
AlphaFold Accurately Predicts the Structure of Ribosomally Synthesized and Post-Translationally Modified Peptide Biosynthetic Enzymes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Modeling ATP-Grasp Ligase Family Enzymes
2.2. Set Parameters
2.3. Application of Pipeline to Other RiPP Biosynthetic Enzymes
2.4. Evaluation of Models
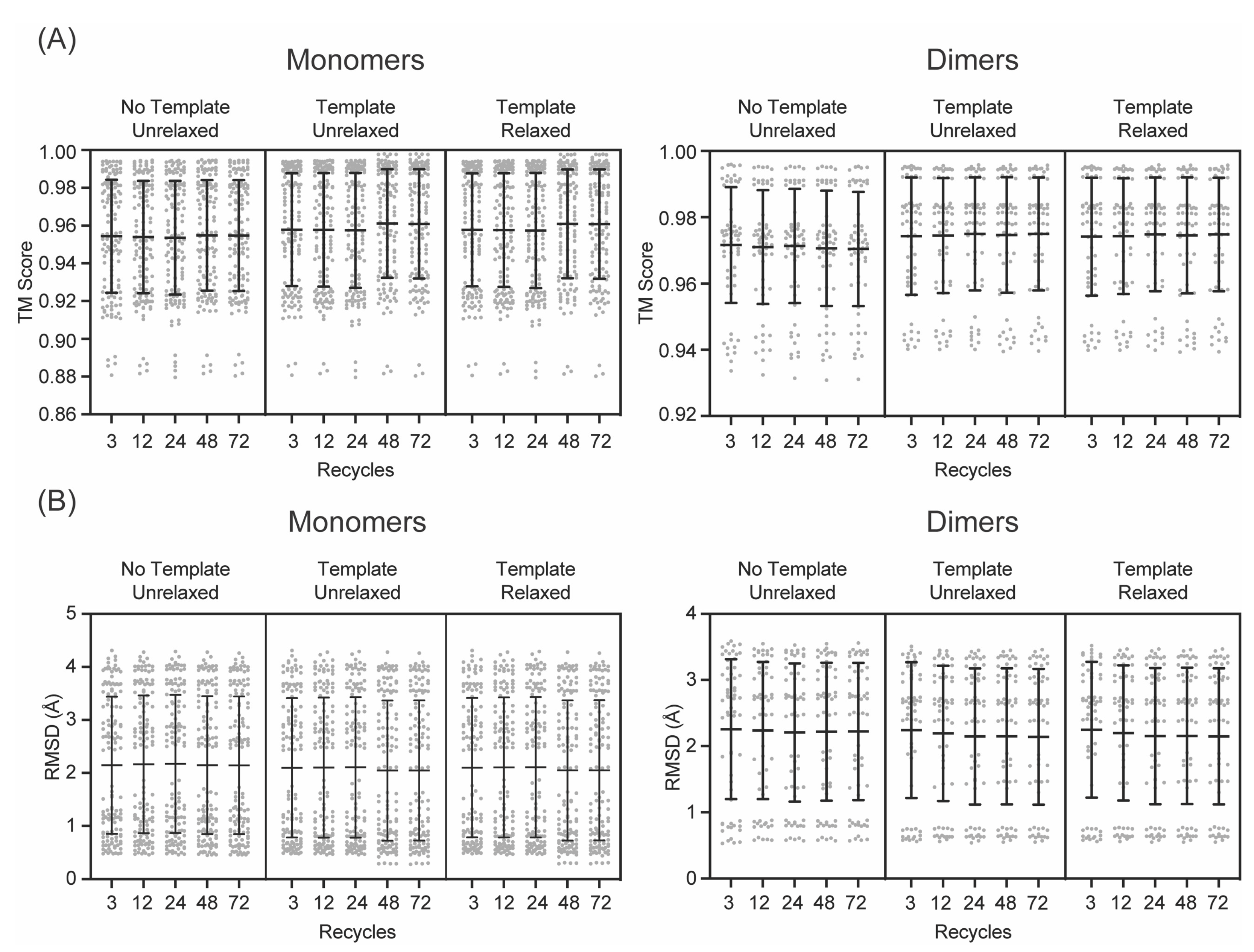
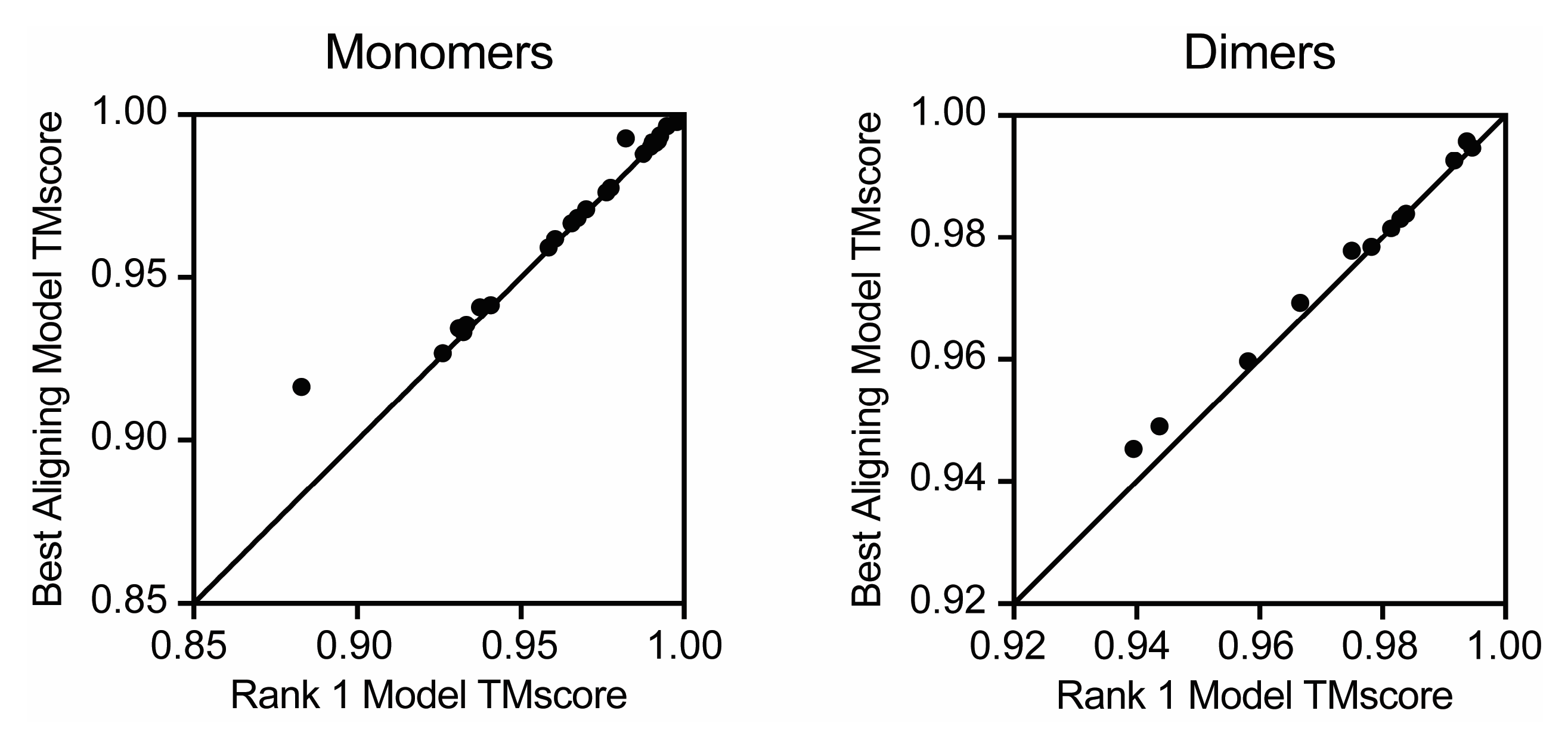
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Ongpipattanakul, C.; Desormeaux, E.K.; DiCaprio, A.; van der Donk, W.A.; Mitchell, D.A.; Nair, S.K. Mechanism of action of ribosomally synthesized and post- translationally modified peptides. Chem. Rev. 2022, 122, 14722–14814. [Google Scholar] [CrossRef]
- Montalban-Lopez, M.; Scott, T.A.; Ramesh, S.; Rahman, I.R.; van Heel, A.J.; Viel, J.H.; Bandarian, V.; Dittmann, E.; Genilloud, O.; Goto, Y.; et al. New developments in RiPP discovery, enzymology and engineering. Nat. Prod. Rep. 2021, 38, 130–239. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; van der Donk, W.A. Ribosomally synthesized and post-translationally modified peptide natural products: New insights into the role of leader and core peptides during biosynthesis. Chem. Eur. J. 2013, 19, 7662–7677. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lennard, K.R.; He, C.; Walker, M.C.; Ball, A.T.; Doigneaux, C.; Tavassoli, A.; van der Donk, W.A. A lanthipeptide library used to identify a protein-protein interaction inhibitor. Nat. Chem. Biol. 2018, 14, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Urban, J.H.; Moosmeier, M.A.; Aumuller, T.; Thein, M.; Bosma, T.; Rink, R.; Groth, K.; Zulley, M.; Siegers, K.; Tissot, K.; et al. Phage display and selection of lanthipeptides on the carboxy-terminus of the gene-3 minor coat protein. Nat. Commun. 2017, 8, 1500. [Google Scholar] [CrossRef]
- Zhao, X.H.; Li, Z.B.; Kuipers, O.P. Mimicry of a non-ribosomally produced antimicrobial, brevicidine, by ribosomal synthesis and post-translational modification. Cell Chem. Biol. 2020, 27, 1262–1271. [Google Scholar] [CrossRef]
