



Biomolecules of Muscle Fatigue in Metabolic Myopathies



{kind=link}
Abstract
:1. Introduction
1.1. Determinants of Muscle Contractions and Fatigue Definition
1.2. Classification of Peripheral Fatigue Determinants
1.2.1. Classification by Pathophysiology/Pathogenesis
1.2.2. Muscle Contraction Energy Source
- (i)
- Glucose
- (ii)
- Fatty Acids
- (iii)
- Others
1.2.3. Enzymes Involved in Muscle Fatigue
Creatine Kinase (CK)
Lactate Dehydrogenase (LDH)
Phosphofructokinase (PFK)
Mitochondrial Enzymes
1.2.4. Ion Channels
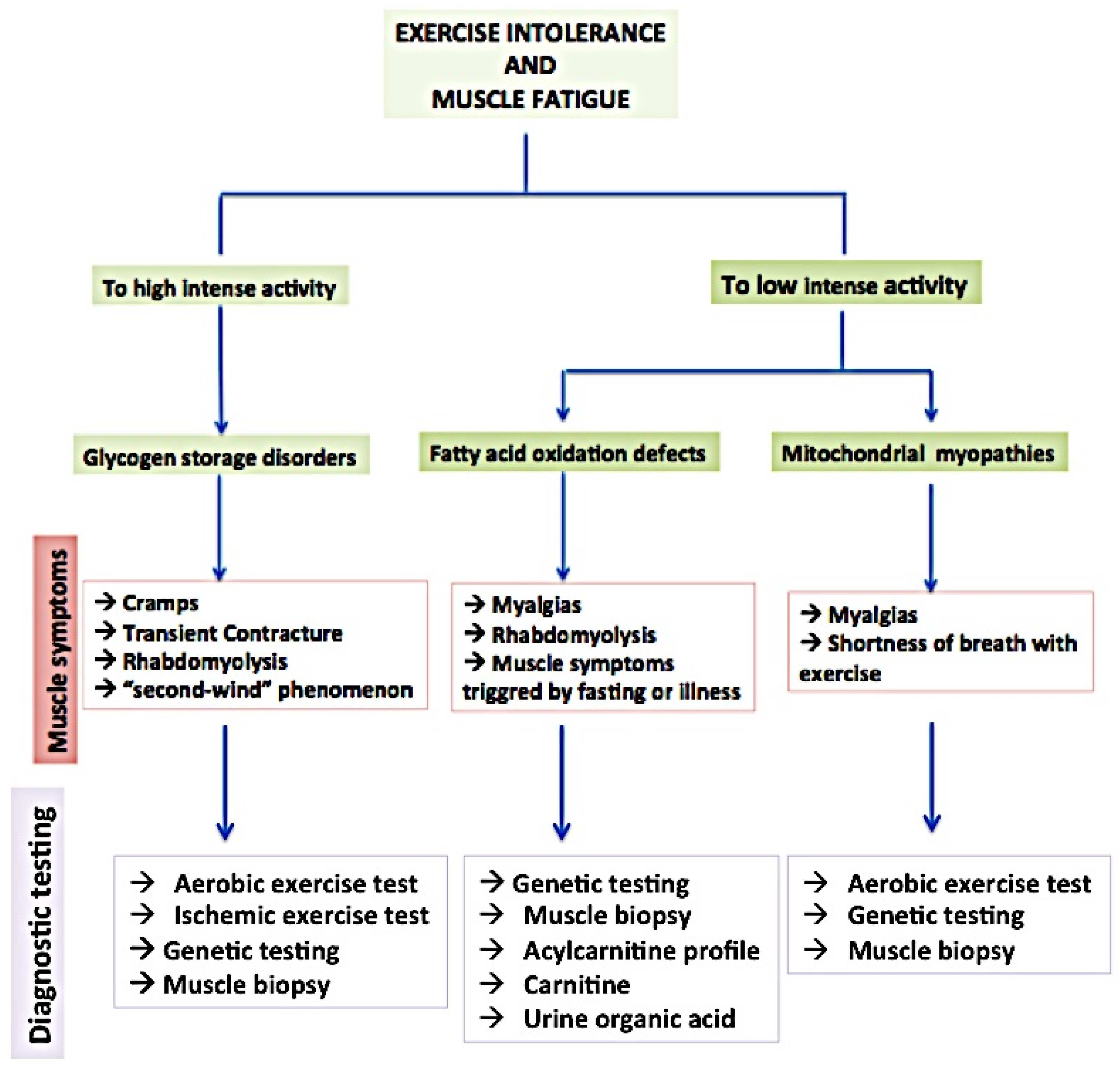
2. Overview of Principal Metabolic Myopathies and Their Impact on Muscle Function
2.1. Glycogen Storage Diseases (GSDs)
2.2. Fatty Acid Oxidation Disorders (FAODs)
2.3. Mitochondrial Myopathies
3. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Finsterer, J.; Mahjoub, S.Z. Fatigue in Healthy and Diseased Individuals. Am. J. Hosp. Palliat. Care 2014, 31, 562–575. [Google Scholar] [CrossRef]
- Kluger, B.M.; Krupp, L.B.; Enoka, R.M. Fatigue and Fatigability in Neurologic Illnesses: Proposal for a Unified Taxonomy. Neurology 2013, 80, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.P.; Walsh, D. Mechanisms of Fatigue. J. Support. Oncol. 2010, 8, 164–174. [Google Scholar] [PubMed]
- Constantin-Teodosiu, D.; Constantin, D. Molecular Mechanisms of Muscle Fatigue. Int. J. Mol. Sci. 2021, 22, 11587. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.V. Current Topics for Teaching Skeletal Muscle Physiology. Adv. Physiol. Educ. 2003, 27, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Gandevia, S.C. Spinal and Supraspinal Factors in Human Muscle Fatigue. Physiol. Rev. 2001, 81, 1725–1789. [Google Scholar] [CrossRef] [PubMed]
- Ament, W.; Verkerke, G.J. Exercise and Fatigue. Sports Med. 2009, 39, 389–422. [Google Scholar] [CrossRef]
- Plotkin, D.L.; Roberts, M.D.; Haun, C.T.; Schoenfeld, B.J. Muscle Fiber Type Transitions with Exercise Training: Shifting Perspectives. Sports 2021, 9, 127. [Google Scholar] [CrossRef]
- Barry, B.K.; Enoka, R.M. The Neurobiology of Muscle Fatigue: 15 Years Later. Integr. Comp. Biol. 2007, 47, 465–473. [Google Scholar] [CrossRef]
- Brooks, G.A.; Osmond, A.D.; Arevalo, J.A.; Duong, J.J.; Curl, C.C.; Moreno-Santillan, D.D.; Leija, R.G. Lactate as a Myokine and Exerkine: Drivers and Signals of Physiology and Metabolism. J. Appl. Physiol. 2023, 134, 529–548. [Google Scholar] [CrossRef]
- Magherini, F.; Fiaschi, T.; Marzocchini, R.; Mannelli, M.; Gamberi, T.; Modesti, P.A.; Modesti, A. Oxidative Stress in Exercise Training: The Involvement of Inflammation and Peripheral Signals. Free Radic. Res. 2019, 53, 1155–1165. [Google Scholar] [CrossRef] [PubMed]
- Tornero-Aguilera, J.F.; Jimenez-Morcillo, J.; Rubio-Zarapuz, A.; Clemente-Suárez, V.J. Central and Peripheral Fatigue in Physical Exercise Explained: A Narrative Review. Int. J. Environ. Res. Public Health 2022, 19, 3909. [Google Scholar] [CrossRef] [PubMed]
