Abstract
The progressive degeneration of the skeletal musculature in Duchenne muscular dystrophy is accompanied by reactive myofibrosis, fat substitution, and chronic inflammation. Fibrotic changes and reduced tissue elasticity correlate with the loss in motor function in this X-chromosomal disorder. Thus, although dystrophinopathies are due to primary abnormalities in the DMD gene causing the almost-complete absence of the cytoskeletal Dp427-M isoform of dystrophin in voluntary muscles, the excessive accumulation of extracellular matrix proteins presents a key histopathological hallmark of muscular dystrophy. Animal model research has been instrumental in the characterization of dystrophic muscles and has contributed to a better understanding of the complex pathogenesis of dystrophinopathies, the discovery of new disease biomarkers, and the testing of novel therapeutic strategies. In this article, we review how mass-spectrometry-based proteomics can be used to study changes in key components of the endomysium, perimysium, and epimysium, such as collagens, proteoglycans, matricellular proteins, and adhesion receptors. The mdx-4cv mouse diaphragm displays severe myofibrosis, making it an ideal model system for large-scale surveys of systematic alterations in the matrisome of dystrophic fibers. Novel biomarkers of myofibrosis can now be tested for their appropriateness in the preclinical and clinical setting as diagnostic, pharmacodynamic, prognostic, and/or therapeutic monitoring indicators.
Keywords:
biglycan; collagen; dystrophin; dystrophinopathy; extracellular matrix; fibronectin; fibrosis; matrisome; mdx; periostin 1. Introduction
Animal model research plays an important role in the field of biomedicine and, particularly, in biomolecular investigations into the basic mechanisms that underlie the cellular pathogenesis of common human diseases [1]. In the case of monogenetic disorders, such as the X-chromosomally inherited muscle-wasting disease named Duchenne muscular dystrophy [2], the animal models should ideally (i) mimic a similar genotype that properly reflects the primary abnormality leading to the complex disease phenotype; (ii) show comparable patterns of disease onset, progression, and severity; (iii) display comparable secondary changes as observed in the human disorder such as multi-system alterations and pathobiochemical crosstalk; (iv) be characterized by complex pathophysiological features that resemble the disease in patients including myonecrosis and myofibrosis; (v) exhibit equivalent effects due to changes in energy metabolism, cellular signaling pathways, physiological adaptations, and natural repair mechanisms; (vi) trigger analogous responses of the innate and/or adaptive immune system; (vii) be relatively cost-effective and easy to breed, handle, and house; (viii) be appropriate for efficient genetic manipulations; (ix) be suitable for standardized physiological, biochemical, cellular, or surgical manipulations, and (x) yield sufficient cellular and biofluid material for meaningful bioanalysis and high-throughput surveys [3,4,5]. Unless too many biological factors and mechanisms cause major differences in the pathophysiological progression between animal models and patients, then the results from studying disease models can at least partially be extrapolated for a better understanding of human disorders [6].
A variety of multifaceted skeletal-muscle-wasting diseases exist which are often characterized by a complex pathogenesis. Because of the high abundance of muscle fibers and their many interactions with other tissues, muscular disorders are often associated with body-wide changes and adaptations [7]. Since fibrosis is generally linked to chronic degeneration, oxidative stress, and inflammatory processes in human disorders [8,9], it is not surprising that myofibrosis is also seen in a variety of neuromuscular diseases [10]. Characteristic hallmarks of fibrotic changes in the skeletal musculature are the excess deposition of collagens and other extracellular matrix components [11]. Myofibrosis can be both of a primary or secondary nature due to reactive processes, such as the primary disorganization of the extracellular matrix in COL6-related myopathies [12,13,14] or reactive fibrosis as a secondary complication of dystrophinopathies [15,16,17], respectively.
X-linked muscular dystrophies that are based on primary abnormalities in the human DMD gene [2] and trigger the almost-complete absence of the full-length Dp427-M isoform of the cytoskeletal protein dystrophin [18] are highly progressive muscle-wasting diseases. Since the DMD gene contains several promoters that produce eight distinct and tissue-specific protein isoforms, different mutations affect individual patients in a disproportionate way. This is reflected by the dissimilar severity and onset of respiratory dysfunction, cardiomyopathy, kidney failure, liver disease, and abnormalities in the central nervous system [19]. Thus, the pattern of altered organ crosstalk, secondary abnormalities, and body-wide adaptations differ within patient cohorts. In the skeletal musculature, dystrophin is closely associated with a sarcolemmal glycoprotein complex, which is intrinsically involved in cellular signaling, the maintenance of plasmalemma integrity, the organization of the cytoskeleton, and the provision of lateral force transmission [20,21]. In the absence of the anchoring function of dystrophin, the protein complex consisting of dystroglycans, sarcoglycans, sarcospan, dystrobrevins, and syntrophins disintegrates and renders dystrophic fibers more susceptible to micro-rupturing. This causes an increased influx of Ca2+ ions into damaged muscle cells and promotes enhanced proteolysis and muscle necrosis [22,23,24]. The Ca2+-dependent damage pathway of myonecrosis is followed by fat substitution, chronic inflammation, and reactive myofibrosis [25,26,27].
This makes the development of novel anti-fibrotic strategies a crucial aim of muscle pharmacotherapy and applied myology in order to prevent and/or reverse fibrosis-related abnormalities that are linked to muscle dysfunction [28]. Animal model research plays a crucial role in the detailed analysis of the genetic, biochemical, and physiological parameters that underlie the precise mechanisms of fibrosis [29]. A critical aspect of this research focuses on how abnormal processes within the three main muscle-associated layers of connective tissue, i.e., the endomysium that surrounds contractile fibers, the perimysium that forms bundles around muscle fibers, and the epimysium that surrounds individual skeletal muscles [30,31,32], relate to chronic cellular damage, inflammatory responses, stem cell exhaustion, and alterations in the signaling pathways that regulate the fibro-adipogenic progenitor cell niche [33,34,35].
This article summarizes mass-spectrometry-based proteomic strategies to study the extracellular matrix in order to carry out unbiased investigations into the mechanisms that underlie skeletal muscle fibrosis. This includes an outline of the general importance and diverse utilization of the mdx-4cv mouse model in Duchenne muscular dystrophy research [36] with a special focus on fibrosis-related changes in the skeletal musculature as judged by mass spectrometric studies [37]. In contrast to the most widely used and naturally occurring mdx-23 mouse, which is characterized by a point mutation in exon-23 of the murine Dmd gene and elevated levels of serum creatine kinase in association with histological lesions and cycles of muscle necrosis and regeneration [38,39,40], the mdx-4cv mouse has been generated by chemical mutagenesis [41]. This mouse model harbors a nonsense mutation that introduces a premature stop codon in exon-53 [42,43,44] and exhibits a considerably lower frequency of dystrophin-positive revertants [45] and different mutation-related alterations in the dystrophin isoform expression patterns [36] compared to the natural mdx-23 mouse [40]. Dystrophin-lacking mdx-type mice exhibit varying degrees of fibrotic changes in the general musculature [46,47,48] but show severe reactive myofibrosis in the diaphragm muscle [49,50,51]. This histopathological hallmark in mdx-type diaphragm muscles is of considerable importance in the field of dystrophinopathy research [52,53], since endomysial fibrosis has been clearly established as a reliable marker that correlates well with motor loss in Duchenne patients [54]. The aged mdx-4cv mouse diaphragm, especially, shows high levels of extracellular matrix deposition [55], making it a suitable surrogate for studying reactive myofibrosis in dystrophinopaty, as discussed in this review.
2. Proteomic Strategies to Study the Extracellular Matrix
Mass spectrometry has been instrumental for the systematic cataloguing and characterization of the human proteome [56,57,58,59,60]. A large number of studies have focused on the establishment of the skeletal muscle proteome with over 10,000 identified protein species in contractile tissue specimens from both humans and a variety of animal species that are frequently used in biomedical research [61,62,63,64,65,66]. Since the extracellular matrix plays a key role in tissue organization, cellular integrity, wound healing, and repair mechanisms, proteomics has been extensively applied to characterize the matrisome and its associated proteins in many cell/tissue/organ types and species [67,68,69]. Cell biological studies in combination with advanced biochemical separation approaches and systematic protein identification by mass spectrometry have been instrumental for the general proteomic cataloguing of the overarching extracellular matrix system that is present in many different cell and tissue types [70,71,72,73]. The bioanalytical advantages versus technical limitations of the main methodological strategies and proteomic analysis pipelines that are routinely used for the characterization of the subproteome that is associated with the extracellular matrix have been outlined in previous reviews [74,75,76]. Figure 1, Figure 2 and Figure 3 summarize (i) direct and extracellular-matrix-enrichment-independent preparations for the bottom-up proteomic profiling of the skeletal muscle proteome including the accessible matrisome, (ii) proteoform-centric and extracellular-matrix-enrichment-independent preparations for the top-down proteomic profiling of the skeletal muscle proteome including the accessible matrisome, and (iii) extracellular-matrix-enrichment-based proteomic-profiling strategies for studying the matrisome and using sequential depletion or decellularization methodology, respectively.
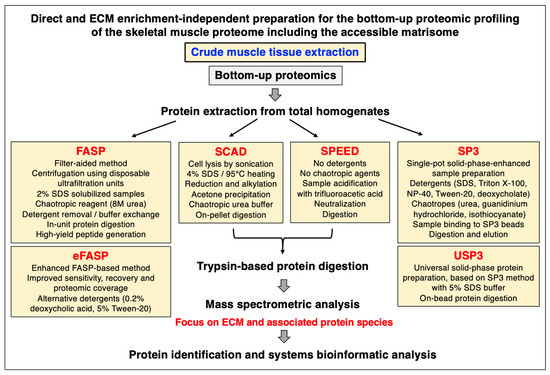
Figure 1.
Overview of frequently used extracellular-matrix-enrichment-independent approaches for the bottom-up proteomic analysis of skeletal muscle specimens. Abbreviations used: ECM, extracellular matrix; FASP, filter-aided sample preparation; SCAD, surfactant-and-chaotropic-agent-assisted sequential extraction/on-pellet digestion; SDS, sodium dodecyl sulfate; SP3, single-pot solid-phase-enhanced sample preparation; SPEED, detergent-free sample preparation by easy extraction and digestion; USP3, universal solid-phase protein preparation.
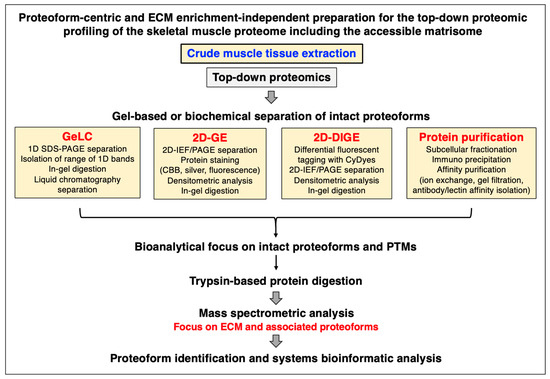
Figure 2.
Outline of extracellular-matrix-enrichment-independent preparations for the proteoform-centric and top-down proteomic analysis of skeletal muscle specimens. Abbreviations used: 1D, one-dimensional; 2D, two-dimensional; DIGE, difference gel electrophoresis; ECM, extracellular matrix; GE, gel electrophoresis; GeLC, gel electrophoresis liquid chromatography; IEF, isoelectric focusing; PAGE, polyacrylamide gel electrophoresis; PTMs, post-translational modifications; SDS, sodium dodecyl sulfate.
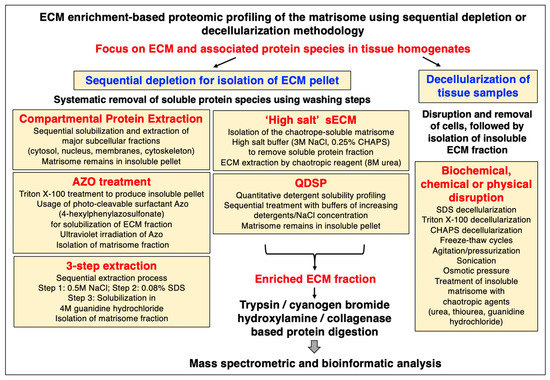
Figure 3.
Outline of extracellular-matrix-enrichment-based approaches for the proteomic profiling of the matrisome. Abbreviations used: Azo, 4-hexylphenylazosulfonate; CHAPS, 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate; ECM, extracellular matrix; QDSP, quantitative detergent solubility profiling; SDS, sodium dodecyl sulfate; sECM, soluble extracellular matrix.
2.1. Bottom-Up Proteomics versus Top-Down Proteomics
In principle, the main proteomic approaches used in the biological sciences can be categorized into two main types, bottom-up proteomics and top-down proteomics [77]. Bottom-up proteomics focuses on the large-scale identification of proteins using crude extracts as the starting material and peptide mass spectrometry for protein detection [78]. Top-down proteomics is usually based on the biochemical or gel electrophoretic separation of intact protein species which is then followed by the mass spectrometric analysis of intact proteoforms and their post-translational modifications [79]. An overview of the main techniques employed in skeletal muscle proteomics, including a description of the main analysis platforms, two-dimensional gel electrophoresis, liquid chromatography, antibody-based approaches, sample preparation, protein digestion protocols, mass spectrometry, data acquisition, single-cell analysis, and aptamer-based proteomics, has recently been published in the context of the proteomic profiling of the contractile apparatus of skeletal muscles [80]. Of note, single-cell proteomics is becoming increasingly important [81] and has been applied for studying individual muscle fibers [64,65]. Studying changes in specialized muscle fibers is crucial since fiber type shifting occurs in the skeletal musculature during physiological adaptations and in many neuromuscular disorders [82]. Figure 1 and Figure 2 provide an overview of commonly used methods for the bottom-up or top-down proteomic analysis of crude protein extracts from total muscle tissue specimens without the prior usage of extensive enrichment processes [79,83].
2.2. Sample Handling and Protein Extraction for Gel-Free and Bottom-Up Proteomics
As summarized in Figure 1, efficient protein extraction for bottom-up muscle proteomics can be performed by various direct sample preparation techniques [83,84,85], such as the widely used detergent-based and filter-aided sample preparation (FASP) method [86,87], enhanced eFASP using alternative detergents [88], sample processing from cell lysis through the elution of purified peptides by the In-StageTip (iST) method [89,90], the single-pot solid-phase-enhanced sample preparation (SP3) method [91], the universal solid-phase protein preparation (USP3) method [92], pressure-cycling technology (PCT) [93], surfactant-and-chaotropic-agent-assisted sequential extraction/on-pellet digestion (SCAD) [94], or by detergent-free sample preparation by easy extraction and digestion (SPEED) [95]. A comprehensive description of a standardized bottom-up proteomic workflow, incorporating the FASP method, has recently been described in detail [96].
2.3. Sample Handling and Protein Extraction for Gel-Based and Top-Down Proteomics
Although the majority of mass spectrometric surveys are carried out with peptides and proteins that are isolated by gel-free systems, top-down proteomics routinely employs one-dimensional or two-dimensional gel electrophoretic techniques, or biochemical isolation methods such as affinity chromatography, for the preparation of intact proteoforms. As summarized in Figure 2, gel-based top-down proteomics can be based on GeLC-MS/MS using one-dimensional gradient gels in combination with liquid chromatography [97,98,99] or classical two-dimensional gel electrophoresis using isoelectric focusing in the first dimension and slab gel electrophoresis in the second dimension [100,101,102]. A gel-based method that has overcome the technical limitation of gel-to-gel variations is represented by two-dimensional fluorescence difference gel electrophoresis (2D-DIGE) [103,104,105]. This advanced gel technique employs differential proteome tagging with the CyDyes Cy2, Cy3, and Cy5 in combination with various software analysis packages to determine protein changes in comparative proteomics [79].
2.4. Extracellular-Matrix-Enrichment-Based Proteomic-Profiling Approaches
The main enrichment strategies for mass spectrometric studies of the isolated extracellular matrix are summarized in Figure 3, including both sequential depletion methods and a list of decellularization methods. The matrisome can be divided into components that belong to the insoluble versus the soluble extracellular matrix, often referred to as the iECM versus the sECM [74,75,76]. Sequential depletion approaches focus on the isolation of an insoluble pellet consisting of key extracellular matrix proteins via the systematic removal of soluble protein species by various washing steps. Commercially available ‘Compartmental Protein Extraction’ kits represent a convenient way to carry out the sequential solubilization and extraction of major subcellular fractions, such as the cytosol, nucleus, membranes, and cytoskeletal networks, and thereby enrich the iECM part of the matrisome as an insoluble pellet [75,106]. In contrast, the sECM can be prepared by a ‘high salt’ method that aims to isolate the chaotropic-agent-soluble matrisome. High-salt buffers are employed to remove the soluble protein fraction, followed by extracellular matrix extraction using chaotropic reagents [75,107]. Quantitative detergent solubility profiling (QDSP) is based on the sequential treatment with buffers of increasing detergent and salt concentration and harvesting of the matrisome as an insoluble pellet [75,108]. An interesting approach is the usage of the photo-cleavable surfactant 4-hexylphenylazosulfonate (Azo). Following treatment with the nonionic detergent Triton X-100 to produce an insoluble pellet, the Azo detergent is then used for the specific solubilization and isolation of the extracellular matrix fraction in association with the ultraviolet irradiation of Azo [75,109,110]. The ‘three-step extraction’ method is a sequential extraction process that is based on a high-salt step, a detergent solubilization step, and a final chaotropic-agent-based solubilization of the matrisome fraction [75,111].
Decellularization methods to treat tissue samples usually start with the disruption and removal of cellular material using various biochemical, chemical, or physical techniques, followed then by the isolation of the mostly insoluble extracellular matrix fraction [74,75,76]. Freeze–thaw cycles, agitation, pressurization, sonication, and osmotic pressure are typical methods used for cellular disruption. Biochemical approaches used for decellularization often use chemicals such as SDS, Triton X-100, and CHAPS, followed by the treatment of the insoluble matrisome with chaotropic agents, including high concentrations of urea, thiourea, or guanidine hydrochloride [111,112,113]. Prior to peptide mass spectrometry, the enriched extracellular matrix fraction is often digested by trypsin and LysC, but also with the help of cyanogen bromide or hydroxylamine [74,75,76].
