Abstract
Immunotoxins (ITXs) are chimeric molecules that combine the specificity of a targeting domain, usually derived from an antibody, and the cytotoxic potency of a toxin, leading to the selective death of tumor cells. However, several issues must be addressed and optimized in order to use ITXs as therapeutic tools, such as the selection of a suitable tumor-associated antigen (TAA), high tumor penetration and retention, low kidney elimination, or low immunogenicity of foreign proteins. To this end, we produced and characterized several ITX designs, using a nanobody against EGFR (VHH 7D12) as the targeting domain. First, we generated a nanoITX, combining VHH 7D12 and the fungal ribotoxin α-sarcin (αS) as the toxic moiety (VHHEGFRαS). Then, we incorporated a trimerization domain (TIEXVIII) into the construct, obtaining a trimeric nanoITX (TriVHHEGFRαS). Finally, we designed and characterized a bispecific ITX, combining the VHH 7D12 and the scFv against GPA33 as targeting domains, and a deimmunized (DI) variant of α-sarcin (BsITXαSDI). The results confirm the therapeutic potential of α-sarcin-based nanoITXs. The incorporation of nanobodies as target domains improves their therapeutic use due to their lower molecular size and binding features. The enhanced avidity and toxic load in the trimeric nanoITX and the combination of two different target domains in the bispecific nanoITX allow for increased antitumor effectiveness.
1. Introduction
Colorectal cancer currently occupies the third position in incidence (1.9 million new cases per year) and second place in terms of mortality (935.000 annual deaths) among the different cancer types [1]. Current therapies consist mainly of surgery, followed by chemotherapy based on fluoropyrimidines [2] or radiotherapy [3] as adjuvant treatments. However, these are non-specific therapies, leading to off-target toxicities and directly affecting the quality of life of patients. In this context, immunotherapy and the advent of monoclonal antibodies have arisen as a breakthrough in the treatment of this disease, as they are targeted therapies with great specificity and promising clinical results [4,5].
With regard to passive immunotherapy based on monoclonal antibodies, immunotoxins (ITXs) are a type of immunoconjugate that can be used in the treatment of both tumoral and infectious diseases [6,7]. ITXs are chimeric molecules with a target domain, usually an antibody fragment such as Fab (fragment antigen-binding) or scFv (single-chain variable fragment), but also cytokines or growth factors that specifically direct the action of a toxic protein of bacterial or plant origin towards the cancerous cell, leading to its death [8].
As target domains, nanobodies (VHH) are the single variable domains of heavy-chain-only antibodies obtained from camelids [9]. These nanobodies retain the full antigen-binding potential and are considered the smallest naturally-derived antigen-binding fragments. Nanobodies exhibit several advantages over full-length antibodies or scFvs, such as smaller size (~15 kDa), allowing deeper penetration in solid tumors, the ability to cross the blood–brain barrier [10], or their increased stability and resistance to high temperatures (60–80 °C) and pressures (500–700 MPa), extreme pHs (3.0–9.0), and chemical denaturants, such as 2–3 M guanidinium chloride or 6–8 M urea [11,12]. In addition, they exhibit a longer complementary determining region loop 3 (CDR3), which enables nanobodies to bind with high affinity to traditionally inaccessible epitopes [13,14]. Regarding immunogenicity, nanobodies are mostly considered non-immunogenic proteins, since they do not trigger either T-cell proliferation or anti-VHH antibody production, possibly due to the lack of Fc fragments and the high homology sequence with human VH domains [15]. All these features have led to the medical use of nanobodies in imaging and treatment applications, including the treatment of viral infections such as HIV or SARS-CoV2 [16,17], or several cancer types [18,19].
The main critical issue for the therapeutic effect of immunoconjugates and ITXs is the selection of target tumor-associated antigens (TAAs) [8,20]. Ideal TAAs should be expressed only in tumoral tissue and be absent from healthy cells to avoid off-target toxicities. In addition, the internalization rate of the receptor after ITX binding may be a more determinant step than the receptor expression level [21,22]. In this sense, nanoITXs targeting HER2 [23], the transferrin receptor [24], IL2R [25], or mesothelin [26], among others, have already been described.
GPA33 is a transmembrane glycoprotein that is overexpressed in 95% of primary and metastatic colorectal cancers, while it is mostly absent in healthy tissues [27,28]. It is believed to be involved in cell adhesion functions and the development of the intestinal mucosa during the embryonic early stages [29]. GPA33 remains constitutively attached to the membrane cell, showing a high degree of internalization after the binding of the corresponding antibody, and is thus an ideal TAA for targeting [30].
