
Short Link N Modulates Inflammasome Activity in Intervertebral Discs Through Interaction with CD14

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Peptide Synthesis
2.2. Antibodies
2.3. Human Nucleus Pulposus Cell
2.4. Murine Macrophages
2.5. Rabbit Annular Puncture Model of IVDD
2.6. Histology
2.7. Inflammasome Signaling
2.8. Assessment of Inflammasome Markers in hNP Cells
2.9. Caspase-1 and IL-1β Maturation
2.10. RNA Extraction and Quantitative Real-Time PCR
2.11. Macrophage Polarization Induced by hNP
2.12. Immunoprecipitation
2.13. Statistical Analysis
3. Results
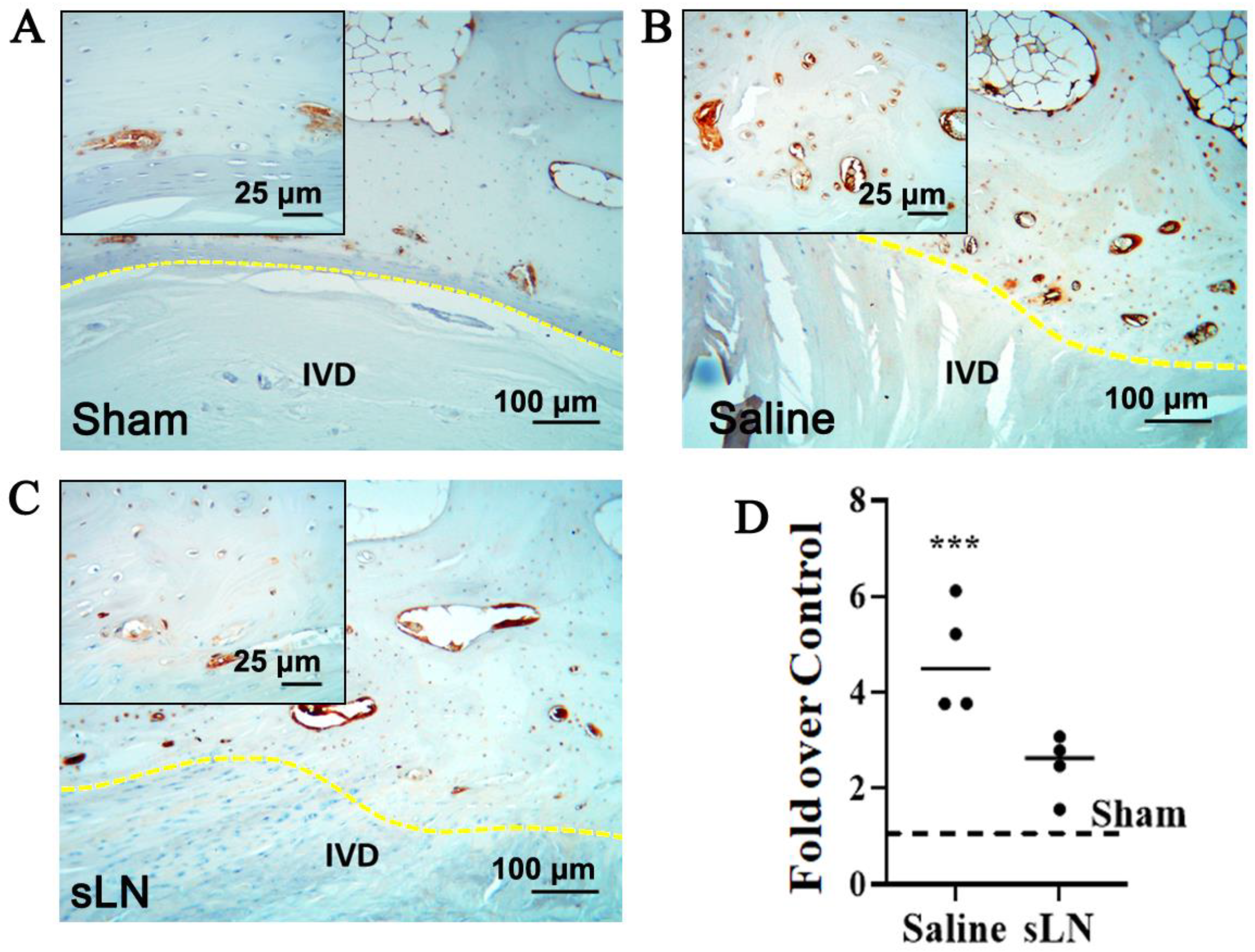
3.1. Impact of sLN on NLRP3 Expression in a Rabbit Annular Puncture Model of IVDD
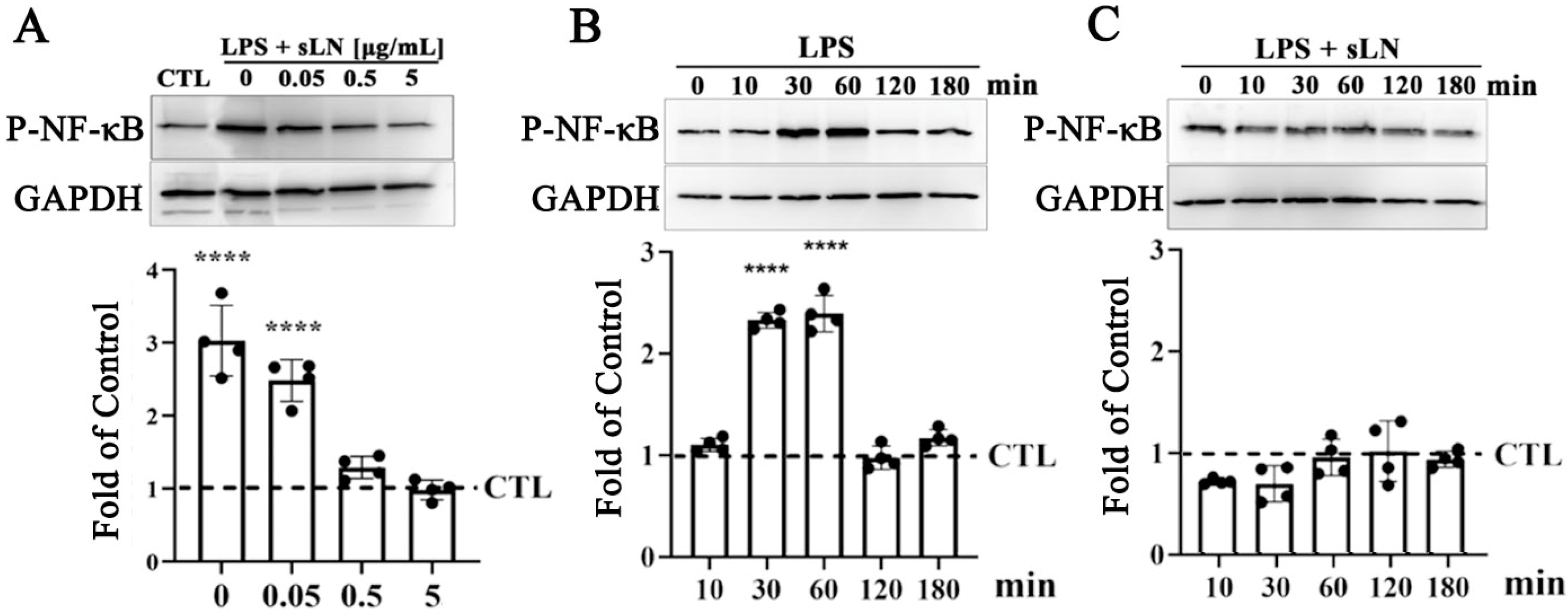
3.2. Regulation of Inflammasome Signaling by sLN in Human Nucleus Pulposus Cells
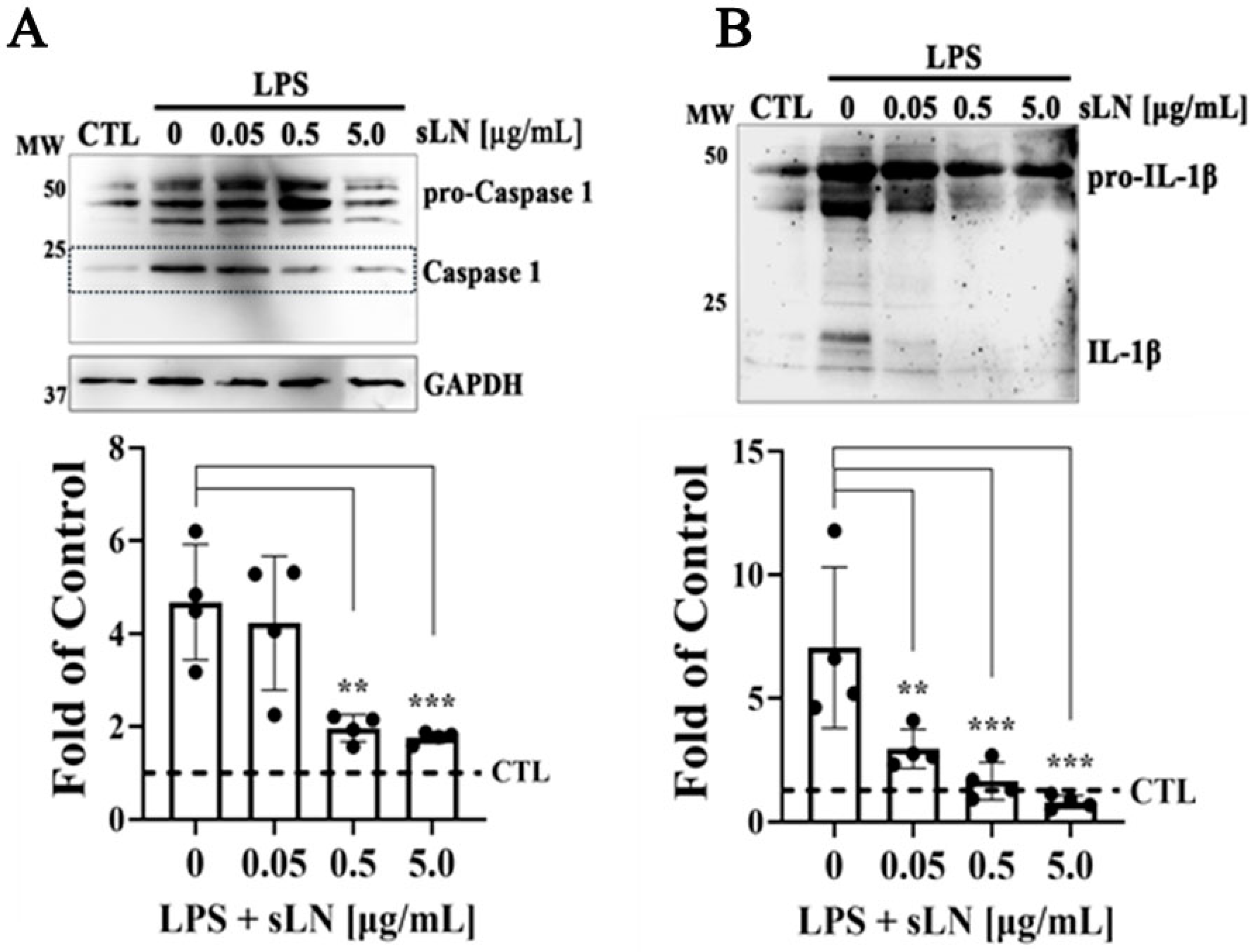
3.3. Modulation of Inflammasome Activation Markers by sLN
3.4. Combined Inhibition of Caspase-1 Activation and IL-1β Secretion by sLN in LPS-Stimulated
3.5. Inflammasome Activation and Macrophage Polarization
3.6. Interaction of sLN with CD14
3.7. Schematic Mechanisms of sLN Inhibition of Inflammasome Activation in IVD Cells
3.7.1. Pathway of Activation and Inhibition
3.7.2. Role of sLN
3.7.3. Outcome
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2017 DALYs and HALE Collaborators; Global Burden of Disease Study 2017. Global, regional, and national disability-adjusted life-years (DALYs) for 359 diseases and injuries and healthy life expectancy (HALE) for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1859–1922. [Google Scholar] [CrossRef] [PubMed]
- Gureje, O.; Von Korff, M.; Simon, G.E.; Gater, R. Persistent pain and well-being: A World Health Organization Study in Primary Care. Jama 1998, 280, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Risbud, M.V.; Shapiro, I.M. Role of cytokines in intervertebral disc degeneration: Pain and disc content. Nat. Rev. Rheumatol. 2014, 10, 44–56. [Google Scholar] [CrossRef]