- Meeusen, R.; Van Cutsem, J.; Roelands, B. Endurance Exercise-Induced and Mental Fatigue and the Brain. Exp. Physiol. 2021, 106, 2294–2298. [Google Scholar] [CrossRef] [PubMed]
- Lorist, M.M.; Bezdan, E.; ten Caat, M.; Span, M.M.; Roerdink, J.B.T.M.; Maurits, N.M. The Influence of Mental Fatigue and Motivation on Neural Network Dynamics; an EEG Coherence Study. Brain Res. 2009, 1270, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Marcora, S.M.; Staiano, W.; Manning, V. Mental Fatigue Impairs Physical Performance in Humans. J. Appl. Physiol. 2009, 106, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Sandrone, S.; Bacigaluppi, M.; Galloni, M.R.; Martino, G. Angelo Mosso (1846–1910). J. Neurol. 2012, 259, 2513–2514. [Google Scholar] [CrossRef] [PubMed]
- Di Giulio, C.; Daniele, F.; Tipton, C.M. Angelo Mosso and Muscular Fatigue: 116 Years after the First Congress of Physiologists: IUPS Commemoration. Adv. Physiol. Educ. 2006, 30, 51–57. [Google Scholar] [CrossRef]
- Tarnopolsky, M.A. Metabolic Myopathies. Continuum 2022, 28, 1752–1777. [Google Scholar] [CrossRef]
- Gehlert, S.; Bloch, W.; Suhr, F. Ca2+-Dependent Regulations and Signaling in Skeletal Muscle: From Electro-Mechanical Coupling to Adaptation. Int. J. Mol. Sci. 2015, 16, 1066–1095. [Google Scholar] [CrossRef]
- Barclay, C.J.; Curtin, N.A. Advances in Understanding the Energetics of Muscle Contraction. J. Biomech. 2023, 156, 111669. [Google Scholar] [CrossRef]
- Allen, D.G.; Lamb, G.D.; Westerblad, H. Skeletal Muscle Fatigue: Cellular Mechanisms. Physiol. Rev. 2008, 88, 287–332. [Google Scholar] [CrossRef] [PubMed]
- Baumert, P.; Temple, S.; Stanley, J.M.; Cocks, M.; Strauss, J.A.; Shepherd, S.O.; Drust, B.; Lake, M.J.; Stewart, C.E.; Erskine, R.M. Neuromuscular Fatigue and Recovery after Strenuous Exercise Depends on Skeletal Muscle Size and Stem Cell Characteristics. Sci. Rep. 2021, 11, 7733. [Google Scholar] [CrossRef] [PubMed]
- Grefte, S.; Kuijpers-Jagtman, A.M.; Torensma, R.; Von den Hoff, J.W. Skeletal Muscle Development and Regeneration. Stem Cells Dev. 2007, 16, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Karalaki, M.; Fili, S.; Philippou, A.; Koutsilieris, M. Muscle Regeneration: Cellular and Molecular Events. In Vivo 2009, 23, 779–796. [Google Scholar] [PubMed]
- Sahlin, K. Muscle Glucose Metabolism during Exercise. Ann. Med. 1990, 22, 85–89. [Google Scholar] [PubMed]
- Gollnick, P.D. Metabolism of Substrates: Energy Substrate Metabolism during Exercise and as Modified by Training. Fed. Proc. 1985, 44, 353–357. [Google Scholar]
- Gollnick, P.D.; Matoba, H. Role of Carbohydrate in Exercise. Clin. Sports Med. 1984, 3, 583–593. [Google Scholar] [CrossRef]
- Caruel, M.; Truskinovsky, L. Physics of Muscle Contraction. Rep. Prog. Phys. 2018, 81, 036602. [Google Scholar] [CrossRef]
