
Unveiling the Complexities of Hereditary Angioedema


 ,
,  and
and 
Abstract
1. Introduction
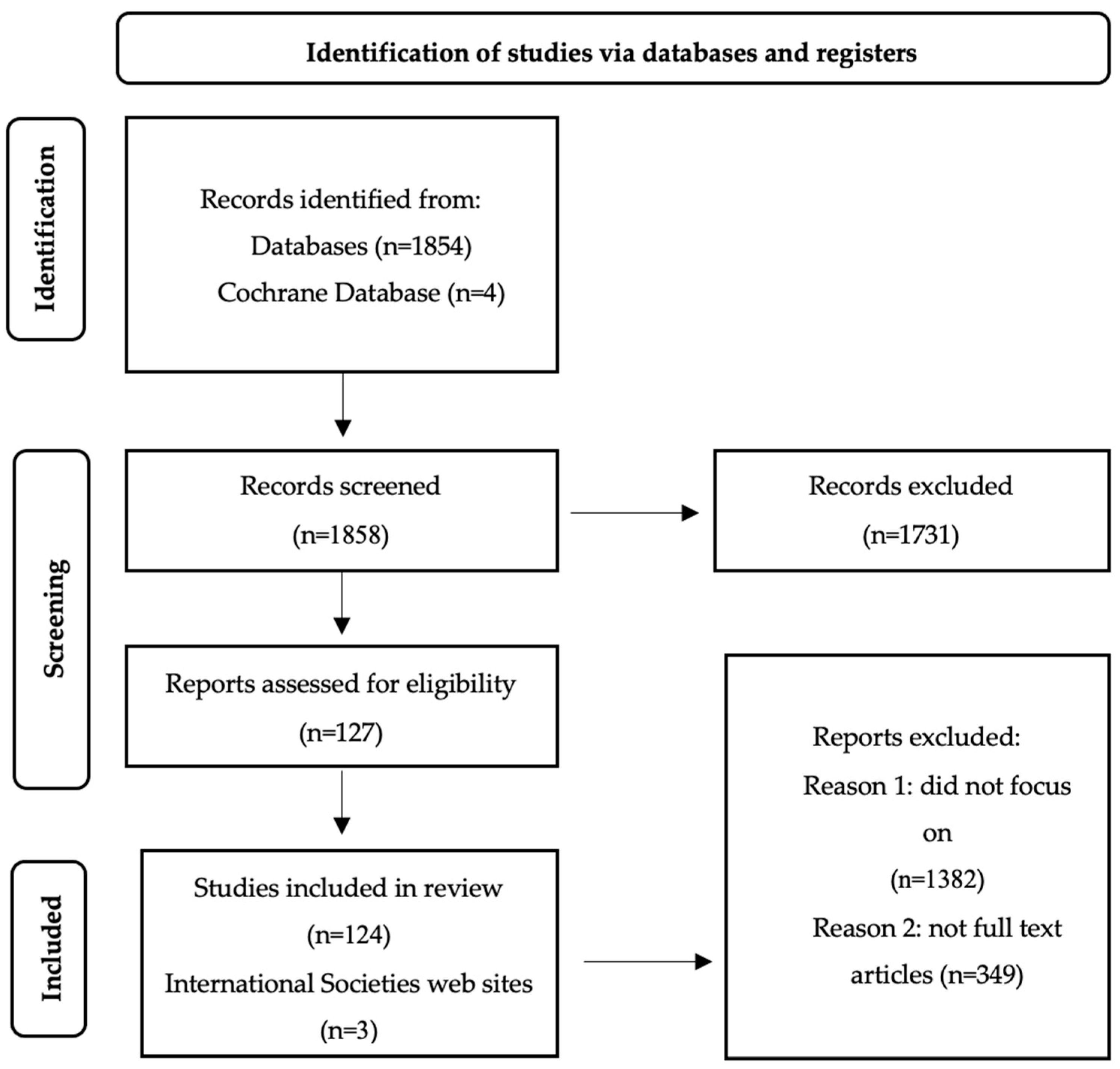
2. Materials and Methods
3. Discussions and Results
3.1. Epidemiological Data
3.2. Etiopathogenesis
3.3. Signs and Symptoms
3.4. Diagnosis
3.4.1. Laboratory Testing
3.4.2. Imaging Studies
3.4.3. Histology
3.5. Biomarkers
3.6. Differential Diagnosis
3.7. Prognosis
3.8. Standard Treatment
3.9. Prophylaxis
3.9.1. Short-Term Prophylaxis
3.9.2. Long-Term Prophylaxis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sinnathamby, E.S.; Issa, P.P.; Roberts, L.; Norwood, H.; Malone, K.; Vemulapalli, H.; Ahmadzadeh, S.; Cornett, E.M.; Shekoohi, S.; Kaye, A.D. Hereditary Angioedema: Diagnosis, Clinical Implications, and Pathophysiology. Adv Ther. 2023, 40, 814–827. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wilkerson, R.G.; Moellman, J.J. Hereditary Angioedema. Emerg. Med. Clin. 2022, 40, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Andrejević, S.; Korošec, P.; Šilar, M.; Košnik, M.; Mijanović, R.; Bonači-Nikolić, B.; Rijavec, M. Hereditary angioedema due to C1 inhibitor deficiency in Serbia: Two novel mutations and evidence of genotype–phenotype association. PLoS ONE 2015, 10, e0142174. [Google Scholar] [CrossRef] [PubMed]
- Nasr, I.H.; Manson, A.L.; Al Wahshi, H.A.; Longhurst, H.J. Optimizing hereditary angioedema management through tailored treatment approaches. Expert. Rev. Clin. Immunol. 2016, 12, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Longhurst, H.J.; Bork, K. Hereditary angioedema: An update on causes, manifestations and treatment. Br. J. Hosp. Med. (Lond.) 2019, 80, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Bova, M.; De Feo, G.; Parente, R.; De Pasquale, T.; Gravante, C.; Pucci, S.; Nettis, E.; Triggiani, M. Hereditary and Acquired Angioedema: Heterogeneity of Pathogenesis and Clinical Phenotypes. Int. Arch. Allergy Immunol. 2018, 175, 126–135. [Google Scholar] [CrossRef]
- Santacroce, R.; D’Andrea, G.; Maffione, A.B.; Margaglione, M.; d’Apolito, M. The Genetics of Hereditary Angioedema: A Review. J. Clin. Med. 2021, 10, 2023. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kaplan, A.P.; Joseph, K. Pathogenic mechanisms of bradykinin mediated diseases: Dysregulation of an innate inflammatory pathway. Adv. Immunol. 2014, 121, 41–89. [Google Scholar]
- Maurer, M.; Bader, M.; Bas, M.; Bossi, F.; Cicardi, M.; Cugno, M.; Howarth, P.; Kaplan, A.; Kojda, G.; Leeb-Lundberg, F.; et al. New topics in bradykinin research. Allergy 2011, 66, 1397–1406. [Google Scholar] [CrossRef]
- Porebski, G.; Kwitniewski, M.; Reshef, A. Biomarkers in Hereditary Angioedema. Clin. Rev. Allergy Immunol. 2021, 60, 404–415. [Google Scholar] [CrossRef]
- Memon, R.J.; Tiwari, V. Angioedema. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK538489 (accessed on 8 August 2022).