- Emanuel, K.S.; Mader, K.T.; Peeters, M.; Kingma, I.; Rustenburg, C.M.E.; Vergroesen, P.-P.A.; Sammon, C.; Smit, T.H. Early changes in the extracellular matrix of the degenerating intervertebral disc, assessed by Fourier transform infrared imaging. Osteoarthr. Cartil. 2018, 26, 1400–1408. [Google Scholar] [CrossRef]
- Diwan, A.D.; Melrose, J. Intervertebral disc degeneration and how it leads to low back pain. JOR Spine 2023, 6, e1231. [Google Scholar] [CrossRef]
- Lyu, F.J.; Cui, H.; Pan, H.; Mc Cheung, K.; Cao, X.; Iatridis, J.C.; Zheng, Z. Painful intervertebral disc degeneration and inflammation: From laboratory evidence to clinical interventions. Bone Res. 2021, 9, 7. [Google Scholar] [CrossRef]
- Pedersen, H.E.; Blunck, C.F.; Gardner, E. The anatomy of lumbosacral posterior rami and meningeal branches of spinal nerve (sinu-vertebral nerves); with an experimental study of their functions. J. Bone Jt. Surg. Am. 1956, 38, 377–391. [Google Scholar] [CrossRef]
- Colombini, A.; Lombardi, G.; Corsi, M.M.; Banfi, G. Pathophysiology of the human intervertebral disc. Int. J. Biochem. Cell Biol. 2008, 40, 837–842. [Google Scholar] [CrossRef]
- Hadjipavlou, A.G.; Tzermiadianos, M.N.; Bogduk, N.; Zindrick, M.R. The pathophysiology of disc degeneration: A critical review. J. Bone Jt. Surg. Br. 2008, 90, 1261–1270. [Google Scholar] [CrossRef]
- Edgar, M.A. The nerve supply of the lumbar intervertebral disc. J. Bone Jt. Surg. Br. 2007, 89, 1135–1139. [Google Scholar] [CrossRef]
- Raj, P.P. Intervertebral disc: Anatomy-physiology-pathophysiology-treatment. Pain Pract. 2008, 8, 18–44. [Google Scholar] [CrossRef] [PubMed]
- Coppes, M.H.; Marani, E.; Thomeer, R.T.; Groen, G.J. Innervation of “painful” lumbar discs. Spine 1997, 22, 2342–2349; discussion 2349–2350. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E.; Sivan, S.; Wright, K.T.; Eisenstein, S.M.; Maroudas, A.; Roberts, S. Human intervertebral disc cells promote nerve growth over substrata of human intervertebral disc aggrecan. Spine 2006, 31, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Delcroix, J.D.; Valletta, J.S.; Wu, C.; Hunt, S.J.; Kowal, A.S.; Mobley, W.C. NGF signaling in sensory neurons: Evidence that early endosomes carry NGF retrograde signals. Neuron 2003, 39, 69–84. [Google Scholar] [CrossRef]
