
An Integrated In Silico and In Vitro Approach for the Identification of Natural Products Active against SARS-CoV-2

 ,
, 
 ,
,  ,
, 

Abstract
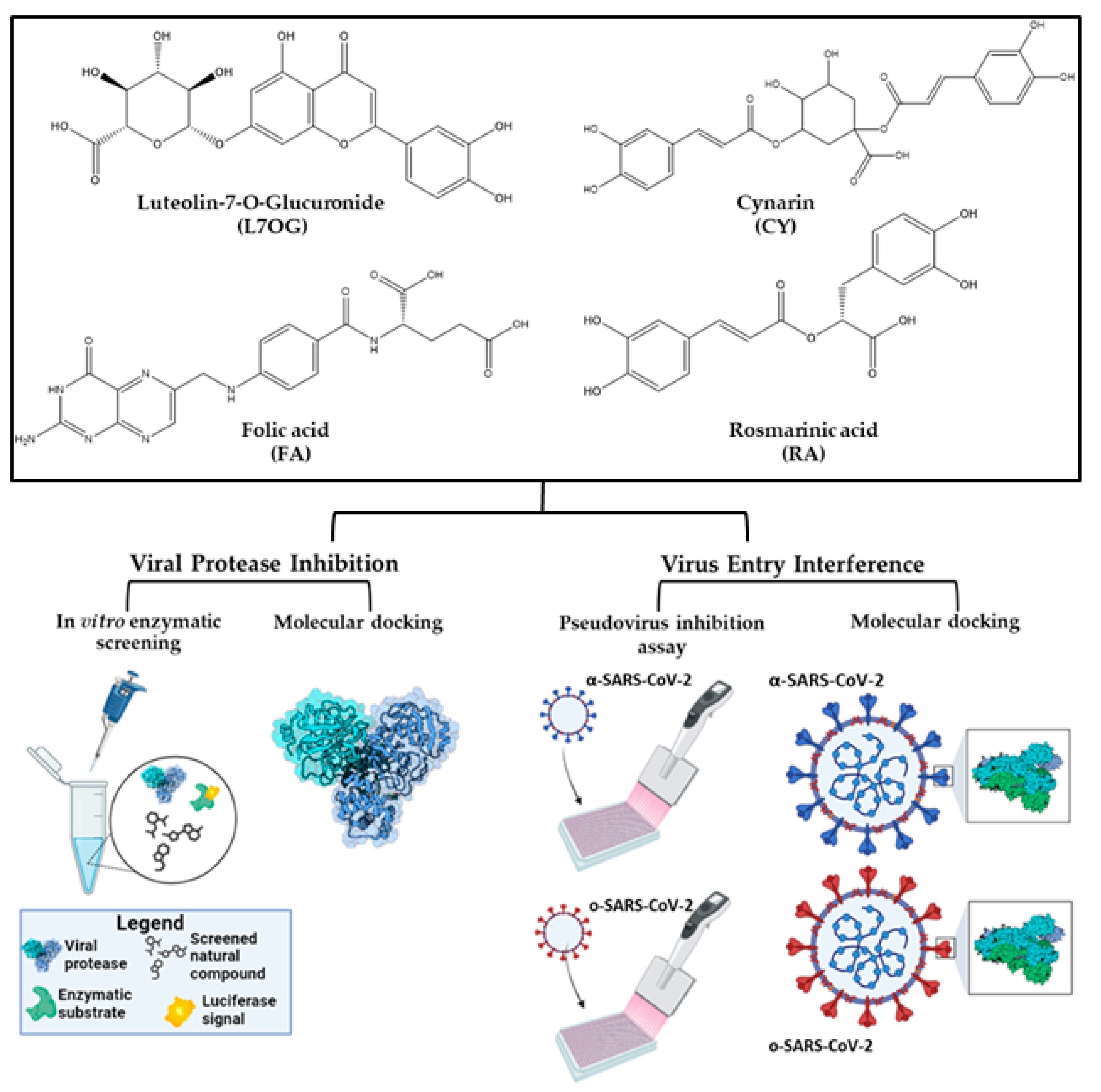
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Materials
2.3. SARS-CoV-2 3CLpro and PLpro Luminescent Assays
2.4. Viability Assay
2.5. Production of SARS-CoV-2 Pseudotyped Particles
2.6. In Vitro Pseudovirus Entry Inhibition Assay
2.7. Molecular Docking
2.8. Molecular Dynamics Simulations
2.9. Statistical Analysis
3. Results
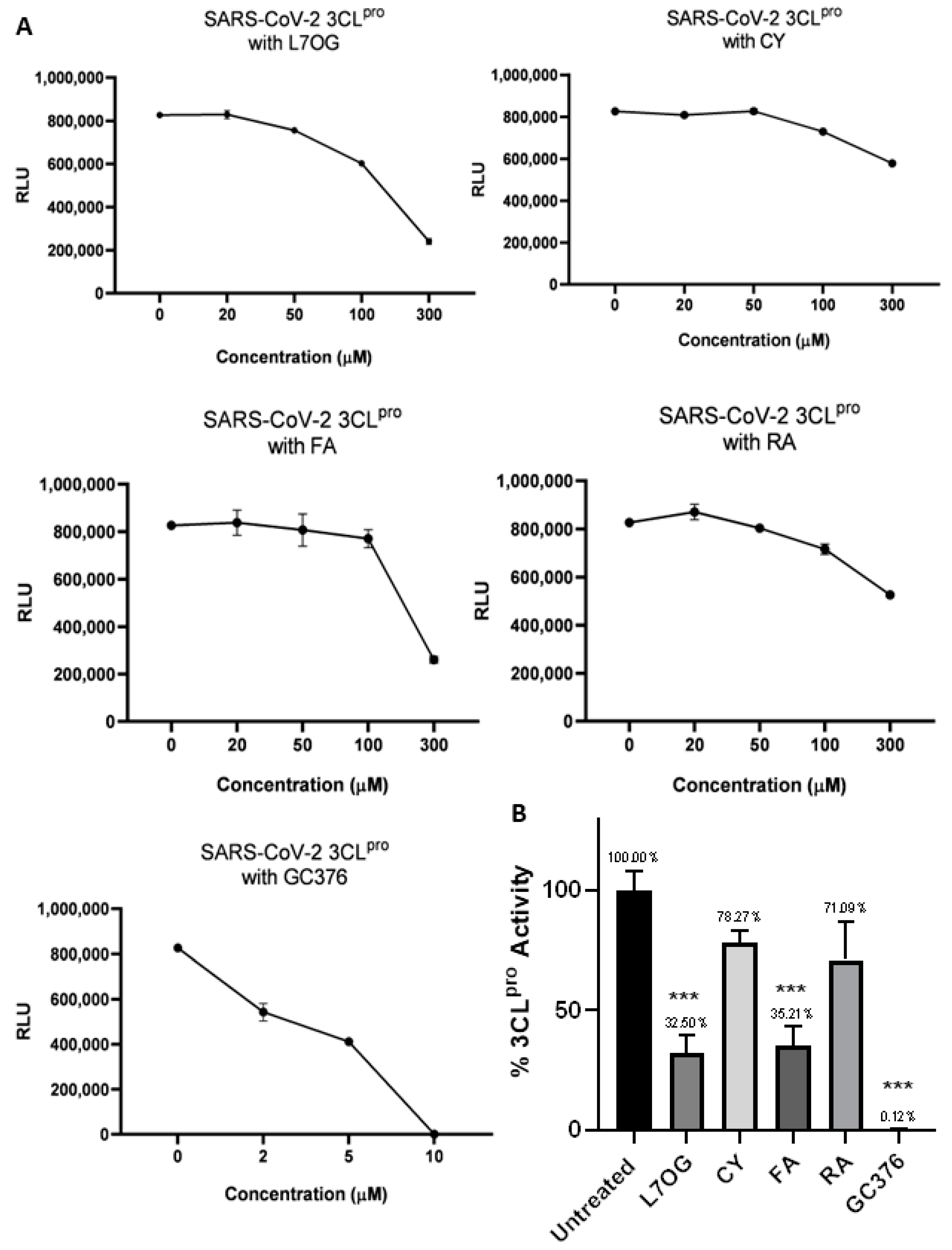
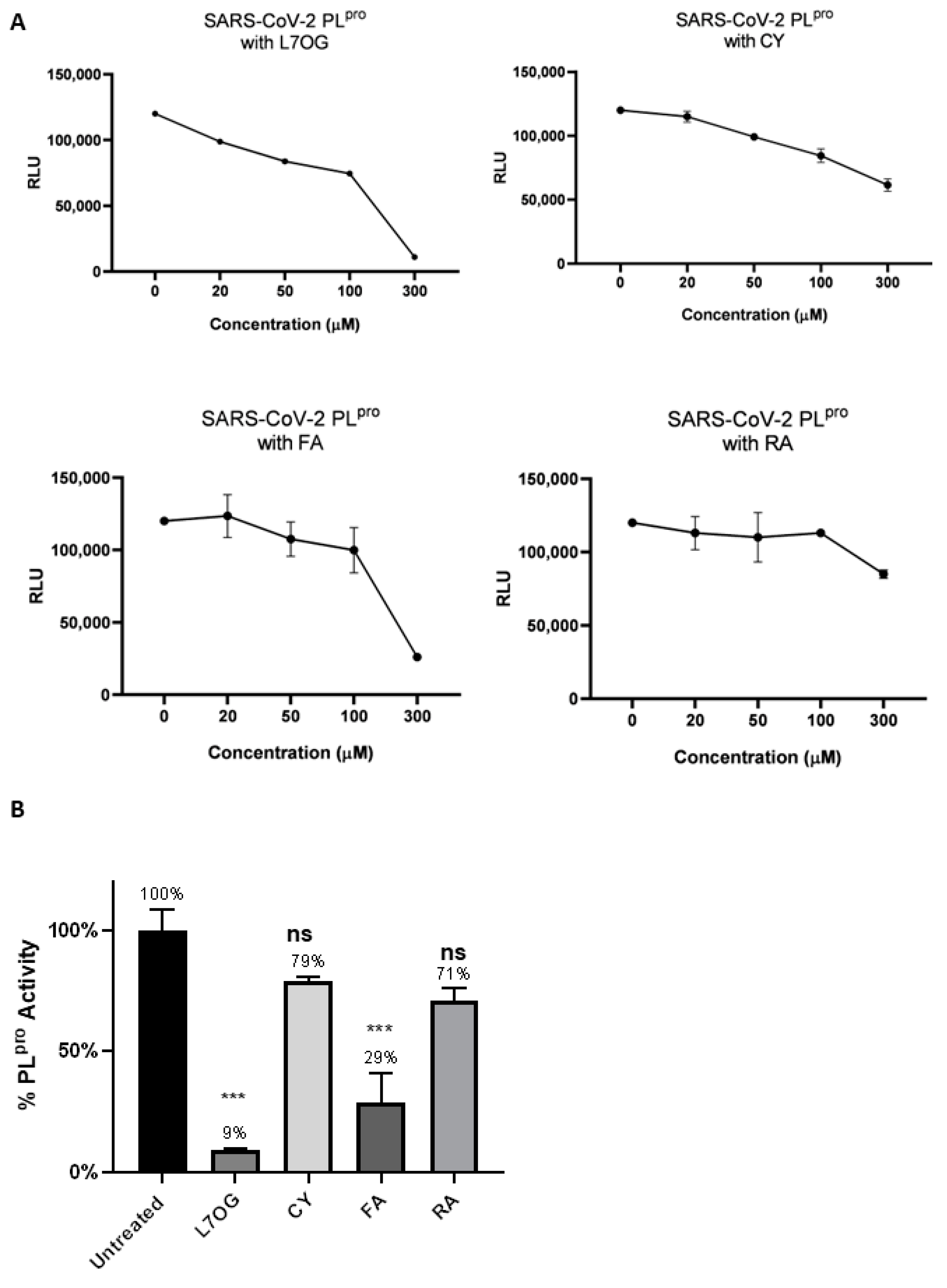
3.1. In Vitro Screening for 3CLpro and PLpro Inhibition by Natural Compounds
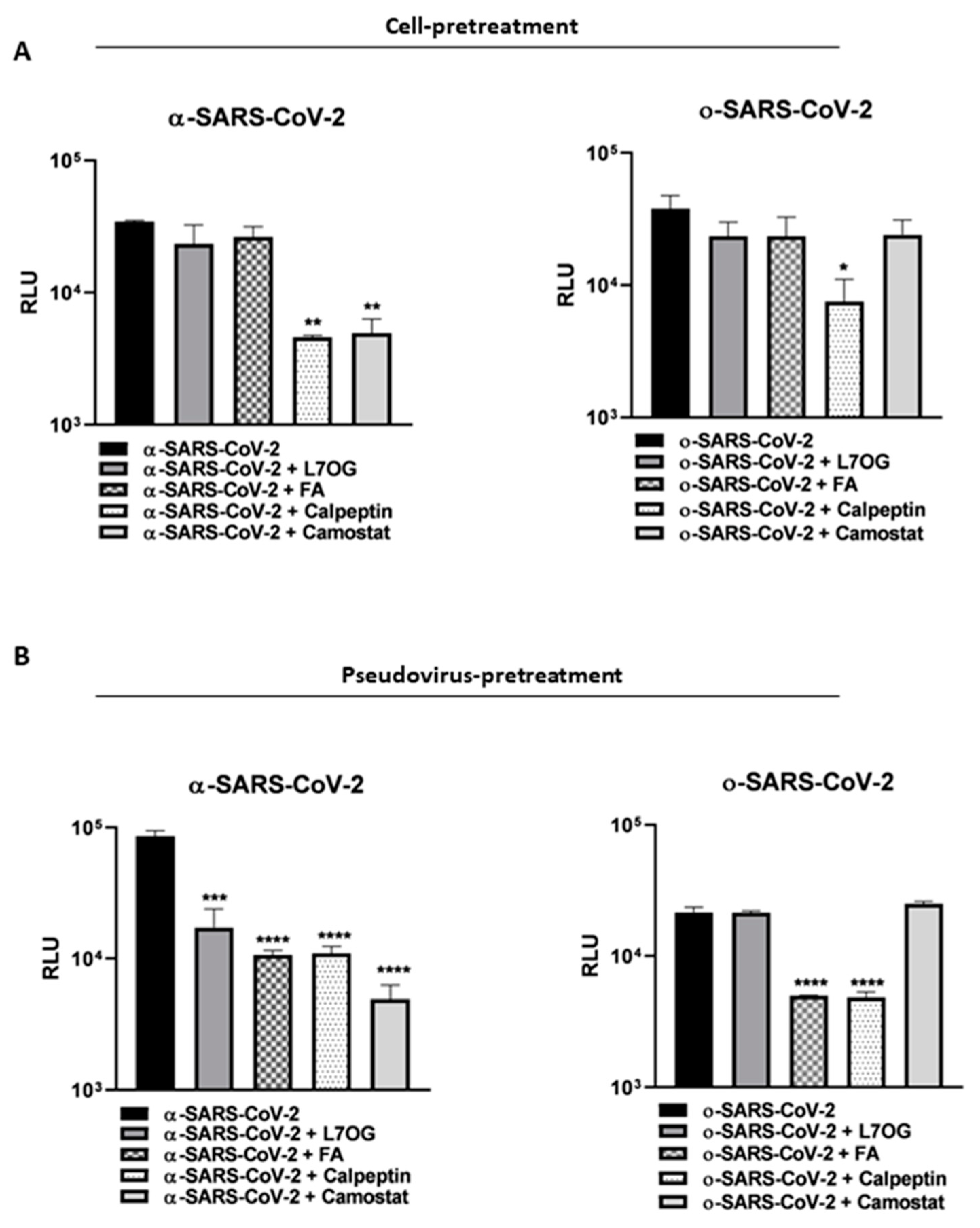
3.2. Pseudoviruses Entry Inhibition Screening of L7OG and FA
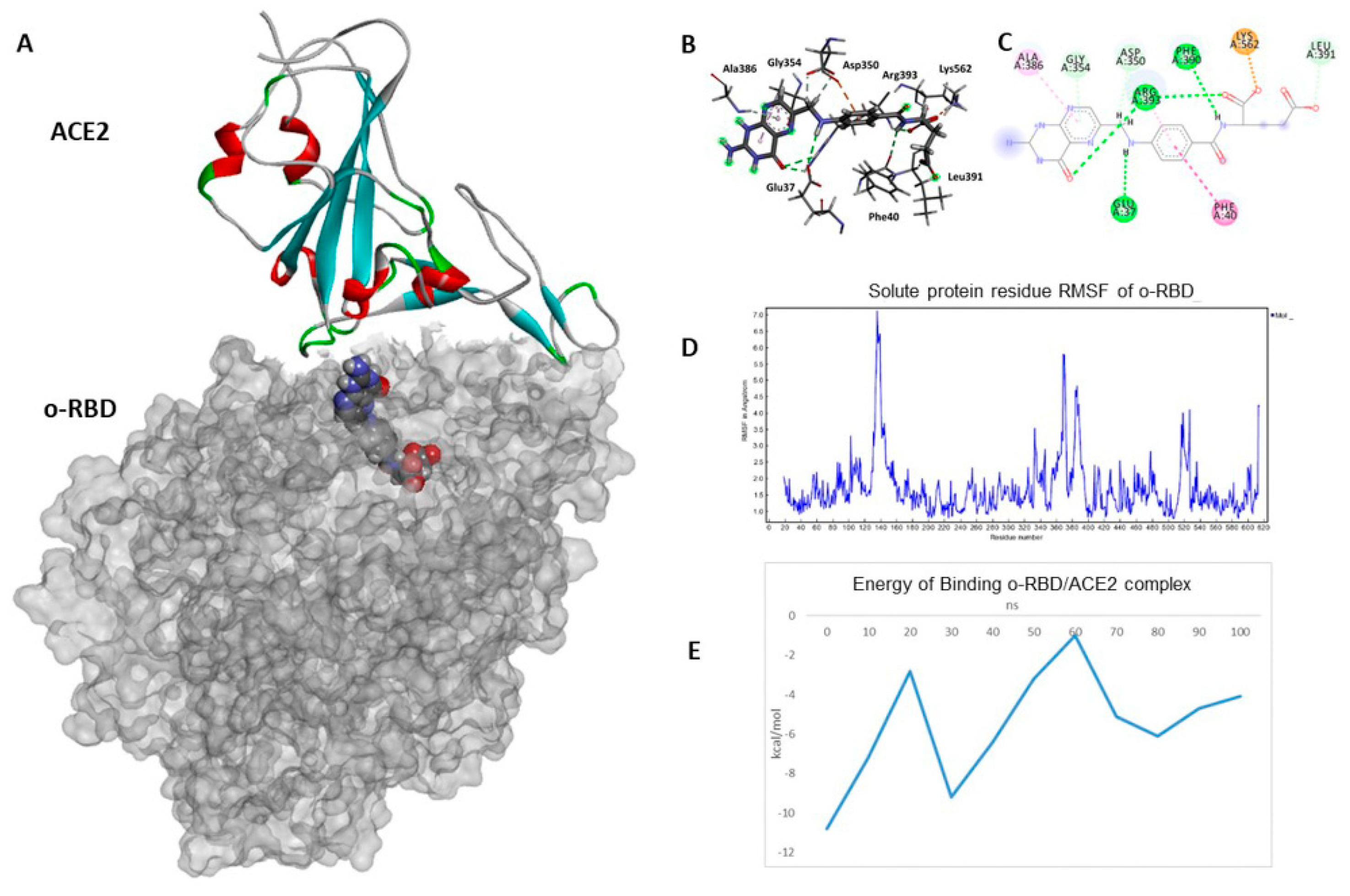
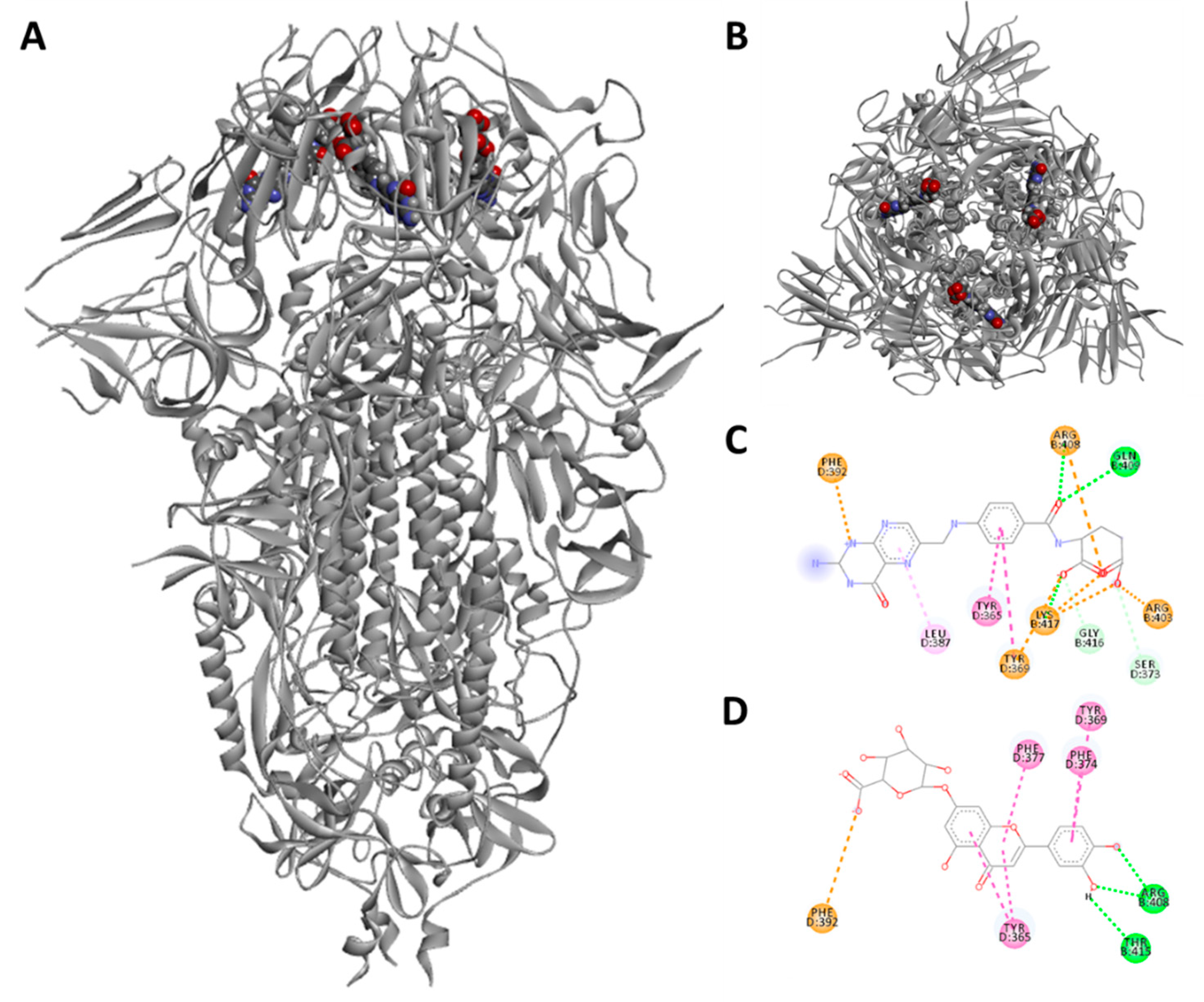
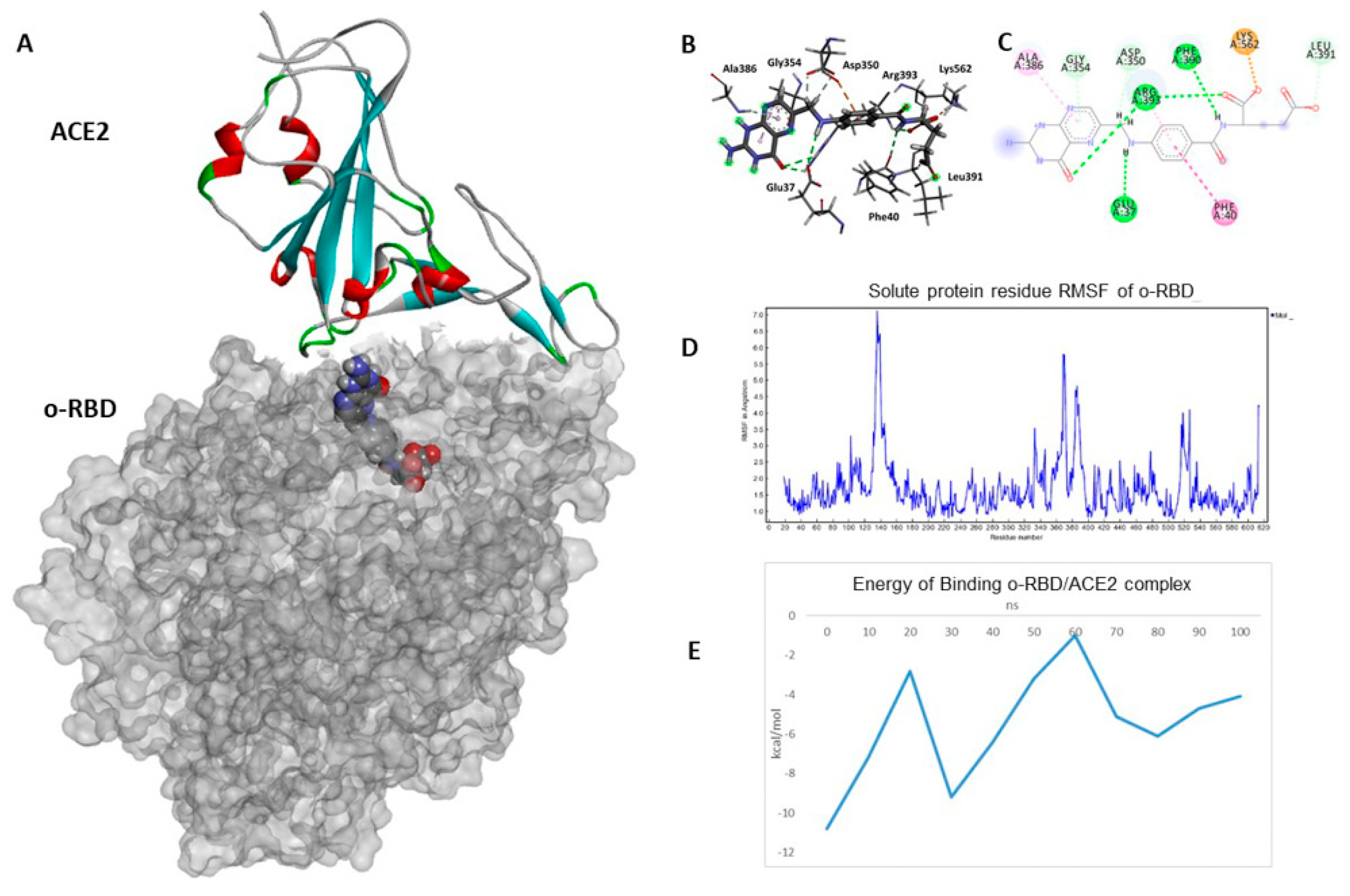
3.3. Docking and Molecular Dynamics Simulations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mukherjee, R.; Dikic, I. Proteases of SARS Coronaviruses. In Encyclopedia of Cell Biology; Elsevier: Amsterdam, The Netherlands, 2023; pp. 930–941. [Google Scholar]
- Lv, Z.; Cano, K.E.; Jia, L.; Drag, M.; Huang, T.T.; Olsen, S.K. Targeting SARS-CoV-2 Proteases for COVID-19 Antiviral Development. Front. Chem. 2022, 9, 1221. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A.; Narwal, M.; Majowicz, S.A.; Varricchio, C.; Toner, S.A.; Ballatore, C.; Brancale, A.; Murakami, K.S.; Jose, J. Identification of SARS-CoV-2 Inhibitors Targeting Mpro and PLpro Using in-Cell-Protease Assay. Commun. Biol. 2022, 5, 169. [Google Scholar] [CrossRef] [PubMed]
- Citarella, A.; Gentile, D.; Rescifina, A.; Piperno, A.; Mognetti, B.; Gribaudo, G.; Sciortino, M.T.; Holzer, W.; Pace, V.; Micale, N. Pseudo-Dipeptide Bearing α,α-Difluoromethyl Ketone Moiety as Electrophilic Warhead with Activity against Coronaviruses. Int. J. Mol. Sci. 2021, 22, 1398. [Google Scholar] [CrossRef] [PubMed]