- Moellman, J.J.; Bernstein, J.A.; Lindsell, C.; Banerji, A.; Busse, P.J.; Camargo, C.A., Jr.; Collins, S.P.; Craig, T.J.; Lumry, W.R.; Nowak, R.; et al. A consensus parameter for the evaluation and management of angioedema in the emergency department. Acad. Emerg. Med. 2014, 21, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Zuraw, B.L. Hereditary angioedema. N. Engl. J. Med. 2008, 359, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Gábos, G.; Dobru, D.; Mihály, E.; Bara, N.; Dumitrache, C.; Popa, R.; Nădășan, V.; Moldovan, D. Recurrent ascites: A need to evaluate for hereditary angio-oedema. Lancet 2017, 390, 2119–2120. [Google Scholar] [CrossRef] [PubMed]
- Keeney, S.; Halalau, A. Anchoring bias in a case of recurrent abdominal pain. BMJ Case Rep. 2017, 2017, 2017221027. [Google Scholar] [CrossRef] [PubMed]
- Elenburg, S.N.; Assa’ad, A.H.; Bernstein, J.A.; Nanda, M. Clinical features of pediatric hereditary angioedema. J. Allergy Clin. Immunol. 2014, 133, AB32. [Google Scholar] [CrossRef]
- Sarkar, A.; Nwagwu, C.; Craig, T. Hereditary Angioedema: A Disease Often Misdiagnosed and Mistreated. Primary Care 2023, 50, 295–303. [Google Scholar] [CrossRef]
- Maurer, M.; Magerl, M.; Betschel, S.; Aberer, W.; Ansotegui, I.J.; Aygören-Pürsün, E.; Banerji, A.; Bara, N.A.; Boccon-Gibod, I.; Bork, K.; et al. The international WAO/EAACI guideline for the management of hereditary angioedema-The 2021 revision and update. Allergy 2022, 77, 1961–1990. [Google Scholar] [CrossRef]
- Ghazi, A.; Grant, J.A. Hereditary angioedema: Epidemiology, management, and role of icatibant. Biologics 2013, 7, 103–113. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cicardi, M.; Agostoni, A. Hereditary angioedema. N. Engl. J. Med. 1996, 334, 1666–1667. [Google Scholar] [CrossRef]
- Quincke, H. Concerning the acute localized edema of the skin. Monatsh Prakt Derm 1882, 1, 129–131. [Google Scholar]
- Osler, W. Hereditary angioneurotic edema. Am. J. Med. Sci. 1888, 95, 362–367. [Google Scholar] [CrossRef]
- Donaldson, V.H.; Evans, R.R. A biochemical abnormality in hereditary angioneurotic edema: Absence of serum inhibitor of C’ 1-esterase. Am. J. Med. 1963, 35, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Nzeako, U.C.; Frigas, E.; Tremaine, W.J. Hereditary angioedema: A broad review for clinicians. Arch. Intern. Med. 2001, 161, 2417–2429. [Google Scholar] [CrossRef] [PubMed]
- Dreskin, S. Urticaria and angioedema. In Cecil Medicine, 24th ed.; Goldman, L., Schafer, A.I., Eds.; Saunders Elsevier: Philadelphia, PA, USA, 2011; Chap 260. [Google Scholar]
- Talavera, A.; Larraona, J.L.; Ramos, J.L.; López, T.; Maraver, A.; Arias, J.; Barrios, A. Hereditary angioedema: An infrequent cause of abdominal pain with ascites. Am. J. Gastroenterol. 1995, 90, 471–474. [Google Scholar] [PubMed]
- Agostoni, A.; Aygören-Pürsün, E.; Binkley, K.E.; Blanch, A.; Bork, K.; Bouillet, L.; Bucher, C.; Castaldo, A.J.; Cicardi, M.; Davis, A.E.; et al. Hereditary and acquired angioedema: Problems and progress: Proceedings of the third C1 esterase inhibitor deficiency workshop and beyond. J. Allergy Clin. Immunol. 2004, 114 (Suppl. S3), S51–S131. [Google Scholar] [CrossRef]
- Frank, M.M.; Gelfand, J.A.; Atkinson, J.P. Hereditary angioedema: The clinical syndrome and its management. Ann. Intern. Med. 1976, 84, 580–593. [Google Scholar] [CrossRef]
- Kesh, S.; Bernstein, J.A. Isolated angioedema: A review of classification and update on management. Ann. Allergy Asthma Immunol. 2022, 129, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Longhurst, H.J.; Zanichelli, A.; Caballero, T.; Bouillet, L.; Aberer, W.; Maurer, M.; Fain, O.; Fabien, V.; Andresen, I.; IOS Study Group. Comparing acquired angioedema with hereditary angioedema (types I/II): Findings from the Icatibant Outcome Survey. Clin. Exp. Immunol. 2017, 188, 148–153. [Google Scholar] [CrossRef]
- Levi, M.; Cohn, D.M. The Role of Complement in Hereditary Angioedema. Transfus. Med. Rev. 2019, 33, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Caccia, S.; Suffritti, C.; Cicardi, M. Pathophysiology of hereditary angioedema. Pediatr. Allergy Immunol. Pulmonol. 2014, 27, 159–163. [Google Scholar] [CrossRef]
- Betschel, S.D.; Banerji, A.; Busse, P.J.; Cohn, D.M.; Magerl, M. Hereditary Angioedema: A Review of the Current and Evolving Treatment Landscape. The journal of allergy and clinical immunology. In Practice 2023, 11, 2315–2325. [Google Scholar] [CrossRef] [PubMed]
- Bork, K.; Wulff, K.; Steinmüller-Magin, L.; Braenne, I.; Staubach-Renz, P.; Witzke, G.; Hardt, J. Hereditary angioedema with a mutation in the plasminogen gene. Allergy 2018, 73, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Fijen, L.M.; Bork, K.; Cohn, D.M. Current and Prospective Targets of Pharmacologic Treatment of Hereditary Angioedema Types 1 and 2. Clinic. Rev. Allerg. Immunol. 2021, 61, 66–76. [Google Scholar] [CrossRef]
