

APOL1 Modulates Renin–Angiotensin System
 ,
,  and
and 
Abstract
1. Introduction
2. Methods
2.1. Renal Histology and Immunofluorescence Studies
2.2. Immunoprecipitation Studies
2.3. ANG II ELISA
2.4. RNA Isolation and qRTPCR Studies
2.5. MicroRNA Assay
2.6. Transfection of APOL1 and miR193a Expression Plasmid
2.7. VDR Overexpression with the Treatment of Vitamin D Receptor Agonist
2.8. Silencing of BASP1
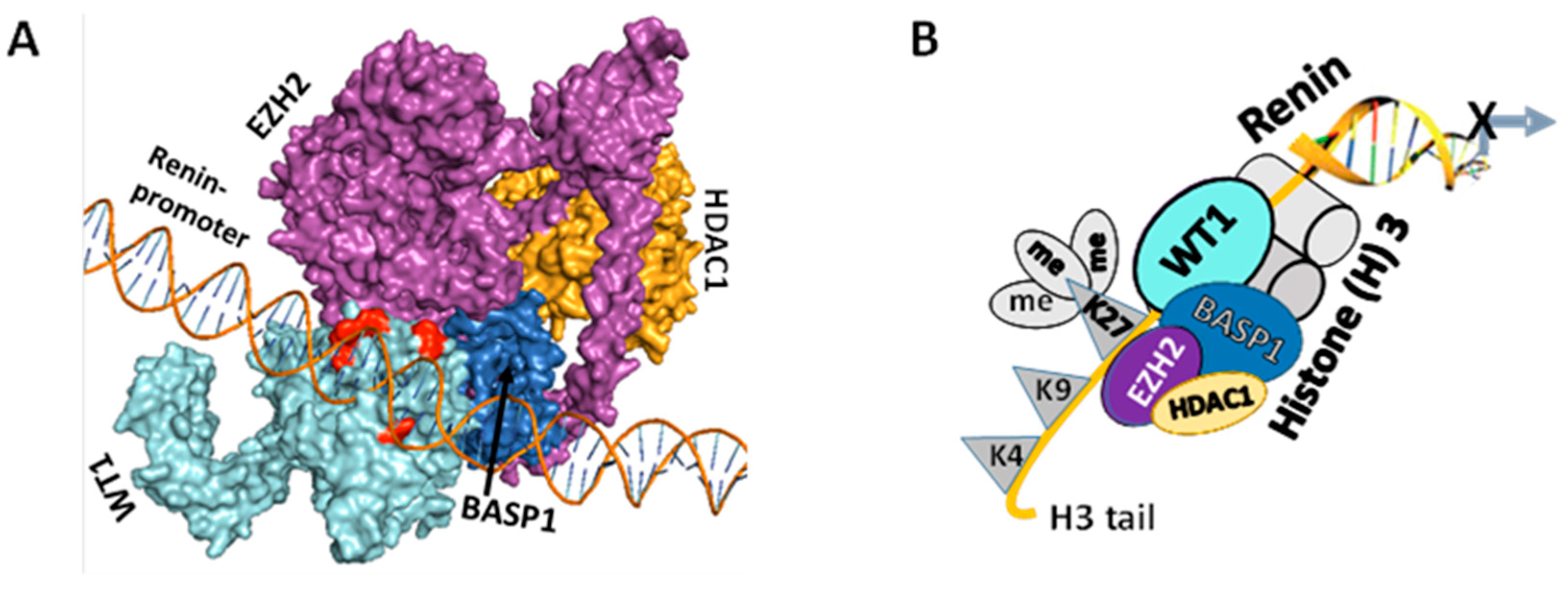
2.9. Renin Promoter Identification
2.10. Homology Modeling and Docking
2.11. Protein-Protein Interactions
2.12. Visualization
2.13. Statistical Analysis
3. Results
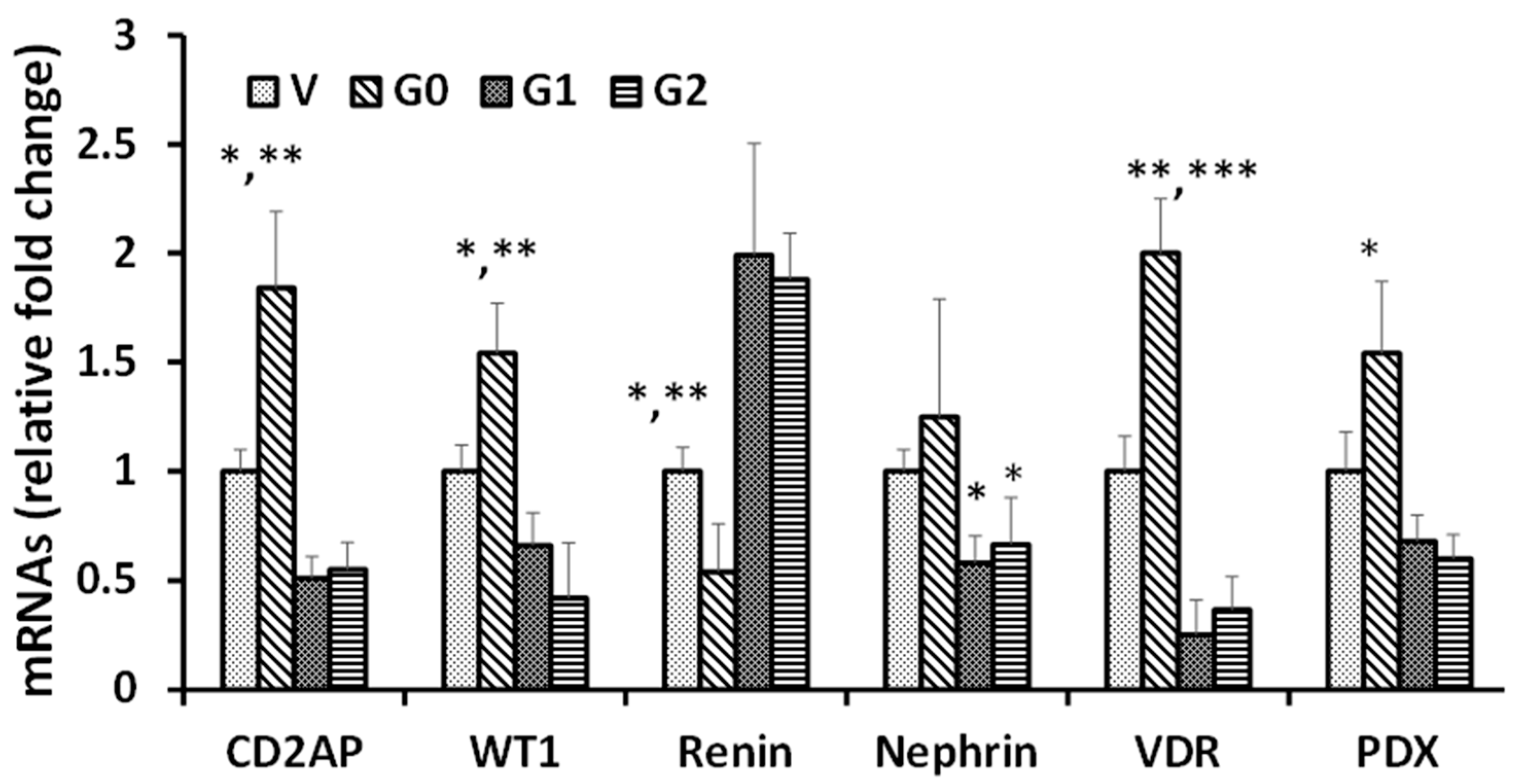
3.1. Effect of APOL1 Risk and Non-Risk Alleles on Podocyte Renin, VDR, Podocyte Molecular Markers (PDMMs), and miR193a Expression
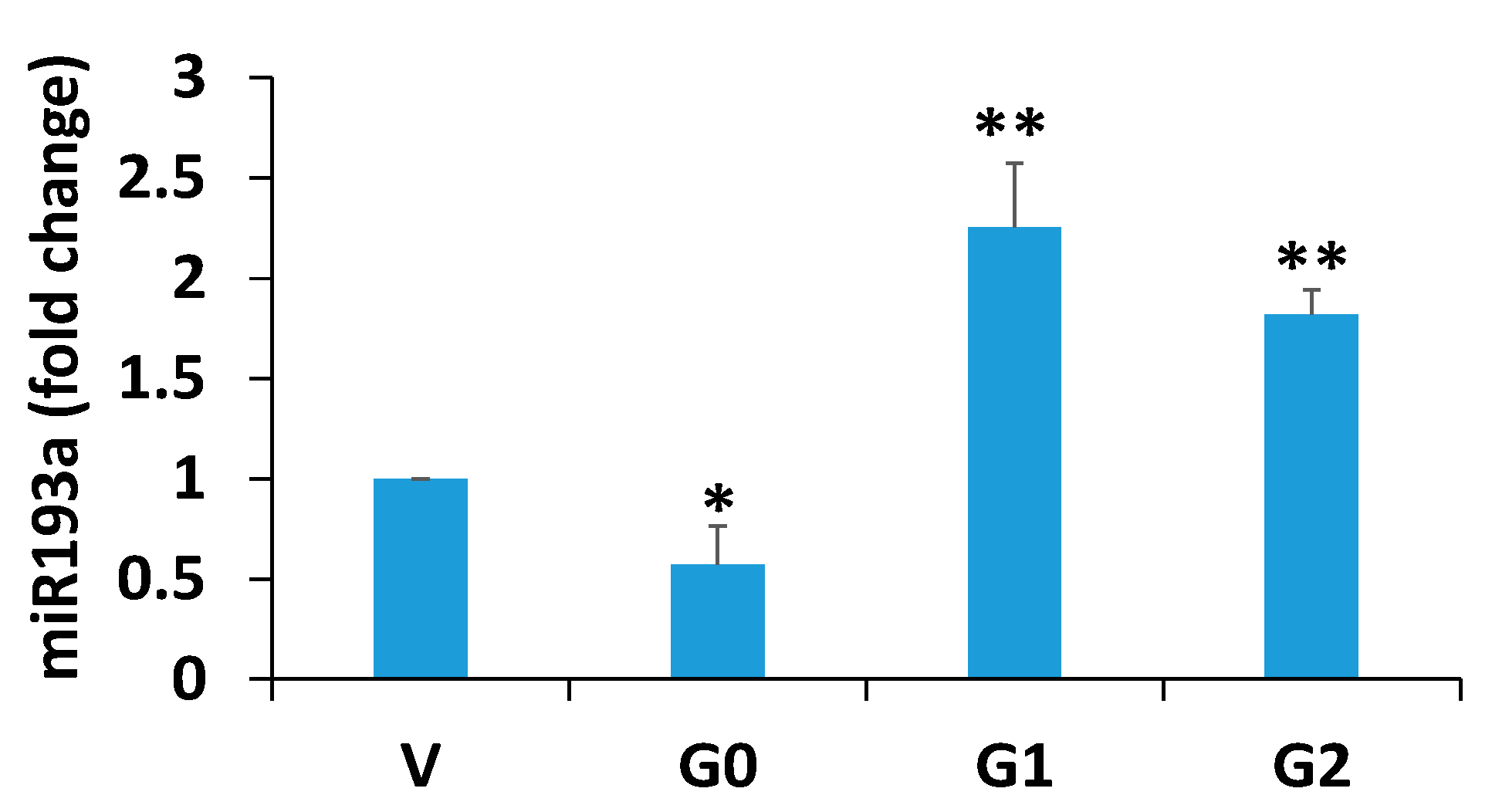
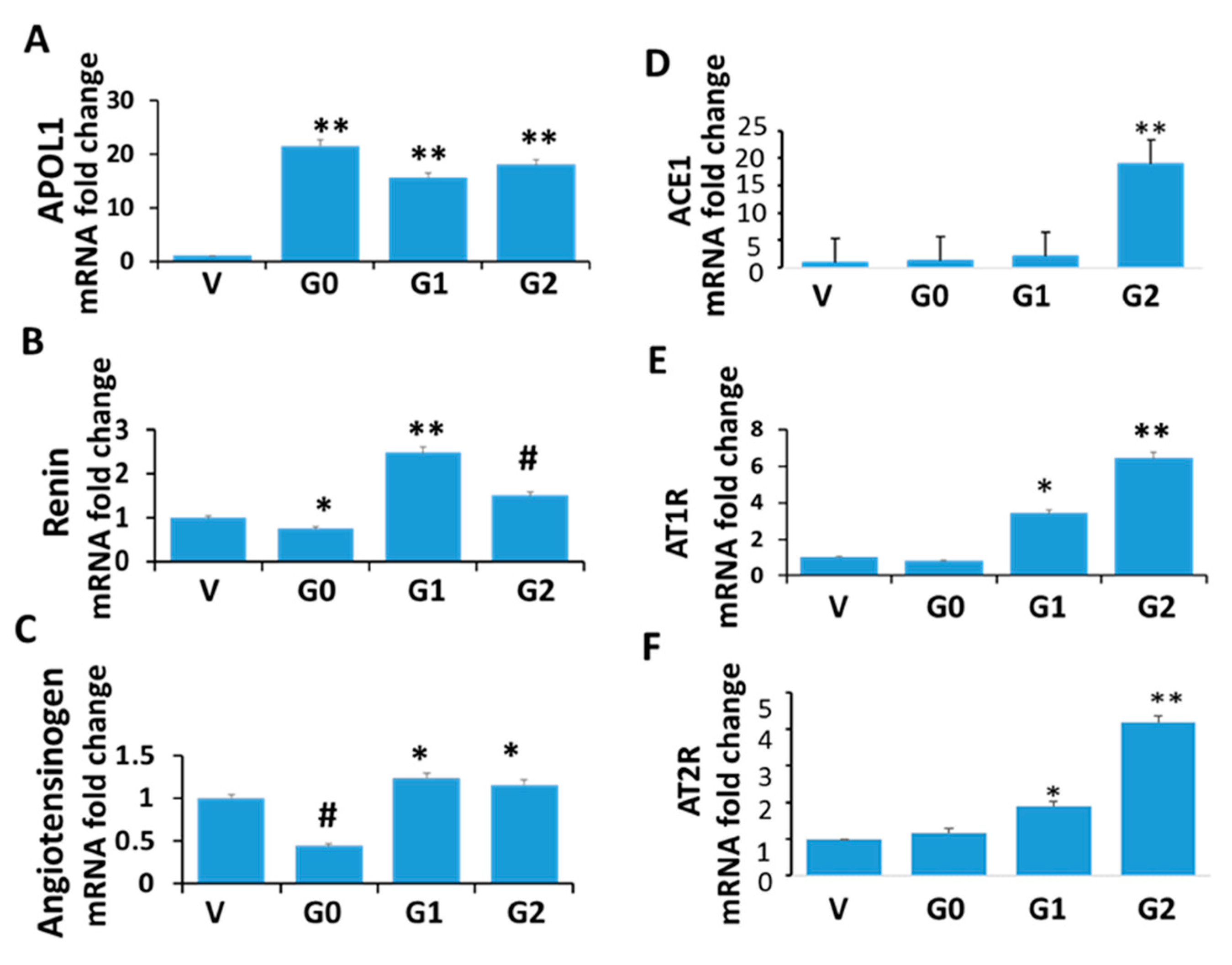
3.2. Renin-Angiotensin System in APOL1 Milieus
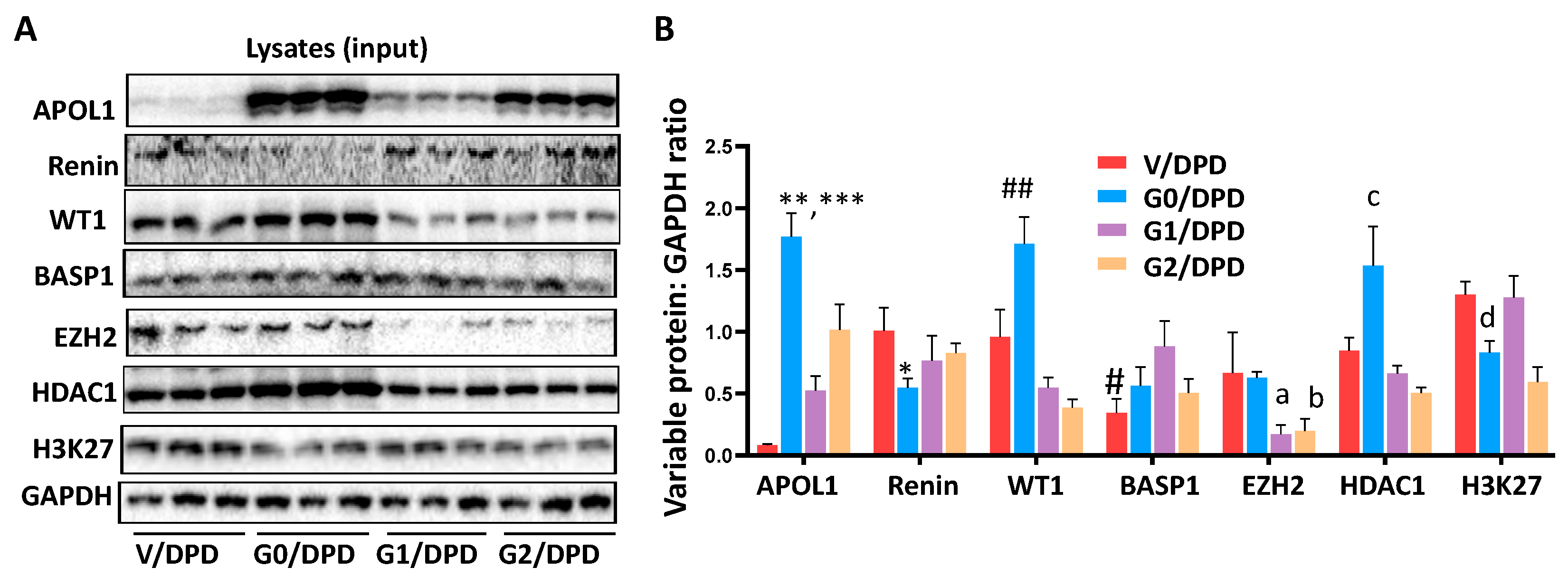
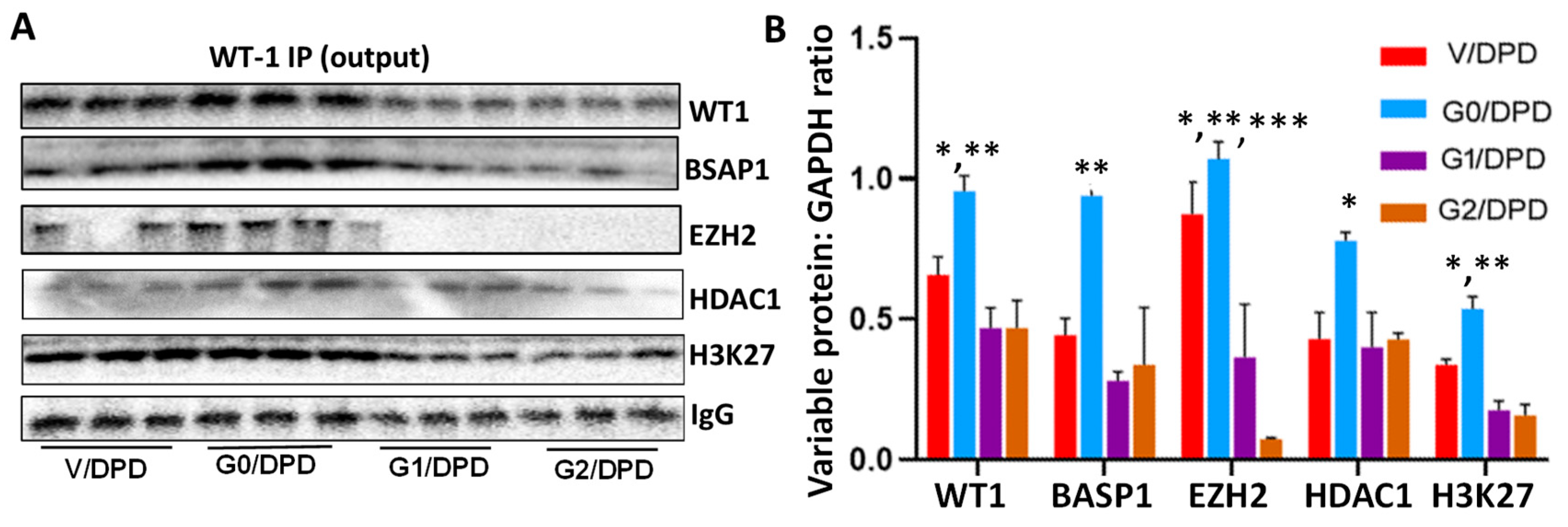
3.3. WT1-BASP1 Repressor Complex Modulates Renin Expression
3.4. Podocytes Overexpressing VDR Showed a Reduction in Renin Expression but Enhanced Expression of Podocyte Molecular Markers (PDMMs)
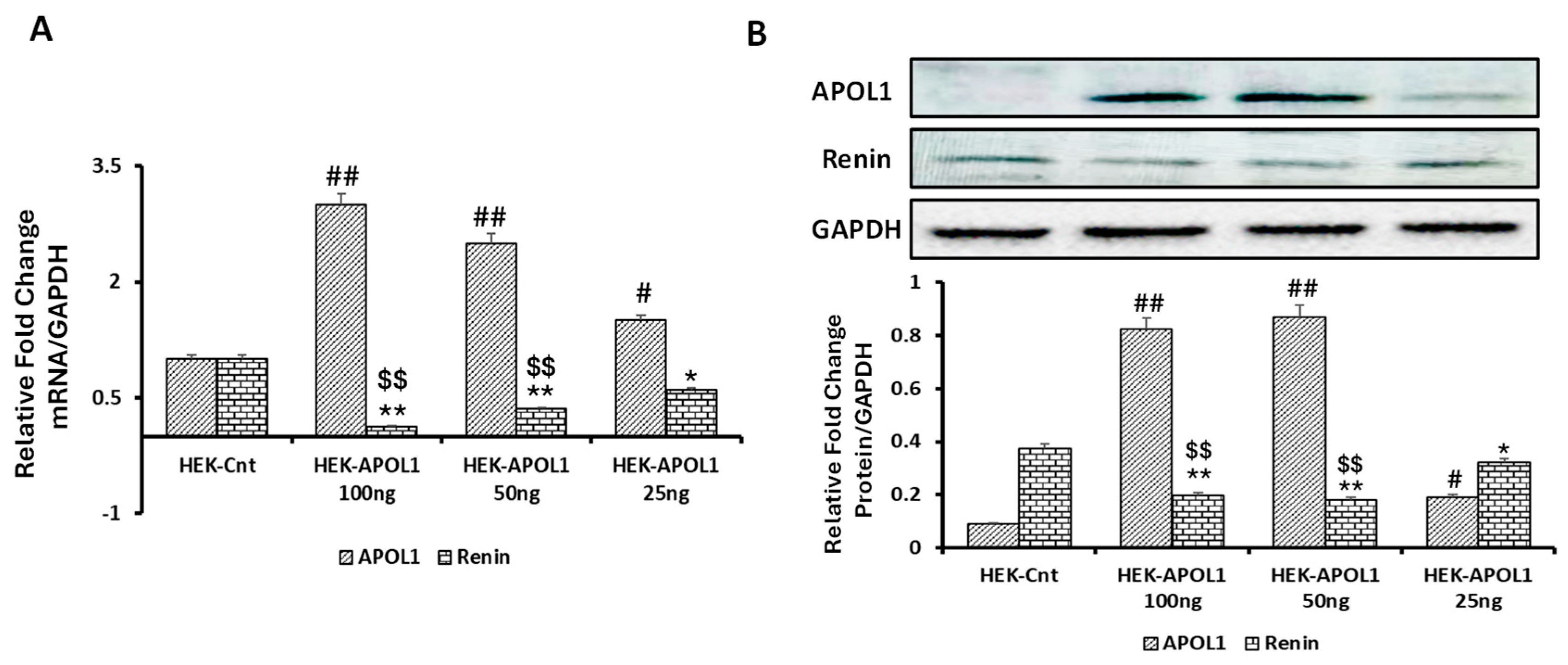
3.5. Dose-Response Effect of APOL1G0 on Renin Expression
3.6. Relationship Between miR193a and VDR
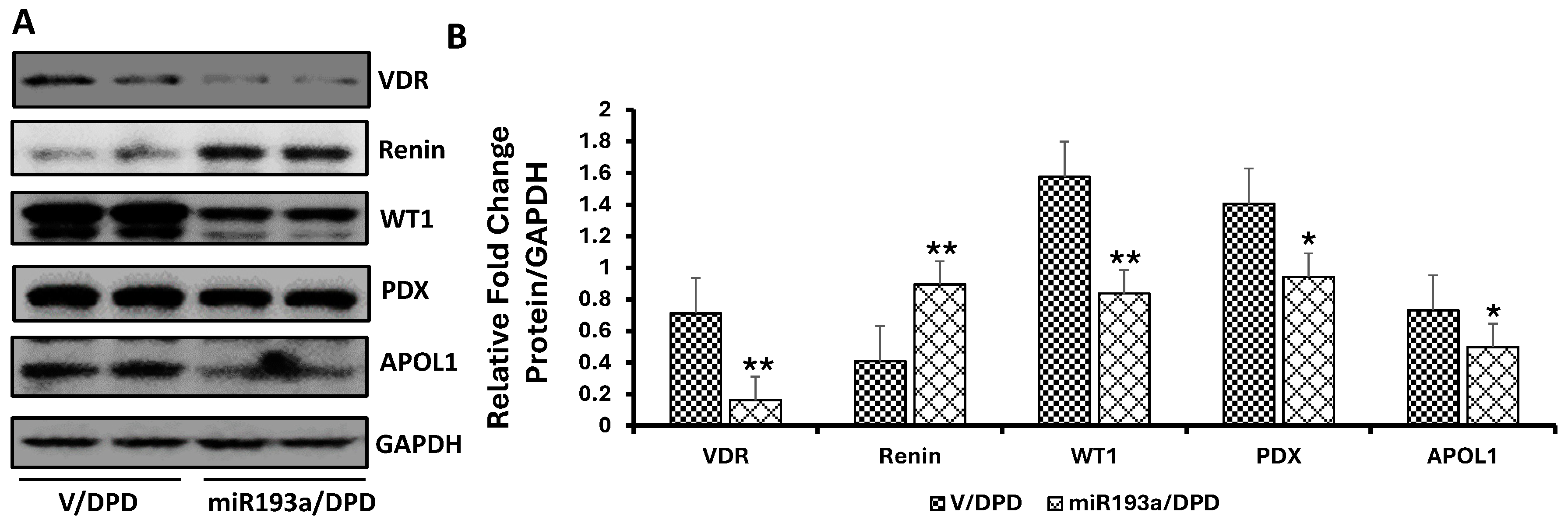
3.7. Effect of miR193a on the Expression of Podocyte and VDR, Renin, and Podocyte Molecular Markers
3.8. Effect of miR193a on the Expression of Podocyte and VDR, Renin, and Podocyte Molecular Markers in Renal Tissues of Control and miR193aTr Mice
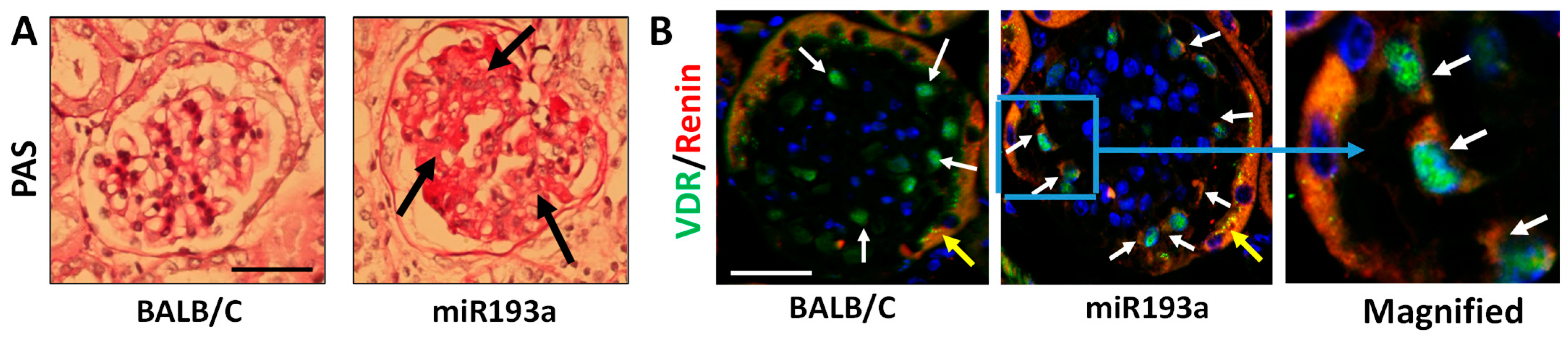
3.9. Renal Histology and Immunofluorescence Labeling for VDR and Renin in Renal Cortical Sections of Control and miR193aTr Mice
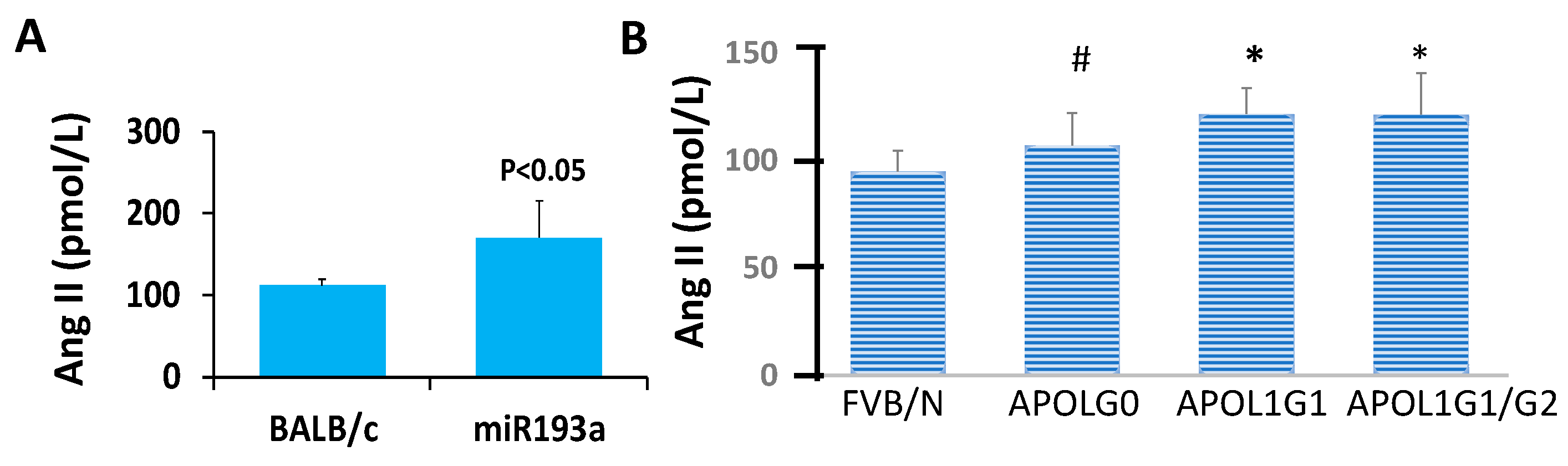
3.10. Ang II Blood Levels in Control and Experimental (miR193aTr and APOL1 G0, APOL1G1, and APOL1 G1/G2) Mice
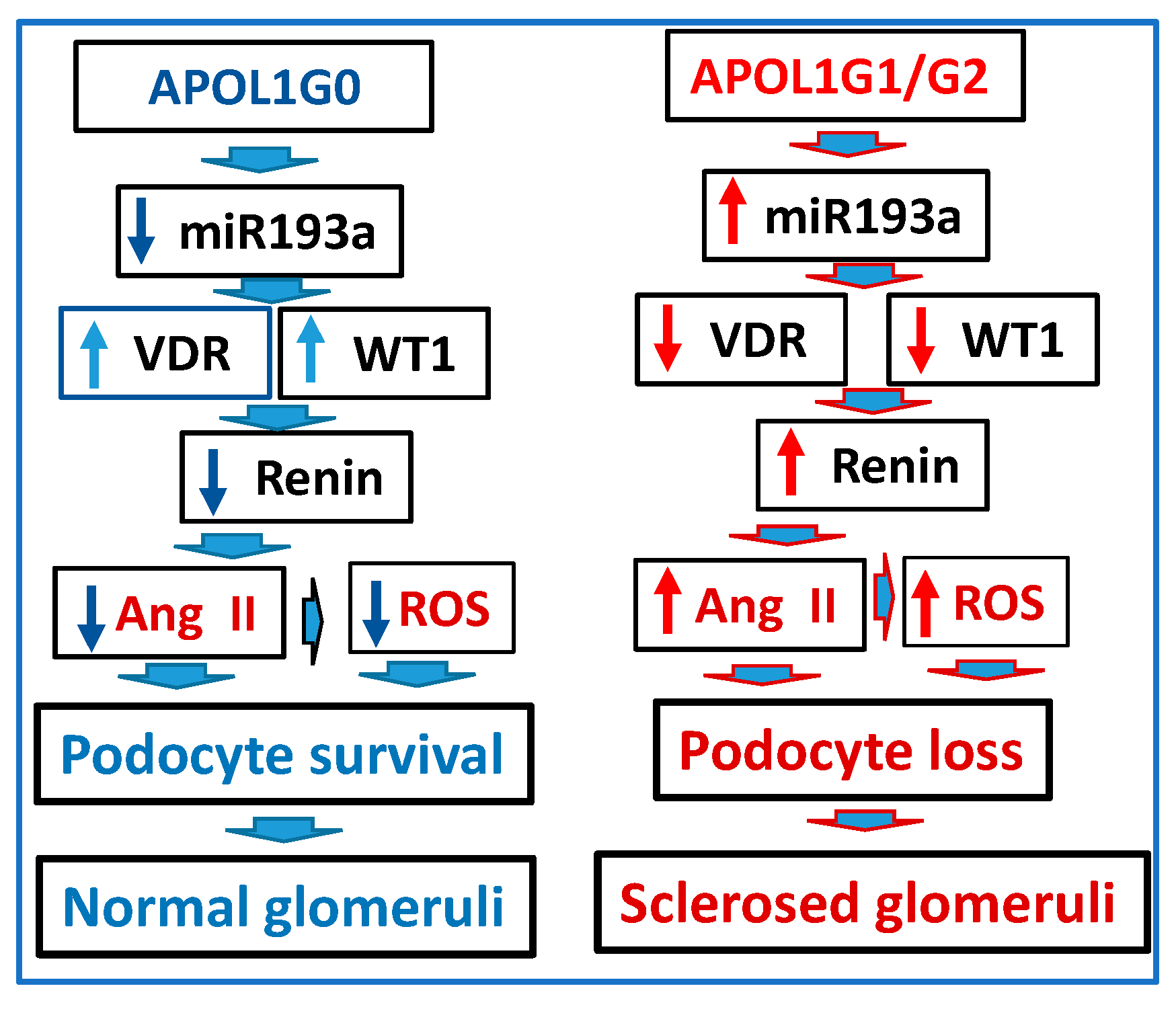
3.11. Role of APOL1 Dynamics in the RAS Activation and Glomerular Sclerosis
4. Discussion