EGFR, also named ErbB1, is a tyrosine kinase receptor that belongs to a family of four receptors: ErbB1 (EGFR), ErbB2 (HER2), ErbB3 (HER3), and ErbB4 (HER4). They are transmembrane glycoproteins that upon the binding of their ligand, EGF or TGF-α, homo- or hetero-dimerize and activate downstream signaling routes, such as RAS/RAF/MEK/ERK, PI3K/AKT, and JAK/STAT3, involved in the proliferation and survival of a great number of different cell types [31,32]. Because of these functions, the overexpression of EGFR confers great advantages on tumoral cells in epithelial cancers, such as in 15–30% of breast cancers, 60% of lung cancers, and 80% of colorectal cancers [33].
Regarding the toxic domain, toxins from different bacteria and plants have been routinely used, such as Pseudomonas exotoxin A, Diphtheria toxin, gelonin, and the plant toxin ricin, among others [34,35]. However, they present some drawbacks that limit their use as therapeutic drugs, such as immunogenicity. These toxins trigger an immune response from the patient, who thereby develops anti-drug antibodies (ADAs). These ADAs neutralize the immunotoxin, preventing further treatment cycles [36,37].
Ribotoxins, a protein family that belongs to the fungal extracellular RNase superfamily, have emerged as promising toxic domains due to their small size, high thermostability and resistance to proteases, low immunogenicity, and efficient cytotoxic activity. Ribotoxins recognize a conserved motif in the large rRNA subunit, namely the sarcin/ricin loop (SRL), and produce a single cleavage, leading to protein biosynthesis inhibition and cell death by apoptosis [38,39]. Among ribotoxins, the most characterized member is α-sarcin, a small basic ribotoxin of 150 amino acids that has been employed as the toxic domain in the design of several ITXs against allergies [40] and cancers, showing potent antitumoral activity both in vitro and in vivo [41,42,43,44].
In order to overcome the versatility and extreme adaptability of colorectal cancer, we produced and characterized several designs of ITXs, based on the VHH 7D12 [45] that binds to the EGFR extracellular domain III, preventing its dimerization and subsequent signaling activity and thus taking advantage of the unique features of nanobodies as the target domain. First, we designed the nanoITX VHHEGFRαS, combining the VHH7D12 and α-sarcin as the toxic domain. Second, we designed a trimeric version, TriVHHEGFRαS, to increase the avidity and toxic load of the nanoITX. In addition, its larger size should avoid fast renal clearance, leading to increased treatment efficacy. TriVHHEGFRαS includes the collagen XVIII trimerization domain (TIEXVIII) flanked by 18 amino acid linkers, leading to a trimeric nanoITX [46,47,48]. Finally, we produced a bispecific immunotoxin, BsITXαSDI, combining the VHH 7D12 and the scFvA33 against GPA33 [49,50], along with a non-immunogenic variant of α-sarcin (αSDI) [51]. This bispecific design allows the recognition of two different TAAs and therefore would maintain its efficacy in case of antigen loss during tumor progression.
2. Materials and Methods
2.1. Plasmid Design
Plasmids containing the cDNA sequence encoding VHH 7D12 and TIEXVIII domains were provided by Laura Sanz’s group from Hospital Puerta del Hierro. cDNA corresponding to scFvA33, α-sarcin, and αSDI were previously obtained by our group. The desired cDNA sequences were amplified by PCR, including the restriction sites needed for cloning, and then cloned in pPICZαA (Invitrogen, Carlsbad, CA, USA), to finally obtain the plasmids pPICZαAVHHEGFRαS, pPICZαATriVHHEGFRαS, and pPICZαABsITXαSDI. All three plasmids contained a Zeocin resistance gene, and the plasmid construction included an α-factor signal at the N-terminal site, to allow extracellular secretion, and a six-histidine tag at the C-terminal site, to allow their purification. The three plasmids were sequenced by the Genomic Unit at the Universidad Complutense de Madrid, to confirm the desired sequences.
2.2. Protein Production and Purification
Electrocompetent Pichia pastoris KM71H strain cells were electroporated with 10 μg of linearized plasmid after digestion with the restriction enzyme PmeI, using a Bio-Rad Gene Pulser device. After the electroporation pulse, the cells were seeded in YPDS plates containing increasing zeocin concentrations (100 to 750 μg/mL). Multiple clones were selected to test the optimal conditions for their expression, modifying the induction time (24–72 h) and temperature (15–25 °C). Protein expression was analyzed using 0.1% (w/v) sodium dodecyl sulfate (SDS)–15% polyacrylamide gel electrophoresis (PAGE) and Western blot using an anti-α-sarcin antibody.