- Bernstein, J.A. Severity of hereditary angioedema, prevalence, and diagnostic considerations. Am. J. Manag. Care 2018, 24 (Suppl. S14), S292–S298. [Google Scholar]
- Henao, M.P.; Kraschnewski, J.L.; Kelbel, T.; Craig, T.J. Diagnosis and screening of patients with hereditary angioedema in primary care. Ther. Clin. Risk. Manag. 2016, 12, 701. [Google Scholar] [CrossRef]
- Iwanami, K.; Okano, T.; Ohara, O.; Morio, T. Recurrent acute abdomen as the main manifestation of hereditary angioedema. Intern. Med. 2019, 58, 213–216. Available online: http://www.ncbi.nlm.nih.gov/pubmed/30146609 (accessed on 28 July 2024). [CrossRef]
- Bork, K.; Hardt, J.; Witzke, G. Fatal laryngeal attacks and mortality in hereditary angioedema due to C1-INH deficiency. J. Allergy Clin. Immunol. 2012, 130, 692–697. [Google Scholar] [CrossRef]
- Jindal, A.K.; Bishnoi, A.; Dogra, S. Hereditary angioedema: Diagnostic algorithm and current treatment concepts. Indian Dermatol Online J. 2021, 12, 796. [Google Scholar] [CrossRef]
- Jacobs, J.; Neeno, T. The importance of recognizing and managing a rare form of angioedema: Hereditary angioedema due to C1-inhibitor deficiency. Postgrad. Med. 2021, 133, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Caballero, T.; Baeza, M.L.; Cabañas, R.; Campos, A.; Cimbollek, S.; Gómez-Traseira, C.; González-Quevedo, T.; Guilarte, M.; Jurado-Palomo, G.J.; Larco, J.I.; et al. Consensus statement on the diagnosis, management, and treatment of angioedema mediated by bradykinin. Part I. Classification, epidemiology, pathophysiology, genetics, clinical symptoms, and diagnosis. J. Investig. Allergol. Clin. Immunol. 2011, 21, 333–347, quiz follow 347. [Google Scholar]
- Cicardi, M.; Zanichelli, A. Diagnosing angioedema. Immunol. Allergy Clin. N. Am. 2013, 33, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Nowicki, R.J.; Grubska-Suchanek, E.; Porêbski, G.; Kowalski, M.L.; Jahnz-Różyk, K.; Matuszewski, T.; Rudnicka, L.; Kulus, M.; Barañska-Rybak, W.; Czajkowski, R.; et al. Angioedema. Interdisciplinary diagnostic and therapeutic recommendations of the Polish Dermatological Society (PTD) and Polish Society of Allergology (PTA). Postepy Dermatol. Alergol. 2020, 37, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Karim, Y.; Griffiths, H.; Deacock, S. Normal complement C4 values do not exclude hereditary angioedema. J. Clin. Pathol. 2004, 57, 213–214. [Google Scholar] [CrossRef] [PubMed]
- Tarzi, M.D.; Hickey, A.; Forster, T.; Mohammadi, M.; Longhurst, H.J. An evaluation of tests used for the diagnosis and monitoring of C1 inhibitor deficiency: Normal serum C4 does not exclude hereditary angio-oedema. Clin. Exp. Immunol. 2007, 149, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Wagenaar-Bos, I.G.; Drouet, C.; Aygoren-Pursun, E.; Bork, K.; Bucher, C.; Bygum, A.; Farkas, H.; Fust, G.; Gregorek, H.; Hack, C.E.; et al. Functional C1-inhibitor diagnostics in hereditary angioedema: Assay evaluation and recommendations. J. Immunol. Methods 2008, 338, 14–20. [Google Scholar] [CrossRef]
- Aabom, A.; Bygum, A.; Koch, C. Complement factor C4 activation in patients with hereditary angioedema. Clin. Biochem. 2017, 50, 816–821. [Google Scholar] [CrossRef]
- Jindal, A.K.; Reshef, A.; Longhurst, H.; GEHM workgroup (Global Equity in HAE Management). Mitigating disparity in health-care resources between countries for management of hereditary angioedema. Clin. Rev. Allergy Immunol. 2021, 61, 84–97. [Google Scholar] [CrossRef]
- Johnston, D.T. Diagnosis and management of hereditary angioedema. J. Osteopath. Med. 2011, 111, 28–36. [Google Scholar]
- Germenis, A.E.; Margaglione, M.; Pesquero, J.B.; Farkas, H.; Cichon, S.; Csuka, D.; López Lera, A.; Rijavec, M.; Jolles, S.; Szilagyi, A.; et al. International consensus on the use of genetics in the management of hereditary angioedema. J. Allergy Clin. Immunol. Pract. 2020, 8, 901–911. [Google Scholar] [CrossRef]
- Veronez, C.L.; Aabom, A.; Martin, R.P.; Filippelli-Silva, R.; Gonçalves, R.F.; Nicolicht, P.; Mendes, A.R.; Da Silva, J.; Guilarte, M.; Grumach, A.S.; et al. Genetic Variation of Kallikrein-Kinin System and Related Genes in Patients with Hereditary Angioedema. Front. Med. 2019, 6, 6–28. [Google Scholar] [CrossRef]
- Csuka, D.; Füst, G.; Farkas, H.; Varga, L. Parameters of the classical complement pathway predict disease severity in hereditary angioedema. Clin. Immunol. 2011, 139, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Germenis, A.E.; Cicardi, M. Driving towards precision medicine for angioedema without wheals. J. Autoimmun. 2019, 104, 102312. [Google Scholar] [CrossRef] [PubMed]
