/biom14121626
Chicago/Turabian StyleFeresin, Agnese, Mathilde Lefebvre, Emilie Sjøstrøm, Caterina Zanus, Elisa Paccagnella, Irene Bruno, Erica Valencic, Anna Morgan, Alberto Tommasini, Christel Thauvin, and et al. 2024. "In-Depth Phenotyping of PIGW-Related Disease and Its Role in 17q12 Genomic Disorder" Biomolecules 14, no. 12: 1626. https://doi.org/10.3390/biom14121626
APA StyleFeresin, A., Lefebvre, M., Sjøstrøm, E., Zanus, C., Paccagnella, E., Bruno, I., Valencic, E., Morgan, A., Tommasini, A., Thauvin, C., Bayat, A., Girotto, G., & Musante, L. (2024). In-Depth Phenotyping of PIGW-Related Disease and Its Role in 17q12 Genomic Disorder. Biomolecules, 14(12), 1626. https://doi.org/10.3390/biom14121626