Once the optimal conditions were selected for each nanoITX, large-scale expression was carried out. First, the selected yeast clone was grown in 2 L of BMGY medium using baffled Erlenmeyer flasks, at 30 °C with vigorous agitation. After 24 h, the cells were harvested by centrifugation, and resuspended in 1 L of BMMY for induction, inducing the protein expression at the previously optimized temperature and time. Once the induction was finished, the extracellular medium was dialyzed several times against 50 mM sodium phosphate, 0.1 M NaCl, and pH 7.5 buffer.
The three nanoITXs were purified by following the same protocol, including affinity chromatography using a Ni2+-NTA agarose column (GE Healthcare). First, the medium was applied to the column at a flow rate of 1 mL/min. Then, the column was washed with 50 mM sodium phosphate, 0.1 M NaCl, and pH 7.5 buffer, and then washed again with the same buffer containing 20 mM imidazole. The nanoITXs were eluted by the addition of the same buffer containing 250 mM imidazole. Fractions containing the desired protein were pooled, and dialyzed against a sodium phosphate buffer, to remove the imidazole.
2.3. Structural Characterization
Structural characterization was carried out by several means. Absorbance measurements were taken using a UV-1800 spectrophotometer device (Shimadzu, Shimadzu Europa GmbH, Duisburg, Germany). Secondary structure information was obtained from the record of far-UV circular dichroism (CD) spectra, using a Jasco J-715 spectropolarimeter (Jasco Analítica, Madrid, Spain). NanoITX samples were resuspended in 50 mM sodium phosphate buffer, 0.1 M NaCl, and pH 7.5 at a final concentration of 0.2 mg/mL. Cells with an optical path of 0.1 cm were employed, and eight spectra were averaged to obtain the final spectra. To evaluate the thermal stability of the nanoITXs, thermal denaturation profiles (Tm) were obtained by measuring the temperature dependence of the molar ellipticity at 220 nm of a 0.2 mg/mL nanoITX solution in the 20–85 °C temperature range, with a temperature increase rate of 30 °C/hour. FPLC was performed in an AKTA purifier device (GE Healthcare Lifescience) using a Superdex 200 column to analyze the trimeric format and the molecular size in the solution of the trimeric nanoITX (TriVHHEGFRαS).
2.4. Ribonucleolytic Activity Assays
The highly specific ribonucleolytic activity of α-sarcin or its deimmunized variant, α-sarcin DI (αSDI), was assayed, as previously described [52,53], against ribosomes from a rabbit cell-free reticulocyte lysate. The subsequent release of the characteristic 400 nt rRNA fragment, known as α-fragment, was used to confirm the ribonucleolytic activity of α-sarcin. Briefly, a rabbit cell-free reticulocyte lysate (50 μL) was incubated with different amounts of nanoITXs. The ribonucleolytic reaction was stopped by the addition of 250 μL of Tris 50 mM, SDS 5%, and pH 7.5 buffer; RNA was isolated with a phenol:chloroform:isoamyl alcohol (25:24:1) extraction, and then precipitated by the addition of isopropanol. The RNA pellet was washed with −20 °C ethanol 70% and resuspended in 10 μL of DEPC H2O. α-Fragment presence was visualized by electrophoresis in a 2% agarose, 16% paraformaldehyde gel, prestained with ethidium bromide. Images were obtained using a Universal Hood II Transilluminator device.
2.5. Cell Line Cultures
The A431 cell line (ATCC CRL-1555, Rockville MD, USA) was used as a tumoral EGFR+ epidermoid carcinoma cell line [54], whereas the SW1222 cell line (ATCC HB-11028, Rockville, MD, USA) was used as a GPA33+ and EGFR- colorectal cancer cell line. Both cell lines were cultured in RPMI medium, supplemented with 300 mg/mL of L-glutamine, 50 μg/mL of penicillin, 50 mg/mL of streptomycin, and 10% of fetal bovine serum. Cells were cultured at 37 °C in a humidified atmosphere (CO2:air, 1:19 v:v). Harvesting and propagation of both cell lines were performed each 2–3 days through trypsinization, and the number of cells was calculated with a Neubauer chamber.