2.5. Mass Spectrometry and Data Acquisition
The identification and characterization of individual proteoforms in crude tissue samples, subcellular fractions, enriched preparations of the matrisome, or isolated protein assemblies can be carried out by a variety of mass spectrometric methods [114,115,116,117], such as liquid chromatography–tandem mass spectrometry (LC-MS/MS) [115,118] or matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) [114,119]. Both labelling and label-free approaches are routinely used for the detection of protein species. The bioanalytical advantages versus technical limitation of these frequently used methods have been reviewed in detail [120]. Stable heavy isotopes can be incorporated into peptides/proteins using chemical or metabolic labelling and then be used for the quantitation of protein abundance [121]. Examples of well-established proteomic-labelling methods are stable isotope labelling by amino acids in cell culture (SILAC) [122], the tandem mass tagging (TMT) technique [123], the isotope-coded affinity tagging (ICAT) approach [124], and the isobaric tagging for relative and absolute quantitation (iTRAQ) technique [125]. Alternatively, mass spectrometric analyses can be performed with label-free quantitation techniques that focus on features from fragment-ion analysis (MS2), such as spectral counting, or measure the integrated peak intensity from the parent-ion mass analysis (MS1) [126,127]. Of note, the application of artificial intelligence (AI), machine learning (ML), and deep learning (DL) algorithms has been increasingly used to evaluate complex spectral data [128].
Optimized data acquisition plays a key role in mass spectrometric analyses. This can be achieved by data-dependent acquisition (DDA) [129], data-independent acquisition (DIA) [130], or targeted data acquisition (TDA) [131]. In the context of DIA, the sequential window acquisition of all theoretical mass spectra mass spectrometry (SWATH-MS) method, which divides the mass range into small mass windows, has been successfully applied to studying extracellular matrix proteins [132]. Immunoprecipitation combined with SWATH-MS was successfully employed to investigate fukutin-related protein (FKRP)-associated dystroglycanopathy [133]. This methodology revealed that the loss of FKRP function disrupts the localization and tethering of fibronectin to myosin-10, essential for fibronectin maturation and subsequent collagen binding. The evidence accumulated demonstrates the utility of combining SWATH-MS and enrichment methods to investigate distinct phenotypes associated with muscular disorders.
Important methods for focusing on the abundance of target peptides include selected/multiple reaction monitoring (SRM/MRM) [134] or parallel reaction monitoring (PRM) [135], which are based on the measurement of precursor peptides in predefined m/z ranges taking into account retention time. The MRM-based measurement of targeted peptides was recently applied for the detailed analysis of extracellular matrix proteins [136]. Importantly, the absolute quantification (AQUA) workflow involves the usage of stable-isotope-labelled peptides that are spiked into the samples at predetermined concentrations and are then quantified simultaneously with the corresponding endogenous peptides. This greatly facilitates better accuracy, sensitivity, and reproducibility of protein detection [137].
3. The Extracellular Matrix of Skeletal Muscles
The extracellular matrix of skeletal muscle fibers plays a key role in the provision of force transmission and the external support for the continued stabilization, maintenance, and repair of contractile fibers [30,31,32], which is reflected by complex structure–function relationships [138,139]. Importantly, the processes that regulate and support cell adhesion and cell migration during development, wound healing, and regeneration involve extensive cell–matrix interactions, which warrants detailed omics-type studies into the composition of the matrisome. In the field of proteomics including extracellular matrix proteomics, it is important to stress the now generally accepted concept of the proteoform being the basic unit of the dynamic proteome [140,141].
Thus, the detailed biochemical information on individual proteoforms and their post-translational modifications (PTMs), as detected by mass spectrometry, should be used to establish detailed proteomic maps of the matrisome. Importantly, PTM profiles of extracellular matrix proteins can provide important biochemical characteristics for detecting and understanding the complex pathways associated with skeletal muscle physiology and pathophysiology. Mounting evidence exists showing how the existence of various PTMs are essential for matrisomal protein secretion and functionality within the extracellular milieu. The thrombospondin type-1 domain (TSR) is a common molecular feature present in the thrombospondin family, where O-fucosylation has a crucial stabilizing effect on the TSR structure [142]. Methods for detecting hydroxylation and glycosylation sites in collagen IV have previously been documented [143]. Such methods are important steps for understanding the role of specific PTMs in health and disease, as hydroxylation is essential for the stability of collagen molecules, and associated fibril structures. Disulfide bonds, assembled through the action of protein disulfide isomerases (PDIs), are found in thrombospondins and fibronectin, playing important roles in the establishment of subunit stability [144]. Citrullination, in which the conversion of the amino acid arginine into the amino acid citrulline is established, is associated with several extracellular matrix proteins, including fibronectin, having important roles in protein clustering, focal adhesion stability, and signaling [145].
Table 1 lists the representative protein hit list using the standardized mass spectrometric identification of proteins belonging to the various layers of the extracellular matrix from wild-type mouse diaphragm muscle [37,146]. This proteomic analysis of matrisomal proteins was carried out by bottom-up proteomics [96] incorporating the detergent-based and filter-aided sample preparation (FASP) method [86,87]. The proteins listed in Table 1 cover proteoforms that are found throughout the endomysium, perimysium, and epimysium, as well as protein species that are enriched in the basal lamina, myotendinous junctions, tendons, and cartilage [147]. Major members of the proteoglycan family, matricellular proteins, and adhesion receptors of the sarcolemma were clearly identified by proteomics [17,37,146].

Table 1.
Mass-spectrometry-based proteomic identification of key components of the various layers of the matrisome from diaphragm muscle *.
Load bearing, fiber stretching, and continuous cycles of excitation, contraction, and relaxation put enormous physical strain on individual skeletal muscles. The surrounding layers of the extracellular matrix protect the muscular system from excess damage. The physical scaffolds provided by collagens and proteoglycans act as a supporting structure that embeds individual motor units with their motor neurons and corresponding muscle fibers, as well as capillaries and satellite cells [30,31,32]. Of special importance for the integrity of the sarcolemma is the linkage between the intracellular cytoskeleton and the extracellular basement membrane. Listed in Table 1 as a key component of the basal lamina is the heterotrimeric glycoprotein named laminin-211 [147], a multi-functional member of the large laminin family of basement membrane proteins [148]. Laminin-211, whose name refers to its composition consisting of the merosin alpha-2 (LAMA2) subunit, the beta-1 (LAMB1) subunit, and the gamma-1 (LAMC1) subunit, was shown to be involved in fiber stabilization and cell adhesion, as well as cell differentiation and proliferation mechanisms [148]. Laminin-211 can interact with collagens, integrins, the dystroglycan complex, nidogen, and heparan sulfate at the level of the plasma membrane [149] and also plays a central role in the organization of the neuromuscular junction [150].
An important binding partner for the laminin complex is the heparan sulfate proteoglycan named agrin, which mediates the aggregation of the nicotinic acetylcholine receptors at the post-synaptic structures during synaptogenesis [151]. This extracellular matrix component of the neuromuscular junction was identified by mass spectrometry in crude muscle extracts (accession number A2ASQ1-3; Agrn gene; 2 peptides; 1.6% sequence coverage; 198.2 kDa molecular mass) despite its low abundance. Additional basal-lamina-enriched extracellular matrix proteins were detected as collagen isoforms IV (COL4A1), XV (COL15A1), and XVIII (COL18A1) [152], perlecan (HSPG-2) [153], and the nidogens named entactin (NID1) [154] and osteonidogen (NID2) [155], as listed in Table 1. Collagen IV is a major member of the muscle collagen family [156] and is highly enriched in the basal lamina [157]. Perlecan of 398 kDa is an abundant component of the basal lamina and also known as the basement-membrane-specific heparan sulfate proteoglycan core protein (HSPG-2) [153]. The nidogens are intrinsically involved in the assembly of the basal lamina and provide a linkage between the laminin complexes and the collagen IV system, as well as perlecan in the basement membrane [156,157].
Endomysium-enriched extracellular matrix proteins are represented by specific collagens, as listed in Table 1, including collagen V (Col5a1) and collagen VI (Col6a1, Col6a2, Col6a5, Col6a6) [158]. The collagen lattice and associated proteoglycans form an intricate extracellular maintenance system that supports muscle tissue integrity and fiber elasticity, as well as binding sites for trans-plasmalemmal linkage to the cytoskeletal networks to stabilize excitation–contraction–relaxation cycles. Of note, collagen VI plays a key role in the structural stabilization of the neuromuscular junction [159]. Via costameres, the extracellular matrix mediates the transduction of muscle force towards the surrounding tissues. Extracellular matrix components support crucial signaling pathways and thereby preserve neuromuscular homeostasis [160]. Extracellular matrix proteins that can be found throughout the endomysium, perimysium, and epimysium include prolargin (PRELP), dermatopontin (DPT), vitronectin (VTN), fibronectin (FN1), fibrinogen (FGA, FGB, FGG), the microfibril-associated glycoprotein MFAP-4, and fibrillin (FBN1) [32,138]. The large group of muscle-associated small leucine-rich proteoglycans (SLRP type) was represented by asporin (ASPN), biglycan (BGN), decorin (DCN), fibromodulin (FMOD), lumican (LUM), and mimecan/osteoglycin (OGN) [32,161]. As listed in Table 1, mass spectrometry detected key members of the family of non-structural matricellular proteins, including periostin (POSTN), thrombospondin (THBS1, THBS4), and fibulin isoform FBLN5 [162,163].
Major sarcolemmal adhesion proteins that provide cell–matrix linkages are the alpha-7/beta-1 integrin (ITGA7, ITGB1) complex and the dystroglycan (alpha/beta-DAG1) complex, which are both involved in force transmission and signaling mechanisms. See Table 1 for the proteomic identification of these sarcolemmal protein receptors. The alpha-7/beta-1 integrins form an abundant trans-sarcolemmal adhesion site at costameres and myotendious junctions that links laminin and fibronectin of the extracellular matrix to the intracellular cytoskeleton [164]. The dystroglycans form the core of the sarcolemmal dystrophin–glycoprotein complex and mediate the linkage between laminin and cortical actin [165,166,167] via tight interactions between the laminin-binding protein alpha-dystroglycan [168], the integral glycoprotein beta-dystroglycan [169], and the actin-binding domain of the Dp427-M isoform of the membrane cytoskeletal protein dystrophin [20]. The organization of the various layers of the extracellular matrix and the distribution of individual protein components is diagrammatically summarized in Figure 4.
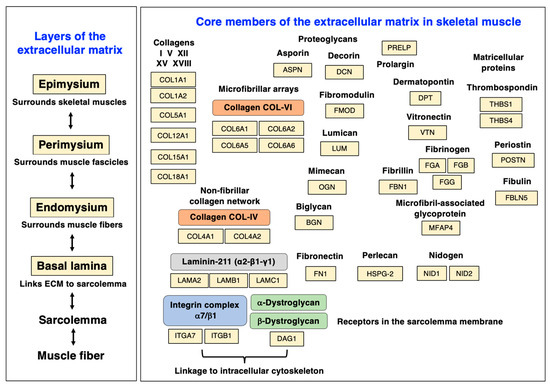
Figure 4.
Diagram of the various layers of the extracellular matrix and distribution of major protein components of the matrisome in skeletal muscle tissues. Abbreviations used: ECM, extracellular matrix.
Myotendinous-junction-enriched proteins listed in Table 1 are collagen XII (COL12A1) and FERM-domain-containing kindlin 2 (FERMT-2) [170,171,172], and the abundant collagens of the tendon are represented by the microfibrillar isoform collagen I (COL1A1, COL1A2) [173]. The group of cartilage-enriched proteins was detected in the form of the cartilage oligomeric matrix protein COMP and the cartilage intermediate layer protein (CILP, CILP2). Extracellular matrix proteins involved in cellular signaling are the transforming growth factor-beta-induced protein ig-h3 (TGFBI) and protein-glutamine gamma-glutamyltransferase TGM2. Extracellular-matrix-associated proteins that belong to the annexin family of proteins were identified as the annexin isoforms ANXA2 and ANXA6, whereby Annexin-6 is a key protein involved in membrane repair [174]. Extracellular matrix regulators belonging to the serpin family include the alpha-antitrypsins A1AT4 (SERPINA1D), A1AT2 (SERPINA1B), A1AT3 (SERPINA1C), and A1AT5 (SERPINA1E), as well as Serpin-H1 (SERPINH1), serine protease inhibitor A3K (SERPINA3K), and cathepsin-B (CTSB) [175,176].
The above-listed components of the extracellular matrix form a highly adaptable, dynamic, and acellular microenvironmental system that supports, embeds, and stabilizes skeletal muscle fibers and their physiological functions [177]. The complex meshwork of extracellular laminins, non-fibrillar collagens, collagen microfibrils, proteoglycans, matricellular proteins, and regulating enzymes, and their linkage to cell-associated adhesion receptors in the sarcolemma membrane, is involved in large numbers of cell biological processes in contractile tissues, including:
- The stabilization of skeletal muscle fibers, and the provision of tissue strength and elasticity;
- Structural support via physical scaffolding, and the provision of an embedding medium for motor units, capillaries, and pools of satellite cells;
- The provision of neuromuscular homeostasis;
- Mechanical force transduction from muscle fibers to the surrounding tissues;
- The provision of cell–matrix interactions for the support of cell adhesion and cell migration during embryonic myogenesis, adult myogenesis, and tissue repair;
- Cell–matrix support during cell differentiation, maturation, remodeling, fiber transitions, and muscle aging.
The bioinformatic STRING analysis [178] of the muscle matrisome is shown in Figure 5, which was carried out with the proteins listed in Table 1 above. The interaction map illustrates the close protein–protein linkage patterns within the extracellular matrix of skeletal muscles.
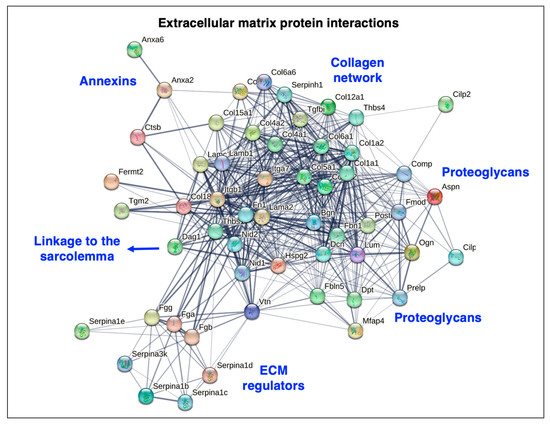
Figure 5.
Bioinformatic STRING analysis of potential protein–protein interactions in the extracellular matrix of skeletal muscle. Matrisomal proteins that were identified by mass spectrometry, as listed in above Table 1, were analyzed by the method of Szklarczyk et al. [178]. The search list consisted of the matrisomal proteins that are encoded by the genes Lama2, Lamb1, Lamc1, Col4a1, Col4a2, Col15a1, Col18a1, Hspg2, Nid1, Nid2, Col5a1, Col6a1, Col6a2, Col6a5, Col6a6, Prelp, Dpt, Vtn, Fn1, Fga, Fgb, Fgg, Mfap4, Fbn1, Aspn, Bgn, Dcn, Fmod, Lum, Ogn, Postn, Thbs1, Thbs4, Fbln5, Itga7, Itgb1, Dag1, Col12a1, Fermt2, Col1a1, Col1a2, Comp, Cilp, Cilp2, Tgfbi, Tgm2, Anxa2, Anxa6, Serpina1d, Serpina1b, Serpina1c, Serpina1e, Serpinh1, Serpina3k, and Ctsb. Species was set to ‘Mus musculus’ with the following parameters: (i) full STRING network analysis where the edges indicate both functional and physical protein associations; (ii) full set of active interaction sources consisting of text mining, experiments, databases, co-expression, neighborhood, fusion, and co-occurrence; (iii) network edges using confidence whereby line thickness indicates the strength of data support; and (iv) network display mode with interactive scalable vector graphic. Abbreviations used: ECM, extracellular matrix.
4. The mdx-4cv Mouse Model and Dystrophinopathy-Associated Myofibrosis
4.1. Duchenne Muscular Dystrophy and Fibrosis
The most severe form of dystrophinopathy, Duchenne muscular dystrophy, is a primary-muscle-wasting disorder of early childhood [179] with a prevalence of approximately 1 in 5000 live male births [180], which is triggered by a variety of genetic abnormalities in the X-chromosomal DMD gene [181]. Initial clinical signs include developmental delays in Duchenne patients with proximal muscle weakness, temporal and spatial gait variations, decreased walking speed, and Gower’s sign [182,183,184]. Characteristic patterns of toe walking and difficulties with climbing stairs progress at more advanced stages of the disease towards respiratory insufficiency, cardiomyopathy, scoliosis, and severe limitations in general mobility [185,186,187,188,189,190], which eventually results in the loss of unassisted ambulation and upper body weakness [191]. Approximately one-fifth of Duchenne patients suffer from intellectual developmental disorder that can be associated with learning difficulties, behavioral deficits, emotional problems, attention deficits, impaired language and speech development, cognitive deficiencies, and mental retardation [192,193,194]. Of note, secondary multi-system abnormalities in dystrophinopathy negatively affect whole-body homeostasis, including the proper functioning of the kidneys, the bladder, the gastrointestinal tract, and the liver [19].
Detailed descriptions of crucial aspects of dystrophinopathy can be found in extensive review articles that focus on the epidemiology [180], X-linked genetics [2], cellular pathogenesis [179], differential diagnosis [195], rehabilitation strategies [196], and emergency management [197] of Duchenne muscular dystrophy. Following the identification of the DMD gene and its involvement in X-linked Duchenne muscular dystrophy [198,199,200] and the discovery of the full-length muscle dystrophin protein (Dp427-M) and its many isoforms (Dp427-B, Dp427-P, Dp260-R, Dp140-B/K, Dp116-S, Dp71, and Dp45) [18,201,202,203], detailed biochemical, biophysical, and physiological studies have established that full-length dystrophin acts as a sub-sarcolemmal anchoring node that is involved in cytoskeletal organization, the provision of lateral force transmission, the maintenance of muscle fiber stability, and the support and integration of cellular signaling mechanisms in skeletal muscles [20]. The loss of the dystrophin isoform Dp427-M destabilizes the entire dystrophin-associated glycoprotein complex and causes a drastic reduction in the core dystroglycan sub-complex at the sarcolemma membrane, as well as lower levels of sarcoglycans, sarcospan, dystrobrevins, and syntrophins [204]. The dystrophin-deficient muscle membrane system was shown to be more prone to contraction-induced rupturing and this, in turn, results in a pathophysiological Ca2+ influx into muscle fibers. The chronically elevated Ca2+ concentration increases the rate of calpain-mediated proteolysis and tissue destruction [22,23,24].