- Vinogradov, A.A.; Zhang, Y.; Hamada, K.; Chang, J.S.; Okada, C.; Nishimura, H.; Terasaka, N.; Goto, Y.; Ogata, K.; Sengoku, T.; et al. De novo discovery of thiopeptide pseudo-natural products acting as potent and selective TNIK kinase inhibitors. J. Am. Chem. Soc. 2022, 144, 20332–20341. [Google Scholar] [CrossRef]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. Charmm—A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Liwo, A.; Ołdziej, S.; Pincus, M.R.; Wawak, R.J.; Rackovsky, S.; Scheraga, H.A. A united-residue force field for off-lattice protein-structure simulations. I. Functional forms and parameters of long-range side-chain interaction potentials from protein crystal data. J. Comput. Chem. 1997, 18, 849–873. [Google Scholar]
- He, Y.; Mozolewska, M.A.; Krupa, P.; Sieradzan, A.K.; Wirecki, T.K.; Liwo, A.; Kachlishvili, K.; Rackovsky, S.; Jagiela, D.; Slusarz, R.; et al. Lessons from application of the UNRES force field to predictions of structures of CASP10 targets. Proc. Natl. Acad. Sci. USA 2013, 110, 14936–14941. [Google Scholar] [CrossRef]
- He, Y.; Xiao, Y.; Liwo, A.; Scheraga, H.A. Exploring the Parameter Space of the Coarse-Grained UNRES Force Field by Random Search: Selecting a Transferable Medium-Resolution Force Field. J. Comput. Chem. 2009, 30, 2127–2135. [Google Scholar] [CrossRef] [PubMed]
- Sander, C.; Schneider, R. Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins 1991, 9, 56–68. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2010. [Google Scholar] [CrossRef]
- Ruff, K.M.; Pappu, R.V. AlphaFold and Implications for Intrinsically Disordered Proteins. J. Mol. Biol. 2021, 433, 167208. [Google Scholar] [CrossRef] [PubMed]
- Bagdonas, H.; Fogarty, C.A.; Fadda, E.; Agirre, J. The case for post-predictional modifications in the AlphaFold Protein Structure Database. Nat. Struct. Mol. Biol. 2021, 28, 869–870. [Google Scholar] [CrossRef]
- Binder, J.L.; Berendzen, J.; Stevens, A.O.; He, Y.; Wang, J.; Dokholyan, N.V.; Oprea, T.I. AlphaFold illuminates half of the dark human proteins. Curr. Opin. Struct. Biol. 2022, 74, 102372. [Google Scholar] [CrossRef]