- Levi-Montalcini, R. The nerve growth factor 35 years later. Science 1987, 237, 1154–1162. [Google Scholar] [CrossRef]
- Noorwali, H.; Grant, M.P.; Epure, L.M.; Madiraju, P.; Sampen, H.J.; Antoniou, J.; Mwale, F. Link N as a therapeutic agent for discogenic pain. JOR Spine 2018, 1, e1008. [Google Scholar] [CrossRef]
- Pezet, S.; McMahon, S.B. Neurotrophins: Mediators and modulators of pain. Annu. Rev. Neurosci. 2006, 29, 507–538. [Google Scholar] [CrossRef]
- Burke, J.G.; Watson, R.W.; McCormack, D.; Dowling, F.E.; Walsh, M.G.; Fitzpatrick, J.M. Intervertebral discs which cause low back pain secrete high levels of proinflammatory mediators. J. Bone Jt. Surg. Br. 2002, 84, 196–201. [Google Scholar] [CrossRef]
- Hiroshi, T.; Toru, S.; Yukikazu, O.; Mituo, M.; Yayoi, O.; Kakiuchi, T. Inflammatory cytokines in the herniated disc of the lumbar spine. Spine 1996, 21, 218–224. [Google Scholar]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Lee, C.C.; Avalos, A.M.; Ploegh, H.L. Accessory molecules for Toll-like receptors and their function. Nat. Rev. Immunol. 2012, 12, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhang, Z.; Lei, Z.; Lei, P. CD14: Biology and role in the pathogenesis of disease. Cytokine Growth Factor. Rev. 2019, 48, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Knezevic, N.N.; Mandalia, S.; Raasch, J.; Knezevic, I.; Candido, K.D. Treatment of chronic low back pain—New approaches on the horizon. J. Pain. Res. 2017, 10, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- AlGarni, N.; Grant, M.P.; Epure, L.M.; Salem, O.; Bokhari, R.; Antoniou, J.; Mwale, F. Short Link N Stimulates Intervertebral Disc Repair in a Novel Long-Term Organ Culture Model that Includes the Bony Vertebrae. Tissue Eng. Part A 2016, 22, 1252–1257. [Google Scholar] [CrossRef]
- Gawri, R.; Ouellet, J.; Önnerfjord, P.; Alkhatib, B.; Steffen, T.; Heinegård, D.; Roughley, P.; Antoniou, J.; Mwale, F.; Haglund, L. Link N is cleaved by human annulus fibrosus cells generating a fragment with retained biological activity. J. Orthop. Res. 2014, 32, 1189–1197. [Google Scholar] [CrossRef]