- Fiorenza, M.; Hostrup, M.; Gunnarsson, T.P.; Shirai, Y.; Schena, F.; Iaia, F.M.; Bangsbo, J. Neuromuscular Fatigue and Metabolism during High-Intensity Intermittent Exercise. Med. Sci. Sports Exerc. 2019, 51, 1642–1652. [Google Scholar] [CrossRef]
- Gastin, P.B. Energy System Interaction and Relative Contribution During Maximal Exercise. Sports Med. 2001, 31, 725–741. [Google Scholar] [CrossRef]
- Jeukendrup, A.E.; Saris, W.H.; Wagenmakers, A.J. Fat Metabolism during Exercise: A Review. Part I: Fatty Acid Mobilization and Muscle Metabolism. Int. J. Sports Med. 1998, 19, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Adler, M.; Shieh, P.B. Metabolic Myopathies. Semin. Neurol. 2015, 35, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.; Cogan, K.E.; Egan, B. Metabolism of Ketone Bodies during Exercise and Training: Physiological Basis for Exogenous Supplementation. J. Physiol. 2017, 595, 2857–2871. [Google Scholar] [CrossRef] [PubMed]
- Kamei, Y.; Hatazawa, Y.; Uchitomi, R.; Yoshimura, R.; Miura, S. Regulation of Skeletal Muscle Function by Amino Acids. Nutrients 2020, 12, 261. [Google Scholar] [CrossRef] [PubMed]
- Tesch, P.; Sjödin, B.; Thorstensson, A.; Karlsson, J. Muscle Fatigue and Its Relation to Lactate Accumulation and LDH Activity in Man. Acta Physiol. Scand. 1978, 103, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Tesch, P.; Sjödin, B.; Karlsson, J. Relationship between Lactate Accumulation, LDH Activity, LDH Isozyme and Fibre Type Distribution in Human Skeletal Muscle. Acta Physiol. Scand. 1978, 103, 40–46. [Google Scholar] [CrossRef]
- Vora, S. Isozymes of Human Phosphofructokinase: Biochemical and Genetic Aspects. Isozymes Curr. Top. Biol. Med. Res. 1983, 11, 3–23. [Google Scholar]
- Smits, B.; van den Heuvel, L.; Knoop, H.; Küsters, B.; Janssen, A.; Borm, G.; Bleijenberg, G.; Rodenburg, R.; van Engelen, B. Mitochondrial Enzymes Discriminate between Mitochondrial Disorders and Chronic Fatigue Syndrome. Mitochondrion 2011, 11, 735–738. [Google Scholar] [CrossRef]
- Filler, K.; Lyon, D.; Bennett, J.; McCain, N.; Elswick, R.; Lukkahatai, N.; Saligan, L.N. Association of Mitochondrial Dysfunction and Fatigue: A Review of the Literature. BBA Clin. 2014, 1, 12–23. [Google Scholar] [CrossRef]
- Kent-Braun, J.A.; Fitts, R.H.; Christie, A. Skeletal Muscle Fatigue. Compr. Physiol. 2012, 2, 997–1044. [Google Scholar] [CrossRef]