Dystrophic skeletal muscles are characterized by highly progressive myonecrosis [19] and chronic inflammation [27], which is accompanied by reactive myofibrosis [17,26,37,50]. On the histological level, the drastic decline in muscle strength is characterized by fiber branching, alterations in fiber diameter, roundly shaped myofibers, hypercontractility, a high level of central myonucleation, intra-muscular fatty accumulation, muscle infiltration by inflammatory cells, and high levels of collagen deposition. In general, muscle-degeneration-induced myofibrosis is usually characterized by increased mechanical stiffness in contractile fibers with extensive levels of fibrillar disorganization and drastically increased deposition of collagens, as well as high levels of myofibroblasts and macrophages [25,26,27,205]. The histopathological changes that result in endomysial fibrosis reflect these changes and they correlate with the loss in motor function in Duchenne patients [54], making the degree of reactive myofibrosis a useful indicator of disease progression in dystrophinopathy [17]. A variety of muscle-associated cell types, including resident fibroblasts, the important category of fibro-adipogenic progenitors, inflammatory cells, and pericytes, can be activated to transform towards the myofibroblast phenotype [33,34,35]. This complex network of activated cells and drastically increased deposition of extracellular matrix components reflects the pathophysiological impact of reactive myofibrosis on the cellular pathogenesis of Duchenne muscular dystrophy [11]. This gives the development of novel anti-fibrotic pharmacotherapies a central position in the design of new treatment options to counteract dystrophinopathy-associated muscle pathology [28,206,207,208,209].
4.2. Characterization of the mdx-4cv Mouse within the Context of Other Dystrophic Models
Both spontaneous and bioengineered mouse models that lack the dystrophin protein isoform Dp427-M have been instrumental in the detailed elucidation of the molecular and cellular pathogenesis of Duchenne muscular dystrophy, as well as the evaluation of experimental treatment strategies [210,211,212]. A comparative listing of major genetic mouse models of dystrophinopathy is provided in Table 2. This includes the original mdx-23 mouse compared to a variety of modified models including mdx/Dtna, mdx/Cmah, mdx/Utr, mdx/α7, mdx/Myod1, mdx-2cv, mdx-3cv, mdx-4cv, mdx-4cv/mTR-G2, mdx-5cv, mdx-52, mdx-βgeo, Dmd-null, hDMD/mdx-45, and hDMD/mdx-52. The table lists information on the genetic abnormalities that characterize the various mouse models and the observed severity of the dystrophic phenotype in individual models of Duchenne muscular dystrophy. Importantly, Table 2 gives an overview of the effect of mutations on the expression of distinct dystrophin isoforms ranging from Dp71 to Dp427. Depending on the genetic abnormality within the Dmd gene, impaired expression patterns might only cause the loss of the full-length dystrophins Dp427-M, Dp427-B, and Dp427-P, or also additional effects on the tissue-specific density of the shorter dystrophins Dp260-R, Dp140-B/K, Dp116-S, Dp71, and/or Dp45. Detailed comparisons of dystrophic mouse models have been carried out in recent reviews [213,214,215].
Table 2.
Comparison of major genetic mouse models of Duchenne muscular dystrophy *.
Compared to the onset of dystrophic changes in the skeletal musculature at approximately 1 month of age in the mdx-23 mouse and a maximum lifespan of approximately 2 years, the modified mdx-type mice differ considerably in the onset of histopathological changes, ranging from 2 to 8 weeks, and their longevity, ranging in lifespan from 1 to 23 months [216]. Both the spontaneously mutated mdx-23 mouse and the chemically mutated mdx-4cv mouse have comparable survival rates and similar onsets of moderate hindlimb degeneration, as judged by histopathology, at 3–4 weeks of age in association with physiological dysfunction such as reduced muscle force. The mdx-23 mouse is characterized by extensive cycles of degeneration and regeneration from 3 to 8 weeks, illustrated by high levels of central myonucleation, and some stabilization with age [38,39,40]. The mdx-4cv musculature shows large variation in skeletal muscle fiber sizes. In both model systems, the dystrophin-deficient diaphragm is severely affected starting at 3–4 weeks of age and shows high levels of reactive myofibrosis during aging [213,214,215]. Analogous to the skeletal musculature, the reduced presence of the cardiac dystrophin–glycoprotein complex causes sarcolemmal disintegration, myonecrosis, fatty tissue replacement, fibrotic scarring, and interstitial inflammation in the mdx-type heart [217]. The onset of cardiomyopathy is at approximately 6–9 months of age and associated with considerable proteome-wide changes during aging [218].
The range of modified mdx-type mouse models of dystrophinopathy with usually more severe phenotypes includes double mutants that also affect, besides the dystrophin levels, the expression of proteins such as the dystrophin-associated component dystrobrevin, the autosomal dystrophin homologue utrophin, myogenic differentiation protein MOD1, and α7-integrin [213,214,215,216]. In addition to the most frequently used murine models with their rapid disease progression, large animal models of Duchenne muscular dystrophy exist in the form of canine and porcine mutants [5,219,220,221]. The Golden Retriever muscular dystrophy (GRMD) model exhibits a splice site mutation in intron-6, which causes the skipping of exon-7 and a resulting out-of-frame DMD transcript in exon-8 [220]. The complexity and progression of the clinical syndrome is severe in the GRMD model including cardiomyopathic complications, making dystrophic dogs good model systems for translational studies [222,223]. The porcine model of dystrophinopathy (DMD pig) is characterized by a deletion in exon-52 of the DMD gene and exhibits progressive cardiomyopathy [224]. In conjunction with mouse models, larger animals can be utilized to study pathophysiological mechanisms, validate the suitability and robustness of novel biomarker candidates, and be helpful during the preclinical testing phase of new therapeutic approaches such as gene editing [219,221]. Large disease-model animals are highly useful for facilitating the efficient translation of novel diagnostic methods to the clinical setting, such as imaging technology for the monitoring of myofibrosis [225].
Skeletal muscle fibrosis is seen in different mdx-type muscles to varying degrees [46,47,48,226], with the diaphragm being the most structurally affected and fibrotic muscle tissue [49,50,51,52,227,228]. However, one of the most widely employed dystrophic animal models, the mdx-23 mouse [38], exhibits a relatively high frequency of revertant fibers [39] and a relatively mild dystrophic phenotype [40]. In contrast, the chemically mutated mdx-4cv mouse which harbors a nonsense mutation in exon-53 [41,42,43] has a drastically reduced number of dystrophin-positive revertants [45]. Figure 6 provides a comparison of the main genetic, biochemical, physiological, and histological features of muscular abnormalities in Duchenne muscular dystrophy versus the widely used mdx-23 and mdx-4cv mouse models of dystrophinopathy.
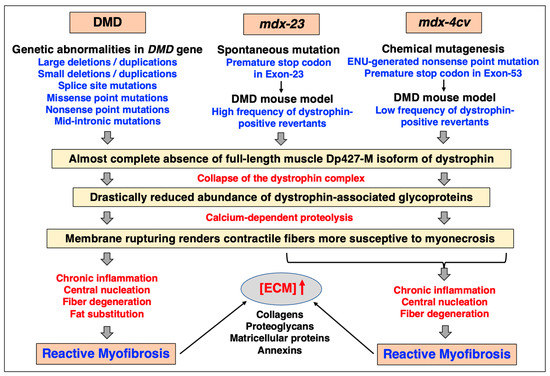
Figure 6.
Comparative overview of the main features of Duchenne muscular dystrophy versus the mdx-23 and the mdx-4cv models of dystrophinopathy that are due to a spontaneous mutation versus being generated by chemical mutagenesis, respectively. Analogous to Duchenne patients, the skeletal musculature of both the mdx-23 and the mdx-4cv mouse harbor mutations in the Dmd gene, and exhibit progressive myonecrosis and chronic inflammation in combination with reactive myofibrosis, which is especially prominent in the aged diaphragm muscle. In contrast to the spontaneous mdx-23 mouse, the mdx-4cv skeletal musculature is characterized by a low frequency of dystrophin-positive revertant fibers. Abbreviations used: DMD, Duchenne muscular dystrophy; ECM, extracellular matrix; ENU, N-ethyl-N-nitrosourea.
Although the mdx-4cv mouse with a nonsense point mutation that introduces a premature stop codon in exon-53 [41] does not reflect the genetic heterogeneity seen in large cohorts of human Duchenne patients [2], consisting of a variety of splice site mutations, missense point mutations, nonsense point mutations, large deletions, large duplications, small deletions, small duplications, and mid-intronic mutations [181], the pathobiochemical down-stream effects due to dystrophin deficiency are relatively comparable. The almost-complete absence of the Dp427-M isoform of full-length dystrophin in skeletal muscle fibers initiates the reduced anchoring of the alpha/beta-dystroglycan subcomplex, alpha/beta/gamma/delta-sarcoglycans, sarcospan, syntrophins, and dystrobrevins [146,229,230,231]. The collapse of the dystrophin-associated glycoprotein complex renders the muscle plasma membrane more susceptible to contraction-induced rupturing followed by the chronic influx of Ca2+ ions both through the leaky sarcolemma and Ca2+-leak channels. The resulting elevation of sarcosolic Ca2+ levels and concomitant impairment of the luminal Ca2+-buffering capacity of the sarcoplasmic reticulum negatively affect the fine regulation of the excitation–contraction coupling mechanism and overwhelm the Ca2+-extrusion system of damaged muscle fibers resulting in Ca2+-dependent proteolysis [22,23,24]. The histopathological consequences of the interplay between progressive fiber degeneration and abnormal ion homeostasis causes increased levels of inflammatory cells, impaired myoblast cell survival, central nucleation in mature fibers due to ongoing cycles of degeneration and regeneration, and increased levels of extracellular proteins [52,229,232]. One histopathological feature that is not observed in mdx-4cv muscles, compared to dystrophic skeletal muscles from both Duchenne and Becker’s muscular dystrophy patients [233,234,235,236], are high levels of fat substitution.
However, progressive myonecrosis, chronic inflammation, and reactive myofibrosis with increased levels of collagens, proteoglycans, matricellular proteins, and annexins are clearly observed in the mdx-4cv model with increasing severity during aging [55]. Table 3 lists major studies that have been carried out to characterize the mdx-4cv mouse model in the context of (i) chemical mutagenesis, genotyping, and phenotyping [41,42,43,44,231,237]; (ii) the testing of novel therapeutic strategies to treat X-linked Duchenne muscular dystrophy, including exon-skipping therapy [238], virus- or nanocarrier-mediated micro-dystrophin gene therapy [239,240,241,242,243], cell-based therapy [244,245,246], and gene editing [247,248]; (iii) the biochemical profiling of proteome-wide changes due to multi-systemic changes, including various skeletal muscles [55,146,229,230,249,250,251,252], heart [253], liver [254], kidney [255,256], spleen [96,257], stomach wall and pancreas [258], and central nervous system [259]; and (iv) the biochemical profiling of proteome-wide changes in biofluids including serum [260,261], urine [262], and saliva [261,263]. The mdx-4cv mouse has also been used to produce an immunodeficient muscular dystrophy model, NSG-mdx-4cv [264,265,266,267], and an mdx-4cv/mTR-G2 model with humanized telomere lengths [216,268], as listed in above Table 2. The testing of experimental therapies with suitable animal models is crucial for the establishment of novel therapeutic options to treat Duchenne muscular dystrophy [5,215,269], such as cell transfer therapy, genomic editing, exon-skipping therapy, and multi-drug combination pharmacotherapy [270,271,272,273,274,275], including anti-fibrosis strategies [28].
Table 3.
List of major studies on the generation and characterization of the mdx-4cv mouse model of Duchenne muscular dystrophy.
4.3. Histological and Biochemical Characterization of Fibrosis in the mdx-4cv Mouse Diaphragm
The diaphragm muscle is an important type of tissue for the focused analysis of pathophysiological changes since it is majorly affected in Duchenne patients that suffer from impaired cardiorespiratory function. A reduced thoracic cavity area and altered chest wall contraction patterns have been established to occur during cycles of inspiration and expiration in dystrophic respiratory muscles by imaging studies [276]. Severe diaphragmatic dysfunction, excessive fat infiltration in respiratory muscles, and cardiomyopathic complications occur mostly in the second decade of life in inherited muscular dystrophy, which frequently necessitates both the management of respiratory insufficiency by mechanical ventilatory support [277] and targeted drug therapy of the weakened and dystrophin-deficient heart muscle [185,278]. However, respiratory decline can already be observed during the early ambulatory phase in dystrophic boys [279] with diaphragmatic motions being drastically impaired following inspiration [280]. Analogous to the severely degenerative muscle phenotype seen in Duchenne patients, the aged mdx-4cv mouse diaphragm muscle shows the main histopathological hallmarks of dystrophinopathy [49,52]. This is summarized in the histological and immunofluorescence microscopical analysis depicted in Figure 7.
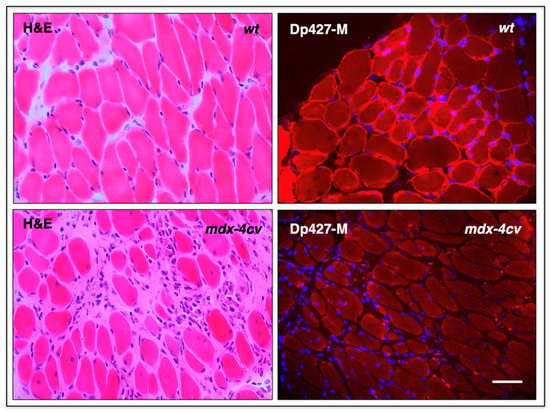
Figure 7.
Histological and immunofluorescence microscopical analysis of dystrophic mdx-4cv mouse diaphragm muscle. Shown are transverse sections of 12-month-old diaphragm muscles from wild-type (wt) versus age-matched dystrophic mdx-4cv mice. Cryosections were stained with haematoxylin and eosin (H&E), as well as immuno-labelled with a monoclonal antibody to full-length dystrophin isoform Dp427-M. Hoechst-33342 staining was used to visualize myo-nuclei. Tissue preparation and immunofluorescence microscopy were carried out by optimized methods [146]. Bar equals 40 μm.
The illustration highlights myonecrosis, reactive myofibrosis, and zones of chronic inflammation. The dystrophic diaphragm exhibits more roundly shaped myofibers and a high frequency of central myo-nucleation, which is due to ongoing cycles of fiber degeneration and regeneration. These histopathological changes clearly correlate with skeletal muscle dysfunction and severe force deficits in the dystrophic mdx-type diaphragm [281]. However, one additional sign of muscular changes, which is often observed in human patients, is not present in dystrophic mdx-4cv mice, i.e., fat substitution. Immuno-labelling of dystrophin isoform Dp427-M confirmed the sarcolemmal localization of this membrane cytoskeletal protein in wild-type diaphragm and its almost-complete loss in the dystrophic and highly fibrotic mdx-4cv diaphragm. To complement microscopical studies, a comparative immunoblot analysis is routinely used to independently verify findings from proteomic studies. Western blotting surveys have clearly confirmed the drastic increase in many extracellular matrix proteins in muscular dystrophy, including various fibrosis markers such as collagens, fibronectin, dermatopontin, biglycan, and periostin in mdx-type muscles [282,283,284]. The glycoprotein fibronectin [285] is an exciting fibrosis-related biomarker candidate since its levels are also significantly increased in the serum of Duchenne patients compared to age-matched controls [286]. The immunoblots shown in Figure 8 illustrate the elevated levels of collagen isoform COL-VI, which, in muscle tissue, consists of three alpha chains [158], the annexin isoform ANXA2, and the matricellular protein periostin in the mdx-4cv diaphragm. It is not surprising that enhanced collagen deposition and a high concentration of fibronectin in fibrotic muscle tissue are closely related to increased dermatopontin levels since this tyrosine-rich acidic matrix protein plays a central role in cell–matrix interactions and matrix assembly [287,288,289].
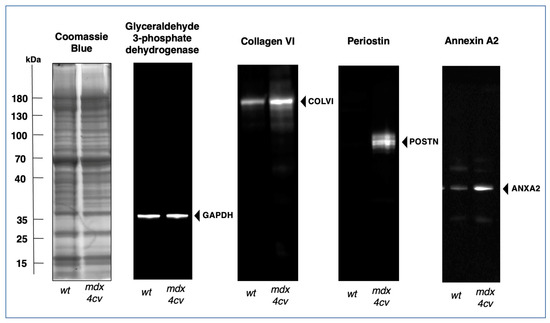
Figure 8.
Comparative immunoblot analysis of the dystrophic mdx-4cv mouse diaphragm muscle. Shown is the immunoblotting of the glycolytic enzyme glyceraldehyde-3-phopshate dehydrogenase (GAPDH), used here as a house-keeping protein and loading control, alpha chain-containing collagen VI (COL-VI) of the endomysium as an abundant marker of the extracellular matrix, the matricellular protein periostin (POSTN), and the membrane repair protein annexin A2 (ANXA2). Gel electrophoretic separation was carried out with total protein extracts from aged wild-type (wt) versus aged and dystrophic mdx-4cv diaphragm. Lanes 1 and 2 contain protein extracts from 15-month-old wt and age-matched mdx-4cv muscle, respectively. On the left is shown a Coomassie Brilliant Blue-stained protein gel. The other images are identical immunoblots generated by electrophoretic transfer and then labelled with monoclonal antibodies to GAPDH, COL-VI, POSTN, and ANXA2, as previously described in detail [55]. The position of immuno-labelled protein bands is indicated by arrowheads. Molecular weight standards are marked on the left.
4.4. Proteomic Profiling of Fibrosis in the mdx-4cv Mouse Diaphragm
Because myofibrosis does not only directly impair tissue elasticity and the contractile functions of skeletal muscle fibers but also has a negative impact on the regenerative capacity of motor units following muscle degeneration and augments the susceptibility for muscular re-injury [10,15,16,17], it is a major contributing factor to muscle weakness in dystrophinopathy [290]. It was therefore of interest to determine biological-system-wide changes in X-linked muscular dystrophy using multi-omics approaches [291]. Proteomic profiling of dystrophic skeletal muscle has been carried out with a large number of proteomic approaches using both patient biopsy specimens and a variety of animal models of dystrophinopathy [292,293,294,295,296,297,298]. Proteome-wide changes include alterations in the abundance of muscle-associated proteins that are involved in the regulation of excitation–contraction coupling, ion homeostasis, cellular signaling, the contraction–relaxation cycle, cytoskeletal networks, the extracellular matrix, bioenergetic pathways, and the cellular stress response [299,300,301]. The extensive findings that have been generated by animal model research underline the critical importance of preclinical studies for increasing our mechanistic knowledge of muscle pathogenesis, establishing robust and reliable biomarker candidates, and developing novel treatment strategies for X-linked Duchenne muscular dystrophy [6,302,303,304].