- Mwale, F.; Masuda, K.; Grant, M.P.; Epure, L.M.; Kato, K.; Miyazaki, S.; Cheng, K.; Yamada, J.; Bae, W.C.; Muehleman, C.; et al. Short Link N promotes disc repair in a rabbit model of disc degeneration. Arthritis Res. Ther. 2018, 20, 201. [Google Scholar] [CrossRef]
- Petit, A.; Yao, G.; Rowas, S.A.; Gawri, R.; Epure, L.; Antoniou, J.; Mwale, F. Effect of synthetic link N peptide on the expression of type I and type II collagens in human intervertebral disc cells. Tissue Eng. Part A 2011, 17, 899–904. [Google Scholar] [CrossRef]
- Liu, H.; McKenna, L.A.; Dean, M.F. The macromolecular characteristics of cartilage proteoglycans do not change when synthesis is up-regulated by link protein peptide. Biochim. Biophys. Acta 1999, 1428, 191–200. [Google Scholar] [CrossRef]
- Antoniou, J.; Wang, H.T.; Alaseem, A.M.; Haglund, L.; Roughley, P.J.; Mwale, F. The effect of Link N on differentiation of human bone marrow-derived mesenchymal stem cells. Arthritis Res. Ther. 2012, 14, R267. [Google Scholar] [CrossRef]
- Liu, H.; McKenna, L.A.; Dean, M.F. An N-terminal peptide from link protein can stimulate biosynthesis of collagen by human articular cartilage. Arch. Biochem. Biophys. 2000, 378, 116–122. [Google Scholar] [CrossRef]
- Mwale, F.; Demers, C.N.; Petit, A.; Roughley, P.; Poole, A.R.; Steffen, T.; Aebi, M.; Antoniou, J. A synthetic peptide of link protein stimulates the biosynthesis of collagens II, IX and proteoglycan by cells of the intervertebral disc. J. Cell Biochem. 2003, 88, 1202–1213. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Weitzmann, M.N.; Sangadala, S.; Hutton, W.C.; Yoon, S.T. Link protein N-terminal peptide binds to bone morphogenetic protein (BMP) type II receptor and drives matrix protein expression in rabbit intervertebral disc cells. J. Biol. Chem. 2013, 288, 28243–28253. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.Z.; Xu, Z.Q.; Han, B.Z.; Su, D.F.; Liu, C. NLRP3 inflammasome and its inhibitors: A review. Front. Pharmacol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Phillips, K.L.; Cullen, K.; Chiverton, N.; Michael, A.L.; Cole, A.A.; Breakwell, L.M.; Haddock, G.; Bunning, R.A.; Cross, A.K.; Le Maitre, C.L. Potential roles of cytokines and chemokines in human intervertebral disc degeneration: Interleukin-1 is a master regulator of catabolic processes. Osteoarthr. Cartil. 2015, 23, 1165–1177. [Google Scholar] [CrossRef]