- Stevens, A.O.; He, Y. Benchmarking the Accuracy of AlphaFold 2 in Loop Structure Prediction. Biomolecules 2022, 12, 985. [Google Scholar] [CrossRef]
- Javed, R.; Jain, A.; Duque, T.; Hendrix, E.; Paddar, M.A.; Khan, S.; Claude-Taupin, A.; Jia, J.; Allers, L.; Wang, F.; et al. Mammalian ATG8 proteins maintain autophagosomal membrane integrity through ESCRTs. EMBO J. 2023, 42, e112845. [Google Scholar] [CrossRef]
- Yin, R.; Feng, B.Y.; Varshney, A.; Pierce, B.G. Benchmarking AlphaFold for protein complex modeling reveals accuracy determinants. Protein Sci. 2022, 31, e4379. [Google Scholar] [CrossRef]
- Chen, X.; Morehead, A.; Liu, J.; Cheng, J. A gated graph transformer for protein complex structure quality assessment and its performance in CASP15. Bioinformatics 2023, 39, i308–i317. [Google Scholar] [CrossRef]
- Zhu, W.; Shenoy, A.; Kundrotas, P.; Elofsson, A. Evaluation of AlphaFold-Multimer prediction on multi-chain protein complexes. Bioinformatics 2023, 39, btad424. [Google Scholar] [CrossRef]
- Jumper, J.; Hassabis, D. Protein structure predictions to atomic accuracy with AlphaFold. Nat. Methods 2022, 19, 11–12. [Google Scholar] [CrossRef]
- Mirdita, M.; Schutze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Condurso, H.L.; Li, G.; Ding, Y.; Bruner, S.D. Structural basis for precursor protein–directed ribosomal peptide macrocyclization. Nat. Chem. Biol. 2016, 12, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Patel, K.; Zhang, Y.; Pugmire, J.K.; Ding, Y.; Bruner, S.D. Structural and biochemical studies of an iterative ribosomal peptide macrocyclase. Proteins 2021, 90, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Kosek, D.; Liu, H.-B.; Ohlemacher, S.I.; Blackburne, B.; Nikolskaya, A.; Makarova, K.S.; Sun, J.; Barry, C.E., III.; Koonin, E.V.; et al. Structural basis for a dual function ATP grasp ligase that installs single and bicyclic ω-ester macrocycles in a new multicore RiPP natural product. J. Am. Chem. Soc. 2021, 143, 8056–8068. [Google Scholar] [CrossRef]