Myofibrosis-associated changes in dystrophin-deficient muscle fibers are diagrammatically summarized in Figure 9, which highlights characteristic alterations in distinct protein species at the level of the sarcolemma, basal lamina, endomysium, perimysium, and epimysium. The figure is based on the proteomic profiling of the dystrophic mdx-4cv diaphragm muscle [55] and relates to the information given in Figure 4 above on the distribution of key extracellular matrix proteins. The almost-complete loss of dystrophin isoform Dp427-M was shown to affect all layers of the matrisome, including the basal lamina with the significant reduction in the laminin-binding protein alpha-dystroglycan. The loss of the trans-sarcolemmal linkage via the dystrophin/dystroglycan sub-complex is the key trigger of membrane micro-rupturing in dystrophinopathies. The elevated levels of matrisomal proteins at the level of the basal lamina are characterized by the increased abundance of collagen IV and nidogen-2. Changes in the endomysium are reflected by an increased concentration of collagen V and collagen VI. Additional extracellular matrix proteins are found throughout the endomysium, perimysium, and epimysium. Fibrosis-related increases were established for the matrisomal proteins fibronectin, vitronectin, and dermatopontin, as well as the non-structural matricellular protein periostin and the small leucine-rich proteoglycans asporin, biglycan, decorin, lumican, and mimecan/osteoglycin. Higher expression levels were also established for the extracellular-matrix-associated repair proteins annexin-2 and annexin-6 and the sarcolemmal integrin adhesion receptor subunits alpha-7 and beta-1.
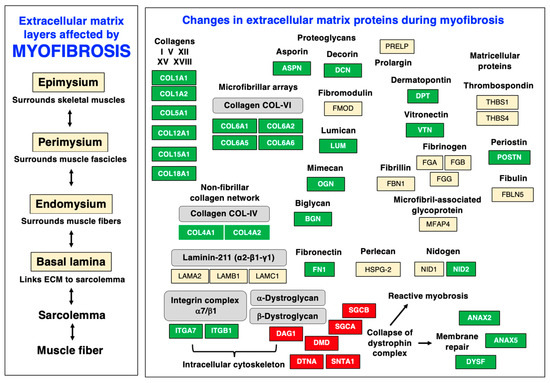
Figure 9.
Diagram of myofibrosis-related changes within the various layers of the extracellular matrix and associated structures in dystrophin-deficient skeletal muscle. Increased versus decreased protein isoforms are shown with a green versus red background, respectively. The collapse of the dystrophin–glycoprotein complex results in compensatory mechanisms, such as the activation of membrane repair mechanisms and reactive myofibrosis, which is characterized by drastic increases in various collagens, matricellular proteins, and proteoglycans.
Especially interesting is the drastic increase of periostin in the fibrotic mdx-4cv diaphragm, a matricellular protein that is involved in cellular signaling and the regulation of collagen fibrillogenesis [163,305]. In the extracellular matrix of skeletal muscles, periostin is only temporally expressed during regeneration and differentiation [306] but drastically increased during periods of fiber regeneration [307], including X-linked Duchenne muscular dystrophy [37,284,308]. This makes periostin a suitable fibrosis marker that can be used to judge the degree of myofibrotic changes in dystrophinopathy [284]. Changes in the annexin isoform ANAX2 [309,310,311] also have the potential to be highly useful for monitoring the level of membrane repair in fibrotic muscle fibers [312,313,314].
In summary, elevated levels of the below-listed major matrisomal marker proteins, which all play crucial roles in the extracellular matrix [73,315,316,317], were identified to occur during extended periods of myofibrosis. The detection and characterization of these protein species were carried out by various mass-spectrometry-based proteomic techniques in combination with biochemical, cell biological, and physiological analyses.
The following proteomic markers [37,55,284] could therefore be useful for the future establishment of an improved biomarker signature of dystrophinopathy-associated myofibrosis:
- (i)
- Basal-lamina-enriched extracellular matrix proteins:
- Collagen IV (COL4A1, COL4A2, COL4A3);
- Collagen XV (COL15A1);
- Collagen XVIII (COL18A1);
- Nidogen-2/Osteonidogen (NID2).
- (ii)
- Endomysium-enriched extracellular matrix proteins:
- Collagen V (COL5A1, COL5A2);
- Collagen VI (COL6A1, COL6A2, COL6A5, COL6A6).
- (iii)
- Matrisomal proteins of the endomysium, perimysium, and epimysium:
- Fibronectin (FN1);
- Vitronectin (VTN);
- Dermatopontin (DPT).
- (iv)
- Small leucine-rich proteoglycans:
- Asporin (ASPN);
- Biglycan (BGN);
- Decorin (DCN);
- Lumican (LUM);
- Mimecan/Osteoglycin (OGN).
- (v)
- Non-structural matricellular proteins:
- Periostin (POSTN).
- (vi)
- Extracellular-matrix-associated repair proteins:
- Annexin-2 (ANXA2);
- Annexin-6 (ANXA6).
- (vii)
- Adhesion receptors:
- Integrin, alpha-7 (ITGA7);
- Integrin, beta-1 (ITGB1);
- Dystroglycan, alpha/beta (DAG1).
- (viii)
- Myotendinous-junction-enriched proteins:
- Collagen XII (COL12A1).
- (ix)
- Tendon-enriched proteins:
- Collagen I (COL1A1, COL1A2).
5. Conclusions
The suitability of the severely dystrophic and fibrotic mdx-4cv diaphragm as a pathophysiological surrogate to study the progressive nature of dystrophinopathies has been outlined in this article. Histological, cell biological, and mass-spectrometry-based proteomic studies of the aged mdx-4cv mouse diaphragm have confirmed the presence of extensive reactive myofibrosis in this genetic mouse model of Duchenne muscular dystrophy. In conjunction with progressive myonecrosis, central myonuleation, and chronic inflammation, the fibrotic changes in the senescent mdx-4cv diaphragm make it an excellent model to study X-linked muscular dystrophy. The mdx-4cv mouse model was shown to be suitable for high-throughput surveys of complex changes in the expression levels of collagens, proteoglycans, matricellular proteins, regulatory enzymes, and adhesion receptors of the extracellular matrix. Following independent verification of their sensitivity, specificity, and robustness, proteomic biomarkers can now be used to improve differential diagnostics, prognostics, and therapeutic monitoring of X-linked Duchenne muscular dystrophy.
Author Contributions
Conceptualization, P.D. and K.O.; writing—original draft preparation, P.D. and K.O.; writing—review and editing, P.D., S.G., M.Z., D.S. and K.O. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a project grant from the Kathleen Lonsdale Institute for Human Health Research, Maynooth University, and equipment funding was provided under the Research Infrastructure Call 2012 by Science Foundation Ireland (SFI-12/RI/2346/3).
Data Availability Statement
The mass spectrometric raw data from studies of the extracellular matrix from skeletal muscle shown in tables are available upon request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mukherjee, P.; Roy, S.; Ghosh, D.; Nandi, S.K. Role of animal models in biomedical research: A review. Lab. Anim. Res. 2022, 38, 18. [Google Scholar] [CrossRef]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef]
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. Dis. Model. Mech. 2015, 8, 195–213. [Google Scholar] [CrossRef]
- Swiderski, K.; Lynch, G.S. Murine models of Duchenne muscular dystrophy: Is there a best model? Am. J. Physiol.-Cell Physiol. 2021, 321, C409–C412. [Google Scholar] [CrossRef]
- Stirm, M.; Fonteyne, L.M.; Shashikadze, B.; Stöckl, J.B.; Kurome, M.; Keßler, B.; Zakhartchenko, V.; Kemter, E.; Blum, H.; Arnold, G.J.; et al. Pig models for Duchenne muscular dystrophy—From disease mechanisms to validation of new diagnostic and therapeutic concepts. Neuromuscul. Disord. 2022, 32, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Henrot, P.; Blervaque, L.; Dupin, I.; Zysman, M.; Esteves, P.; Gouzi, F.; Hayot, M.; Pomiès, P.; Berger, P. Cellular interplay in skeletal muscle regeneration and wasting: Insights from animal models. J. Cachexia Sarcopenia Muscle 2023, 14, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Nathan, J.A.; Goldberg, A.L. Muscle wasting in disease: Molecular mechanisms and promising therapies. Nat. Rev. Drug Discov. 2015, 14, 58–74. [Google Scholar] [CrossRef]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Antar, S.A.; Ashour, N.A.; Marawan, M.E.; Al-Karmalawy, A.A. Fibrosis: Types, Effects, Markers, Mechanisms for Disease Progression, and Its Relation with Oxidative Stress, Immunity, and Inflammation. Int. J. Mol. Sci. 2023, 24, 4004. [Google Scholar] [CrossRef]
- Mahdy, M.A.A. Skeletal muscle fibrosis: An overview. Cell Tissue Res. 2019, 375, 575–588. [Google Scholar] [CrossRef]
- Smith, L.R.; Barton, E.R. Regulation of fibrosis in muscular dystrophy. Matrix Biol. 2018, 68–69, 602–615. [Google Scholar] [CrossRef] [PubMed]
- Lamandé, S.R.; Bateman, J.F. Collagen VI disorders: Insights on form and function in the extracellular matrix and beyond. Matrix Biol. 2018, 71–72, 348–367. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, S.; Magri, F.; Salani, S.; Napoli, L.; Ripolone, M.; Ronchi, D.; Fortunato, F.; Ciscato, P.; Velardo, D.; D’Angelo, M.G.; et al. Extracellular Matrix Disorganization and Sarcolemmal Alterations in COL6-Related Myopathy Patients with New Variants of COL6 Genes. Int. J. Mol. Sci. 2023, 24, 5551. [Google Scholar] [CrossRef] [PubMed]
- Villar-Quiles, R.N.; Donkervoort, S.; de Becdelièvre, A.; Gartioux, C.; Jobic, V.; Foley, A.R.; McCarty, R.M.; Hu, Y.; Menassa, R.; Michel, L.; et al. Clinical and Molecular Spectrum Associated with COL6A3 c.7447A>G p.(Lys2483Glu) Variant: Elucidating its Role in Collagen VI-related Myopathies. J. Neuromuscul. Dis. 2021, 8, 633–645. [Google Scholar] [CrossRef]
- Klingler, W.; Jurkat-Rott, K.; Lehmann-Horn, F.; Schleip, R. The role of fibrosis in Duchenne muscular dystrophy. Acta Myol. 2012, 31, 184–195. [Google Scholar]
- Kharraz, Y.; Guerra, J.; Pessina, P.; Serrano, A.L.; Muñoz-Cánoves, P. Understanding the process of fibrosis in Duchenne muscular dystrophy. Biomed. Res. Int. 2014, 2014, 965631. [Google Scholar] [CrossRef]
- Ohlendieck, K.; Swandulla, D. Molecular pathogenesis of Duchenne muscular dystrophy-related fibrosis. Pathologe 2017, 38, 21–29. [Google Scholar] [CrossRef]
- Hoffman, E.P. The discovery of dystrophin, the protein product of the Duchenne muscular dystrophy gene. FEBS J. 2020, 287, 3879–3887. [Google Scholar] [CrossRef]
- Ohlendieck, K.; Swandulla, D. Complexity of skeletal muscle degeneration: Multi-systems pathophysiology and organ crosstalk in dystrophinopathy. Pflügers Arch. 2021, 473, 1813–1839. [Google Scholar] [CrossRef]
- Dowling, P.; Gargan, S.; Murphy, S.; Zweyer, M.; Sabir, H.; Swandulla, D.; Ohlendieck, K. The Dystrophin Node as Integrator of Cytoskeletal Organization, Lateral Force Transmission, Fiber Stability and Cellular Signaling in Skeletal Muscle. Proteomes 2021, 9, 9. [Google Scholar] [CrossRef]
- Wilson, D.G.S.; Tinker, A.; Iskratsch, T. The role of the dystrophin glycoprotein complex in muscle cell mechanotransduction. Commun. Biol. 2022, 5, 1022. [Google Scholar] [CrossRef] [PubMed]
- Zabłocka, B.; Górecki, D.C.; Zabłocki, K. Disrupted Calcium Homeostasis in Duchenne Muscular Dystrophy: A Common Mechanism behind Diverse Consequences. Int. J. Mol. Sci. 2021, 22, 11040. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Gargan, S.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of impaired excitation-contraction coupling and abnormal calcium handling in muscular dystrophy. Proteomics 2022, 22, e2200003. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Belosludtsev, K.N. Ion Channels of the Sarcolemma and Intracellular Organelles in Duchenne Muscular Dystrophy: A Role in the Dysregulation of Ion Homeostasis and a Possible Target for Therapy. Int. J. Mol. Sci. 2023, 24, 2229. [Google Scholar] [CrossRef]
- Rosenberg, A.S.; Puig, M.; Nagaraju, K.; Hoffman, E.P.; Villalta, S.A.; Rao, V.A.; Wakefield, L.M.; Woodcock, J. Immune-mediated pathology in Duchenne muscular dystrophy. Sci. Transl. Med. 2015, 7, 299rv4. [Google Scholar] [CrossRef]
- Serrano, A.L.; Muñoz-Cánoves, P. Fibrosis development in early-onset muscular dystrophies: Mechanisms and translational implications. Semin. Cell Dev. Biol. 2017, 64, 181–190. [Google Scholar] [CrossRef]
- Tidball, J.G.; Welc, S.S.; Wehling-Henricks, M. Immunobiology of Inherited Muscular Dystrophies. Compr. Physiol. 2018, 8, 1313–1356. [Google Scholar]
- Muraine, L.; Bensalah, M.; Butler-Browne, G.; Bigot, A.; Trollet, C.; Mouly, V.; Negroni, E. Update on anti-fibrotic pharmacotherapies in skeletal muscle disease. Curr. Opin. Pharmacol. 2023, 68, 102332. [Google Scholar] [CrossRef]
- Serrano, A.L.; Muñoz-Cánoves, P. Mouse Models of Muscle Fibrosis. Methods Mol. Biol. 2021, 2299, 357–370. [Google Scholar] [PubMed]
- Sleboda, D.A.; Stover, K.K.; Roberts, T.J. Diversity of extracellular matrix morphology in vertebrate skeletal muscle. J. Morphol. 2020, 281, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Purslow, P.P. The Structure and Role of Intramuscular Connective Tissue in Muscle Function. Front. Physiol. 2020, 11, 495. [Google Scholar] [CrossRef] [PubMed]
- Schüler, S.C.; Liu, Y.; Dumontier, S.; Grandbois, M.; Le Moal, E.; Cornelison, D.; Bentzinger, C.F. Extracellular matrix: Brick and mortar in the skeletal muscle stem cell niche. Front. Cell Dev. Biol. 2022, 10, 1056523. [Google Scholar] [CrossRef]
- Moratal, C.; Arrighi, N.; Dechesne, C.A.; Dani, C. Control of Muscle Fibro-Adipogenic Progenitors by Myogenic Lineage is Altered in Aging and Duchenne Muscular Dystrophy. Cell. Physiol. Biochem. 2019, 53, 1029–1045. [Google Scholar]
- Molina, T.; Fabre, P.; Dumont, N.A. Fibro-adipogenic progenitors in skeletal muscle homeostasis, regeneration and diseases. Open Biol. 2021, 11, 210110. [Google Scholar] [CrossRef]
- Giuliani, G.; Rosina, M.; Reggio, A. Signaling pathways regulating the fate of fibro/adipogenic progenitors (FAPs) in skeletal muscle regeneration and disease. FEBS J. 2022, 289, 6484–6517. [Google Scholar] [CrossRef]
- Im, W.B.; Phelps, S.F.; Copen, E.H.; Adams, E.G.; Slightom, J.L.; Chamberlain, J.S. Differential expression of dystrophin isoforms in strains of mdx mice with different mutations. Hum. Mol. Genet. 1996, 5, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Murphy, S.; Dowling, P.; Ohlendieck, K. Pathoproteomic profiling of the skeletal muscle matrisome in dystrophinopathy associated myofibrosis. Proteomics 2016, 16, 345–366. [Google Scholar] [CrossRef] [PubMed]
- Bulfield, G.; Siller, W.G.; Wight, P.A.; Moore, K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef]
- Tanabe, Y.; Esaki, K.; Nomura, T. Skeletal muscle pathology in X chromosome-linked muscular dystrophy (mdx) mouse. Acta Neuropathol. 1986, 69, 91–95. [Google Scholar] [CrossRef]
- Partridge, T.A. The mdx mouse model as a surrogate for Duchenne muscular dystrophy. FEBS J. 2013, 280, 4177–4186. [Google Scholar] [CrossRef] [PubMed]
- Chapman, V.M.; Miller, D.R.; Armstrong, D.; Caskey, C.T. Recovery of induced mutations for X chromosome-linked muscular dystrophy in mice. Proc. Natl. Acad. Sci. USA 1989, 86, 1292–1296. [Google Scholar] [CrossRef] [PubMed]
- Pearson-White, S.H. DMD(mdx3Cv) and DMD(mdx4Cv) dystrophin mutations in mice: Rapid polymerase chain reaction genotyping. Neuromuscul. Disord. 2002, 12, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Banks, G.B.; Combs, A.C.; Chamberlain, J.S. Sequencing protocols to genotype mdx, mdx(4cv), and mdx(5cv) mice. Muscle Nerve 2010, 42, 268–270. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Hakim, C.H.; Zhang, K.; Duan, D. Genotyping mdx, mdx3cv, and mdx4cv mice by primer competition polymerase chain reaction. Muscle Nerve 2011, 43, 283–286. [Google Scholar] [CrossRef]
- Danko, I.; Chapman, V.; Wolff, J.A. The frequency of revertants in mdx mouse genetic models for Duchenne muscular dystrophy. Pediatr. Res. 1992, 32, 128–131. [Google Scholar] [CrossRef]