- Cugno, M.; Nuijens, J.; Hack, E.; Eerenberg, A.; Frangi, D.; Agostoni, A.; Cicardi, M. Plasma levels of C1− inhibitor complexes and cleaved C1− inhibitor in patients with hereditary angioneurotic edema. J. Clin. Investig. 1990, 85, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Cugno, M.; Hack, C.E.; de Boer, J.P.; Eerenberg, A.J.; Agostoni, A.; Cicardi, M. Generation of plasmin during acute attacks of hereditary angioedema. J. Lab. Clin. Med. 1993, 121, 38–43. [Google Scholar]
- Cicardi, M.; Bergamaschini, L.; Cugno, M.; Hack, E.; Agostoni, G.; Agostoni, A. Long-term treatment of hereditary angioedema with attenuated androgens: A survey of a 13-year experience. J. Allergy Clin. Immunol. 1991, 87, 768–773. [Google Scholar] [CrossRef]
- Li, H.H.; Busse, P.; Lumry, W.R.; Frazer-Abel, A.; Levy, H.; Steele, T.; Dayno, J.; Riedl, M. Comparison of chromogenic and ELISA functional C1 inhibitor tests in diagnosing hereditary angioedema. J. Allergy Clin. Immunol. Pract. 2015, 3, 200–205. [Google Scholar] [CrossRef]
- Gompels, M.M.; Lock, R.J.; Morgan, J.E.; Osborne, J.; Brown, A.; Virgo, P.F. A multicentre evaluation of the diagnostic efficiency of serological investigations for C1 inhibitor deficiency. J. Clin. Pathol. 2002, 55, 145–147. [Google Scholar] [CrossRef]
- Kelemen, Z.; Moldovan, D.; Mihály, E.; Visy, B.; Széplaki, G.; Csuka, D.; Füst, G.; Farkas, H.; Varga, L. Baseline level of functional C1-inhibitor correlates with disease severity scores in hereditary angioedema. Clin. Immunol. 2010, 134, 354–358. [Google Scholar] [CrossRef]
- Longhurst, H.; Cicardi, M.; Craig, T.; Bork, K.; Grattan, C.; Baker, J.; Li, H.H.; Reshef, A.; Bonner, J.; Bernstein, J.A.; et al. Prevention of hereditary angioedema attacks with a subcutaneous C1 inhibitor. N. Engl. J. Med. 2017, 376, 1131–1140. [Google Scholar] [CrossRef]
- Hack, C.E.; Relan, A.; van Amersfoort, E.S.; Cicardi, M. Target levels of functional C1-inhibitor in hereditary angioedema. Allergy 2012, 67, 123–130. [Google Scholar] [CrossRef]
- Kaplan, A.P.; Pawaskar, D.; Chiao, J. C1 inhibitor activity and angioedema attacks in patients with hereditary angioedema. J. Allergy Clin. Immunol. Pract. 2020, 8, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Betschel, S.; Badiou, J.; Binkley, K.; Borici-Mazi, R.; Hébert, J.; Kanani, A.; Keith, P.; Lacuesta, G.; Waserman, S.; Yang, B.; et al. The International/Canadian Hereditary Angioedema Guideline. Allergy Asthma Clin. Immunol. 2019, 15, 72. [Google Scholar] [CrossRef] [PubMed]
- Varga, L.; Széplaki, G.; Visy, B.; Füst, G.; Harmat, G.; Miklós, K.; Németh, J.; Cervenak, L.; Karádi, I.; Farkas, H. C1-inhibitor (C1-INH) autoantibodies in hereditary angioedema. Strong correlation with the severity of disease in C1-INH concentrate naïve patients. Mol. Immunol. 2007, 44, 1454–1460. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.B.; Csuka, D.; Munthe-Fog, L.; Varga, L.; Farkas, H.; Hansen, K.M.; Koch, C.; Skjødt, K.; Garred, P.; Skjoedt, M.O. The levels of the lectin pathway serine protease MASP-1 and its complex formation with C1 inhibitor are linked to the severity of hereditary angioedema. J. Immunol. 2015, 195, 3596–3604. [Google Scholar] [CrossRef] [PubMed]
- Csuka, D.; Munthe-Fog, L.; Hein, E.; Zotter, Z.; Prohászka, Z.; Farkas, H.; Varga, L.; Garred, P. Activation of the ficolin-lectin pathway during attacks of hereditary angioedema. J. Allergy Clin. Immunol. 2014, 134, 1388–1393.e1. [Google Scholar] [CrossRef]
- Nussberger, J.; Cugno, M.; Amstutz, C.; Cicardi, M.; Pellacani, A.; Agostoni, A. Plasma bradykinin in angio-oedema. Lancet 1998, 351, 1693–1697. [Google Scholar] [CrossRef]
- Nussberger, J.; Cugno, M.; Cicardi, M.; Agostoni, A. Local bradykinin generation in hereditary angioedema. J. Allergy Clin. Immunol. 1999, 104, 1321–1322. [Google Scholar] [CrossRef]
- Suffritti, C.; Zanichelli, A.; Maggioni, L.; Bonanni, E.; Cugno, M.; Cicardi, M. High-molecular-weight kininogen cleavage correlates with disease states in the bradykinin-mediated angioedema due to hereditary C1-inhibitor deficiency. Clin. Exp. Allergy 2014, 44, 1503–1514. [Google Scholar] [CrossRef]
- Cugno, M.; Cicardi, M.; Coppola, R.; Agostoni, A. Activation of the factor XII and cleavage of high molecular weight kininogen during acute attacks in hereditary and acquired C1-inhibitor deficiencies. Immunopharmacology 1996, 33, 361–364. [Google Scholar] [CrossRef]
- Joseph, K.; Tuscano, T.B.; Kaplan, A.P. Studies of the mechanisms of bradykinin generation in hereditary angioedema plasma. Ann. Allergy Asthma Immunol. 2008, 101, 279–286. [Google Scholar] [CrossRef]