2.6. ELISA Assay
The binding ability of the nanoITXs towards its antigen, EGFR, was first analyzed by ELISA. Plates were coated with EGFR (0.5 µg/mL) at 4 °C, overnight. Then, after three phosphate-buffered saline (PBS) washes, the wells were blocked with BSA 5% PBS, and incubated with the different nanoITXs (1 μM) for 1 h at RT. After three more PBS washes, an anti-Histag-HRP secondary antibody diluted 1/2000 was added to each well and incubated for 2 h at RT. After incubation, the wells were washed again with PBS and 100 μL of substrate solution was added. The reaction was stopped by the addition of 100 μL of H2SO4 2M. The VHHEGFR nanobody was used as a positive binding control, and BSA-coated wells were used as a negative control. Triplicates of each condition were tested. The results, represented as mean ± standard deviation, were calculated as the subtraction of the absorbance at 450 nm of EGFR-coated wells from those of the BSA-coated wells.
2.7. Flow Cytometry Studies
Trypsinized cells were distributed into aliquots of 3 × 105 cells/mL and washed three times with 300 μL of BSA 1% (w/v) PBS. Then, cells were incubated with different concentrations of nanoITXs for 45 min at room temperature with gentle agitation. After another three washes with BSA 1% (w/v) PBS, a second incubation was carried out, using a diluted 1/100 anti-Histag Alexa 488 antibody (Sigma Aldrich, St. Louis, MO, USA) for 45 min at room temperature with gentle agitation. Then, the cells were finally washed three times with BSA 1% (w/v) PBS, and fluorescence was measured using a FACScan (Becton Dickinson, NJ, USA), at the Centro de Apoyo a la Investigación of the Universidad Complutense de Madrid. The results were analyzed using the FlowJo software (FlowJo v10, Oregon, OR, USA).
2.8. MTT Viability Assay
To analyze the cytotoxicity of the nanoITXs, MTT viability assays were performed. Cells were trypsinized and seeded into 96-well plates at a cellular density according to the cell line, in a culture medium, and maintained under standard culture conditions for 24 h. Then, different concentrations of immunotoxin diluted in an FBS-free medium were added to the cells and kept in culture conditions for 24, 48, and 72 h. After the incubation, each well was incubated with 20 μL of MTT (5 mg/mL) for 4 h. Then, 100 μL of DMSO:methanol (1:1, v/v) was added to each well, to dissolve the formazan crystals formed by the reduction of MTT carried out by the enzymatic activity of live cells. The results were obtained by colorimetric quantification, measuring the optical density at 570 nm and expressed as a percentage of viability. Triplicates of each condition were tested. Medium-cultured cells were used as the 100% viability control and the IC50 was determined as the ITX concentration that led to a 50% decrease in the cell viability.
2.9. In Vivo Antitumor Assay
All animal procedures were carried out according to the guidelines of the Universidad Complutense Animal Experimentation Committee, and the Community of Madrid official regulations (Royal Decree 53/2013). Balb/c nude male mice (7 weeks old) were purchased from Harlan Laboratories (Barcelona, Spain) to analyze the in vivo effect of VHHEGFRαS against human epidermoid cancer xenografts. Assays were performed at the Animal Facility of the Centro de Investigaciones Biológicas-Consejo Superior de Investigaciones Científicas (CIB-CSIC) in Madrid.
Mice were split into three experimental groups (n = 5): PBS (phosphate-buffered saline) and VHHEGFRαS 25 or VHHEGFRαS 50, according to the dose administered (25 or 50 μg of nanoITX per injection). Prior to the experimental procedure, the animals were given a 7-day adaptation period with free access to food and water. Each mouse received a subcutaneous injection into the right flank of 2 × 106 A431 cells, resuspended in 200 μL of 1:1(v/v) PBS–Matrigel (BD Biosciences, San Jose, CA, USA) mixture. Once the tumor volume reached 50–100 mm3, the mice were injected intravenously either with PBS or different doses of VHHEGFRαS. Five doses, either of PBS or of the two doses (25–50 μg) of VHHEGFRαS were given every 48 h. Tumors were measured each 48 h using an external caliper, and volume was calculated with the formula: tumoral volume = length × width2 × 0.52. After the administration of 5 doses (day 10), the drug administration stopped, but the tumoral volume measurement was continued until the volume was too high (2500 mm3) and the animals had to be sacrificed. Survival analysis of the experimental mice was carried out using the Kaplan–Meier representation. ANOVA with a post hoc analysis using the Student–Newman–Keuls test was used for statistical analysis within each test to compare the results obtained with the different doses administered. All values were expressed as arithmetic means ± sem (standard error of the mean). Differences between experimental groups were considered statistically significant at p < 0.05.