- Gutpell, K.M.; Hrinivich, W.T.; Hoffman, L.M. Skeletal muscle fibrosis in the mdx/utrn+/- mouse validates its suitability as a murine model of Duchenne muscular dystrophy. PLoS ONE 2015, 10, e0117306. [Google Scholar] [CrossRef]
- Giovarelli, M.; Arnaboldi, F.; Zecchini, S.; Cornaghi, L.B.; Nava, A.; Sommariva, M.; Clementi, E.G.I.; Gagliano, N. Characterisation of Progressive Skeletal Muscle Fibrosis in the Mdx Mouse Model of Duchenne Muscular Dystrophy: An In Vivo and In Vitro Study. Int. J. Mol. Sci. 2022, 23, 8735. [Google Scholar] [CrossRef]
- Lazzarin, M.C.; Dos Santos, J.F.; Quintana, H.T.; Pidone, F.A.M.; de Oliveira, F. Duchenne muscular dystrophy progression induced by downhill running is accompanied by increased endomysial fibrosis and oxidative damage DNA in muscle of mdx mice. J. Mol. Histol. 2023, 54, 41–54. [Google Scholar] [CrossRef]
- Stedman, H.H.; Sweeney, H.L.; Shrager, J.B.; Maguire, H.C.; Panettieri, R.A.; Petrof, B.; Narusawa, M.; Leferovich, J.M.; Sladky, J.T.; Kelly, A.M. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 1991, 352, 536–539. [Google Scholar] [CrossRef]
- Sahani, R.; Wallace, C.H.; Jones, B.K.; Blemker, S.S. Diaphragm muscle fibrosis involves changes in collagen organization with mechanical implications in Duchenne muscular dystrophy. J. Appl. Physiol. 2022, 132, 653–672. [Google Scholar] [CrossRef]
- Wohlgemuth, R.P.; Feitzinger, R.M.; Henricson, K.E.; Dinh, D.T.; Brashear, S.E.; Smith, L.R. The extracellular matrix of dystrophic mouse diaphragm accounts for the majority of its passive stiffness and is resistant to collagenase digestion. Matrix Biol. Plus. 2023, 18, 100131. [Google Scholar] [CrossRef] [PubMed]
- Zweyer, M.; Sabir, H.; Dowling, P.; Gargan, S.; Murphy, S.; Swandulla, D.; Ohlendieck, K. Histopathology of Duchenne muscular dystrophy in correlation with changes in proteomic biomarkers. Histol. Histopathol. 2022, 37, 101–116. [Google Scholar]
- Dubuisson, N.; Versele, R.; Planchon, C.; Selvais, C.M.; Noel, L.; Abou-Samra, M.; Davis-López de Carrizosa, M.A. Histological Methods to Assess Skeletal Muscle Degeneration and Regeneration in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2022, 23, 16080. [Google Scholar] [CrossRef]
- Desguerre, I.; Mayer, M.; Leturcq, F.; Barbet, J.P.; Gherardi, R.K.; Christov, C. Endomysial fibrosis in Duchenne muscular dystrophy: A marker of poor outcome associated with macrophage alternative activation. J. Neuropathol. Exp. Neurol. 2009, 68, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Gargan, S.; Dowling, P.; Zweyer, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic Identification of Markers of Membrane Repair, Regeneration and Fibrosis in the Aged and Dystrophic Diaphragm. Life 2022, 12, 1679. [Google Scholar] [CrossRef] [PubMed]
- Omenn, G.S. Reflections on the HUPO Human Proteome Project, the Flagship Project of the Human Proteome Organization, at 10 Years. Mol. Cell. Proteom. 2021, 20, 100062. [Google Scholar] [CrossRef] [PubMed]
- Omenn, G.S.; Lane, L.; Overall, C.M.; Pineau, C.; Packer, N.H.; Cristea, I.M.; Lindskog, C.; Weintraub, S.T.; Orchard, S.; Roehrl, M.H.A.; et al. The 2022 Report on the Human Proteome from the HUPO Human Proteome Project. J. Proteome Res. 2022, 22, 1024–1042. [Google Scholar] [CrossRef]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef]
- Adhikari, S.; Nice, E.C.; Deutsch, E.W.; Lane, L.; Omenn, G.S.; Pennington, S.R.; Paik, Y.K.; Overall, C.M.; Corrales, F.J.; Cristea, I.M.; et al. A high-stringency blueprint of the human proteome. Nat. Commun. 2020, 11, 5301. [Google Scholar] [CrossRef]
- Højlund, K.; Yi, Z.; Hwang, H.; Bowen, B.; Lefort, N.; Flynn, C.R.; Langlais, P.; Weintraub, S.T.; Mandarino, L.J. Characterization of the human skeletal muscle proteome by one-dimensional gel electrophoresis and HPLC-ESI-MS/MS. Mol. Cell. Proteom. 2008, 7, 257–267. [Google Scholar] [CrossRef]
- Parker, K.C.; Walsh, R.J.; Salajegheh, M.; Amato, A.A.; Krastins, B.; Sarracino, D.A.; Greenberg, S.A. Characterization of human skeletal muscle biopsy samples using shotgun proteomics. J. Proteome Res. 2009, 8, 3265–3277. [Google Scholar] [CrossRef] [PubMed]
- Capitanio, D.; Moriggi, M.; Gelfi, C. Mapping the human skeletal muscle proteome: Progress and potential. Expert Rev. Proteom. 2017, 14, 825–839. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C.; Murgia, M. Fiber type diversity in skeletal muscle explored by mass spectrometry-based single fiber proteomics. Histol. Histopathol. 2020, 35, 239–246. [Google Scholar]
- Murgia, M.; Nogara, L.; Baraldo, M.; Reggiani, C.; Mann, M.; Schiaffino, S. Protein profile of fiber types in human skeletal muscle: A single-fiber proteomics study. Skelet. Muscle 2021, 11, 24. [Google Scholar] [CrossRef]
- Dowling, P.; Gargan, S.; Swandulla, D.; Ohlendieck, K. Identification of Subproteomic Markers for Skeletal Muscle Profiling. Methods Mol. Biol. 2023, 2596, 291–302. [Google Scholar]
- Randles, M.; Lennon, R. Applying Proteomics to Investigate Extracellular Matrix in Health and Disease. Curr. Top. Membr. 2015, 76, 171–196. [Google Scholar]
- Taha, I.N.; Naba, A. Exploring the extracellular matrix in health and disease using proteomics. Essays Biochem. 2019, 63, 417–432. [Google Scholar]
- Naba, A. Ten Years of Extracellular Matrix Proteomics: Accomplishments, Challenges, and Future Perspectives. Mol. Cell. Proteom. 2023, 22, 100528. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O.; Naba, A. Overview of the matrisome—An inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 2016, 49, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Taha, I.N.; Clauser, K.R.; Gao, Y.T.; Naba, A. MatrisomeDB: The ECM-protein knowledge database. Nucleic Acids Res. 2020, 48, D1136–D1144. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Gomez, C.D.; Kapoor, N.; Considine, J.M.; Grams, C.; Gao, Y.T.; Naba, A. MatrisomeDB 2.0: 2023 updates to the ECM-protein knowledge database. Nucleic Acids Res. 2023, 51, D1519–D1530. [Google Scholar] [CrossRef] [PubMed]
- Krasny, L.; Paul, A.; Wai, P.; Howard, B.A.; Natrajan, R.C.; Huang, P.H. Comparative proteomic assessment of matrisome enrichment methodologies. Biochem. J. 2016, 473, 3979–3995. [Google Scholar] [CrossRef]
- Krasny, L.; Huang, P.H. Advances in the proteomic profiling of the matrisome and adhesome. Expert Rev. Proteom. 2021, 18, 781–794. [Google Scholar] [CrossRef]
- Dussoyer, M.; Page, A.; Delolme, F.; Rousselle, P.; Nyström, A.; Moali, C. Comparison of extracellular matrix enrichment protocols for the improved characterization of the skin matrisome by mass spectrometry. J. Proteom. 2022, 251, 104397. [Google Scholar] [CrossRef]
- Ercan, H.; Resch, U.; Hsu, F.; Mitulovic, G.; Bileck, A.; Gerner, C.; Yang, J.W.; Geiger, M.; Miller, I.; Zellner, M. A Practical and Analytical Comparative Study of Gel-Based Top-Down and Gel-Free Bottom-Up Proteomics Including Unbiased Proteoform Detection. Cells 2023, 12, 747. [Google Scholar] [CrossRef]
- Manes, N.P.; Nita-Lazar, A. Application of targeted mass spectrometry in bottom-up proteomics for systems biology research. J. Proteom. 2018, 189, 75–90. [Google Scholar] [CrossRef]
- Ohlendieck, K. Top-Down Proteomics and Comparative 2D-DIGE Analysis. Methods Mol. Biol. 2023, 2596, 19–38. [Google Scholar] [PubMed]
- Dowling, P.; Gargan, S.; Swandulla, D.; Ohlendieck, K. Fiber-Type Shifting in Sarcopenia of Old Age: Proteomic Profiling of the Contractile Apparatus of Skeletal Muscles. Int. J. Mol. Sci. 2023, 24, 2415. [Google Scholar] [CrossRef]
- Mansuri, M.S.; Williams, K.; Nairn, A.C. Uncovering biology by single-cell proteomics. Commun. Biol. 2023, 6, 381. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Murphy, S.; Ohlendieck, K. Proteomic profiling of muscle fibre type shifting in neuromuscular diseases. Expert Rev. Proteom. 2016, 13, 783–799. [Google Scholar] [CrossRef] [PubMed]
- Sielaff, M.; Kuharev, J.; Bohn, T.; Hahlbrock, J.; Bopp, T.; Tenzer, S.; Distler, U. Evaluation of FASP, SP3, and iST Protocols for Proteomic Sample Preparation in the Low Microgram Range. J. Proteome Res. 2017, 16, 4060–4072. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Mann, M. Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J. Proteome Res. 2009, 8, 5674–5678. [Google Scholar] [CrossRef]
- Xie, Z.; Feng, Q.; Zhang, S.; Yan, Y.; Deng, C.; Ding, C.F. Advances in proteomics sample preparation and enrichment for phosphorylation and glycosylation analysis. Proteomics 2022, 22, e2200070. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Wiśniewski, J.R. Filter Aided Sample Preparation—A tutorial. Anal. Chim. Acta 2019, 1090, 23–30. [Google Scholar] [CrossRef]
- Erde, J.; Loo, R.R.; Loo, J.A. Improving Proteome Coverage and Sample Recovery with Enhanced FASP (eFASP) for Quantitative Proteomic Experiments. Methods Mol. Biol. 2017, 1550, 11–18. [Google Scholar]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Kostas, J.C.; Greguš, M.; Schejbal, J.; Ray, S.; Ivanov, A.R. Simple and Efficient Microsolid-Phase Extraction Tip-Based Sample Preparation Workflow to Enable Sensitive Proteomic Profiling of Limited Samples (200 to 10,000 Cells). J. Proteome Res. 2021, 20, 1676–1688. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.S.; Moggridge, S.; Müller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef]
- Dagley, L.F.; Infusini, G.; Larsen, R.H.; Sandow, J.J.; Webb, A.I. Universal Solid-Phase Protein Preparation (USP3) for Bottom-up and Top-down Proteomics. J. Proteome Res. 2019, 18, 2915–2924. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Xue, Z.; Wu, C.; Sun, R.; Qian, L.; Yue, L.; Ge, W.; Yi, X.; Liu, W.; Chen, C.; et al. High-throughput proteomic sample preparation using pressure cycling technology. Nat. Protoc. 2022, 17, 2307–2325. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Liu, F.; Xu, W.; Li, L. Surfactant and Chaotropic Agent Assisted Sequential Extraction/On-Pellet Digestion (SCAD) for Enhanced Proteomics. J. Proteome Res. 2018, 17, 2744–2754. [Google Scholar] [CrossRef] [PubMed]
- Doellinger, J.; Schneider, A.; Hoeller, M.; Lasch, P. Sample Preparation by Easy Extraction and Digestion (SPEED)—A Universal, Rapid, and Detergent-free Protocol for Proteomics Based on Acid Extraction. Mol. Cell. Proteom. 2020, 19, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Gargan, S.; Zweyer, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Protocol for the Bottom-Up Proteomic Analysis of Mouse Spleen. STAR Protoc. 2020, 1, 100196. [Google Scholar] [CrossRef]
- Murphy, S.; Ohlendieck, K. Proteomic profiling of large myofibrillar proteins from dried and long-term stored polyacrylamide gels. Anal. Biochem. 2018, 543, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Wen, M.; Yuan, Q.; Zhang, J.; Tan, W. Beneficial effects of Coomassie staining on proteomic analysis employing PAGE separation followed with whole-gel slicing, in-gel digestion and quantitative LC-MS/MS. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2019, 1110–1111, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wen, M.; Jin, Y. 1DE-MS Profiling for Proteoform-Correlated Proteomic Analysis, by Combining SDS-PAGE, Whole-Gel Slicing, Quantitative LC-MS/MS, and Reconstruction of Gel Distributions of Several Thousands of Proteins. J. Proteome Res. 2022, 21, 2311–2330. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Characterization of Contractile Proteins from Skeletal Muscle Using Gel-Based Top-Down Proteomics. Proteomes 2019, 7, 25. [Google Scholar] [CrossRef]
- Zhan, X.; Li, B.; Zhan, X.; Schlüter, H.; Jungblut, P.R.; Coorssen, J.R. Innovating the Concept and Practice of Two-Dimensional Gel Electrophoresis in the Analysis of Proteomes at the Proteoform Level. Proteomes 2019, 7, 36. [Google Scholar] [CrossRef]
- Carbonara, K.; Padula, M.P.; Coorssen, J.R. Quantitative assessment confirms deep proteome analysis by integrative top-down proteomics. Electrophoresis 2023, 44, 472–480. [Google Scholar] [CrossRef]
- Arentz, G.; Weiland, F.; Oehler, M.K.; Hoffmann, P. State of the art of 2D DIGE. Proteom. Clin. Appl. 2015, 9, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Blundon, M.; Ganesan, V.; Redler, B.; Van, P.T.; Minden, J.S. Two-Dimensional Difference Gel Electrophoresis. Methods Mol. Biol. 2019, 1855, 229–247. [Google Scholar]
- Ohlendieck, K. Comparative 3-Sample 2D-DIGE Analysis of Skeletal Muscles. Methods Mol. Biol. 2023, 2596, 127–146. [Google Scholar] [PubMed]
- Naba, A.; Clauser, K.R.; Hoersch, S.; Liu, H.; Carr, S.A.; Hynes, R.O. The matrisome: In silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell. Proteom. 2012, 11, M111.014647. [Google Scholar] [CrossRef]
- Hill, R.C.; Calle, E.A.; Dzieciatkowska, M.; Niklason, L.E.; Hansen, K.C. Quantification of extracellular matrix proteins from a rat lung scaffold to provide a molecular readout for tissue engineering. Mol. Cell. Proteom. 2015, 14, 961–973. [Google Scholar] [CrossRef]
- Schiller, H.B.; Fernandez, I.E.; Burgstaller, G.; Schaab, C.; Scheltema, R.A.; Schwarzmayr, T.; Strom, T.M.; Eickelberg, O.; Mann, M. Time- and compartment-resolved proteome profiling of the extracellular niche in lung injury and repair. Mol. Syst. Biol. 2015, 11, 819. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.A.; Chen, B.; Guardado-Alvarez, T.M.; Lin, Z.; Hwang, L.; Ayaz-Guner, S.; Jin, S.; Ge, Y. A photocleavable surfactant for top-down proteomics. Nat. Methods 2019, 16, 417–420. [Google Scholar] [CrossRef]
- Knott, S.J.; Brown, K.A.; Josyer, H.; Carr, A.; Inman, D.; Jin, S.; Friedl, A.; Ponik, S.M.; Ge, Y. Photocleavable Surfactant-Enabled Extracellular Matrix Proteomics. Anal. Chem. 2020, 92, 15693–15698. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, T.W.; Sellaro, T.L.; Badylak, S.F. Decellularization of tissues and organs. Biomaterials 2006, 27, 3675–3683. [Google Scholar] [CrossRef]
- Fernández-Pérez, J.; Ahearne, M. The impact of decellularization methods on extracellular matrix derived hydrogels. Sci. Rep. 2019, 9, 14933. [Google Scholar] [CrossRef]
- Mendibil, U.; Ruiz-Hernandez, R.; Retegi-Carrion, S.; Garcia-Urquia, N.; Olalde-Graells, B.; Abarrategi, A. Tissue-Specific Decellularization Methods: Rationale and Strategies to Achieve Regenerative Compounds. Int. J. Mol. Sci. 2020, 21, 5447. [Google Scholar] [CrossRef] [PubMed]
- Domon, B.; Aebersold, R. Mass spectrometry and protein analysis. Science 2006, 312, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.C.; Yates, J.R., 3rd. Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R.; Ruse, C.I.; Nakorchevsky, A. Proteomics by mass spectrometry: Approaches, advances, and applications. Annu. Rev. Biomed. Eng. 2009, 11, 49–79. [Google Scholar] [CrossRef]
- Yates, J.R., 3rd. Recent technical advances in proteomics. F1000Research 2019, 8, 351. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I.; Vitek, O.; Aebersold, R. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat. Methods 2007, 4, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, A.J.; Ferreira, T.L. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry in the diagnosis of microorganisms. Future Microbiol. 2022, 17, 1409–1419. [Google Scholar] [CrossRef]
- Sabidó, E.; Selevsek, N.; Aebersold, R. Mass spectrometry-based proteomics for systems biology. Curr. Opin. Biotechnol. 2012, 23, 591–597. [Google Scholar] [CrossRef]
- Rauniyar, N.; Yates, J.R., 3rd. Isobaric labeling-based relative quantification in shotgun proteomics. J. Proteome Res. 2014, 13, 5293–5309. [Google Scholar] [CrossRef]