- Purmessur, D.; Walter, B.A.; Roughley, P.J.; Laudier, D.M.; Hecht, A.C.; Iatridis, J. A role for TNFα in intervertebral disc degeneration: A non-recoverable catabolic shift. Biochem. Biophys. Res. Commun. 2013, 433, 151–156. [Google Scholar] [CrossRef]
- Kang, R.; Chen, R.; Zhang, Q.; Hou, W.; Wu, S.; Cao, L.; Huang, J.; Yu, Y.; Fan, X.G.; Yan, Z.; et al. HMGB1 in health and disease. Mol. Asp. Med. 2014, 40, 1–116. [Google Scholar]
- Tang, Y.L.; Jiang, J.H.; Wang, S.; Liu, Z.; Tang, X.Q.; Peng, J.; Yang, Y.Z.; Gu, H.F. TLR4/NF-κB Signaling Contributes to Chronic Unpredictable Mild Stress-Induced Atherosclerosis in ApoE-/- Mice. PLoS ONE 2015, 10, e0123685. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Jo, E.K.; Kim, J.K.; Shin, D.M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 2012, 28, 137–161. [Google Scholar] [CrossRef] [PubMed]
- Séguin, C.A.; Pilliar, R.M.; Roughley, P.J.; Kandel, R.A. Tumor necrosis factor-alpha modulates matrix production and catabolism in nucleus pulposus tissue. Spine 2005, 30, 1940–1948. [Google Scholar] [CrossRef]
- Zhao, W.; Ma, L.; Deng, D.; Zhang, T.; Han, L.; Xu, F.; Huang, S.; Ding, Y.; Chen, X. M2 macrophage polarization: A potential target in pain relief. Front. Immunol. 2023, 14, 1243149. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediators Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef]
- Opal, S.M.; DePalo, V.A. Anti-inflammatory cytokines. Chest 2000, 117, 1162–1172. [Google Scholar] [CrossRef]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef]
- Johnson, Z.I.; Schoepflin, Z.R.; Choi, H.; Shapiro, I.M.; Risbud, M.V. Disc in flames: Roles of TNF-α and IL-1β in intervertebral disc degeneration. Eur. Cell Mater. 2015, 30, 104–116; discussion 116–117. [Google Scholar] [CrossRef] [PubMed]