- Song, I.; Kim, Y.; Yu, J.; Go, S.Y.; Lee, H.G.; Song, W.J.; Kim, S. Molecular mechanism underlying substrate recognition of the peptide macrocyclase PsnB. Nat. Chem. Biol. 2021, 17, 1123–1131. [Google Scholar] [CrossRef]
- Ouchi, T.; Tomita, T.; Horie, A.; Yoshida, A.; Takahashi, K.; Nishida, H.; Lassak, K.; Taka, H.; Mineki, R.; Fujimura, T.; et al. Lysine and arginine biosyntheses mediated by a common carrier protein in Sulfolobus. Nat. Chem. Biol. 2013, 9, 277–283. [Google Scholar] [CrossRef]
- Ortega, M.A.; Hao, Y.; Zhang, Q.; Walker, M.C.; van der Donk, W.A.; Nair, S.K. Structure and mechanism of the tRNA-dependent lantibiotic dehydratase NisB. Nature 2015, 517, 509–512. [Google Scholar] [CrossRef]
- Dong, S.H.; Tang, W.X.; Lukk, T.; Yu, Y.; Nair, S.K.; van der Donk, W.A. The enterococcal cytolysin synthetase has an unanticipated lipid kinase fold. eLife 2015, 4, e07607. [Google Scholar] [CrossRef] [PubMed]
- Cogan, D.P.; Hudson, G.A.; Zhang, Z.G.; Pogorelov, T.V.; van der Donk, W.A.; Mitchell, D.A.; Nair, S.K. Structural insights into enzymatic [4+2] aza-cycloaddition in thiopeptide antibiotic biosynthesis. Proc. Natl. Acad. Sci. USA 2017, 114, 12928–12933. [Google Scholar] [CrossRef] [PubMed]
- Bothwell, I.R.; Cogan, D.P.; Kim, T.; Reinhardt, C.J.; van der Donk, W.A.; Nair, S.K. Characterization of glutamyl-tRNA-dependent dehydratases using nonreactive substrate mimics. Proc. Natl. Acad. Sci. USA 2019, 116, 17245–17250. [Google Scholar] [CrossRef] [PubMed]
- Koehnke, J.; Bent, A.F.; Zollman, D.; Smith, K.; Houssen, W.E.; Zhu, X.F.; Mann, G.; Lebl, T.; Scharff, R.; Shirran, S.; et al. The cyanobactin heterocyclase enzyme: A processive adenylase that operates with a defined order of reaction. Angew. Chem. Int. Ed. 2013, 52, 13991–13996. [Google Scholar] [CrossRef]
- Dong, S.H.; Liu, A.D.; Mahanta, N.; Mitchell, D.A.; Nair, S.K. Mechanistic basis for ribosomal peptide backbone modifications. ACS Cent. Sci. 2019, 5, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Pierce, E.; McIntosh, J.; Schmidt, E.W.; Nair, S.K. Structures of cyanobactin maturation enzymes define a family of transamidating proteases. Chem. Biol. 2012, 19, 1411–1422. [Google Scholar] [CrossRef]