- Citarella, A.; Dimasi, A.; Moi, D.; Passarella, D.; Scala, A.; Piperno, A.; Micale, N. Recent Advances in SARS-CoV-2 Main Protease Inhibitors: From Nirmatrelvir to Future Perspectives. Biomolecules 2023, 13, 1339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, H.; Liu, J.; He, L.; Yu, R.; Kang, C. Design of SARS-CoV-2 Mpro, PLpro Dual-Target Inhibitors Based on Deep Reinforcement Learning and Virtual Screening. Future Med. Chem. 2022, 14, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Sacco, M.D.; Ma, C.; Lagarias, P.; Gao, A.; Townsend, J.A.; Meng, X.; Dube, P.; Zhang, X.; Hu, Y.; Kitamura, N.; et al. Structure and Inhibition of the SARS-CoV-2 Main Protease Reveal Strategy for Developing Dual Inhibitors against Mpro and Cathepsin L. Sci. Adv. 2020, 6, eabe0751. [Google Scholar] [CrossRef] [PubMed]
- Daniloski, Z.; Jordan, T.X.; Wessels, H.-H.; Hoagland, D.A.; Kasela, S.; Legut, M.; Maniatis, S.; Mimitou, E.P.; Lu, L.; Geller, E.; et al. Identification of Required Host Factors for SARS-CoV-2 Infection in Human Cells. Cell 2021, 184, 92–105.e16. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Pišlar, A.; Mitrović, A.; Sabotič, J.; Pečar Fonović, U.; Perišić Nanut, M.; Jakoš, T.; Senjor, E.; Kos, J. The Role of Cysteine Peptidases in Coronavirus Cell Entry and Replication: The Therapeutic Potential of Cathepsin Inhibitors. PLoS Pathog. 2020, 16, e1009013. [Google Scholar] [CrossRef]
- Tang, T.; Jaimes, J.A.; Bidon, M.K.; Straus, M.R.; Daniel, S.; Whittaker, G.R. Proteolytic Activation of SARS-CoV-2 Spike at the S1/S2 Boundary: Potential Role of Proteases beyond Furin. ACS Infect. Dis. 2021, 7, 264–272. [Google Scholar] [CrossRef]
- Seth, S.; Batra, J.; Srinivasan, S. COVID-19: Targeting Proteases in Viral Invasion and Host Immune Response. Front. Mol. Biosci. 2020, 7, 215. [Google Scholar] [CrossRef] [PubMed]
- Bontempo, P.; De Masi, L.; Rigano, D. Functional Properties of Natural Products and Human Health. Nutrients 2023, 15, 2961. [Google Scholar] [CrossRef] [PubMed]