- Csuka, D.; Veszeli, N.; Imreh, É.; Zotter, Z.; Skopál, J.; Prohászka, Z.; Varga, L.; Farkas, H. Comprehensive study into the activation of the plasma enzyme systems during attacks of hereditary angioedema due to C1-inhibitor deficiency. Orphanet. J. Rare Dis. 2015, 10, 132. [Google Scholar] [CrossRef] [PubMed]
- Dessart, P.; Defendi, F.; Humeau, H.; Nicolie, B.; Sarre, M.E.; Charignon, D.; Ponard, D.; Cichon, S.; Drouet, C.; Martin, L. Distinct conditions support a novel classification for bradykinin-mediated angio-oedema. Dermatology 2015, 230, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Charignon, D.; Ghannam, A.; Defendi, F.; Ponard, D.; Monnier, N.; López Trascasa, M.; Launay, D.; Caballero, T.; Djenouhat, K.; Fain, O.; et al. Hereditary angioedema with F12 mutation: Factors modifying the clinical phenotype. Allergy 2014, 69, 1659–1665. [Google Scholar] [CrossRef] [PubMed]
- Defendi, F.; Charignon, D.; Ghannam, A.; Baroso, R.; Csopaki, F.; Allegret-Cadet, M.; Ponard, D.; Favier, B.; Cichon, S.; Nicolie, B.; et al. Enzymatic assays for the diagnosis of bradykinin-dependent angioedema. PLoS ONE 2013, 8, e70140. [Google Scholar] [CrossRef]
- Cugno, M.; Zanichelli, A.; Bellatorre, A.G.; Griffini, S.; Cicardi, M. Plasma biomarkers of acute attacks in patients with angioedema due to C1-inhibitor deficiency. Allergy 2009, 64, 254–257. [Google Scholar] [CrossRef]
- Reshef, A.; Zanichelli, A.; Longhurst, H.; Relan, A.; Hack, C.E. Elevated D-dimers in attacks of hereditary angioedema are not associated with increased thrombotic risk. Allergy 2015, 70, 506–513. [Google Scholar] [CrossRef]
- Bouillet, L.; Mannic, T.; Arboleas, M.; Subileau, M.; Massot, C.; Drouet, C.; Huber, P.; Vilgrain, I. Hereditary angioedema: Key role for kallikrein and bradykinin in vascular endothelial-cadherin cleavage and edema formation. J. Allergy Clin Immunol. 2011, 128, 232–234. [Google Scholar] [CrossRef]
- Kajdácsi, E.; Jani, P.K.; Csuka, D.; Varga, L.Á.; Prohászka, Z.; Farkas, H.; Cervenak, L. Endothelial cell activation during edematous attacks of hereditary angioedema types I and II. J. Allergy Clin. Immunol. 2014, 133, 1686–1691. [Google Scholar] [CrossRef]
- Czúcz, J.; Schaffer, G.; Csuka, D.; Walentin, S.; Kunde, J.; Prohászka, Z.; Farkas, H.; Cervenak, L. Endothelial cell function in patients with hereditary angioedema: Elevated soluble E-selectin level during inter-attack periods. J. Clin. Immunol. 2012, 32, 61–69. [Google Scholar] [CrossRef]
- Kajdácsi, E.; Jani, P.K.; Csuka, D.; Varga, L.; Prohászka, Z.; Farkas, H.; Cervenak, L. Novel vasoregulatory aspects of hereditary angioedema: The role of arginine vasopressin, adrenomedullin and endothelin-1. J. Clin. Immunol. 2016, 36, 160–170. [Google Scholar] [CrossRef]
- Kajdácsi, E.; Varga, L.; Prohászka, Z.; Farkas, H.; Cervenak, L. Atrial natriuretic peptide as a novel biomarker of hereditary angioedema. Clin. Immunol. 2016, 165, 45–46. [Google Scholar] [CrossRef] [PubMed]
- Demirturk, M.; Akpinar, T.S.; Kose, M.; Gelincik, A.; Colakoğlu, B.; Buyukozturk, S. Endocan: A novel marker of endothelial dysfunction in C1-inhibitor-deficient hereditary angioedema. Int. Arch. Allergy Immunol. 2017, 174, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, S.; Bova, M.; Suffritti, C.; Borriello, F.; Zanichelli, A.; Petraroli, A.; Varricchi, G.; Triggiani, M.; Cicardi, M.; Marone, G. Elevated plasma levels of vascular permeability factors in C1 inhibitor-deficient hereditary angioedema. Allergy 2016, 71, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, S.; Ferrara, A.L.; Bova, M.; Borriello, F.; Suffritti, C.; Veszeli, N.; Petraroli, A.; Galdiero, M.R.; Varricchi, G.; Granata, F.; et al. Secreted phospholipases A2 in hereditary angioedema with C1-inhibitor deficiency. Front. Immunol. 2018, 9, 1721. [Google Scholar] [CrossRef]
- Bova, M.; Suffritti, C.; Bafunno, V.; Loffredo, S.; Cordisco, G.; Del Giacco, S.; De Pasquale, T.M.A.; Firinu, D.; Margaglione, M.; Montinaro, V.; et al. Impaired control of the contact system in hereditary angioedema with normal C1-inhibitor. Allergy 2020, 75, 1394–1403. [Google Scholar] [CrossRef]
- Ferrara, A.L.; Bova, M.; Petraroli, A.; Veszeli, N.; Galdiero, M.R.; Braile, M.; Marone, G.; Cristinziano, L.; Marcella, S.; Modestino, L.; et al. Hereditary angioedema attack: What happens to vasoactive mediators? Int. Immunopharmacol. 2020, 78, 106079. [Google Scholar] [CrossRef]
- Márkus, B.; Veszeli, N.; Temesszentandrási, G.; Farkas, H.; Kalabay, L. Serum fetuin-A, tumor necrosis factor alpha and C-reactive protein concentrations in patients with hereditary angioedema with C1-inhibitor deficiency. Orphanet. J. Rare Dis. 2019, 14, 67. [Google Scholar] [CrossRef]