- Song, H.; van der Velden, N.S.; Shiran, S.L.; Bleiziffer, P.; Zach, C.; Sieber, R.; Imani, A.S.; Krausbeck, F.; Aebi, M.; Freeman, M.F.; et al. A molecular mechanism for the enzymatic methylation of nitrogen atoms within peptide bonds. Sci. Adv. 2018, 4, eaat2720. [Google Scholar] [CrossRef]
- Chekan, J.R.; Estrada, P.; Covello, P.S.; Nair, S.K. Characterization of the macrocyclase involved in the biosynthesis of RiPP cyclic peptides in plants. Proc. Natl. Acad. Sci. USA 2017, 114, 6551–6556. [Google Scholar] [CrossRef]
- Ghodge, S.V.; Biernat, K.A.; Bassett, S.J.; Redinbo, M.R.; Bowers, A.A. Post-translational claisen condensation and decarboxylation en route to the bicyclic core of pantocin A. J. Am. Chem. Soc. 2016, 138, 5487–5490. [Google Scholar] [CrossRef]
- Sumida, T.; Dubiley, S.; Wilcox, B.; Severinov, K.; Tagami, S. Structural basis of leader peptide recognition in lasso peptide biosynthesis pathway. ACS Chem. Biol. 2019, 14, 1619–1627. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.H.; Kulikovsky, A.; Zukher, I.; Estrada, P.; Dubiley, S.; Severinov, K.; Nair, S.K. Biosynthesis of the RiPP trojan horse nucleotide antibiotic microcin C is directed by the N-formyl of the peptide precursor. Chem. Sci. 2019, 10, 2391–2395. [Google Scholar] [CrossRef]
- Lee, J.; Hao, Y.; Blair, P.M.; Melby, J.O.; Agarwal, V.; Burkhart, B.J.; Nair, S.K.; Mitchell, D.A. Structural and functional insight into an unexpectedly selective N-methyltransferase involved in plantazolicin biosynthesis. Proc. Natl. Acad. Sci. USA. 2013, 110, 12954–12959. [Google Scholar] [CrossRef] [PubMed]
- Mo, T.L.; Yuan, H.; Wang, F.T.; Ma, S.Z.; Wang, J.X.; Li, T.; Liu, G.F.; Yu, S.N.; Tan, X.S.; Ding, W.; et al. Convergent evolution of the Cys decarboxylases involved in aminovinyl-cysteine (AviCys) biosynthesis. FEBS Lett. 2019, 593, 573–580. [Google Scholar] [CrossRef]