- Gorth, D.J.; Shapiro, I.M.; Risbud, M.V. A New Understanding of the Role of IL-1 in Age-Related Intervertebral Disc Degeneration in a Murine Model. J. Bone Min. Res. 2019, 34, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- Hodges, P.W.; James, G.; Blomster, L.; Hall, L.; Schmid, A.; Shu, C.; Little, C.; Melrose, J. Multifidus Muscle Changes After Back Injury Are Characterized by Structural Remodeling of Muscle, Adipose and Connective Tissue, but Not Muscle Atrophy: Molecular and Morphological Evidence. Spine 2015, 40, 1057–1071. [Google Scholar] [CrossRef]
- James, G.; Sluka, K.A.; Blomster, L.; Hall, L.; Schmid, A.B.; Shu, C.C.; Little, C.B.; Melrose, J.; Hodges, P.W. Macrophage polarization contributes to local inflammation and structural change in the multifidus muscle after intervertebral disc injury. Eur. Spine J. 2018, 27, 1744–1756. [Google Scholar] [CrossRef]
- James, G.; Stecco, C.; Blomster, L.; Hall, L.; Schmid, A.B.; Shu, C.C.; Little, C.B.; Melrose, J.; Hodges, P.W. Muscle spindles of the multifidus muscle undergo structural change after intervertebral disc degeneration. Eur. Spine J. 2022, 31, 1879–1888. [Google Scholar] [CrossRef]
- Pavlicevic, M.; Marmiroli, N.; Maestri, E. Immunomodulatory peptides—A promising source for novel functional food production and drug discovery. Peptides 2022, 148, 170696. [Google Scholar] [CrossRef]
- La Manna, S.; Di Natale, C.; Florio, D.; Marasco, D. Peptides as Therapeutic Agents for Inflammatory-Related Diseases. Int. J. Mol. Sci. 2018, 19, 2714. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Otero, M. Inflammation in osteoarthritis. Curr. Opin. Rheumatol. 2011, 23, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Sokolove, J.; Lepus, C.M. Role of inflammation in the pathogenesis of osteoarthritis: Latest findings and interpretations. Ther. Adv. Musculoskelet. Dis. 2013, 5, 77–94. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef]
- So, A.K.; Martinon, F. Inflammation in gout: Mechanisms and therapeutic targets. Nat. Rev. Rheumatol. 2017, 13, 639–647. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S. Rheumatoid arthritis. Nat. Rev. Dis. Primers 2018, 4, 18001. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human Genes | Primer Sequence |