3. Results
3.1. Generation, Production, and Purification of NanoITXs
The expression vectors for the production of the three nanoITX variants (Figure 1) were electroporated into KM71H Pichia pastoris cells, and the proteins were successfully secreted to the extracellular medium by the addition of methanol.
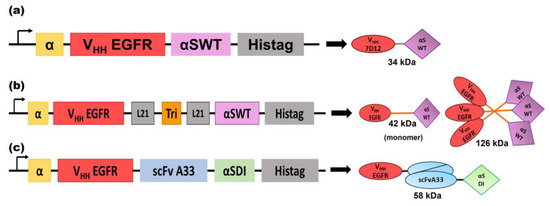
Figure 1.
Schematic diagrams showing the genetic and protein domain arrangements of the nanoITXs VHHEGFRαS (a), TriVHHEGFRαS (b), and BsITXαSDI (c). The cDNA constructions appear on the left side of the figure. The schematic representation of the protein motifs of the nanoITXs appears on the right side of the figure, with its molecular size underneath. In both cases, structural and functional domains are highlighted with different colors: α-factor secretion signal peptide (α, yellow), VHHEGFR (corresponds to VHH 7D12 [45]) (red), scFvA33 (blue), the 21 aa flexible linkers (gray), the trimerization domain derived from the collagen XVIII (TIEVIII, orange), the wild-type α-sarcin (αSWT, purple), the non-immunogenic α-sarcin (αSDI, green), and histidine-tag (Histag, black).
The three nanoITXs were purified from the extracellular medium by immobilized metal affinity chromatography and were analyzed by SDS-PAGE electrophoresis, followed by Coomassie blue staining or Western blot immunodetection using an anti-α-sarcin antibody (Figure 2). VHHEGFRαS and BsITXαSDI showed an expected mass of 34 and 58 kDa, respectively, showing a high degree of purity. TriVHHEGFRαS purification resulted in two different bands, 45 and 30 kDa, with only the 45 kDa band reacting with the anti-α-sarcin serum (Figure 2e). The purification yields of VHHEGFRαS, TriVHHEGFRαS, and BsITXαSDI were around 4, 0.5, and 3 mg/L of induction media, respectively.
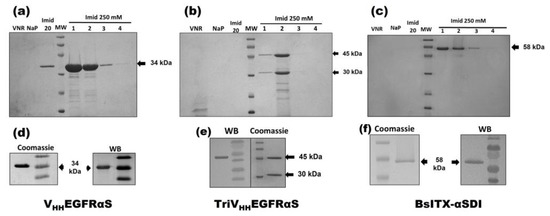
Figure 2.
Coomassie-blue-stained SDS-PAGE analysis after affinity purification of VHHEGFRαS (a), TriVHHEGFRαS (b), and BsITXαSDI (c) and Coomassie blue staining and Western blot analysis (WB) of the purified final fraction of VHHEGFRαS (d), TriVHHEGFRαS (e), and BsITXαSDI (f). Western blot analysis was carried out using rabbit anti-α-serum. Notes in gels correspond to the following: MW, prestained molecular weight standard (kDa); VNR, not retained fraction; NaP, washed fraction eluted with sodium phosphate buffer; Imidazole 20 mM, washed fraction eluted with sodium phosphate buffer containing imidazole 20 mM; and different 1 mL fractions eluted with 250 mM imidazole. The original full-length gels and uncropped Western Blot images can be found in Figures S1 and S2.
3.2. Structural Characterization
All nanoITX circular dichroism (CD) spectra were compatible with water-soluble globular proteins with a high degree of β-sheet secondary structure (Figure 3a–c). TriVHHEGFRαS spectra showed a high percentage of random structure, due to the contribution of the 18 residue linkers that flank both sides of the trimerization motif and the TIEXVIII domain inserted between the target and the toxic domain in the trimeric nanoITX. FPLC size exclusion chromatography was carried out in order to confirm the trimeric conformation of TriVHHEGFRαS in native conditions (Figure 3e). The size exclusion chromatography elution profile (Figure 3e) showed a main peak at an elution volume corresponding to 130 KDa, as expected for the trimeric ITX. The thermal denaturation profiles of VHHEGFRαS and BsITXαSDI showed Tms of 50 and 55 °C, respectively (Figure 3d,e), indicating that both immunotoxins are highly stable at high temperatures, which is consistent with the high stability of both the nanobodies and α-sarcin.