- Beller, N.C.; Hummon, A.B. Advances in stable isotope labeling: Dynamic labeling for spatial and temporal proteomic analysis. Mol. Omics 2022, 18, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sun, Y.; Zhang, T.; Shu, L.; Roepstorff, P.; Yang, F. Quantitative Proteomics Using Isobaric Labeling: A Practical Guide. Genom. Proteom. Bioinform. 2021, 19, 689–706. [Google Scholar] [CrossRef]
- Chahrour, O.; Cobice, D.; Malone, J. Stable isotope labelling methods in mass spectrometry-based quantitative proteomics. J. Pharm. Biomed. Anal. 2015, 113, 2–20. [Google Scholar] [CrossRef] [PubMed]
- Xing, T.; Wang, C.; Zhao, X.; Dai, C.; Zhou, G.; Xu, X. Proteome Analysis Using Isobaric Tags for Relative and Absolute Analysis Quantitation (iTRAQ) Reveals Alterations in Stress-Induced Dysfunctional Chicken Muscle. J. Agric. Food Chem. 2017, 65, 2913–2922. [Google Scholar] [CrossRef]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Sobsey, C.A.; Ibrahim, S.; Richard, V.R.; Gaspar, V.; Mitsa, G.; Lacasse, V.; Zahedi, R.P.; Batist, G.; Borchers, C.H. Targeted and Untargeted Proteomics Approaches in Biomarker Development. Proteomics 2020, 20, e1900029. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Kumar, C.; Zeng, W.F.; Strauss, M.T. Artificial intelligence for proteomics and biomarker discovery. Cell Syst. 2021, 12, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Fenaille, F.; Barbier Saint-Hilaire, P.; Rousseau, K.; Junot, C. Data acquisition workflows in liquid chromatography coupled to high resolution mass spectrometry-based metabolomics: Where do we stand? J. Chromatogr. A 2017, 1526, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kitata, R.B.; Yang, J.C.; Chen, Y.J. Advances in data-independent acquisition mass spectrometry towards comprehensive digital proteome landscape. Mass Spectrom. Rev. 2022, e21781. [Google Scholar] [CrossRef]
- Abdollahi, M.; Segura, P.A.; Beaudry, F. Is nontargeted data acquisition for target analysis (nDATA) in mass spectrometry a forward-thinking analytical approach? Biomed. Chromatogr. 2023, 37, e5531. [Google Scholar] [CrossRef] [PubMed]
- Krasny, L.; Bland, P.; Kogata, N.; Wai, P.; Howard, B.A.; Natrajan, R.C.; Huang, P.H. SWATH mass spectrometry as a tool for quantitative profiling of the matrisome. J. Proteom. 2018, 189, 11–22. [Google Scholar] [CrossRef]
- Wood, A.J.; Lin, C.H.; Li, M.; Nishtala, K.; Alaei, S.; Rossello, F.; Sonntag, C.; Hersey, L.; Miles, L.B.; Krisp, C.; et al. FKRP-dependent glycosylation of fibronectin regulates muscle pathology in muscular dystrophy. Nat. Commun. 2021, 12, 2951. [Google Scholar] [CrossRef] [PubMed]
- Kontostathi, G.; Makridakis, M.; Bitsika, V.; Tsolakos, N.; Vlahou, A.; Zoidakis, J. Development and Validation of Multiple Reaction Monitoring (MRM) Assays for Clinical Applications. Methods Mol. Biol. 2019, 1959, 205–223. [Google Scholar] [PubMed]
- Cho, B.G.; Gutierrez Reyes, C.D.; Goli, M.; Gautam, S.; Banazadeh, A.; Mechref, Y. Targeted N-Glycan Analysis with Parallel Reaction Monitoring Using a Quadrupole-Orbitrap Hybrid Mass Spectrometer. Anal. Chem. 2022, 94, 15215–15222. [Google Scholar] [CrossRef]
- Thorsen, A.F.; Riber, L.P.S.; Rasmussen, L.M.; Overgaard, M. A targeted multiplex mass spectrometry method for quantitation of abundant matrix and cellular proteins in formalin-fixed paraffin embedded arterial tissue. J. Proteom. 2023, 272, 104775. [Google Scholar] [CrossRef]
- Maaß, S. Absolute Protein Quantification Using AQUA-Calibrated 2D-PAGE. Methods Mol. Biol. 2018, 1841, 141–162. [Google Scholar]
- Gillies, A.R.; Lieber, R.L. Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve 2011, 44, 318–331. [Google Scholar] [CrossRef]
- Lieber, R.L.; Meyer, G. Structure-Function relationships in the skeletal muscle extracellular matrix. J. Biomech. 2023, 152, 111593. [Google Scholar] [CrossRef]
- Carbonara, K.; Andonovski, M.; Coorssen, J.R. Proteomes Are of Proteoforms: Embracing the Complexity. Proteomes 2021, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, L.V.; Millikin, R.J.; Miller, R.M.; Anderson, L.C.; Fellers, R.T.; Ge, Y.; Kelleher, N.L.; LeDuc, R.D.; Liu, X.; Payne, S.H.; et al. Identification and Quantification of Proteoforms by Mass Spectrometry. Proteomics 2019, 19, e1800361. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Al-Shareffi, E.; Haltiwanger, R.S. Biological functions of fucose in mammals. Glycobiology 2017, 27, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Basak, T.; Vega-Montoto, L.; Zimmerman, L.J.; Tabb, D.L.; Hudson, B.G.; Vanacore, R.M. Comprehensive Characterization of Glycosylation and Hydroxylation of Basement Membrane Collagen IV by High-Resolution Mass Spectrometry. J. Proteome Res. 2016, 15, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Galligan, J.J.; Petersen, D.R. The human protein disulfide isomerase gene family. Hum. Genom. 2012, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Stefanelli, V.L.; Choudhury, S.; Hu, P.; Liu, Y.; Schwenzer, A.; Yeh, C.R.; Chambers, D.M.; von Beck, K.; Li, W.; Segura, T.; et al. Citrullination of fibronectin alters integrin clustering and focal adhesion stability promoting stromal cell invasion. Matrix Biol. 2019, 82, 86–104. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Zweyerm, M.; Raucamp, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of the mouse diaphragm and refined mass spectrometric analysis of the dystrophic phenotype. J. Muscle Res. Cell Motil. 2019, 40, 9–28. [Google Scholar] [CrossRef] [PubMed]
- Halper, J. Basic Components of Connective Tissues and Extracellular Matrix: Fibronectin, Fibrinogen, Laminin, Elastin, Fibrillins, Fibulins, Matrilins, Tenascins and Thrombospondins. Adv. Exp. Med. Biol. 2021, 1348, 105–126. [Google Scholar] [PubMed]
- Holmberg, J.; Durbeej, M. Laminin-211 in skeletal muscle function. Cell Adhes. Migr. 2013, 7, 111–121. [Google Scholar] [CrossRef]
- Goddi, A.; Schroedl, L.; Brey, E.M.; Cohen, R.N. Laminins in metabolic tissues. Metabolism 2021, 120, 154775. [Google Scholar] [CrossRef]
- Rogers, R.S.; Nishimune, H. The role of laminins in the organization and function of neuromuscular junctions. Matrix Biol. 2017, 57–58, 86–105. [Google Scholar] [CrossRef]
- Singhal, N.; Martin, P.T. Role of extracellular matrix proteins and their receptors in the development of the vertebrate neuromuscular junction. Dev. Neurobiol. 2011, 71, 982–1005. [Google Scholar] [CrossRef] [PubMed]
- Kadler, K.E.; Baldock, C.; Bella, J.; Boot-Handford, R.P. Collagens at a glance. J. Cell Sci. 2007, 120, 1955–1958. [Google Scholar] [CrossRef] [PubMed]
- Goody, M.F.; Sher, R.B.; Henry, C.A. Hanging on for the ride: Adhesion to the extracellular matrix mediates cellular responses in skeletal muscle morphogenesis and disease. Dev. Biol. 2015, 401, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Grefte, S.; Adjobo-Hermans, M.J.W.; Versteeg, E.M.M.; Koopman, W.J.H.; Daamen, W.F. Impaired primary mouse myotube formation on crosslinked type I collagen films is enhanced by laminin and entactin. Acta Biomater. 2016, 30, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Fox, M.A.; Ho, M.S.; Smyth, N.; Sanes, J.R. A synaptic nidogen: Developmental regulation and role of nidogen-2 at the neuromuscular junction. Neural Dev. 2008, 3, 24. [Google Scholar] [CrossRef]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef]
- Mienaltowski, M.J.; Gonzales, N.L.; Beall, J.M.; Pechanec, M.Y. Basic Structure, Physiology, and Biochemistry of Connective Tissues and Extracellular Matrix Collagens. Adv. Exp. Med. Biol. 2021, 1348, 5–43. [Google Scholar]
- Cescon, M.; Gattazzo, F.; Chen, P.; Bonaldo, P. Collagen VI at a glance. J. Cell Sci. 2015, 128, 3525–3531. [Google Scholar] [CrossRef]
- Cescon, M.; Gregorio, I.; Eiber, N.; Borgia, D.; Fusto, A.; Sabatelli, P.; Scorzeto, M.; Megighian, A.; Pegoraro, E.; Hashemolhosseini, S.; et al. Collagen VI is required for the structural and functional integrity of the neuromuscular junction. Acta Neuropathol. 2018, 136, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Ling, Z.; Ren, X. Extracellular matrix dynamics: Tracking in biological systems and their implications. J. Biol. Eng. 2022, 16, 13. [Google Scholar] [CrossRef]
- Zheng, Z.; Granado, H.S.; Li, C. Fibromodulin, a Multifunctional Matricellular Modulator. J. Dent. Res. 2023, 102, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, D.; Schips, T.G.; Kwong, J.Q.; Davis, J.; Tjondrokoesoemo, A.; Brody, M.J.; Sargent, M.A.; Kanisicak, O.; Yi, H.; Gao, Q.Q.; et al. Thrombospondin expression in myofibers stabilizes muscle membranes. Elife 2016, 5, e17589. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; An, J.; Zhu, D.; Chen, H.; Lin, A.; Kang, J.; Liu, W.; Kang, X. Periostin: An emerging activator of multiple signaling pathways. J. Cell. Commun. Signal. 2022, 16, 515–530. [Google Scholar] [CrossRef]
- Boppart, M.D.; Mahmassani, Z.S. Integrin signaling: Linking mechanical stimulation to skeletal muscle hypertrophy. Am. J. Physiol.-Cell Physiol. 2019, 317, C629–C641. [Google Scholar] [CrossRef]
- Ibraghimov-Beskrovnaya, O.; Ervasti, J.M.; Leveille, C.J.; Slaughter, C.A.; Sernett, S.W.; Campbell, K.P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 1992, 355, 696–702. [Google Scholar] [CrossRef]
- Ohlendieck, K. Towards an understanding of the dystrophin-glycoprotein complex: Linkage between the extracellular matrix and the membrane cytoskeleton in muscle fibers. Eur. J. Cell Biol. 1996, 69, 1–10. [Google Scholar]
- Murphy, S.; Ohlendieck, K. The biochemical and mass spectrometric profiling of the dystrophin complexome from skeletal muscle. Comput. Struct. Biotechnol. J. 2015, 14, 20–27. [Google Scholar] [CrossRef]
- Sciandra, F.; Bozzi, M.; Bigotti, M.G.; Brancaccio, A. The multiple affinities of α-dystroglycan. Curr. Protein Pept. Sci. 2013, 14, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Sciandra, F.; Gawlik, K.I.; Brancaccio, A.; Durbeej, M. Dystroglycan: A possible mediator for reducing congenital muscular dystrophy? Trends Biotechnol. 2007, 25, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.R.; Lipp, S.; Acuna, A.; Leng, Y.; Bu, Y.; Calve, S. Comparative Analysis of the Extracellular Matrix Proteome across the Myotendinous Junction. J. Proteome Res. 2020, 19, 3955–3967. [Google Scholar] [CrossRef] [PubMed]
- Izu, Y.; Adams, S.M.; Connizzo, B.K.; Beason, D.P.; Soslowsky, L.J.; Koch, M.; Birk, D.E. Collagen XII mediated cellular and extracellular mechanisms regulate establishment of tendon structure and function. Matrix Biol. 2021, 95, 52–67. [Google Scholar] [CrossRef]
- Karlsen, A.; Gonzalez-Franquesa, A.; Jakobsen, J.R.; Krogsgaard, M.R.; Koch, M.; Kjaer, M.; Schiaffino, S.; Mackey, A.L.; Deshmukh, A.S. The proteomic profile of the human myotendinous junction. iScience 2022, 25, 103836. [Google Scholar] [CrossRef]
- Franchi, M.; Trirè, A.; Quaranta, M.; Orsini, E.; Ottani, V. Collagen structure of tendon relates to function. Sci. World J. 2007, 7, 404–420. [Google Scholar] [CrossRef]
- Croissant, C.; Gounou, C.; Bouvet, F.; Tan, S.; Bouter, A. Annexin-A6 in Membrane Repair of Human Skeletal Muscle Cell: A Role in the Cap Subdomain. Cells 2020, 9, 1742. [Google Scholar] [CrossRef]
- Vidak, E.; Javoršek, U.; Vizovišek, M.; Turk, B. Cysteine Cathepsins and their Extracellular Roles: Shaping the Microenvironment. Cells 2019, 8, 264. [Google Scholar] [CrossRef]
- Spence, M.A.; Mortimer, M.D.; Buckle, A.M.; Minh, B.Q.; Jackson, C.J. A Comprehensive Phylogenetic Analysis of the Serpin Superfamily. Mol. Biol. Evol. 2021, 38, 2915–2929. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, Y.; Zhang, H. Extracellular matrix: An important regulator of cell functions and skeletal muscle development. Cell Biosci. 2021, 11, 65. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Guiraud, S.; Aartsma-Rus, A.; Vieira, N.M.; Davies, K.E.; van Ommen, G.J.; Kunkel, L.M. The Pathogenesis and Therapy of Muscular Dystrophies. Annu. Rev. Genom. Hum. Genet. 2015, 16, 281–308. [Google Scholar] [CrossRef]
- Crisafulli, S.; Sultana, J.; Fontana, A.; Salvo, F.; Messina, S.; Trifirò, G. Global epidemiology of Duchenne muscular dystrophy: An updated systematic review and meta-analysis. Orphanet J. Rare Dis. 2020, 15, 141. [Google Scholar] [CrossRef]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef]
- Doglio, L.; Pavan, E.; Pernigotti, I.; Petralia, P.; Frigo, C.; Minetti, C. Early signs of gait deviation in Duchenne muscular dystrophy. Eur. J. Phys. Rehabil. Med. 2011, 47, 587–594. [Google Scholar] [PubMed]
- Migliorelli, C.; Gómez-Martinez, M.; Subías-Beltrán, P.; Claramunt-Molet, M.; Idelsohn-Zielonka, S.; Mas-Hurtado, E.; Miralles, F.; Montolio, M.; Roselló-Ruano, M.; Medina-Cantillo, J. Multidimensional Biomechanics-Based Score to Assess Disease Progression in Duchenne Muscular Dystrophy. Sensors 2023, 23, 831. [Google Scholar] [CrossRef]
- Sienko, S.; Buckon, C.; Bagley, A.; Fowler, E.; Heberer, K.; Staudt, L.; Sison-Williamson, M.; McDonald, C.; Sussman, M. Kinematic changes in gait in boys with Duchenne Muscular Dystrophy: Utility of the Gait Deviation Index, the Gait Profile Score and the Gait Variable Scores. Gait Posture 2023, 100, 157–164. [Google Scholar] [CrossRef]
- Meyers, T.A.; Townsend, D. Cardiac Pathophysiology and the Future of Cardiac Therapies in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2019, 20, 4098. [Google Scholar] [CrossRef] [PubMed]
- Power, L.C.; O’Grady, G.L.; Hornung, T.S.; Jefferies, C.; Gusso, S.; Hofman, P.L. Imaging the heart to detect cardiomyopathy in Duchenne muscular dystrophy: A review. Neuromuscul. Disord. 2018, 28, 717–730. [Google Scholar] [CrossRef]
- Vianello, A.; Arcaro, G.; Guarnieri, G.; Sukthi, A.; Molena, B.; Turato, C.; Braccioni, F.; Gallan, F.; Lugato, F.; Turrin, M.; et al. Non-Invasive Ventilation for Acute Respiratory Failure in Duchenne Muscular Dystrophy Patients. Arch. Bronconeumol. 2021, 57, 666–668. [Google Scholar] [CrossRef] [PubMed]
- Birnkrant, D.J.; Bello, L.; Butterfield, R.J.; Carter, J.C.; Cripe, L.H.; Cripe, T.P.; McKim, D.A.; Nandi, D.; Pegoraro, E. Cardiorespiratory management of Duchenne muscular dystrophy: Emerging therapies, neuromuscular genetics, and new clinical challenges. Lancet Respir. Med. 2022, 10, 403–420. [Google Scholar] [CrossRef]
- Tsaknakis, K.; Jäckle, K.; Lüders, K.A.; Lorenz, H.M.; Braunschweig, L.; Hell, A.K. Reduced bone mineral density in adolescents with Duchenne Muscular Dystrophy (DMD) and scoliosis. Osteoporos. Int. 2022, 33, 2011–2018. [Google Scholar] [CrossRef]
- Conway, K.M.; Gedlinske, A.; Mathews, K.D.; Perlman, S.; Johnson, N.; Butterfield, R.; Hung, M.; Bounsanga, J.; Matthews, D.; Oleszek, J.; et al. A population-based study of scoliosis among males diagnosed with a dystrophinopathy identified by the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet). Muscle Nerve 2022, 65, 193–202. [Google Scholar] [CrossRef]
- Messina, S.; Vita, G.L. Clinical management of Duchenne muscular dystrophy: The state of the art. Neurol. Sci. 2018, 39, 1837–1845. [Google Scholar] [CrossRef]
- Snow, W.M.; Anderson, J.E.; Jakobson, L.S. Neuropsychological and neurobehavioral functioning in Duchenne muscular dystrophy: A review. Neurosci. Biobehav. Rev. 2013, 37, 743–752. [Google Scholar] [CrossRef]
- Chieffo, D.P.R.; Moriconi, F.; Pane, M.; Lucibello, S.; Ferraroli, E.; Norcia, G.; Ricci, M.; Capasso, A.; Cicala, G.; Buchignani, B.; et al. A Longitudinal Follow-Up Study of Intellectual Function in Duchenne Muscular Dystrophy over Age: Is It Really Stable? J. Clin. Med. 2023, 12, 403. [Google Scholar] [CrossRef]