|---|---|
| h-IL1β | F: 5′-ACCTATCTTCTTCGACACATG-3′ R: 5′-ACCACTTGTTGCTCCATATCC-3′ |
| h-GAPDH | F: 5′-TGTAAAACGACGGCCAGT-3′ R: 5′-CAGGAAACAGCTATGACC-3′ |
| h-NLRP3 | F: 5′-GGGTCTCCTCTCTCATCCA-3′ R: 5′-AGCCTCCTGAACCAGGTCTTA-3′ |
| h-PYCARD | F: 5′-ACATCCAGCAGGCTAGAAG-3′ R: 5′-AAGATGCGGAAGCTCTICAGTT-3′ |
| h-Caspase-1 | F: 5′-AAAGAAAGGTCCAATAGCCAGTTT-3′ R: 5′-CTTCTTCTGGTCAGTGCAGAC-3′ |
| Murine Genes | Primer Sequence |
|---|---|
| m-IL1β | F: 5′-TGGACCTTCCAGGATGAGGACA-3′ R: 5′-GTTCATCTCGGAGCCTGTAGTG-3′ |
| m-ARG1 | F: 5′-TGTAATGAAAGACGGCACACC-3′ R: 5′-TCTTCTTTGGGTATTGCTTGG-3′ |
| m-CD86 | F: 5′-ACGTATTGGAAGGAGATTACAGCT-3′ R: 5′-TCTGTCAGCGTTACTATCCCGC-3′ |
| m-CD80 | F: 5′-CCTCAAGTTTCCATGTCCAAGGC-3′ R: 5′-GAGGAGAGTTGTAACGGCAAGG-3′ |
| m-IL10 | F: 5′-CGGGAAGACAATAACTGCACCC-3′ R: 5′-CGGTTAGCAGTATGTTGTCCAGC-3′ |
| m-TNFA | F: 5′-ATGAGCACAGAAAGCATGATCCG-3′ R: 5′-CCTTGTCCCTTGAAGAGAACCTG-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alad, M.; Grant, M.P.; Epure, L.M.; Shih, S.Y.; Merle, G.; Im, H.-J.; Antoniou, J.; Mwale, F. Short Link N Modulates Inflammasome Activity in Intervertebral Discs Through Interaction with CD14. Biomolecules 2024, 14, 1312. https://doi.org/10.3390/biom14101312
Alad M, Grant MP, Epure LM, Shih SY, Merle G, Im H-J, Antoniou J, Mwale F. Short Link N Modulates Inflammasome Activity in Intervertebral Discs Through Interaction with CD14. Biomolecules. 2024; 14(10):1312. https://doi.org/10.3390/biom14101312
Chicago/Turabian StyleAlad, Muskan, Michael P. Grant, Laura M. Epure, Sunny Y. Shih, Geraldine Merle, Hee-Jeong Im, John Antoniou, and Fackson Mwale. 2024. "Short Link N Modulates Inflammasome Activity in Intervertebral Discs Through Interaction with CD14" Biomolecules 14, no. 10: 1312. https://doi.org/10.3390/biom14101312
APA StyleAlad, M., Grant, M. P., Epure, L. M., Shih, S. Y., Merle, G., Im, H.-J., Antoniou, J., & Mwale, F. (2024). Short Link N Modulates Inflammasome Activity in Intervertebral Discs Through Interaction with CD14. Biomolecules, 14(10), 1312. https://doi.org/10.3390/biom14101312