Strengths and Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kopp, J.B.; Nelson, G.W.; Sampath, K.; Johnson, R.C.; Genovese, G.; An, P.; Friedman, D.; Briggs, W.; Dart, R.; Korbet, S.; et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropath. J. Am. Soc. Nephrol. 2011, 22, 2129–2137. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.J.; Kozlitina, J.; Genovese, G.; Jog, P.; Pollak, M.R. Population-based risk assessment of APOL1 on renal disease. J. Am. Soc. Nephrol. 2011, 22, 2098–2105. [Google Scholar] [CrossRef] [PubMed]
- Kasembeli, A.N.; Duarte, R.; Ramsay, M.; Mosiane, P.; Dickens, C.; Dix-Peek, T.; Limou, S.; Sezgin, E.; Nelson, G.W.; Fogo, A.B.; et al. APOL1 Risk Variants Are Strongly Associated with HIV-Associated Nephropathy in Black South African. J. Am. Soc. Nephrol. 2015, 26, 2882–2890. [Google Scholar] [CrossRef]
- Kopp, J.B.; Heymann, J.; Winkler, C.A. APOL1 Renal Risk Variants: Fertile Soil for HIV-Associated Nephropathy. Semin. Nephrol. 2017, 37, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Kasembeli, A.N.; Duarte, R.; Ramsay, M.; Naicker, S. African origins and chronic kidney disease susceptibility in the human immunodeficiency virus era. World J. Nephrol. 2015, 4, 295–306. [Google Scholar] [CrossRef]
- Weir, M.R.; Lakkis, J.I.; Jaar, B.; Rocco, M.V.; Choi, M.J.; Kramer, H.J.; Ku, E. Use of Renin-Angiotensin System Blockade in Advanced CKD: An NKF-KDOQI Controversies Report. Am. J. Kidney Dis. 2018, 72, 873–888. [Google Scholar] [CrossRef]
- Schunkert, H.; Ingelfinger, J.R.; Dzau, V.J. Evolving concepts of the intrarenal renin-angiotensin system in health and disease: Contributions of molecular biology. Kidney Blood Press. Res. 1991, 14, 146–154. [Google Scholar] [CrossRef]
- Johnston, C.I.; Fabris, B.; Jandeleit, K. Intrarenal renin-angiotensin system in renal physiology and pathophysiology. Kidney Int. Suppl. 1993, 42, S59–S63. [Google Scholar]
- He, F.J.; Markandu, N.D.; Sagnella, G.A.; MacGregor, G. Importance of the renin system in determining blood pressure fall with salt restriction in black and white hypertensive. Hypertension 1998, 32, 820–824. [Google Scholar] [CrossRef]
- Cushman, W.C.; Reda, D.J.; Perry, H.M.; Williams, D.; Abdellatif, M.; Materson, B. Regional and racial differences in response to antihypertensive medication use in a randomized controlled trial of men with hypertension in the United States. Department of Veterans Affairs Cooperative Study Group on antihypertensive agent. Arch. Intern. Med. 2000, 160, 825–883. [Google Scholar] [CrossRef]