- Magi, G.; Marini, E.; Brenciani, A.; Di Lodovico, S.; Gentile, D.; Ruberto, G.; Cellini, L.; Nostro, A.; Facinelli, B.; Napoli, E. Chemical Composition of Pistacia vera L. Oleoresin and Its Antibacterial, Anti-Virulence and Anti-Biofilm Activities against Oral Streptococci, Including Streptococcus Mutans. Arch. Oral Biol. 2018, 96, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Musarra-Pizzo, M.; Pennisi, R.; Ben-Amor, I.; Mandalari, G.; Sciortino, M.T. Antiviral Activity Exerted by Natural Products against Human Viruses. Viruses 2021, 13, 828. [Google Scholar] [CrossRef] [PubMed]
- Prasansuklab, A.; Theerasri, A.; Rangsinth, P.; Sillapachaiyaporn, C.; Chuchawankul, S.; Tencomnao, T. Anti-COVID-19 Drug Candidates: A Review on Potential Biological Activities of Natural Products in the Management of New Coronavirus Infection. J. Tradit. Complement. Med. 2021, 11, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Muruganathan, N.; Dhanapal, A.R.; Baskar, V.; Muthuramalingam, P.; Selvaraj, D.; Aara, H.; Shiek Abdullah, M.Z.; Sivanesan, I. Recent Updates on Source, Biosynthesis, and Therapeutic Potential of Natural Flavonoid Luteolin: A Review. Metabolites 2022, 12, 1145. [Google Scholar] [CrossRef]
- Zhu, J.; Yan, H.; Shi, M.; Zhang, M.; Lu, J.; Wang, J.; Chen, L.; Wang, Y.; Li, L.; Miao, L.; et al. Luteolin Inhibits Spike Protein of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) Binding to Angiotensin-converting Enzyme 2. Phytother. Res. 2023, 37, 3508–3521. [Google Scholar] [CrossRef]
- Gezer, C. Potential Health Effects of the Popular Compound of Artichoke: Cynarin (CYN). Prog. Nutr. 2017, 19, 5–9. [Google Scholar]
- Heleno, S.A.; Carocho, M.; Reis, F.S.; Pires, T.C.S.P.; Pintado, M.; Ferreira, I.C.F.R.; Barros, L. Plant Extracts and SARS-CoV-2: Research and Applications. Life 2023, 13, 386. [Google Scholar] [CrossRef]
- Karakousis, N.D.; Gourgoulianis, K.I.; Kotsiou, O.S. The Role of Folic Acid in SARS-CoV-2 Infection: An Intriguing Linkage under Investigation. J. Pers. Med. 2023, 13, 561. [Google Scholar] [CrossRef]