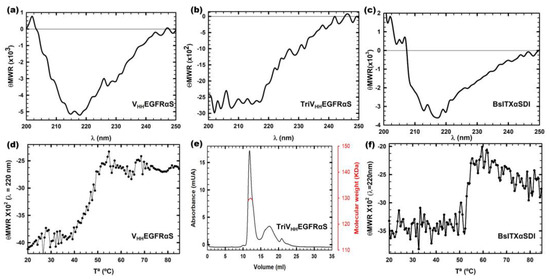
Figure 3.
Structural characterization by far-UV circular dichroism (CD) spectra of VHHEGFRαS (a), TriVHHEGFRαS (b), and BsITXαSDI (c). θMRW represents the mean residue weight ellipticities as degree × cm2 × dmol−1. The thermal denaturation profiles of VHHEGFRαS (d) and BsITXαSDI (f) by means of the temperature dependence of the ellipticity at 220 nm. All spectra were carried out at a protein concentration of 0.15 mg/mL in 50 mM sodium phosphate, 0.1 M NaCl, and pH 7.4. Analysis of the trimeric nature of TriVHHEGFRαS by Superdex 200 FPLC chromatography analysis (e). The eluted protein shows a major symmetric elution peak at the expected volume corresponding to its trimeric size (126 kDa), with the indicated molecular weight measured at the center of the chromatography peak (red curve).
3.3. Ribonucleolytic Activity
Functional characterization of the three nanoITXs was first carried out by analyzing the function of each domain separately. In all functional assays, the concentration of TriVHHEGFRαS was calculated as a monomer, to highlight the advantages of the trimeric over the monomeric format. The ribonucleolytic activity of the toxic domain was assayed using the reticulocyte assay as described in Materials and Methods, by the specific release of the α-fragment from the larger subunit of the rRNA. The release of the α-fragment was observed with all nanoITXs demonstrating that ribonucleolytic α-sarcin activity was preserved (Figure 4); this was also the case with non-immunogenic α-sarcin (αSDI) (Figure 4b), showing small differences depending on the amount of protein assayed, within 80–100% of the activity compared to wild-type α-sarcin (Figure 4c).
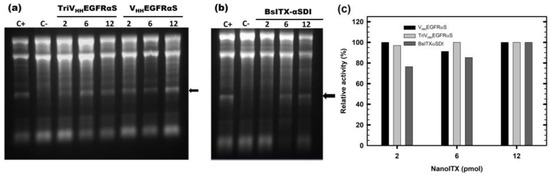
Figure 4.
In vitro functional characterization. The ribonucleolytic activity of the toxic domain of VHHEGFRαS and TriVHHEGFRαS (a), and BsITXαSDI (b). The arrow indicates the release of the α-fragment, produced by the cleavage of the SRL due to the α-sarcin. In both gels, 2, 6, and 12 pmoles of all three nanoITXs were assayed. C+ represents 2 pmoles of fungal wild-type α-sarcin, whereas in C-, the protein sample was replaced by a buffer. Images were acquired and analyzed using the Gel Doc XR Imaging System and the Quantity One software (BioRad). (c) Quantitation of specific ribonucleolytic activity of the three nanoITXs, expressed as a percentage of α-fragment/RNA 18S ratio, considering 100% to be the ratio obtained by 2 pmol of α-sarcin. Band intensities were quantitated with Quantity One software. The original full-length gels can be found in Figure S3.
3.4. Binding Activity
First, an ELISA using immobilized EGFR was carried out, in order to confirm the correct binding of VHHEGFRαS, TriVHHEGFRαS, and BsITXαSDI to its antigen. As observed in Figure 5a, the three nanoITXs were able to bind to immobilized EGFR, in a similar way as the positive control (VHHEGFR), retaining at least 80% of the binding ability of the control. These differences can be explained in terms of possible steric impairments due to the presence of the toxic domain or the second target domain. In addition, in order to consider the native conformation of EGFR in a cellular context, flow cytometry assays were carried out, using the EGFR+ A431 cell line. VHHEGFRαS was able to bind specifically to A431 cell lines, in a dose-dependent manner (Figure 5b).
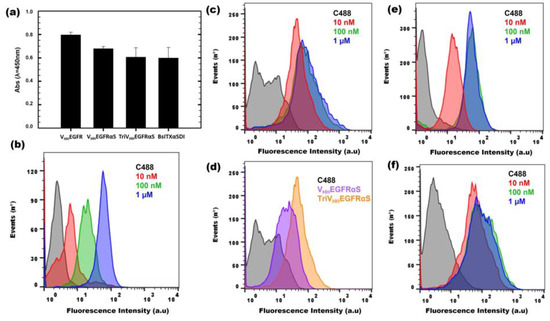
Figure 5.