- Gong, Y.; McDonough, C.W.; Wang, Z.; Hou, W.; Cooper-DeHoff, R.M.; Langaee, T.Y.; Beitelshees, A.L.; Chapman, A.B.; Gums, J.G.; Bailey, K.R.; et al. Hypertension susceptibility loci and blood pressure response to antihypertensives: Results from the pharmacogenomic evaluation of antihypertensive responses study. Circ. Cardiovasc. Genet. 2012, 5, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.T.; Schwartz, G.L.; Chapman, A.B.; Beitelshees, A.L.; Gums, J.G.; Cooper-DeHoff, R.M.; Boerwinkle, E.; Johnson, J.A.; Bailey, K.R. Plasma renin activity predicts blood pressure responses to beta-blockers and thiazide diuretics as monotherapy and add-on therapy for hypertension. Am. J. Hypertens. 2010, 23, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Flack, J.; Hamat, M. Difficult-to-treat hypertensive populations: Focus on African-Americans and people with type 2 diabetes. J. Hypertens. 1999, 17 (Suppl 1), S19–S24. [Google Scholar]
- Mishra, A.; Ayasolla, K.; Kumar, V.; Lan, X.; Vashistha, H.; Aslam, R.; Hussain, A.; Chowdhary, S.; Marashi Shoshtari, S.; Paliwal, N.; et al. Modulation of the apolipoprotein L1-microRNA-193a axis prevents podocyte dedifferentiation in a high-glucose milieu. Am. J. Physiol.-Ren. Physiol. 2018, 314, F832–F843. [Google Scholar] [CrossRef]
- Jha, A.; Saha, S.; Ayasolla, K.; Vashistha, H.; Malhotra, A.; Skorecki, K.; Singhal, P.C. MiR193a Modulation and Podocyte Phenotype. Cell 2020, 9, 1004. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Paliwal, N.; Ayasolla, K.; Vashistha, H.; Jha, A.; Chandel, N.; Chowdhary, S.; Saleem, M.A.; Malhotra, A.; Chander, P.N.; et al. Disruption of APOL1-miR193a Axis Induces Disorganization of Podocyte Actin Cytoskeleton. Sci. Rep. 2019, 9, 3582. [Google Scholar] [CrossRef]
- Gebeshuber, C.A.; Kornauth, C.; Dong, L.; Sierig, R.; Seibler, J.; Reiss, M.; Tauber, S.; Bilban, M.; Wang, S.; Kain, R.; et al. Focal segmental glomerulosclerosis is induced by microRNA-193a and its downregulation of WT 1. Nat. Med. 2013, 19, 481–487. [Google Scholar] [CrossRef]