- Veszeli, N.; Csuka, D.; Zotter, Z.; Imreh, É.; Józsi, M.; Benedek, S.; Varga, L.; Farkas, H. Neutrophil activation during attacks in patients with hereditary angioedema due to C1-inhibitor deficiency. Orphanet. J. Rare Dis. 2015, 10, 156. [Google Scholar] [CrossRef]
- Arcoleo, F.; Salemi, M.; la Porta, A.; Selvaggio, V.; Mandalà, V.; Muggeo, V.; Misiano, G.; Milano, S.; Romano, G.C.; Cillari, E. Upregulation of cytokines and IL-17 in patients with hereditary angioedema. Clin. Chem. Lab Med. 2014, 52, e91–e93. [Google Scholar] [CrossRef]
- Salemi, M.; Mandalà, V.; Muggeo, V.; Misiano, G.; Milano, S.; Colonna-Romano, G.; Arcoleo, F.; Cillari, E. Growth factors and IL-17 in hereditary angioedema. Clin. Exp. Med. 2016, 16, 213–218. [Google Scholar] [CrossRef]
- Hofman, Z.L.M.; Relan, A.; Hack, C.E. C-reactive protein levels in hereditary angioedema. Clin. Exp. Immunol. 2014, 177, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration, HHS. International Conference on Harmonisation; Guidance on E15 Pharmacogenomics Definitions and Sample Coding; Availability. Notice. Fed. Regist. 2008, 73, 19074–19076. [Google Scholar]
- Demirtürk, M.; Gelincik, A.; Çinar, S.; Kilercik, M.; Onay-Ucar, E.; Çolakoğlu, B.; Arda, N.; Büyüköztürk, S.; Deniz, G. Increased eNOS levels in hereditary angioedema. Int. Immunopharmacol. 2014, 20, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Germenis, A.E.; Speletas, M. Genetics of hereditary angioedema revisited. Clin. Rev. Allergy Immunol. 2016, 51, 170–182. [Google Scholar] [CrossRef]
- Beard, N.; Frese, M.; Smertina, E.; Mere, P.; Katelaris, C.; Mills, K. Interventions for the long-term prevention of hereditary angioedema attacks. Cochrane Database Syst. Rev. 2022, 11, CD013403. [Google Scholar] [CrossRef]
- Spath, P.J.; Wuthrich, B.; Butler, R. Quantification of C1-inhibitor functional activities by immunodiffusion assay in plasma of patients with hereditary angioedema—evidence of a functionally critical level of C1- inhibitor concentration. Complement 1984, 1, 147–159. [Google Scholar] [CrossRef]
- Cichon, S.; Martin, L.; Hennies, H.C.; Müller, F.; Van Driessche, K.; Karpushova, A.; Stevens, W.; Colombo, R.; Renné, T.; Drouet, C.; et al. Increased activity of coagulation factor XII (Hageman Factor) causes hereditary angioedema type III. Am. J. Hum. Genet. 2006, 79, 1098–1104. [Google Scholar] [CrossRef]
- Bork, K.; Kleist, R.; Hardt, J.; Witzke, G. Kallikrein-kinin system and fibrinolysis in hereditary angioedema due to factor XII gene mutation Thr309Lys. Blood Coagul. Fibrinolysis 2009, 20, 325–332. [Google Scholar] [CrossRef]
- Konings, J.; Cugno, M.; Suffritti, C.; Ten Cate, H.; Cicardi, M.; Govers-Riemslag, J.W. Ongoing contact activation in patients with hereditary angioedema. PLoS ONE 2013, 8, e74043. [Google Scholar] [CrossRef]
- Marlu, R.; Deroux, A.; Du-Thanh, A.; Boccon-Gibod, I.; Launay, D.; Bouillet, L. Normal PAI-2 level in French FXII-HAE patients. J. Allergy Clin. Immunol. 2017, 139, 1719–1720. [Google Scholar] [CrossRef]
- Bas, M.; Storck, K.; Strassen, U. Potential biomarkers for the diagnosis of angiotensin-converting enzyme inhibitor-induced angioedema. ORL J. Otorhinolaryngol. Relat. Spec. 2017, 79, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Caballero, T. Treatment of Hereditary Angioedema. J. Investig. Allergol. Clin. Immunol. 2021, 31, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Agboola, F.; Lubinga, S.; Carlson, J.; Lin, G.A.; Dreitlein, W.B.; Pearson, S.D. The Effectiveness and Value of Lanadelumab and C1 Esterase Inhibitors for Prophylaxis of Hereditary Angioedema Attacks. J. Manag. Care Spec. Pharm. 2019, 25, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Riedl, M.A.; Banerji, A.; Manning, M.E.; Burrell, E.; Joshi, N.; Patel, D.; Machnig, T.; Tai, M.H.; Watson, D.J. Treatment patterns and healthcare resource utilization among patients with hereditary angioedema in the United States. Orphanet. J. Rare Dis. 2018, 13, 180. [Google Scholar] [CrossRef]
- Cardarelli, W.J. Economic burden limiting proper healthcare delivery, management, and improvement of patient outcomes. Am. J. Manag. Care 2018, 24 (Suppl. S14), S308–S313. [Google Scholar]
- Murphy, E.; Donahue, C.; Omert, L.; Persons, S.; Tyma, T.J.; Chiao, J.; Lumry, W. Training patients for self-administration of a new subcutaneous C1-inhibitor concentrate for hereditary angioedema. Nurs. Open 2019, 6, 126–135. [Google Scholar] [CrossRef]
- Busse, P.J.; Christiansen, S.C.; Riedl, M.A.; Banerji, A.; Bernstein, J.A.; Castaldo, A.J.; Craig, T.; Davis-Lorton, M.; Frank, M.M.; Li, H.H.; et al. US HAEA medical advisory board 2020 guidelines for the management of hereditary angioedema. J. Allergy Clin. Immunol. Pract. 2021, 9, 132–150. [Google Scholar] [CrossRef]