- Pascual-Morena, C.; Cavero-Redondo, I.; Álvarez-Bueno, C.; Jiménez-López, E.; Saz-Lara, A.; Martínez-García, I.; Martínez-Vizcaíno, V. Global prevalence of intellectual developmental disorder in dystrophinopathies: A systematic review and meta-analysis. Dev. Med. Child Neurol. 2023, 65, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.A.; Apkon, S.D.; Blackwell, A.; Case, L.E.; Cripe, L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018, 17, 347–361. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Colvin, M.K.; Cripe, L.; Herron, A.R.; Kennedy, A.; Kinnett, K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 3: Primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol. 2018, 17, 445–455. [Google Scholar] [CrossRef]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Forrest, S.M.; Cross, G.S.; Flint, T.; Speer, A.; Robson, K.J.; Davies, K.E. Further studies of gene deletions that cause Duchenne and Becker muscular dystrophies. Genomics 1988, 2, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, L.M.; Hoffman, E.P. Duchenne/Becker muscular dystrophy: A short overview of the gene, the protein, and current diagnostics. Br. Med. Bull. 1989, 45, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Hudecki, M.S.; Rosenberg, P.A.; Pollina, C.M.; Kunkel, L.M. Cell and fiber-type distribution of dystrophin. Neuron 1988, 1, 411–420. [Google Scholar] [CrossRef]
- Bonilla, E.; Samitt, C.E.; Miranda, A.F.; Hays, A.P.; Salviati, G.; DiMauro, S.; Kunkel, L.M.; Hoffman, E.P.; Rowland, L.P. Duchenne muscular dystrophy: Deficiency of dystrophin at the muscle cell surface. Cell 1988, 54, 447–452. [Google Scholar] [CrossRef]
- Ohlendieck, K.; Matsumura, K.; Ionasescu, V.V.; Towbin, J.A.; Bosch, E.P.; Weinstein, S.L.; Sernett, S.W.; Campbell, K.P. Duchenne muscular dystrophy: Deficiency of dystrophin-associated proteins in the sarcolemma. Neurology 1993, 43, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Lieber, R.L.; Ward, S.R. Cellular mechanisms of tissue fibrosis. 4. Structural and functional consequences of skeletal muscle fibrosis. Am. J. Physiol.-Cell Physiol. 2013, 305, C241–C252. [Google Scholar] [CrossRef] [PubMed]
- Levi, O.; Genin, O.; Angelini, C.; Halevy, O.; Pines, M. Inhibition of muscle fibrosis results in increases in both utrophin levels and the number of revertant myofibers in Duchenne muscular dystrophy. Oncotarget 2015, 6, 23249–23260. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, S.; Bragato, C.; Zucchella, A.; Maggi, L.; Mantegazza, R.; Morandi, L.; Mora, M. Anti-fibrotic effect of pirfenidone in muscle derived-fibroblasts from Duchenne muscular dystrophy patients. Life Sci. 2016, 145, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, S.; Shimizu-Motohashi, Y.; Takeda, S.; Aoki, Y. Anti-inflammatory drugs for Duchenne muscular dystrophy: Focus on skeletal muscle-releasing factors. Drug Des. Devel. Ther. 2016, 10, 2745–2758. [Google Scholar] [PubMed]
- March, J.T.; Golshirazi, G.; Cernisova, V.; Carr, H.; Leong, Y.; Lu-Nguyen, N.; Popplewell, L.J. Targeting TGFβ Signaling to Address Fibrosis Using Antisense Oligonucleotides. Biomedicines 2018, 6, 74. [Google Scholar] [CrossRef]
- Durbeej, M.; Campbell, K.P. Muscular dystrophies involving the dystrophin-glycoprotein complex: An overview of current mouse models. Curr. Opin. Genet. Dev. 2002, 12, 349–361. [Google Scholar] [CrossRef]
- Sciandra, F.; Bigotti, M.G.; Giardina, B.; Bozzi, M.; Brancaccio, A. Genetic Engineering of Dystroglycan in Animal Models of Muscular Dystrophy. Biomed. Res. Int. 2015, 2015, 635792. [Google Scholar] [CrossRef]
- Wilson, K.; Faelan, C.; Patterson-Kane, J.C.; Rudmann, D.G.; Moore, S.A.; Frank, D.; Charleston, J.; Tinsley, J.; Young, G.D.; Milici, A.J. Duchenne and Becker Muscular Dystrophies: A Review of Animal Models, Clinical End Points, and Biomarker Quantification. Toxicol. Pathol. 2017, 45, 961–976. [Google Scholar] [CrossRef]
- van Putten, M.; Lloyd, E.M.; de Greef, J.C.; Raz, V.; Willmann, R.; Grounds, M.D. Mouse models for muscular dystrophies: An overview. Dis. Models Mech. 2020, 13, dmm043562. [Google Scholar] [CrossRef]
- Sztretye, M.; Szabó, L.; Dobrosi, N.; Fodor, J.; Szentesi, P.; Almássy, J.; Magyar, Z.É.; Dienes, B.; Csernoch, L. From Mice to Humans: An Overview of the Potentials and Limitations of Current Transgenic Mouse Models of Major Muscular Dystrophies and Congenital Myopathies. Int. J. Mol. Sci. 2020, 21, 8935. [Google Scholar] [CrossRef]
- Zaynitdinova, M.I.; Lavrov, A.V.; Smirnikhina, S.A. Animal models for researching approaches to therapy of Duchenne muscular dystrophy. Transgenic Res. 2021, 30, 709–725. [Google Scholar] [CrossRef]
- Yucel, N.; Chang, A.C.; Day, J.W.; Rosenthal, N.; Blau, H.M. Humanizing the mdx mouse model of DMD: The long and the short of it. NPJ Regen. Med. 2018, 3, 4. [Google Scholar] [CrossRef]
- Holland, A.; Ohlendieck, K. Proteomic profiling of the dystrophin-deficient mdx phenocopy of dystrophinopathy-associated cardiomyopathy. Biomed. Res. Int. 2014, 2014, 246195. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Dowling, P.; Zweyer, M.; Swandulla, D.; Henry, M.; Clynes, M.; Ohlendieck, K. Proteomic profiling of cardiomyopathic tissue from the aged mdx model of Duchenne muscular dystrophy reveals a drastic decrease in laminin, nidogen and annexin. Proteomics 2013, 13, 2312–2323. [Google Scholar] [CrossRef]
- Yu, X.; Bao, B.; Echigoya, Y.; Yokota, T. Dystrophin-deficient large animal models: Translational research and exon skipping. Am. J. Transl. Res. 2015, 7, 1314–1331. [Google Scholar] [PubMed]
- Kornegay, J.N. The golden retriever model of Duchenne muscular dystrophy. Skelet. Muscle 2017, 7, 9. [Google Scholar] [CrossRef]
- Duan, D. Duchenne muscular dystrophy gene therapy in the canine model. Hum. Gene Ther. Clin. Dev. 2015, 26, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Cassano, M.; Berardi, E.; Crippa, S.; Toelen, J.; Barthelemy, I.; Micheletti, R.; Chuah, M.; Vandendriessche, T.; Debyser, Z.; Blot, S.; et al. Alteration of cardiac progenitor cell potency in GRMD dogs. Cell Transpl. 2012, 21, 1945–1967. [Google Scholar] [CrossRef] [PubMed]
- Lardenois, A.; Jagot, S.; Lagarrigue, M.; Guével, B.; Ledevin, M.; Larcher, T.; Dubreil, L.; Pineau, C.; Rouger, K.; Guével, L. Quantitative proteome profiling of dystrophic dog skeletal muscle reveals a stabilized muscular architecture and protection against oxidative stress after systemic delivery of MuStem cells. Proteomics 2016, 16, 2028–2042. [Google Scholar] [CrossRef]
- Tamiyakul, H.; Kemter, E.; Kösters, M.; Ebner, S.; Blutke, A.; Klymiuk, N.; Flenkenthaler, F.; Wolf, E.; Arnold, G.J.; Fröhlich, T. Progressive Proteome Changes in the Myocardium of a Pig Model for Duchenne Muscular Dystrophy. iScience 2020, 23, 101516. [Google Scholar] [CrossRef] [PubMed]
- Regensburger, A.P.; Fonteyne, L.M.; Jüngert, J.; Wagner, A.L.; Gerhalter, T.; Nagel, A.M.; Heiss, R.; Flenkenthaler, F.; Qurashi, M.; Neurath, M.F.; et al. Detection of collagens by multispectral optoacoustic tomography as an imaging biomarker for Duchenne muscular dystrophy. Nat. Med. 2019, 25, 1905–1915. [Google Scholar] [CrossRef]
- Smith, L.R.; Hammers, D.W.; Sweeney, H.L.; Barton, E. Increased collagen cross-linking is a signature of dystrophin-deficient muscle. Muscle Nerve 2016, 54, 71–78. [Google Scholar] [CrossRef]
- Dupont-Versteegden, E.E.; McCarter, R.J. Differential expression of muscular dystrophy in diaphragm versus hindlimb muscles of mdx mice. Muscle Nerve 1992, 15, 1105–1110. [Google Scholar] [CrossRef]
- Ritter, P.; Nübler, S.; Buttgereit, A.; Smith, L.R.; Mühlberg, A.; Bauer, J.; Michael, M.; Kreiß, L.; Haug, M.; Barton, E.; et al. Myofibrillar Lattice Remodeling Is a Structural Cytoskeletal Predictor of Diaphragm Muscle Weakness in a Fibrotic mdx (mdx Cmah-/-) Model. Int. J. Mol. Sci. 2022, 23, 10841. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Concurrent Label-Free Mass Spectrometric Analysis of Dystrophin Isoform Dp427 and the Myofibrosis Marker Collagen in Crude Extracts from mdx-4cv Skeletal Muscles. Proteomes 2015, 3, 298–327. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Henry, M.; Meleady, P.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic analysis of the sarcolemma-enriched fraction from dystrophic mdx-4cv skeletal muscle. J. Proteom. 2019, 191, 212–227. [Google Scholar] [CrossRef]
- Judge, L.M.; Haraguchiln, M.; Chamberlain, J.S. Dissecting the signaling and mechanical functions of the dystrophin-glycoprotein complex. J. Cell Sci. 2006, 119, 1537–1546. [Google Scholar] [CrossRef] [PubMed]
- Arecco, N.; Clarke, C.J.; Jones, F.K.; Simpson, D.M.; Mason, D.; Beynon, R.J.; Pisconti, A. Elastase levels and activity are increased in dystrophic muscle and impair myoblast cell survival, proliferation and differentiation. Sci. Rep. 2016, 6, 24708. [Google Scholar] [CrossRef]
- Abdulhady, H.; Sakr, H.M.; Elsayed, N.S.; El-Sobky, T.A.; Fahmy, N.; Saadawy, A.M.; Elsedfy, H. Ambulatory Duchenne muscular dystrophy children: Cross-sectional correlation between function, quantitative muscle ultrasound and MRI. Acta Myol. 2022, 41, 1–14. [Google Scholar] [PubMed]
- Peng, F.; Xu, H.; Song, Y.; Xu, K.; Li, S.; Cai, X.; Guo, Y.; Gong, L. Utilization of T1-Mapping for the pelvic and thigh muscles in Duchenne Muscular Dystrophy: A quantitative biomarker for disease involvement and correlation with clinical assessments. BMC Musculoskelet. Disord. 2022, 23, 681. [Google Scholar] [CrossRef] [PubMed]
- Veeger, T.T.J.; van de Velde, N.M.; Keene, K.R.; Niks, E.H.; Hooijmans, M.T.; Webb, A.G.; de Groot, J.H.; Kan, H.E. Baseline fat fraction is a strong predictor of disease progression in Becker muscular dystrophy. NMR Biomed. 2022, 35, e4691. [Google Scholar] [CrossRef]
- Barnard, A.M.; Hammers, D.W.; Triplett, W.T.; Kim, S.; Forbes, S.C.; Willcocks, R.J.; Daniels, M.J.; Senesac, C.R.; Lott, D.J.; Arpan, I.; et al. Evaluating Genetic Modifiers of Duchenne Muscular Dystrophy Disease Progression Using Modeling and MRI. Neurology 2022, 99, e2406–e2416. [Google Scholar] [CrossRef]
- Tichy, E.D.; Mourkioti, F. A new method of genotyping MDX4CV mice by PCR-RFLP analysis. Muscle Nerve 2017, 56, 522–524. [Google Scholar] [CrossRef]
- Mitrpant, C.; Fletcher, S.; Iversen, P.L.; Wilton, S.D. By-passing the nonsense mutation in the 4 CV mouse model of muscular dystrophy by induced exon skipping. J. Gene Med. 2009, 11, 46–56. [Google Scholar] [CrossRef]
- Li, S.; Kimura, E.; Ng, R.; Fall, B.M.; Meuse, L.; Reyes, M.; Faulkner, J.A.; Chamberlain, J.S. A highly functional mini-dystrophin/GFP fusion gene for cell and gene therapy studies of Duchenne muscular dystrophy. Hum. Mol. Genet. 2006, 15, 1610–1622. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.; Li, S.; Gregorevic, P.; Fall, B.M.; Chamberlain, J.S. Dystrophin delivery to muscles of mdx mice using lentiviral vectors leads to myogenic progenitor targeting and stable gene expression. Mol. Ther. 2010, 18, 206–213. [Google Scholar] [CrossRef]
- Ramos, J.N.; Hollinger, K.; Bengtsson, N.E.; Allen, J.M.; Hauschka, S.D.; Chamberlain, J.S. Development of Novel Micro-dystrophins with Enhanced Functionality. Mol. Ther. 2019, 27, 623–635. [Google Scholar] [CrossRef]
- Hersh, J.; Condor Capcha, J.M.; Iansen Irion, C.; Lambert, G.; Noguera, M.; Singh, M.; Kaur, A.; Dikici, E.; Jiménez, J.J.; Shehadeh, L.A.; et al. Peptide-Functionalized Dendrimer Nanocarriers for Targeted Microdystrophin Gene Delivery. Pharmaceutics 2021, 13, 2159. [Google Scholar] [CrossRef]
- Eren, S.A.; Tastan, C.; Karadeniz, K.B.; Turan, R.D.; Cakirsoy, D.; Kancagi, D.D.; Yilmaz, S.U.; Oztatlici, M.; Oztatlici, H.; Ozer, S.; et al. Lentiviral micro-dystrophin gene treatment into late-stage mdx mice for Duchene Muscular Dystrophy disease. Curr. Gene Ther. 2023, 23, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.; Han, J.J.; Li, S.; Fall, B.; Ra, J.; Haraguchi, M.; Tapscott, S.J.; Chamberlain, J.S. Cell-lineage regulated myogenesis for dystrophin replacement: A novel therapeutic approach for treatment of muscular dystrophy. Hum. Mol. Genet. 2008, 17, 2507–2517. [Google Scholar] [CrossRef] [PubMed]
- Muir, L.A.; Nguyen, Q.G.; Hauschka, S.D.; Chamberlain, J.S. Engraftment potential of dermal fibroblasts following in vivo myogenic conversion in immunocompetent dystrophic skeletal muscle. Mol. Ther. Methods Clin. Dev. 2014, 1, 14025. [Google Scholar] [CrossRef]
- Stuelsatz, P.; Shearer, A.; Li, Y.; Muir, L.A.; Ieronimakis, N.; Shen, Q.W.; Kirillova, I.; Yablonka-Reuveni, Z. Extraocular muscle satellite cells are high performance myo-engines retaining efficient regenerative capacity in dystrophin deficiency. Dev. Biol. 2015, 397, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, N.E.; Hall, J.K.; Odom, G.L.; Phelps, M.P.; Andrus, C.R.; Hawkins, R.D.; Hauschka, S.D.; Chamberlain, J.R.; Chamberlain, J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 14454. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhang, C.; Li, H.; Wang, P.; Gao, Y.; Mokadam, N.A.; Ma, J.; Arnold, W.D.; Han, R. Efficient precise in vivo base editing in adult dystrophic mice. Nat. Commun. 2021, 12, 3719. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Comparative gel-based proteomic analysis of chemically crosslinked complexes in dystrophic skeletal muscle. Electrophoresis 2018, 39, 1735–1744. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Chemical crosslinking analysis of β-dystroglycan in dystrophin-deficient skeletal muscle. HRB Open Res. 2018, 1, 17. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Zweyer, M.; Henry, M.; Meleady, P.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Subproteomic profiling of sarcolemma from dystrophic mdx-4cv skeletal muscle. Data Brief. 2018, 17, 980–993. [Google Scholar] [CrossRef] [PubMed]
- Gargan, S.; Dowling, P.; Zweyer, M.; Reimann, J.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Mass Spectrometric Profiling of Extraocular Muscle and Proteomic Adaptations in the mdx-4cv Model of Duchenne Muscular Dystrophy. Life 2021, 11, 595. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Dowling, P.; Zweyer, M.; Mundegar, R.R.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic analysis of dystrophin deficiency and associated changes in the aged mdx-4cv heart model of dystrophinopathy-related cardiomyopathy. J. Proteom. 2016, 145, 24–36. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Henry, M.; Meleady, P.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of liver tissue from the mdx-4cv mouse model of Duchenne muscular dystrophy. Clin. Proteom. 2018, 15, 34. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Zweyer, M.; Raucamp, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic and cell biological profiling of the renal phenotype of the mdx-4cv mouse model of Duchenne muscular dystrophy. Eur. J. Cell Biol. 2020, 99, 151059. [Google Scholar] [CrossRef]
- Dowling, P.; Zweyer, M.; Raucamp, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Dataset on the mass spectrometry-based proteomic profiling of the kidney from wild type and the dystrophic mdx-4cv mouse model of X-linked muscular dystrophy. Data Brief. 2020, 28, 105067. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Gargan, S.; Zweyer, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteome-wide Changes in the mdx-4cv Spleen due to Pathophysiological Cross Talk with Dystrophin-Deficient Skeletal Muscle. iScience 2020, 23, 101500. [Google Scholar] [CrossRef]
- Dowling, P.; Gargan, S.; Zweyer, M.; Sabir, H.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of the interface between the stomach wall and the pancreas in dystrophinopathy. Eur. J. Transl. Myol. 2021, 31, 9627. [Google Scholar]