- Krüger, N.; Kronenberger, T.; Xie, H.; Rocha, C.; Pöhlmann, S.; Su, H.; Xu, Y.; Laufer, S.A.; Pillaiyar, T. Discovery of Polyphenolic Natural Products as SARS-CoV-2 Mpro Inhibitors for COVID-19. Pharmaceuticals 2023, 16, 190. [Google Scholar] [CrossRef] [PubMed]
- Toelzer, C.; Gupta, K.; Yadav, S.K.N.; Borucu, U.; Davidson, A.D.; Kavanagh Williamson, M.; Shoemark, D.K.; Garzoni, F.; Staufer, O.; Milligan, R.; et al. Free Fatty Acid Binding Pocket in the Locked Structure of SARS-CoV-2 Spike Protein. Science (1979) 2020, 370, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Dichiara, M.; Gentile, D.; Prezzavento, O.; Marrazzo, A.; Rescifina, A.; Amata, E. Morphing of Ibogaine: A Successful Attempt into the Search for Sigma-2 Receptor Ligands. Int. J. Mol. Sci. 2019, 20, 488. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wei, J.; Qin, R.; Hou, J.; Zang, G.; Zhang, G.; Chen, T. Folic Acid: A Potential Inhibitor against SARS-CoV-2 Nucleocapsid Protein. Pharm. Biol. 2022, 60, 862–878. [Google Scholar] [CrossRef] [PubMed]
- Gentile, D.; Patamia, V.; Fuochi, V.; Furneri, P.M.; Rescifina, A. Natural Substances in the Fight of SARS-CoV-2: A Critical Evaluation Resulting from the Cross-Fertilization of Molecular Modeling Data with the Pharmacological Aspects. Curr. Med. Chem. 2021, 28, 8333–8383. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Matsuyama, S.; Shirato, K.; Maejima, M.; Fukushi, S.; Morikawa, S.; Taguchi, F. Entry from the Cell Surface of Severe Acute Respiratory Syndrome Coronavirus with Cleaved S Protein as Revealed by Pseudotype Virus Bearing Cleaved S Protein. J. Virol. 2008, 82, 11985–11991. [Google Scholar] [CrossRef]
- Wang, H.; Yang, P.; Liu, K.; Guo, F.; Zhang, Y.; Zhang, G.; Jiang, C. SARS Coronavirus Entry into Host Cells through a Novel Clathrin- and Caveolae-Independent Endocytic Pathway. Cell Res. 2008, 18, 290–301. [Google Scholar] [CrossRef]
- Xiang, Q.; Li, L.; Wu, J.; Tian, M.; Fu, Y. Application of Pseudovirus System in the Development of Vaccine, Antiviral-Drugs, and Neutralizing Antibodies. Microbiol. Res. 2022, 258, 126993. [Google Scholar] [CrossRef]