- Wentzel, N.; Panieri, A.; Ayazi, M.; Ntshalintshali, S.D.; Pourpak, Z.; Hawarden, D.; Potter, P.; Levin, M.E.; Fazlollahi, M.R.; Peter, J. Fresh frozen plasma for on-demand hereditary angioedema treatment in South Africa and Iran. World Allergy Organ. J. 2019, 12, 100049. [Google Scholar] [CrossRef]
- Gompels, M.M.; Lock, R.J.; Abinun, M. C1 inhibitor deficiency: Consensus document. Clin. Exp. Immunol. 2005, 139, 379–394. [Google Scholar] [CrossRef]
- Gandhi, P.K.; Gentry, W.M.; Bottorff, M.B. Thrombotic events associated with C1 esterase inhibitor products in patients with hereditary angioedema: Investigation from the United States Food and Drug Administration adverse event reporting system database. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2012, 32, 902–909. [Google Scholar] [CrossRef]
- Deeks, E.D. Icatibant. Drugs 2010, 70, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Craig, T.J.; Li, H.H.; Riedl, M.; Bernstein, J.A.; Lumry, W.R.; MacGinnitie, A.J.; Stolz, L.E.; Biedenkapp, J.; Chyung, Y. Characterization of anaphylaxis after ecallantide treatment of hereditary angioedema attacks. J. Allergy Clin. Immunol. Pract. 2015, 3, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Magerl, M.; Frank, M.; Lumry, W.; Bernstein, J.; Busse, P.; Craig, T.; Martinez-Saguer, I.; Riedl, M.A.; Shapiro, R.; Edelman, J.; et al. Short-term prophylactic use of C1-inhibitor concentrate in hereditary angioedema: Findings from an international patient registry. Ann. Allergy Asthma Immunol. 2017, 118, 110–112. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, T.; Hide, M.; Yamashita, K.; Ohsawa, I. The use of tranexamic acid for on-demand and prophylactic treatment of hereditary angioedema: A systematic review. J. Cutan Immunol. Allergy 2018, 1, 126–138. [Google Scholar] [CrossRef]
- Wang, K.; Geiger, H.; McMahon, A. Tranexamic acid for ACE inhibitor induced angioedema. Am. J. Emerg. Med. 2021, 43, 292.e5–292.e7. [Google Scholar] [CrossRef]
- Li, H.H.; Moldovan, D.; Bernstein, J.A.; Reshef, A.; Porebski, G.; Stobiecki, M.; Baker, J.; Levy, R.; Relan, A.; Riedl, M. Recombinant human-C1 inhibitor is effective and safe for repeat hereditary angioedema attacks. J. Allergy Clin. Immunol. Pract. 2015, 3, 417–423. [Google Scholar] [CrossRef]
- Aygören-Pürsün, E.; Soteres, D.; Moldovan, D.; Christensen, J.; Van Leerberghe, A.; Hao, J.; Schranz, J.; Jacobson, K.W.; Martinez-Saguer, I. Preventing hereditary angioedema attacks in children using Cinryze®: Interim efficacy and safety phase 3 findings. Int. Arch. Allergy Immunol. 2017, 173, 114–119. [Google Scholar] [CrossRef]
- Lumry, W.R.; Craig, T.; Zuraw, B.; Longhurst, H.; Baker, J.; Li, H.H.; Bernstein, J.A.; Anderson, J.; Riedl, M.A.; Manning, M.E.; et al. Health-related quality of life with subcutaneous C1-inhibitor for prevention of attacks of hereditary angioedema. J. Allergy Clin. Immunol. Pract. 2018, 6, 1733–1741. [Google Scholar] [CrossRef]
- Caballero, T.; Farkas, H.; Bouillet, L.; Bowen, T.; Gompel, A.; Fagerberg, C.; Bjökander, J.; Bork, K.; Bygum, A.; Cicardi, M.; et al. International consensus and practical guidelines on the gynecologic and obstetric management of female patients with hereditary angioedema caused by C1 inhibitor deficiency. J. Allergy Clin. Immunol. 2012, 129, 308–320. [Google Scholar] [CrossRef]
- Banerji, A.; Riedl, M.A.; Bernstein, J.A.; Cicardi, M.; Longhurst, H.J.; Zuraw, B.L.; Busse, P.J.; Anderson, J.; Magerl, M.; Martinez-Saguer, I.; et al. Effect of lanadelumab compared with placebo on prevention of hereditary angioedema attacks: A randomized clinical trial. JAMA 2018, 320, 2108–2121. [Google Scholar] [CrossRef]
- Hwang, J.R.; Hwang, G.; Johri, A.; Craig, T. Oral plasma kallikrein inhibitor BCX7353 for treatment of hereditary angioedema. Immunotherapy 2019, 11, 1439–1444. [Google Scholar] [CrossRef] [PubMed]
- Zuraw, B.L.; Davis, D.K.; Castaldo, A.J.; Christiansen, S.C. Tolerability and effectiveness of 17-α-alkylated androgen therapy for hereditary angioedema: A re-examination. J. Allergy Clin. Immunol. Pract. 2016, 4, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Wintenberger, C.; Boccon-Gibod, I.; Launay, D.; Fain, O.; Kanny, G.; Jeandel, P.Y.; Martin, L.; Gompel, A.; Bouillet, L. Tranexamic acid as maintenance treatment for non-histaminergic angioedema: Analysis of efficacy and safety in 37 patients. Clin. Exp. Immunol. 2014, 178, 112–117. [Google Scholar] [CrossRef]
- Henry Li, H.; Riedl, M.; Kashkin, J. Update on the Use of C1-Esterase Inhibitor Replacement Therapy in the Acute and Prophylactic Treatment of Hereditary Angioedema. Clin. Rev. Allergy Immunol. 2019, 56, 207–218. [Google Scholar] [CrossRef]