- Murphy, S.; Zweyer, M.; Henry, M.; Meleady, P.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Label-free mass spectrometric analysis reveals complex changes in the brain proteome from the mdx-4cv mouse model of Duchenne muscular dystrophy. Clin. Proteom. 2015, 12, 27. [Google Scholar] [CrossRef]
- Murphy, S.; Dowling, P.; Zweyer, M.; Henry, M.; Meleady, P.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of mdx-4cv serum reveals highly elevated levels of the inflammation-induced plasma marker haptoglobin in muscular dystrophy. Int. J. Mol. Med. 2017, 39, 1357–1370. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Dataset on the comparative proteomic profiling of mouse saliva and serum from wild type versus the dystrophic mdx-4cv mouse model of dystrophinopathy. Data Brief. 2018, 21, 1236–1245. [Google Scholar] [CrossRef] [PubMed]
- Gargan, S.; Dowling, P.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Identification of marker proteins of muscular dystrophy in the urine proteome from the mdx-4cv model of dystrophinopathy. Mol. Omics 2020, 16, 268–278. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic identification of elevated saliva kallikrein levels in the mdx-4cv mouse model of Duchenne muscular dystrophy. Biochem. Biophys. Rep. 2018, 18, 100541. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Matthias, N.; Lo, J.; Ortiz-Vitali, J.L.; Shieh, A.W.; Wang, S.H.; Darabi, R. A Myogenic Double-Reporter Human Pluripotent Stem Cell Line Allows Prospective Isolation of Skeletal Muscle Progenitors. Cell Rep. 2018, 25, 1966–1981.e4. [Google Scholar] [CrossRef] [PubMed]
- Sakai-Takemura, F.; Narita, A.; Masuda, S.; Wakamatsu, T.; Watanabe, N.; Nishiyama, T.; Nogami, K.; Blanc, M.; Takeda, S.; Miyagoe-Suzuki, Y. Premyogenic progenitors derived from human pluripotent stem cells expand in floating culture and differentiate into transplantable myogenic progenitors. Sci. Rep. 2018, 8, 6555. [Google Scholar] [CrossRef] [PubMed]
- Arpke, R.W.; Darabi, R.; Mader, T.L.; Zhang, Y.; Toyama, A.; Lonetree, C.L.; Nash, N.; Lowe, D.A.; Perlingeiro, R.C.; Kyba, M. A new immuno-, dystrophin-deficient model, the NSG-mdx(4Cv) mouse, provides evidence for functional improvement following allogeneic satellite cell transplantation. Stem Cells 2013, 31, 1611–1620. [Google Scholar] [CrossRef]
- Elhussieny, A.; Nogami, K.; Sakai-Takemura, F.; Maruyama, Y.; Takemura, N.; Soliman, W.T.; Takeda, S.; Miyagoe-Suzuki, Y. Mesenchymal stem cells derived from human induced pluripotent stem cells improve the engraftment of myogenic cells by secreting urokinase-type plasminogen activator receptor (uPAR). Stem Cell Res. Ther. 2021, 12, 532. [Google Scholar] [CrossRef]
- Chang, A.C.; Ong, S.G.; LaGory, E.L.; Kraft, P.E.; Giaccia, A.J.; Wu, J.C.; Blau, H.M. Telomere shortening and metabolic compromise underlie dystrophic cardiomyopathy. Proc. Natl. Acad. Sci. USA 2016, 113, 13120–13125. [Google Scholar] [CrossRef]
- Ziemba, M.; Barkhouse, M.; Uaesoontrachoon, K.; Giri, M.; Hathout, Y.; Dang, U.J.; Gordish-Dressman, H.; Nagaraju, K.; Hoffman, E.P. Biomarker-focused multi-drug combination therapy and repurposing trial in mdx mice. PLoS ONE 2021, 16, e0246507. [Google Scholar] [CrossRef]
- Deng, J.; Zhang, J.; Shi, K.; Liu, Z. Drug development progress in Duchenne muscular dystrophy. Front. Pharmacol. 2022, 13, 950651. [Google Scholar] [CrossRef]
- Eser, G.; Topaloğlu, H. Current Outline of Exon Skipping Trials in Duchenne Muscular Dystrophy. Genes 2022, 13, 1241. [Google Scholar] [CrossRef] [PubMed]
- Chung Liang, L.; Sulaiman, N.; Yazid, M.D. A Decade of Progress in Gene Targeted Therapeutic Strategies in Duchenne Muscular Dystrophy: A Systematic Review. Front. Bioeng. Biotechnol. 2022, 10, 833833. [Google Scholar] [CrossRef]
- Chemello, F.; Olson, E.N.; Bassel-Duby, R. Crispr-Editing Therapy For Duchenne Muscular Dystrophy. Hum. Gene Ther. 2023, 34, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Patterson, G.; Conner, H.; Groneman, M.; Blavo, C.; Parmar, M.S. Duchenne muscular dystrophy: Current treatment and emerging exon skipping and gene therapy approach. Eur. J. Pharmacol. 2023, 947, 175675. [Google Scholar] [CrossRef]
- Heydemann, A.; Bieganski, G.; Wachowiak, J.; Czarnota, J.; Niezgoda, A.; Siemionow, K.; Ziemiecka, A.; Sikorska, M.H.; Bozyk, K.; Tullius, S.G.; et al. Dystrophin Expressing Chimeric (DEC) Cell Therapy for Duchenne Muscular Dystrophy: A First-in-Human Study with Minimum 6 Months Follow-up. Stem Cell Rev. Rep. 2023. [Google Scholar] [CrossRef] [PubMed]
- Barnard, A.M.; Lott, D.J.; Batra, A.; Triplett, W.T.; Forbes, S.C.; Riehl, S.L.; Willcocks, R.J.; Smith, B.K.; Vandenborne, K.; Walter, G.A. Imaging respiratory muscle quality and function in Duchenne muscular dystrophy. J. Neurol. 2019, 266, 2752–2763. [Google Scholar] [CrossRef] [PubMed]
- Boussaïd, G.; Stalens, C.; Devaux, C.; Segovia-Kueny, S.; Lofaso, F.; Reveillere, C. Impact of Mechanical Ventilation Methods on the Life Perception of Subjects With Duchenne Muscular Dystrophy: French Cross-Sectional Survey. Respir. Care 2020, 65, 1712–1720. [Google Scholar] [CrossRef]
- Kipke, J.; Birnkrant, D.J.; Jin, J.B.; Aneja, A.; Bahler, R.C. A systematic review of pharmacologic therapies for the cardiomyopathy of Duchenne muscular dystrophy. Pediatr. Pulmonol. 2021, 56, 782–795. [Google Scholar] [CrossRef]
- Ricotti, V.; Selby, V.; Ridout, D.; Domingos, J.; Decostre, V.; Mayhew, A.; Eagle, M.; Butler, J.; Guglieri, M.; Van der Holst, M.; et al. Respiratory and upper limb function as outcome measures in ambulant and non-ambulant subjects with Duchenne muscular dystrophy: A prospective multicentre study. Neuromuscul. Disord. 2019, 29, 261–268. [Google Scholar] [CrossRef]
- Fayssoil, A.; Chaffaut, C.; Ogna, A.; Stojkovic, T.; Lamothe, L.; Mompoint, D.; Meng, P.; Prigent, H.; Clair, B.; Behin, A.; et al. Echographic Assessment of Diaphragmatic Function in Duchenne Muscular Dystrophy from Childhood to Adulthood. J. Neuromuscul. Dis. 2019, 6, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Beastrom, N.; Lu, H.; Macke, A.; Canan, B.D.; Johnson, E.K.; Penton, C.M.; Kaspar, B.K.; Rodino-Klapac, L.R.; Zhou, L.; Janssen, P.M.; et al. mdx(⁵cv) mice manifest more severe muscle dysfunction and diaphragm force deficits than do mdx Mice. Am. J. Pathol. 2011, 179, 2464–2474. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Henry, M.; Meleady, P.; Winkler, C.K.; Krautwald, M.; Brinkmeier, H.; Ohlendieck, K. Comparative Label-Free Mass Spectrometric Analysis of Mildly versus Severely Affected mdx Mouse Skeletal Muscles Identifies Annexin, Lamin, and Vimentin as Universal Dystrophic Markers. Molecules 2015, 20, 11317–11344. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Brinkmeier, H.; Krautwald, M.; Henry, M.; Meleady, P.; Ohlendieck, K. Proteomic profiling of the dystrophin complex and membrane fraction from dystrophic mdx muscle reveals decreases in the cytolinker desmoglein and increases in the extracellular matrix stabilizers biglycan and fibronectin. J. Muscle Res. Cell Motil. 2017, 38, 251–268. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Dowling, P.; Meleady, P.; Henry, M.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Label-free mass spectrometric analysis of the mdx-4cv diaphragm identifies the matricellular protein periostin as a potential factor involved in dystrophinopathy-related fibrosis. Proteomics 2015, 15, 2318–2331. [Google Scholar] [CrossRef] [PubMed]
- Dalton, C.J.; Lemmon, C.A. Fibronectin: Molecular Structure, Fibrillar Structure and Mechanochemical Signaling. Cells 2021, 10, 2443. [Google Scholar] [CrossRef] [PubMed]
- Cynthia Martin, F.; Hiller, M.; Spitali, P.; Oonk, S.; Dalebout, H.; Palmblad, M.; Chaouch, A.; Guglieri, M.; Straub, V.; Lochmüller, H.; et al. Fibronectin is a serum biomarker for Duchenne muscular dystrophy. Proteom. Clin. Appl. 2014, 8, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Carberry, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Proteomics reveals drastic increase of extracellular matrix proteins collagen and dermatopontin in the aged mdx diaphragm model of Duchenne muscular dystrophy. Int. J. Mol. Med. 2012, 30, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, O.; Fujiwara, S. Dermatopontin, a novel player in the biology of the extracellular matrix. Connect. Tissue Res. 2006, 47, 177–189. [Google Scholar] [CrossRef]
- Kato, A.; Okamoto, O.; Ishikawa, K.; Sumiyoshi, H.; Matsuo, N.; Yoshioka, H.; Nomizu, M.; Shimada, T.; Fujiwara, S. Dermatopontin interacts with fibronectin, promotes fibronectin fibril formation, and enhances cell adhesion. J. Biol. Chem. 2011, 286, 14861–14869. [Google Scholar] [CrossRef]
- Sugihara, H.; Teramoto, N.; Nakamura, K.; Shiga, T.; Shirakawa, T.; Matsuo, M.; Ogasawara, M.; Nishino, I.; Matsuwaki, T.; Nishihara, M.; et al. Cellular senescence-mediated exacerbation of Duchenne muscular dystrophy. Sci. Rep. 2020, 10, 16385. [Google Scholar] [CrossRef]
- Signorelli, M.; Tsonaka, R.; Aartsma-Rus, A.; Spitali, P. Multiomic characterization of disease progression in mice lacking dystrophin. PLoS ONE 2023, 18, e0283869. [Google Scholar] [CrossRef]
- Capitanio, D.; Moriggi, M.; Torretta, E.; Barbacini, P.; De Palma, S.; Viganò, A.; Capitanio, D.; Moriggi, M.; Torretta, E.; Barbacini, P.; et al. Comparative proteomic analyses of Duchenne muscular dystrophy and Becker muscular dystrophy muscles: Changes contributing to preserve muscle function in Becker muscular dystrophy patients. J. Cachexia Sarcopenia Muscle 2020, 11, 547–563. [Google Scholar] [CrossRef]
- Capitanio, D.; Moriggi, M.; Barbacini, P.; Torretta, E.; Moroni, I.; Blasevich, F.; Morandi, L.; Mora, M.; Gelfi, C. Molecular fingerprint of BMD patients lacking a portion in the rod domain of dystrophin. Int. J. Mol. Sci. 2020, 23, 2624. [Google Scholar] [CrossRef]
- Doran, P.; Martin, G.; Dowling, P.; Jockusch, H.; Ohlendieck, K. Proteome analysis of the dystrophin-deficient MDX diaphragm reveals a drastic increase in the heat shock protein cvHSP. Proteomics 2006, 6, 4610–4621. [Google Scholar] [CrossRef]
- Guevel, L.; Lavoie, J.R.; Perez-Iratxeta, C.; Rouger, K.; Dubreil, L.; Feron, M.; Talon, S.; Brand, M.; Megeney, L.A. Quantitative proteomic analysis of dystrophic dog muscle. J. Proteome Res. 2011, 10, 2465–2478. [Google Scholar] [CrossRef]
- Rayavarapu, S.; Coley, W.; Cakir, E.; Jahnke, V.; Takeda, S.; Aoki, Y.; Grodish-Dressman, H.; Jaiswal, J.K.; Hoffman, E.P.; Brown, K.J.; et al. Identification of disease specific pathways using in vivo SILAC proteomics in dystrophin deficient mdx mouse. Mol. Cell. Proteom. 2013, 12, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, T.; Kemter, E.; Flenkenthaler, F.; Klymiuk, N.; Otte, K.A.; Blutke, A.; Krause, S.; Walter, M.C.; Wanke, R.; Wolf, E.; et al. Progressive muscle proteome changes in a clinically relevant pig model of Duchenne muscular dystrophy. Sci. Rep. 2016, 6, 33362. [Google Scholar] [CrossRef]
- van Westering, T.L.E.; Johansson, H.J.; Hanson, B.; Coenen-Stass, A.M.L.; Lomonosova, Y.; Tanihata, J.; Motohashi, N.; Yokota, T.; Takeda, S.; Lehtiö, J.; et al. Mutation-independent proteomic signatures of pathological progression in murine models of Duchenne muscular dystrophy. Mol. Cell. Proteom. 2020, 19, 2047–2068. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Carberry, S.; Ohlendieck, K. Proteomics of the dystrophin-glycoprotein complex and dystrophinopathy. Curr. Protein Pept. Sci. 2013, 14, 680–697. [Google Scholar] [CrossRef] [PubMed]
- Carr, S.J.; Zahedi, R.P.; Lochmüller, H.; Roos, A. Mass spectrometry-based protein analysis to unravel the tissue pathophysiology in Duchenne muscular dystrophy. Proteom. Clin. Appl. 2018, 12, 1700071. [Google Scholar] [CrossRef]
- Al-Khalili Szigyarto, C. Duchenne Muscular Dystrophy: Recent advances in protein biomarkers and the clinical application. Expert Rev. Proteom. 2020, 17, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Gawlik, K.I. At the Crossroads of Clinical and Preclinical Research for Muscular Dystrophy—Are We Closer to Effective Treatment for Patients? Int. J. Mol. Sci. 2018, 19, 1490. [Google Scholar] [CrossRef]
- Dowling, P.; Murphy, S.; Zweyer, M.; Raucamp, M.; Swandulla, D.; Ohlendieck, K. Emerging proteomic biomarkers of X-linked muscular dystrophy. Expert Rev. Mol. Diagn. 2019, 19, 739–755. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D.; Terrill, J.R.; Al-Mshhdani, B.A.; Duong, M.N.; Radley-Crabb, H.G.; Arthur, P.G. Biomarkers for Duchenne muscular dystrophy: Myonecrosis, inflammation and oxidative stress. Dis. Model Mech. 2020, 13, dmm043638. [Google Scholar] [CrossRef]
- Norris, R.A.; Damon, B.; Mironov, V.; Kasyanov, V.; Ramamurthi, A.; Moreno-Rodriguez, R.; Trusk, T.; Potts, J.D.; Goodwin, R.L.; Davis, J.; et al. Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J. Cell. Biochem. 2007, 101, 695–711. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, C.; Akpulat, U.; Sharafi, P.; Yıldız, Y.; Onbaşılar, I.; Kocaefe, C. Periostin is temporally expressed as an extracellular matrix component in skeletal muscle regeneration and differentiation. Gene 2014, 553, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; Miyagoe-Suzuki, Y.; Takeda, S.; Kudo, A. Periostin Is Required for the Maintenance of Muscle Fibers during Muscle Regeneration. Int. J. Mol. Sci. 2021, 22, 3627. [Google Scholar] [CrossRef]
- Lorts, A.; Schwanekamp, J.A.; Baudino, T.A.; McNally, E.M.; Molkentin, J.D. Deletion of periostin reduces muscular dystrophy and fibrosis in mice by modulating the transforming growth factor-β pathway. Proc. Natl. Acad. Sci. USA 2012, 109, 10978–10983. [Google Scholar] [CrossRef]
- Demonbreun, A.R.; Quattrocelli, M.; Barefield, D.Y.; Allen, M.V.; Swanson, K.E.; McNally, E.M. An actin-dependent annexin complex mediates plasma membrane repair in muscle. J. Cell Biol. 2016, 213, 705–718. [Google Scholar] [CrossRef]
- Defour, A.; Medikayala, S.; Van der Meulen, J.H.; Hogarth, M.W.; Holdreith, N.; Malatras, A.; Duddy, W.; Boehler, J.; Nagaraju, K.; Jaiswal, J.K. Annexin A2 links poor myofiber repair with inflammation and adipogenic replacement of the injured muscle. Hum. Mol. Genet. 2017, 26, 1979–1991. [Google Scholar] [CrossRef]
- Bittel, D.C.; Chandra, G.; Tirunagri, L.M.S.; Deora, A.B.; Medikayala, S.; Scheffer, L.; Defour, A.; Jaiswal, J.K. Annexin A2 Mediates Dysferlin Accumulation and Muscle Cell Membrane Repair. Cells 2020, 9, 1919. [Google Scholar] [CrossRef] [PubMed]
- Lennon, N.J.; Kho, A.; Bacskai, B.J.; Perlmutter, S.L.; Hyman, B.T.; Brown, R.H., Jr. Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J. Biol. Chem. 2003, 278, 50466–50473. [Google Scholar] [CrossRef] [PubMed]
- Croissant, C.; Carmeille, R.; Brévart, C.; Bouter, A. Annexins and Membrane Repair Dysfunctions in Muscular Dystrophies. Int. J. Mol. Sci. 2021, 22, 5276. [Google Scholar] [CrossRef]
- Croissant, C.; Gounou, C.; Bouvet, F.; Tan, S.; Bouter, A. Trafficking of Annexins during Membrane Repair in Human Skeletal Muscle Cells. Membranes 2022, 12, 153. [Google Scholar] [CrossRef] [PubMed]
- Mouw, J.K.; Ou, G.; Weaver, V.M. Extracellular matrix assembly: A multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Csapo, R.; Gumpenberger, M.; Wessner, B. Skeletal Muscle Extracellular Matrix—What Do We Know About Its Composition, Regulation, and Physiological Roles? A Narrative Review. Front. Physiol. 2020, 11, 253. [Google Scholar] [CrossRef]
- Brightwell, C.R.; Latham, C.M.; Thomas, N.T.; Keeble, A.R.; Murach, K.A.; Fry, C.S. A glitch in the matrix: The pivotal role for extracellular matrix remodeling during muscle hypertrophy. Am. J. Physiol.-Cell Physiol. 2022, 323, C763–C771. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).