- Kietzmann, L.; Guhr, S.S.; Meyer, T.N.; Ni, L.; Sachs, M.; Panzer, U.; Stahl, R.A.; Saleem MAKerjaschki, D.; Gebeshuber, C.A.; Meyer-Schwesinger, C. MicroRNA-193a Regulates the Transdifferentiation of Human Parietal Epithelial Cells toward a Podocyte Phenotype. J. Am. Soc. Nephrol. 2015, 26, 1389–1401. [Google Scholar] [CrossRef]
- Kumar, V.; Vashistha, H.; Lan, X.; Chandel, N.; Ayasolla, K.; Shoshtari, S.S.M.; Aslam, R.; Paliwal, N.; Abbruscato, F.; Mikulak, J.; et al. Role of Apolipoprotein L1 in Human Parietal Epithelial Cell Transition. Am. J. Pathol. 2018, 188, 2508–2528. [Google Scholar] [CrossRef]
- Steege, A.; Fähling, M.; Paliege, A.; Bondke, A.; Kirschner, K.M.; Martinka, P.; Kaps, C.; Patzak, A.; Persson, P.B.; Thiele, B.J.; et al. Kidney Wilms’ tumor protein (-KTS) modulates renin gene transcription. Kidney Int. 2008, 74, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.G.E. Transcriptional regulation by WT1 in development. Curr. Opin. Genet. Dev. 2005, 15, 542–547. [Google Scholar] [CrossRef] [PubMed]
- McKay, L.M.; Carpenter, B.; Roberts, S.G.E. Regulation of the Wilms’ tumor suppressor protein transcriptional activation domain. Oncogene 1999, 18, 6546–6554. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, B.; Hill, K.J.; Charalambous, M.; Wagner, K.J.; Lahiri, D.; James, D.I.; Anderson, J.S.; Schumacher, V.; Royer-Pokora, B.; Mann, M.; et al. BASP1 is a transcriptional cosuppressor for the Wilms’ tumour suppressor protein WT1. Mol. Cell. Biol. 2004, 24, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.H.; Hoffmann, S.; Di Marco, G.S.; Endlich, N.; Peter-Katalinić, J.; Weide, T.; Pavenstädt, H. Downregulation of the antioxidant protein peroxiredoxin 2 contributes to Angiotensin II-mediated podocyte apoptosis. Kidney Int. 2011, 80, 959–969. [Google Scholar] [CrossRef]
- Whaley-Connell, A.T.; Chowdhury, N.A.; Hayden, M.R.; Stump, C.S.; Habibi, J.; Wiedmeyer, C.E.; Gallagher, P.E.; Tallant, E.A.; Cooper, S.A.; Link, C.D.; et al. Oxidative stress and glomerular filtration barrier injury: Role of the renin-angiotensin system in the Ren2 transgenic rat. Am. J. Physiol.-Ren. Physiol. 2006, 291, F1308–F1314. [Google Scholar] [CrossRef]
- Li, Y.C.; Kong, J.; Wei, M.; Chen, Z.F.; Liu, S.Q.; Cao, L.P. 1.25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J. Clin. Investig. 2002, 110, 229–238. [Google Scholar] [CrossRef]