In vitro functional characterization of binding activity. ELISA assay (a) against immobilized EGFR (0.5 μg/well), using the three nanoITXs and VHHEGFR as a positive control (1 µM). Flow cytometry binding assays of VHHEGFRαS (b) and TriVHHEGFRαS (c) to EGFR-positive cells (A431 cell line). Curves correspond to cells incubated with a secondary antibody anti-His-Alexa488 (black), 10 nM (red), 100 nM (green), or 1 μM (blue) of each immunotoxin. (d) Comparison between 10 nM of VHHEGFRαS (purple) or TriVHHEGFRαS (yellow) in the flow cytometry assay. Binding of BsITXαSDI to EGFR-positive cells (A431) (e) or GPA33-positive cells (SW1222) (f). Curves correspond to cells incubated with a secondary antibody anti-His-Alexa488 (black), 10 nM (red), 100 nM (green), or 1 μM (blue).
Flow cytometry assays using A431 cells incubated with TriVHHEGFRαS showed that the trimeric ITX recognizes and binds A431 cells more efficiently than its monomeric counterpart (Figure 5c), reaching binding saturation at the low concentration of 10 nM (Figure 5d). Moreover, BsITXαSDI (Figure 5e,f) was able to bind to both SW1222 (GPA33+, EGFR-) and A431 cells (EGFR+, GPA33-), matching the binding obtained for its monospecific counterparts, scFvA33 for SW1222 cells [41] and VHHEGFRαS for A431 cells (Figure 5b).
3.5. In Vitro Antitumoral Activity
To assess their therapeutic potential, the in vitro antitumoral effect of the different nanoITXs was analyzed. Tumor cells were incubated with different concentrations of nanoITXs for different periods of time, and viability was measured using the MTT viability assay. VHHEGFRαS exhibited a cytotoxic effect on A431 cells, reducing the viability of tumoral cells in a dose- and time-dependent manner, showing an IC50 of 300 nm at 72 h (Figure 6a). Interestingly, TriVHHEGFRαS showed a significantly higher cytotoxic effect on the same cells compared to its monomeric counterpart, with an IC50 at 72 h of 10 nm (Figure 6b), thirty times lower than VHHEGFRαS.
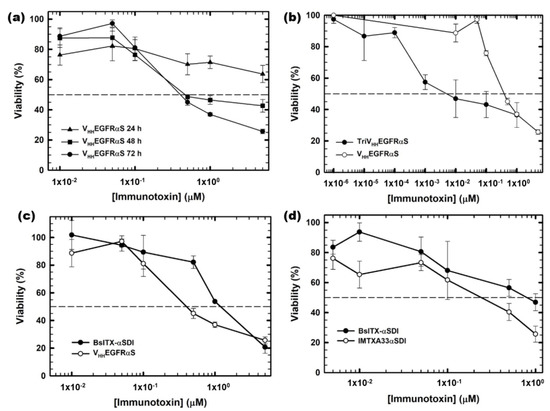
Figure 6.
In vitro cytotoxicity characterization by MTT viability assays. (a) EGFR-positive A431 cells treated with VHHEGFRαS for 24, 48, and 72 h; (b) EGFR-positive A431 cells treated for 72 h with TriVHHEGFRαS (black) compared to VHHEGFRαS (white); (c) EGFR-positive A431 cells treated for 72 h with BsITXαSDI (black) compared to VHHEGFRαS (white) and (d) GPA33-positive cells SW1222 treated with BsITXαSDI (black) and scFv-IMTXA33αSDI (white). Measurements were analyzed and plotted (mean ± SD) against untreated controls. In all cases, triplicate samples were carried out. IC50 values were obtained as the protein concentration leading to 50% viability.
Regarding BsITXαSDI, the bispecific immunotoxin was cytotoxic against both SW1222 and A431 cells, with IC50 values at 72 h of 700 nM and 1 μM, respectively (Figure 6c,d). The IC50 values of BsITXαSDI were higher than those obtained by the monospecific immunotoxins against the respective antigen-positive cell line, with an IC50 of 300 nM for VHHEGFRαS and 700 nm for IMTXA33αS [41].