- Fauler, M.; Jurkat-Rott, K.; Lehmann-Horn, F. Membrane Excitability and Excitation-Contraction Uncoupling in Muscle Fatigue. Neuromuscul. Disord. 2012, 22 (Suppl. S3), S162–S167. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, M.W.; Brinkmeier, H.; Müntener, M. Calcium Ion in Skeletal Muscle: Its Crucial Role for Muscle Function, Plasticity, and Disease. Physiol. Rev. 2000, 80, 1215–1265. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Lamb, G.D.; Westerblad, H. Impaired Calcium Release during Fatigue. J. Appl. Physiol. 2008, 104, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.W.; Standen, N.B.; Stanfield, P.R. ATP-Dependent Potassium Channels of Muscle Cells: Their Properties, Regulation, and Possible Functions. J. Bioenerg. Biomembr. 1991, 23, 509–535. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.M.; Gramolini, A.; Light, P.; Comtois, A. Modulation of Muscle Contractility during Fatigue and Recovery by ATP Sensitive Potassium Channel. Acta Physiol. Scand. 1996, 156, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Torri, F.; Lopriore, P.; Montano, V.; Siciliano, G.; Mancuso, M.; Ricci, G. Pathophysiology and Management of Fatigue in Neuromuscular Diseases. Int. J. Mol. Sci. 2023, 24, 5005. [Google Scholar] [CrossRef] [PubMed]
- Brunk, U.T.; Terman, A. The Mitochondrial-Lysosomal Axis Theory of Aging: Accumulation of Damaged Mitochondria as a Result of Imperfect Autophagocytosis. Eur. J. Biochem. 2002, 269, 1996–2002. [Google Scholar] [CrossRef]
- Palermo, A.T.; Palmer, R.E.; So, K.S.; Oba-Shinjo, S.M.; Zhang, M.; Richards, B.; Madhiwalla, S.T.; Finn, P.F.; Hasegawa, A.; Ciociola, K.M.; et al. Transcriptional Response to GAA Deficiency (Pompe Disease) in Infantile-Onset Patients. Mol. Genet. Metab. 2012, 106, 287–300. [Google Scholar] [CrossRef]
- Batzios, S.P.; Zafeiriou, D.I.; Papakonstantinou, E. Extracellular Matrix Components: An Intricate Network of Possible Biomarkers for Lysosomal Storage Disorders? FEBS Lett. 2013, 587, 1258–1267. [Google Scholar] [CrossRef]
- Morales Corado, J.A.; Lee, C.U.; Enns, G.M. Carnitine-Acylcarnitine Translocase Deficiency. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- An, Y.; Young, S.P.; Kishnani, P.S.; Millington, D.S.; Amalfitano, A.; Corzo, D.; Chen, Y.-T. Glucose Tetrasaccharide as a Biomarker for Monitoring the Therapeutic Response to Enzyme Replacement Therapy for Pompe Disease. Mol. Genet. Metab. 2005, 85, 247–254. [Google Scholar] [CrossRef]
- Young, S.P.; Zhang, H.; Corzo, D.; Thurberg, B.L.; Bali, D.; Kishnani, P.S.; Millington, D.S. Long-Term Monitoring of Patients with Infantile-Onset Pompe Disease on Enzyme Replacement Therapy Using a Urinary Glucose Tetrasaccharide Biomarker. Genet. Med. 2009, 11, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Young, S.P.; Stevens, R.D.; An, Y.; Chen, Y.-T.; Millington, D.S. Analysis of a Glucose Tetrasaccharide Elevated in Pompe Disease by Stable Isotope Dilution–Electrospray Ionization Tandem Mass Spectrometry. Anal. Biochem. 2003, 316, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Tarallo, A.; Carissimo, A.; Gatto, F.; Nusco, E.; Toscano, A.; Musumeci, O.; Coletta, M.; Karali, M.; Acampora, E.; Damiano, C.; et al. microRNAs as Biomarkers in Pompe Disease. Genet. Med. 2019, 21, 591–600. [Google Scholar] [CrossRef]