- Pennisi, R.; Trischitta, P.; Tamburello, M.P.; Barreca, D.; Mandalari, G.; Sciortino, M.T. Mechanistic Understanding of the Antiviral Properties of Pistachios and Zeaxanthin against HSV-1. Viruses 2023, 15, 1651. [Google Scholar] [CrossRef]
- Millet, J.; Whittaker, G. Murine Leukemia Virus (MLV)-Based Coronavirus Spike-Pseudotyped Particle Production and Infection. Bio Protoc. 2016, 6, e2035. [Google Scholar] [CrossRef]
- Millet, J.K.; Tang, T.; Nathan, L.; Jaimes, J.A.; Hsu, H.-L.; Daniel, S.; Whittaker, G.R. Production of Pseudotyped Particles to Study Highly Pathogenic Coronaviruses in a Biosafety Level 2 Setting. J. Vis. Exp. 2019, 145, e59010. [Google Scholar] [CrossRef]
- Gentile, D.; Floresta, G.; Patamia, V.; Nicosia, A.; Mineo, P.G.; Rescifina, A. Cucurbit[7]Uril as a Catalytic Nanoreactor for One-Pot Synthesis of Isoxazolidines in Water. Org. Biomol. Chem. 2020, 18, 1194–1203. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Amata, E.; Barbaraci, C.; Gentile, D.; Turnaturi, R.; Marrazzo, A.; Rescifina, A. A Structure- and Ligand-Based Virtual Screening of a Database of “Small” Marine Natural Products for the Identification of “Blue” Sigma-2 Receptor Ligands. Mar. Drugs 2018, 16, 384. [Google Scholar] [CrossRef] [PubMed]
- Gentile, D.; Floresta, G.; Patamia, V.; Chiaramonte, R.; Mauro, G.L.; Rescifina, A.; Vecchio, M. An Integrated Pharmacophore/Docking/3D-QSAR Approach to Screening a Large Library of Products in Search of Future Botulinum Neurotoxin a Inhibitors. Int. J. Mol. Sci. 2020, 21, 9470. [Google Scholar] [CrossRef]
- Floresta, G.; Patamia, V.; Gentile, D.; Molteni, F.; Santamato, A.; Rescifina, A.; Vecchio, M. Repurposing of FDA-Approved Drugs for Treating Iatrogenic Botulism: A Paired 3D-QSAR/Docking Approach. ChemMedChem 2020, 15, 256–262. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A Point-charge Force Field for Molecular Mechanics Simulations of Proteins Based on Condensed-phase Quantum Mechanical Calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Krieger, E.; Dunbrack, R.L.; Hooft, R.W.W.; Krieger, B. Assignment of Protonation States in Proteins and Ligands: Combining PKa Prediction with Hydrogen Bonding Network Optimization. In Computational Drug Discovery and Design; Springer: Berlin/Heidelberg, Germany, 2012; pp. 405–421. [Google Scholar]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.E.M.; Vriend, G. Fast Empirical PKa Prediction by Ewald Summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, Efficient Generation of High-quality Atomic Charges. AM1-BCC Model: II. Parameterization and Validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New Ways to Boost Molecular Dynamics Simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, M.; Barbera, V.; Guerra, S.; Bernardi, A. Facile functionalization of sp2 carbon allotropes with a biobased janus molecule. Rubber Chem. Technol. 2017, 90, 285–307. [Google Scholar] [CrossRef]
- Ou, J.; Lan, W.; Wu, X.; Zhao, T.; Duan, B.; Yang, P.; Ren, Y.; Quan, L.; Zhao, W.; Seto, D.; et al. Tracking SARS-CoV-2 Omicron Diverse Spike Gene Mutations Identifies Multiple Inter-Variant Recombination Events. Signal Transduct. Target. Ther. 2022, 7, 138. [Google Scholar] [CrossRef] [PubMed]
- Meng, B.; Abdullahi, A.; Ferreira, I.A.T.M.; Goonawardane, N.; Saito, A.; Kimura, I.; Yamasoba, D.; Gerber, P.P.; Fatihi, S.; Rathore, S.; et al. Altered TMPRSS2 Usage by SARS-CoV-2 Omicron Impacts Infectivity and Fusogenicity. Nature 2022, 603, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Metzdorf, K.; Jacobsen, H.; Greweling-Pils, M.C.; Hoffmann, M.; Lüddecke, T.; Miller, F.; Melcher, L.; Kempf, A.M.; Nehlmeier, I.; Bruder, D.; et al. TMPRSS2 Is Essential for SARS-CoV-2 Beta and Omicron Infection. Viruses 2023, 15, 271. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Sarma, P.; Bhattacharyya, A.; Prajapat, M.; Kumar, S.; Prakash, A.; Medhi, B. Folic Acid as Placebo in Controlled Clinical Trials of Hydroxychloroquine Prophylaxis in COVID-19: Is It Scientifically Justifiable? Med. Hypotheses 2021, 149, 110539. [Google Scholar] [CrossRef]