- Chandel, N.; Ayasolla, K.; Wen, H.; Lan, X.; Haque, S.; Saleem, M.A.; Malhotra, A.; Singhal, P.C. Vitamin D receptor deficit induces renin-angiotensin system activation via SIRT1 modulation in podocytes. Exp. Mol. Pathol. 2017, 102, 97–105. [Google Scholar] [CrossRef]
- Lee, T.H.; Pelletier, J. Functional characterization of WT1 binding sites within the human vitamin D receptor gene promoter. Physiol. Genom. 2001, 7, 187–200. [Google Scholar] [CrossRef]
- Saleem, M.A.; O’Hare, M.J.; Reiser, J.; Coward, R.J.; Inward, C.D.; Farren, T.; Xing, C.Y.; Ni, L.; Mathieson, P.W.; Mundel, P. Conditionally immortalized human podocyte cell line demonstrating Nephrin and podocin expression. J. Am. Soc. Nephrol. 2002, 13, 630–638. [Google Scholar] [CrossRef]
- Kruzel-Davila, E.; Shemer, R.; Ofir, A.; Bavli-Kertselli, I.; Darlyuk-Saadon, I.; Oren-Giladi, P.; Wasser, W.G.; Magen, D.; Zaknoun, E.; Schuldiner, M.; et al. APOL1-Mediated Cell Injury Involves Disruption of Conserved Trafficking Processes. J. Am. Soc. Nephrol. 2017, 28, 1117–1130. [Google Scholar] [CrossRef] [PubMed]
- Dreos, R.; Ambrosini, G.; Groux, R.; Périer, R.; Bucher, P. The eukaryotic promoter database, in its 30th Year: Focus on non-vertebrate organisms. Nucleic Acids Res. 2017, 45, D51–D55. [Google Scholar] [CrossRef]
- Lu, X.J.; Olson, W.K. 3DNA: A versatile, integrated software system for the analysis, Rebuilding and visualization of three-dimensional nucleic-acid structures. Nat. Protoc. 2008, 3, 1213–1227. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Method 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S. HDOCK: A web server for protein-protein and protein-DNA/RNA docking is based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef]
- Duhovny, D.S.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid And symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Kristine, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar]
- PyMOL Molecular Graphics System, Version 2.0; Schrodinger, LLC: New York, NY, USA, 2018.
- Wagner, K.D.; Wagner, N.; Wellmann, S.; Schley, G.; Bondke, A.; Theres, H.; Scholz, H. Oxygen-regulated expression of the Wilms’ tumor suppressor Wt1 involves hypoxia-inducible factor-1 (HIF-1). FASEB J. 2003, 17, 1364–1366. [Google Scholar] [CrossRef]