- Molares-Vila, A.; Corbalán-Rivas, A.; Carnero-Gregorio, M.; González-Cespón, J.L.; Rodríguez-Cerdeira, C. Biomarkers in Glycogen Storage Diseases: An Update. Int. J. Mol. Sci. 2021, 22, 4381. [Google Scholar] [CrossRef] [PubMed]
- Khattak, Z.E.; Ashraf, M. McArdle Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Braakhekke, J.P.; de Bruin, M.I.; Stegeman, D.F.; Wevers, R.A.; Binkhorst, R.A.; Joosten, E.M. The Second Wind Phenomenon in McArdle’s Disease. Brain 1986, 109 Pt 6, 1087–1101. [Google Scholar] [CrossRef]
- De Luna, N.; Brull, A.; Lucia, A.; Santalla, A.; Garatachea, N.; Martí, R.; Andreu, A.L.; Pinós, T. PYGM Expression Analysis in White Blood Cells: A Complementary Tool for Diagnosing McArdle Disease? Neuromuscul. Disord. 2014, 24, 1079–1086. [Google Scholar] [CrossRef]
- Howell, J.M.; Walker, K.R.; Creed, K.E.; Dunton, E.; Davies, L.; Quinlivan, R.; Karpati, G. Phosphorylase Re-Expression, Increase in the Force of Contraction and Decreased Fatigue Following Notexin-Induced Muscle Damage and Regeneration in the Ovine Model of McArdle Disease. Neuromuscul. Disord. 2014, 24, 167–177. [Google Scholar] [CrossRef]
- Santacatterina, F.; Chamorro, M.; de Arenas, C.N.; Navarro, C.; Martín, M.A.; Cuezva, J.M.; Sánchez-Aragó, M. Quantitative Analysis of Proteins of Metabolism by Reverse Phase Protein Microarrays Identifies Potential Biomarkers of Rare Neuromuscular Diseases. J. Transl. Med. 2015, 13, 65. [Google Scholar] [CrossRef]
- Nogales-Gadea, G.; Consuegra-García, I.; Rubio, J.C.; Arenas, J.; Cuadros, M.; Camara, Y.; Torres-Torronteras, J.; Fiuza-Luces, C.; Lucia, A.; Martín, M.A.; et al. A Transcriptomic Approach to Search for Novel Phenotypic Regulators in McArdle Disease. PLoS ONE 2012, 7, e31718. [Google Scholar] [CrossRef]
- Ji, L.L.; Yeo, D. Maintenance of NAD+ Homeostasis in Skeletal Muscle during Aging and Exercise. Cells 2022, 11, 710. [Google Scholar] [CrossRef]
- Fiuza-Luces, C.; Santos-Lozano, A.; Llavero, F.; Campo, R.; Nogales-Gadea, G.; Díez-Bermejo, J.; Baladrón, C.; González-Murillo, Á.; Arenas, J.; Martín, M.A.; et al. Muscle Molecular Adaptations to Endurance Exercise Training Are Conditioned by Glycogen Availability: A Proteomics-based Analysis in the McArdle Mouse Model. J. Physiol. 2018, 596, 1035–1061. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Yemul, S.; Wang, J.; Pasinetti, G.M. Grape Seed Polyphenolic Extract as a Potential Novel Therapeutic Agent in Tauopathies. J. Alzheimer’s Dis. 2009, 16, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Hurtado, E.; Núñez-Álvarez, Y.; Muñoz, M.; Gutiérrez-Caballero, C.; Casas, J.; Pendás, A.M.; Peinado, M.A.; Suelves, M. HDAC11 Is a Novel Regulator of Fatty Acid Oxidative Metabolism in Skeletal Muscle. FEBS J. 2021, 288, 902–919. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, B.B.; Wolfe, R.R. Regulation of Fatty Acid Oxidation in Skeletal Muscle. Annu. Rev. Nutr. 1999, 19, 463–484. [Google Scholar] [CrossRef] [PubMed]