- Ugurel, O.M.; Mutlu, O.; Sariyer, E.; Kocer, S.; Ugurel, E.; Inci, T.G.; Ata, O.; Turgut-Balik, D. Evaluation of the Potency of FDA-Approved Drugs on Wild Type and Mutant SARS-CoV-2 Helicase (Nsp13). Int. J. Biol. Macromol. 2020, 163, 1687–1696. [Google Scholar] [CrossRef]
- Zhang, Y.; Pang, Y.; Xu, B.; Chen, X.; Liang, S.; Hu, J.; Luo, X. Folic Acid Restricts SARS-CoV-2 Invasion by Methylating ACE2. Front. Microbiol. 2022, 13, 980903. [Google Scholar] [CrossRef]
- Škrbić, R.; Travar, M.; Stojiljković, M.P.; Djuric, D.M.; Suručić, R. Folic Acid and Leucovorin Have Potential to Prevent SARS-CoV-2-Virus Internalization by Interacting with S-Glycoprotein/Neuropilin-1 Receptor Complex. Molecules 2023, 28, 2294. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yang, C.; Xia, J.; Li, N.; Xiong, W. Luteolin Is a Potential Inhibitor of COVID-19: An in Silico Analysis. Medicine 2023, 102, e35029. [Google Scholar] [CrossRef] [PubMed]
- Júnior, M.L.P.; de Sousa Junior, R.T.; Nze, G.D.A.; Giozza, W.F.; Júnior, L.A.R. Evaluation of Peppermint Leaf Flavonoids as SARS-CoV-2 Spike Receptor-Binding Domain Attachment Inhibitors to the Human ACE2 Receptor: A Molecular Docking Study. Open J. Biophys. 2022, 12, 132–152. [Google Scholar] [CrossRef]
- Shawan, M.M.A.K.; Halder, S.K.; Hasan, M.A. Luteolin and Abyssinone II as Potential Inhibitors of SARS-CoV-2: An in Silico Molecular Modeling Approach in Battling the COVID-19 Outbreak. Bull. Natl. Res. Cent. 2021, 45, 27. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, W.; Perez-Lemus, G.R.; Menéndez, C.A.; Byléhn, F.; de Pablo, J.J. Molecular Characterization of COVID-19 Therapeutics: Luteolin as an Allosteric Modulator of the Spike Protein of SARS-CoV-2. Mol. Syst. Des. Eng. 2022, 7, 58–66. [Google Scholar] [CrossRef]
- Munafò, F.; Donati, E.; Brindani, N.; Ottonello, G.; Armirotti, A.; De Vivo, M. Quercetin and Luteolin Are Single-Digit Micromolar Inhibitors of the SARS-CoV-2 RNA-Dependent RNA Polymerase. Sci. Rep. 2022, 12, 10571. [Google Scholar] [CrossRef]