- Olabisi, O.A.; Zhang, J.Y.; VerPlank, L.; Zahler, N.; DiBartolo, S.; 3rd Heneghan, J.F.; Schlöndorff, J.S.; Suh, J.H.; Yan, P.; Alper, S.L.; et al. APOL1 kidney disease risk variants cause cytotoxicity by depleting cellular potassium and inducing stress-activated protein kinase. Proc. Natl. Acad. Sci. USA 2016, 113, 830–837. [Google Scholar] [CrossRef]
- Schaub, C.; Verdi, J.; Lee, P.; Terra, N.; Limon, G.; Raper, J.; Thomson, R. Cation channel conductance and pH gating of the innate immunity factor APOL1 are governed by pore-lining residues within the C-terminal domain. J. Biol. Chem. 2020, 295, 13138–13149. [Google Scholar] [CrossRef] [PubMed]
- Giovinazzo, J.A.; Thomson, R.P.; Khalizova, N.; Zager, P.J.; Malani, N.; Rodriguez-Boulan, E.; Raper, J.; Schreiner, R. Apolipoprotein L-1 renal risk variants form active channels at the plasma membrane driving cytotoxicity. Elife 2020, 9, e51185. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.; Kumar, V.; Haque, S.; Ayasolla, K.; Saha, S.; Lan, X.; Malhotra, A.; Saleem, M.A.; Skorecki, K.; Singhal, P. Alterations in plasma membrane ion channel structures stimulate NLRP3 inflammasome activation in APOL1 risk milieu. FEBS J. 2020, 287, 2000–2022. [Google Scholar] [CrossRef] [PubMed]
- Latha, M.V.; Borowitz, J.L.; Yim, G.K.; Kanthasamy, A.; Isom, G.E. Plasma membrane hyperpolarization by cyanide in chromaffin cells: Role of potassium channel. Arch. Toxicol. 1994, 68, 370–374. [Google Scholar] [CrossRef]
- Kurtz, A.; Hamann, M.; Götz, K. Role of potassium channels in controlling renin secretion from isolated perfused rat kidneys. Pflugers. Arc. 2000, 440, 889–895. [Google Scholar] [CrossRef]
- Assady, S.; Wanner, N.; Skorecki, K.L.; Huber, T.B.J. New Insights into Podocyte Biology in Glomerular Health and Diseases. J. Am. Soc. Nephrol. 2017, 28, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Shankland, S.J.; Rule, A.D.; Kutz, J.N.; Pippin, J.W.; Wessely, O. Podocyte Senescence and Ageing. Kidney 360 2023, 4, 1784–1793. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, Q.; Yang, J.; Ma, Y.; Ding, G. Angiotensin II induces cholesterol accumulation and injury in podocytes. Sci. Rep. 2017, 7, 10672. [Google Scholar] [CrossRef]