- van Hall, G. The Physiological Regulation of Skeletal Muscle Fatty Acid Supply and Oxidation During Moderate-Intensity Exercise. Sports Med. 2015, 45 (Suppl. S1), S23–S32. [Google Scholar] [CrossRef]
- Engel, A.G.; Angelini, C. Carnitine Deficiency of Human Skeletal Muscle with Associated Lipid Storage Myopathy: A New Syndrome. Science 1973, 179, 899–902. [Google Scholar] [CrossRef]
- Pennisi, E.; Garibaldi, M.; Antonini, G. Lipid Myopathies. JCM 2018, 7, 472. [Google Scholar] [CrossRef]
- Longo, N.; Amat di San Filippo, C.; Pasquali, M. Disorders of Carnitine Transport and the Carnitine Cycle. Am. J. Med. Genet. C Semin. Med. Genet. 2006, 142C, 77–85. [Google Scholar] [CrossRef]
- Joshi, P.R.; Zierz, S. Muscle Carnitine Palmitoyltransferase II (CPT II) Deficiency: A Conceptual Approach. Molecules 2020, 25, 1784. [Google Scholar] [CrossRef]
- Lilleker, J.B.; Keh, Y.S.; Roncaroli, F.; Sharma, R.; Roberts, M. Metabolic Myopathies: A Practical Approach. Pract. Neurol. 2018, 18, 14–26. [Google Scholar] [CrossRef]
- Xia, C.; Lou, B.; Fu, Z.; Mohsen, A.-W.; Shen, A.L.; Vockley, J.; Kim, J.-J.P. Molecular Mechanism of Interactions between ACAD9 and Binding Partners in Mitochondrial Respiratory Complex I Assembly. iScience 2021, 24, 103153. [Google Scholar] [CrossRef] [PubMed]
- Repp, B.M.; Mastantuono, E.; Alston, C.L.; Schiff, M.; Haack, T.B.; Rötig, A.; Ardissone, A.; Lombès, A.; Catarino, C.B.; Diodato, D.; et al. Clinical, Biochemical and Genetic Spectrum of 70 Patients with ACAD9 Deficiency: Is Riboflavin Supplementation Effective? Orphanet J. Rare Dis. 2018, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Adeva-Andany, M.M.; Carneiro-Freire, N.; Seco-Filgueira, M.; Fernández-Fernández, C.; Mouriño-Bayolo, D. Mitochondrial β-Oxidation of Saturated Fatty Acids in Humans. Mitochondrion 2019, 46, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Tein, I.; De Vivo, D.C.; Bierman, F.; Pulver, P.; De Meirleir, L.J.; Cvitanovic-Sojat, L.; Pagon, R.A.; Bertini, E.; Dionisi-Vici, C.; Servidei, S. Impaired Skin Fibroblast Carnitine Uptake in Primary Systemic Carnitine Deficiency Manifested by Childhood Carnitine-Responsive Cardiomyopathy. Pediatr. Res. 1990, 28, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Grünert, S.C. Clinical and Genetical Heterogeneity of Late-Onset Multiple Acyl-Coenzyme A Dehydrogenase Deficiency. Orphanet J. Rare Dis. 2014, 9, 117. [Google Scholar] [CrossRef] [PubMed]
- Lupica, A.; Oteri, R.; Volta, S.; Ghezzi, D.; Drago, S.F.A.; Rodolico, C.; Musumeci, O.; Toscano, A. Diagnostic Challenges in Late Onset Multiple Acyl-CoA Dehydrogenase Deficiency: Clinical, Morphological, and Genetic Aspects. Front. Neurol. 2022, 13, 815523. [Google Scholar] [CrossRef] [PubMed]