- Dissook, S.; Umsumarng, S.; Mapoung, S.; Semmarath, W.; Arjsri, P.; Srisawad, K.; Dejkriengkraikul, P. Luteolin-Rich Fraction from Perilla Frutescens Seed Meal Inhibits Spike Glycoprotein S1 of SARS-CoV-2-Induced NLRP3 Inflammasome Lung Cell Inflammation via Regulation of JAK1/STAT3 Pathway: A Potential Anti-Inflammatory Compound against Inflammation-Induced Long-COVID. Front. Med. 2023, 9, 1072056. [Google Scholar] [CrossRef]
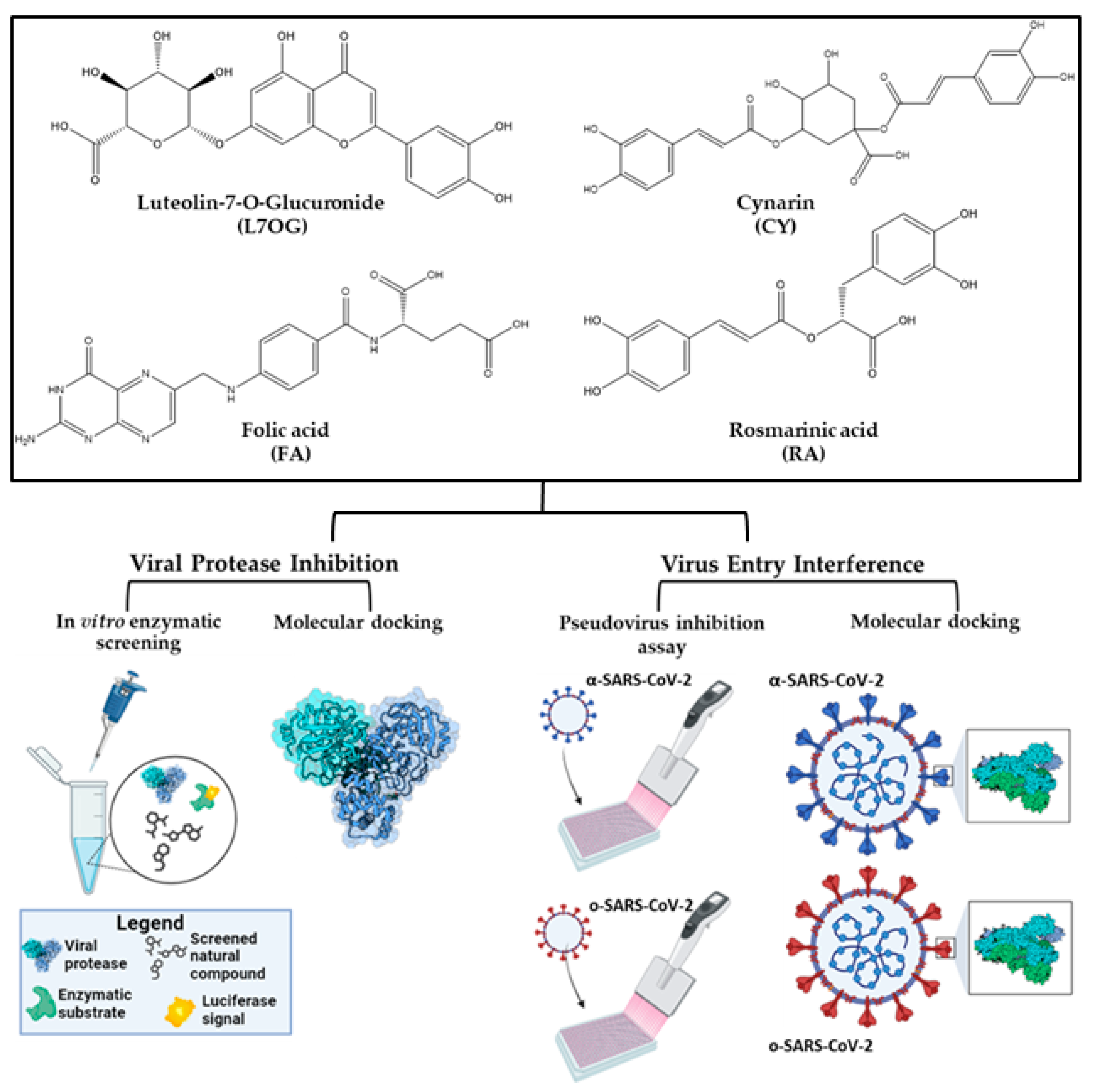
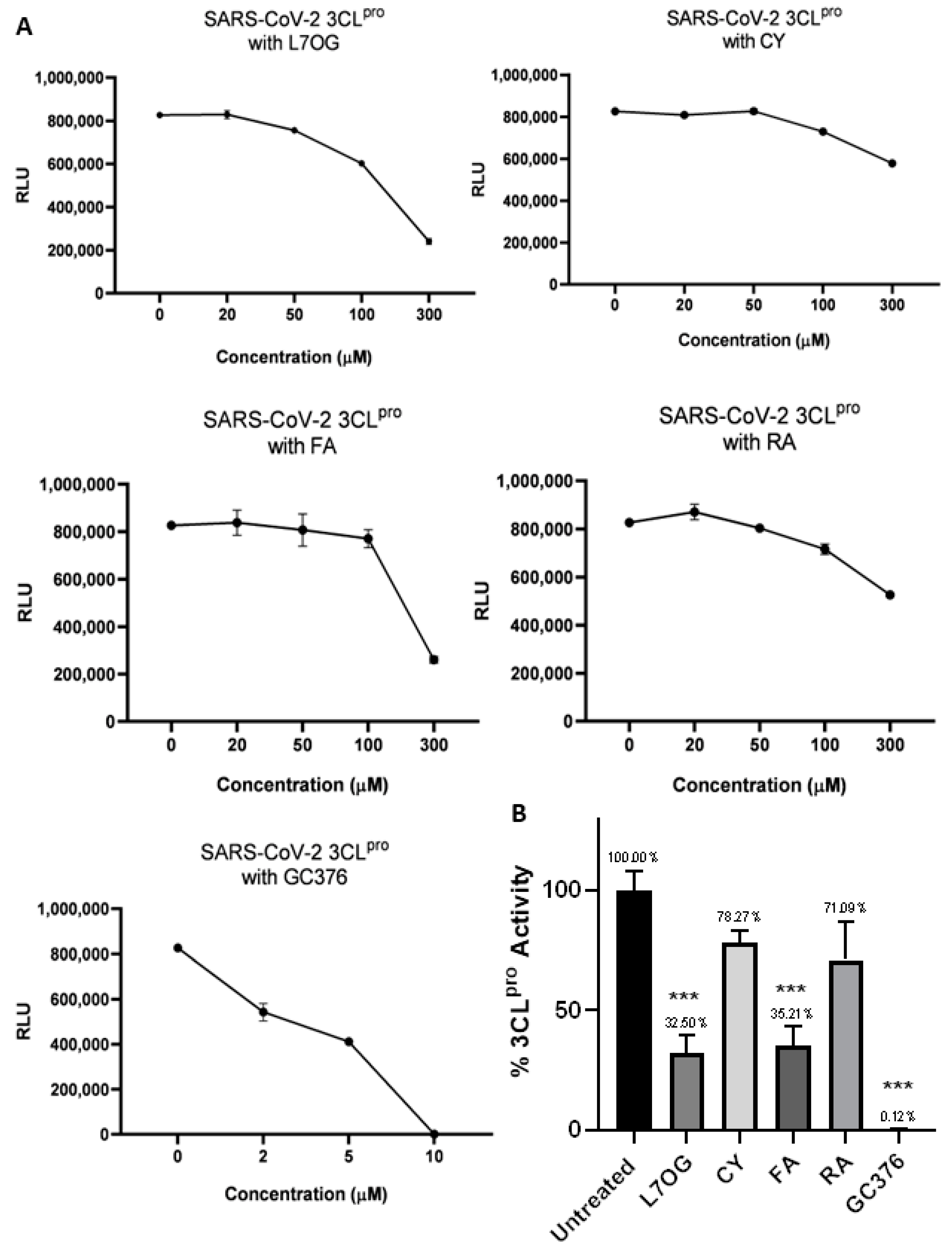
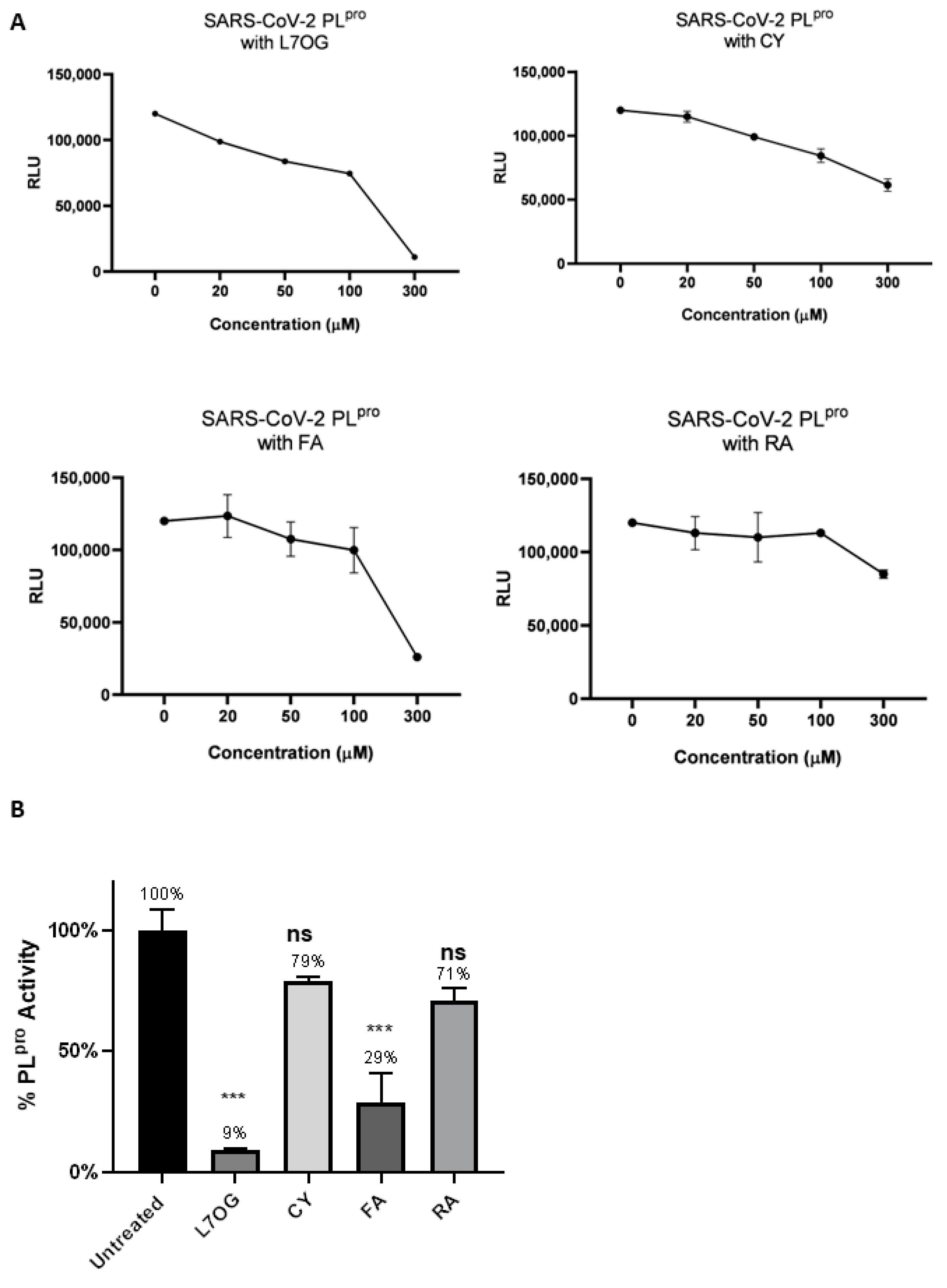

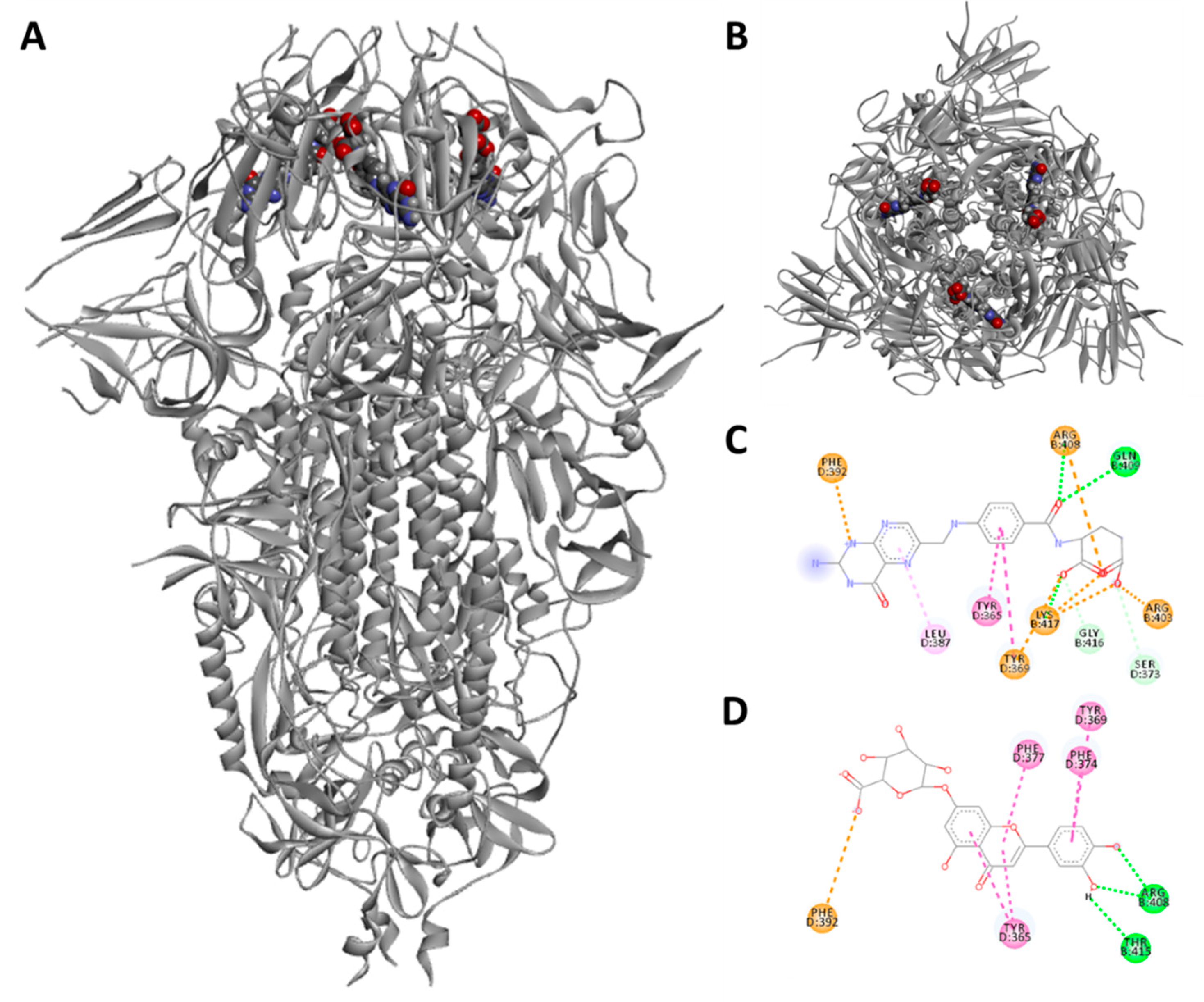

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 3CLpro | PLpro |
|---|---|---|
| L7OG | 170.0 | 152.7 |
| CY | 573.9 | 150.0 |
| FA | 232.0 | 183.0 |
| RA | 394.0 | 338.2 |
| GC376 | 5.5 | — |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pennisi, R.; Gentile, D.; Rescifina, A.; Napoli, E.; Trischitta, P.; Piperno, A.; Sciortino, M.T. An Integrated In Silico and In Vitro Approach for the Identification of Natural Products Active against SARS-CoV-2. Biomolecules 2024, 14, 43. https://doi.org/10.3390/biom14010043
Pennisi R, Gentile D, Rescifina A, Napoli E, Trischitta P, Piperno A, Sciortino MT. An Integrated In Silico and In Vitro Approach for the Identification of Natural Products Active against SARS-CoV-2. Biomolecules. 2024; 14(1):43. https://doi.org/10.3390/biom14010043
Chicago/Turabian StylePennisi, Rosamaria, Davide Gentile, Antonio Rescifina, Edoardo Napoli, Paola Trischitta, Anna Piperno, and Maria Teresa Sciortino. 2024. "An Integrated In Silico and In Vitro Approach for the Identification of Natural Products Active against SARS-CoV-2" Biomolecules 14, no. 1: 43. https://doi.org/10.3390/biom14010043
APA StylePennisi, R., Gentile, D., Rescifina, A., Napoli, E., Trischitta, P., Piperno, A., & Sciortino, M. T. (2024). An Integrated In Silico and In Vitro Approach for the Identification of Natural Products Active against SARS-CoV-2. Biomolecules, 14(1), 43. https://doi.org/10.3390/biom14010043