- Greve, J.; Strassen, U.; Gorczyza, M.; Dominas, N.; Frahm, U.M.; Mühlberg, H.; Wiednig, M.; Zampeli, V.; Magerl, M. Prophylaxis in hereditary angioedema (HAE) with C1 inhibitor deficiency. J. Dtsch. Dermatol. Ges. 2016, 14, 266–275. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Type I HAE | Type II HAE | Type III HAE | Others |
|---|---|---|---|
|
|
|
|
| Type I HAE | Type II HAE | Type III HAE |
|---|---|---|
|
|
|
| Biomarkers | Function | Category | References |
|---|---|---|---|
| AgC1-INH | Regulates complement and contact pathways; deficiency leads to uncontrolled bradykinin production | Established | [10,53,54,55,56,57] |
| C1-INH-C1(r,s) protease-inhibitor complex | These complex forms when C1-INH neutralizes its primary targets—C1r and C1s proteases—which are components of the complement system | Promising | [53,55,56] |
| fC1-INH | Evaluates the actual ability of the protein to regulate complement and contact system activation, which is essential in preventing the formation of bradykinin | Established | [47,53,54,58,59,60,61,62,63] |
| Complement C4 | Low levels during an attack; diagnostic utility | Established | [53,54,60,61] |
| Anti-C1-INH IgM antibody | Binds to C1-INH and reduces its functional activity, contributing to uncontrolled complement activation | Emerging | [64,65,66,67] |
| MASP | Activates the lectin complement pathway | Emerging | [64,65,66,67] |
| Bradykinin | Primary mediator of angioedema in HAE | Established | [10,54,57,60,63,68,69] |
| cHK | Indicates bradykinin activation during acute attacks | Promising | [10,70] |
| PKa | Cleaves high-molecular-weight kininogen to produce bradykinin | Emerging | [10,70] |
| FXIIa | Initiates the contact activation pathway, leading to bradykinin release | Emerging | [10,71,72,73] |
| ACE | Degrades bradykinin; may influence HAE attack frequency | Emerging | [10,74,75,76] |
| Carboxypeptidase N | Involved in bradykinin degradation | Emerging | [10,74,75,76] |
| aPTT | Measures coagulation pathway activity | Emerging | [73] |
| APP | Degrades bradykinin; involved in regulating its levels | Emerging | [10,74] |
| D-dimers | Elevated D-dimer levels indicate increased fibrinolytic activity | Emerging | [55,77,78] |
| VE-cadherin | Involved in endothelial integrity and permeability | Promising | [77,79,80,81,82,83,84] |
| VWF | Plays a role in coagulation; linked to endothelial function | Emerging | [77,79,80,81,82,83,84] |
| VCAM | Modulates leukocyte adhesion and endothelial function | Emerging | [10,79,80,81,82,83,84] |
| VEGF | Promotes angiogenesis and vascular permeability | Promising | [85,86,87,88] |
| PAF-AH | Degrades pro-inflammatory lipids | Promising | [85,86,87,88] |
| ADMA | Modulator of endothelial function and nitric oxide synthesis | Emerging | [10] |
| CRP | Acute-phase reactant; indicative of systemic inflammation | Promising | [89,90,91,92] |
| ESR | Marker of inflammation; elevated in chronic disease | Promising | [89,90,91,92] |
| WBC | Indicator of immune response and infection | Promising | [89,90,91,92] |
| G-CSF | Stimulates neutrophil production; linked to inflammation | Emerging | [92,93,94] |
| ILs | Modulate immune response; IL-6 and IL-1 linked to HAE | Promising | [92,93,94] |
| NO | Modulates vascular tone; related to endothelial function | Emerging | [10,93,95] |
| TNF | Pro-inflammatory cytokine; linked to vascular permeability | Promising | [92,93,94] |
| ANGPTs | Modulate vascular permeability, linked to vascular integrity | Emerging | [10,96,97] |
| Treatment Type | Medication | Administration | Indications/Notes |
|---|---|---|---|
| Acute treatment |
|
|
|
| Treatment Type | Medication | Administration | Indications/Notes |
|---|---|---|---|
| Long-term prophylaxis |
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tutunaru, C.V.; Ică, O.M.; Mitroi, G.G.; Neagoe, C.D.; Mitroi, G.F.; Orzan, O.A.; Bălăceanu-Gurău, B.; Ianoși, S.L. Unveiling the Complexities of Hereditary Angioedema. Biomolecules 2024, 14, 1298. https://doi.org/10.3390/biom14101298
Tutunaru CV, Ică OM, Mitroi GG, Neagoe CD, Mitroi GF, Orzan OA, Bălăceanu-Gurău B, Ianoși SL. Unveiling the Complexities of Hereditary Angioedema. Biomolecules. 2024; 14(10):1298. https://doi.org/10.3390/biom14101298
Chicago/Turabian StyleTutunaru, Cristina Violeta, Oana Maria Ică, George G. Mitroi, Carmen Daniela Neagoe, George F. Mitroi, Olguța Anca Orzan, Beatrice Bălăceanu-Gurău, and Simona Laura Ianoși. 2024. "Unveiling the Complexities of Hereditary Angioedema" Biomolecules 14, no. 10: 1298. https://doi.org/10.3390/biom14101298
APA StyleTutunaru, C. V., Ică, O. M., Mitroi, G. G., Neagoe, C. D., Mitroi, G. F., Orzan, O. A., Bălăceanu-Gurău, B., & Ianoși, S. L. (2024). Unveiling the Complexities of Hereditary Angioedema. Biomolecules, 14(10), 1298. https://doi.org/10.3390/biom14101298