- Erichsen, L.; Thimm, C.; Bohndorf, M.; Rahman, M.S.; Wruck, W.; Adjaye, J. Activation of the Renin-Angiotensin System Disrupts the Cytoskeletal Architecture of Human Urine-Derived Podocytes. Cells 2022, 11, 1095. [Google Scholar] [CrossRef]
- Miceli, I.; Burt, D.; Tarabra, E.; Camussi, G.; Perin, P.C.; Gruden, G. Stretch reduces nephrin expression via an angiotensin II-AT(1)-dependent mechanism in human podocytes: Effect of rosiglitazone. Am. J. Physiol. Renal. Physiol. 2010, 298, F381–F390. [Google Scholar] [CrossRef]
- Durvasula, R.V.; Petermann, A.T.; Hiromura, K.; Blonski, M.; Pippin, J.; Mundel, P.; Pichler, R.; Griffin, S.; Couser, W.G.; Shankland, S.J. Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int. 2004, 65, 30–39. [Google Scholar] [CrossRef]
- Márquez, E.; Riera, M.; Pascual, J.; Soler, M.J. Renin-angiotensin system within the diabetic podocyte. Am. J. Physiol. Renal. Physiol. 2015, 308, F1–F10. [Google Scholar] [CrossRef] [PubMed]
- Salhan, D.; Husain, M.; Subrati, A.; Goyal, R.; Singh, T.; Rai, P.; Malhotra, A.; Singhal, P.C. HIV-induced kidney cell injury: Role of ROS-induced downregulated vitamin D receptor. Am. J. Physiol. Renal. Physiol. 2012, 303, F503–F514. [Google Scholar] [CrossRef]
- Chandel, N.; Sharma, B.; Salhan, D.; Husain, M.; Malhotra, A.; Buch, S.; Singhal, P.C. Vitamin D receptor activation and downregulation of the renin-angiotensin system attenuate morphine-induced T cell apoptosis. Am. J. Physiol. Cell. Physiol. 2012, 303, C607–C615. [Google Scholar] [CrossRef]
- Vaidya, A.; Williams, J.S. The relationship between vitamin D and the renin-angiotensin system in the pathophysiology of hypertension, kidney disease, and diabetes. Metabolism 2012, 61, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Sigmund, C.D. Regulation of renin expression and blood pressure by vitamin D(3). J. Clin. Investig. 2002, 110, 155–156. [Google Scholar] [CrossRef] [PubMed]
- Hayek, S.S.; Koh, K.H.; Grams, M.E.; Wei, C.; Ko, Y.A.; Li, J.; Samelko, B.; Lee, H.; Dande, R.R.; Lee, H.W.; et al. A tripartite complex of suPAR, APOL1 risk variants and alpha(v)beta(3) integrin on podocytes mediates chronic kidney disease. Nat. Med. 2017, 23, 945–953. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, B.; Liu, S.; Xie, S.; Yang, Y.; Ma, J.; Deng, Y.; Wang, W.; Xu, L.; Li, R.; et al. 1,25-Dihydroxyvitamin D(3) Inhibits Podocyte uPAR Expression and Reduces Proteinuria. PLoS ONE 2013, 8, e64912. [Google Scholar] [CrossRef]