- Hao, Y.; Pierce, E.; Roe, D.; Morita, M.; McIntosh, J.A.; Agarwal, V.; Cheatham, T.E.; Schmidt, E.W.; Nair, S.K. Molecular basis for the broad substrate selectivity of a peptide prenyltransferase. Proc. Natl. Acad. Sci. USA 2016, 113, 14037–14042. [Google Scholar] [CrossRef]
- Liu, S.S.; Guo, H.; Zhang, T.L.; Han, L.; Yao, P.F.; Zhang, Y.; Rong, N.Y.; Yu, Y.; Lan, W.X.; Wang, C.X.; et al. Structure-based mechanistic Insights into terminal amide synthase in nosiheptide-represented thiopeptides biosynthesis. Sci. Rep. 2015, 5, 12744. [Google Scholar] [CrossRef]
- An, L.N.; Cogan, D.P.; Navo, C.D.; Jimenez-Oses, G.; Nair, S.K.; van der Donk, W.A. Substrate-assisted enzymatic formation of lysinoalanine in duramycin. Nat. Chem. Biol. 2018, 14, 928–933. [Google Scholar] [CrossRef]
- Zhang, C.X.; Shine, M.; Pyle, A.M.; Zhang, Y. US-align: Universal structure alignments of proteins, nucleic acids, and macromolecular complexes. Nat. Methods 2022, 19, 1109–1115. [Google Scholar] [CrossRef]
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins 2004, 57, 702–710. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gordon, C.H.; Hendrix, E.; He, Y.; Walker, M.C. AlphaFold Accurately Predicts the Structure of Ribosomally Synthesized and Post-Translationally Modified Peptide Biosynthetic Enzymes. Biomolecules 2023, 13, 1243. https://doi.org/10.3390/biom13081243
Gordon CH, Hendrix E, He Y, Walker MC. AlphaFold Accurately Predicts the Structure of Ribosomally Synthesized and Post-Translationally Modified Peptide Biosynthetic Enzymes. Biomolecules. 2023; 13(8):1243. https://doi.org/10.3390/biom13081243
Chicago/Turabian StyleGordon, Catriona H., Emily Hendrix, Yi He, and Mark C. Walker. 2023. "AlphaFold Accurately Predicts the Structure of Ribosomally Synthesized and Post-Translationally Modified Peptide Biosynthetic Enzymes" Biomolecules 13, no. 8: 1243. https://doi.org/10.3390/biom13081243
APA StyleGordon, C. H., Hendrix, E., He, Y., & Walker, M. C. (2023). AlphaFold Accurately Predicts the Structure of Ribosomally Synthesized and Post-Translationally Modified Peptide Biosynthetic Enzymes. Biomolecules, 13(8), 1243. https://doi.org/10.3390/biom13081243