- Missaglia, S.; Tavian, D.; Angelini, C. ETF Dehydrogenase Advances in Molecular Genetics and Impact on Treatment. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 360–372. [Google Scholar] [CrossRef]
- Sopariwala, D.H.; Pant, M.; Shaikh, S.A.; Goonasekera, S.A.; Molkentin, J.D.; Weisleder, N.; Ma, J.; Pan, Z.; Periasamy, M. Sarcolipin Overexpression Improves Muscle Energetics and Reduces Fatigue. J. Appl. Physiol. 2015, 118, 1050–1058. [Google Scholar] [CrossRef]
- Picard, M.; Taivassalo, T.; Gouspillou, G.; Hepple, R.T. Mitochondria: Isolation, Structure and Function. J. Physiol. 2011, 589, 4413–4421. [Google Scholar] [CrossRef]
- Schwerzmann, K.; Hoppeler, H.; Kayar, S.R.; Weibel, E.R. Oxidative Capacity of Muscle and Mitochondria: Correlation of Physiological, Biochemical, and Morphometric Characteristics. Proc. Natl. Acad. Sci. USA 1989, 86, 1583–1587. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial Diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Siciliano, G.; Volpi, L.; Piazza, S.; Ricci, G.; Mancuso, M.; Murri, L. Functional Diagnostics in Mitochondrial Diseases. Biosci. Rep. 2007, 27, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Glover, E.I.; Martin, J.; Maher, A.; Thornhill, R.E.; Moran, G.R.; Tarnopolsky, M.A. A Randomized Trial of Coenzyme Q10 in Mitochondrial Disorders: CoQ10 and Mitochondrial Disease. Muscle Nerve 2010, 42, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Abramov, A.Y. Nrf2 as a Regulator of Mitochondrial Function: Energy Metabolism and Beyond. Free Radic. Biol. Med. 2022, 189, 136–153. [Google Scholar] [CrossRef]
- Almannai, M.; El-Hattab, A.W.; Ali, M.; Soler-Alfonso, C.; Scaglia, F. Clinical Trials in Mitochondrial Disorders, an Update. Mol. Genet. Metab. 2020, 131, 1–13. [Google Scholar] [CrossRef]
- Pitceathly, R.D.S.; Keshavan, N.; Rahman, J.; Rahman, S. Moving towards Clinical Trials for Mitochondrial Diseases. J. Inherit. Metab. Dis. 2021, 44, 22–41. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schirinzi, E.; Ricci, G.; Torri, F.; Mancuso, M.; Siciliano, G. Biomolecules of Muscle Fatigue in Metabolic Myopathies. Biomolecules 2024, 14, 50. https://doi.org/10.3390/biom14010050
Schirinzi E, Ricci G, Torri F, Mancuso M, Siciliano G. Biomolecules of Muscle Fatigue in Metabolic Myopathies. Biomolecules. 2024; 14(1):50. https://doi.org/10.3390/biom14010050
Chicago/Turabian StyleSchirinzi, Erika, Giulia Ricci, Francesca Torri, Michelangelo Mancuso, and Gabriele Siciliano. 2024. "Biomolecules of Muscle Fatigue in Metabolic Myopathies" Biomolecules 14, no. 1: 50. https://doi.org/10.3390/biom14010050
APA StyleSchirinzi, E., Ricci, G., Torri, F., Mancuso, M., & Siciliano, G. (2024). Biomolecules of Muscle Fatigue in Metabolic Myopathies. Biomolecules, 14(1), 50. https://doi.org/10.3390/biom14010050