- Dusso, A.S.; Brown, A.J.; Slatopolsky, E. Vitamin, D. Am. J. Physiol. Renal. Physiol. 2005, 289, F8–F28. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | |
|---|---|
| mNphs1-F | GTGCCCTGAAGGACCCTACT |
| mNphs1-R | CCTGTGGATCCCTTTGACAT |
| mWt1-F | GAGAGCCAGCCTACCATCC |
| mWt1-R | GGGTCCTCGTGTTTGAAGGAA |
| mRenin-F | TGAGCATCAGCAAGACTGACTCC |
| mRenin-R | AGGATGAACCAGTGTCCACCACTAC |
| mAce1-F | CCACTATGGGTCCGAGTACATCAA |
| mAce-R | AGGGCGCCACCAAATCATAG |
| mAgt-F | CTGGATTTATCCACTGACCCAGTTC |
| mAgt-R | TGGACTCCAGGCAGCTGAGA |
| mAgtr1a-F | TGGGCGTCATCCATGACTGTA |
| mAgtr1a-R | TGAGTGCGACTTGGCCTTTG |
| mAgtr1b-F | CTGCTATGCCCATCACCATCTG |
| mAgtr1b-R | GATAACCCTGCATGCGACCTG |
| mAgtr2-F | GTGCATGCGGGAGCTGAGTA |
| mAgtr2-R | ATTGGTGCCAGTTGCGTTGA |
| mVDR-F | CACCTGGCTGATCTTGTCAGT |
| mVDR-R | CTGGTCATCAGAGGTGAGGTC |
| hVDR-F | CCTTCACCATGGACGACATG |
| hVDR-R | CGGCTTTGGTCACGTCACT |
| mCd2ap-F | AGCGAATCAGCACTTATGGA |
| mCd2ap-R | CCACCAGCCTTCTTCTACCT |
| mPodx-F | CCTGAACCTCACAGGAAACACC |
| mPodx-R | TGGAACAGATGCCAGCCGTATG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, V.; Kaur, P.; Ayasolla, K.; Jha, A.; Wiqas, A.; Vashistha, H.; Saleem, M.A.; Popik, W.; Malhotra, A.; Gebeshuber, C.A.; et al. APOL1 Modulates Renin–Angiotensin System. Biomolecules 2024, 14, 1575. https://doi.org/10.3390/biom14121575
Kumar V, Kaur P, Ayasolla K, Jha A, Wiqas A, Vashistha H, Saleem MA, Popik W, Malhotra A, Gebeshuber CA, et al. APOL1 Modulates Renin–Angiotensin System. Biomolecules. 2024; 14(12):1575. https://doi.org/10.3390/biom14121575
Chicago/Turabian StyleKumar, Vinod, Prabhjot Kaur, Kameshwar Ayasolla, Alok Jha, Amen Wiqas, Himanshu Vashistha, Moin A. Saleem, Waldemar Popik, Ashwani Malhotra, Christoph A. Gebeshuber, and et al. 2024. "APOL1 Modulates Renin–Angiotensin System" Biomolecules 14, no. 12: 1575. https://doi.org/10.3390/biom14121575
APA StyleKumar, V., Kaur, P., Ayasolla, K., Jha, A., Wiqas, A., Vashistha, H., Saleem, M. A., Popik, W., Malhotra, A., Gebeshuber, C. A., Skorecki, K., & Singhal, P. C. (2024). APOL1 Modulates Renin–Angiotensin System. Biomolecules, 14(12), 1575. https://doi.org/10.3390/biom14121575