3.6. In Vivo Antitumoral Activity
Finally, the in vivo antitumor activity of VHHEGFRαS was studied, employing for this purpose nude mice bearing A431 (EGFR+) epidermoid cancer xenografts. Two different doses of VHHEGFRαS were assayed (25 or 50 μg) along with a negative control (PBS) (Figure 7a). Treatment with VHHEGFRαS led to significant inhibition of tumor growth (three times in the 25 μg group and four times in the 50 μg group) (Figure 7c). Remarkably, we observed a medium-lasting tumor growth rebound after the last injection, reaching a final volume of 2000 mm3 at day 25.
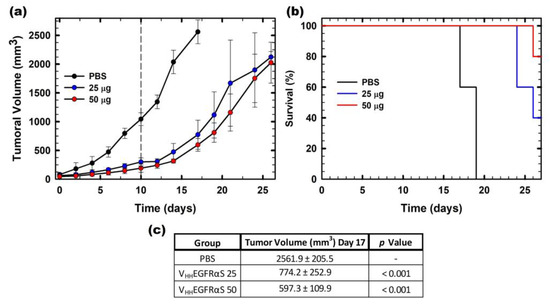
Figure 7.
In vivo antitumoral activity. (a) Time course of tumor volume growth of SW1222-derived xenografts. Mice were non-treated with PBS (black) or treated with two different doses (25 or 50 µg) of VHHEGFRαS (labeled in the graph as 25, blue or 50, red). The different doses were administered each 48 h. The vertical dashed line indicates the end of the administration of the antitumoral treatment. Values are represented as means ± sem (standard error of the mean). (b) Kaplan–Meier survival curves. The Kaplan–Meier representation expresses the time to the experimental endpoint (once tumor volume reaches 2000 mm3 of the in vivo assay). The labels in the graph are the same as those used in (a). (c) Statistical analysis of VHHEGFRαS 25- and VHHEGFRαS 50-treated tumors compared with vehicle-treated tumors at day 17. In all cases, the experimental groups were composed of 5 mice (n = 5).
These data on tumor volume were in agreement with the survival analysis by Kaplan–Meier representation. In Figure 7b, it is shown that the survival rate of the mice treated with 25 μg of VHHEGFRαS at day 27 was 40% and rose to 80% with the 50 μg dose treatment at the end of the experiment.
4. Discussion
Chimeric immunotoxins are promising therapeutic tools that have demonstrated promising activity in pre-clinical and clinical studies, against hematological and solid tumors [55], infectious diseases [6], and autoimmune processes [56]. However, efficacy, stability, tumor penetration, and immunogenicity are still aspects that must be improved, both in the target and the toxic domain of the immunotoxin design, to enable more extended therapeutic use.
To this end, in this work, we successfully designed and characterized different formats of nanoITXs based on the nanobody 7D12, specific against the extracellular domain of EGFR [57]. The binding affinity (Kd) of VHH 7D12 for EGFR was found to be in the range of 219 to 279 nM [58]. Its advantages in epitope recognition, compared to different Fab and complete antibodies directed against the same tumor marker, have been also described. Thus, this nanobody became a very interesting alternative for obtaining different multivalent or multispecific therapeutic designs [58]. First, the monomeric nanoITX, VHHEGFRαS, consisting of the 7D12 nanobody as the targeting domain and the fungal ribotoxin α-sarcin as the toxic domain, was produced. Then, we designed a trimeric format, TriVHHEGFRαS, introducing a collagen-XVIII-derived trimerization domain, TIEXVIII, between the target and the toxic domain. Finally, a bispecific immunotoxin (BsITXαSDI) was generated by combining the nanobody anti-EGFR with an scFv against GPA33 and a non-immunogenic variant of α-sarcin.
VHHEGFRαS was successfully produced in the yeast P. pastoris, with higher yields than those obtained for other ITXs based on scFv possibly due to its smaller size [41,59]. The structural characterization of all nanoITXs showed a high proportion of β-sheet secondary structure, which is consistent with the secondary structure of their components, the VHH 7D12 [60] and the ribotoxin α-sarcin [61,62]. The Tm of VHHEGFRαS was 50 °C, lower than that obtained for the VHH alone, which is around 80 °C [19,63], suggesting that the incorporation of α-sarcin could diminish the stability of the construction. However, the Tm at 50 °C is high above the physiological temperature of 37 °C, at which the VHHEGFRαS should exert its therapeutic effect.
The functional characterization of VHHEGFRαS showed that both the toxic and the target domains were able to exert their ribonucleolytic activity and specific binding to EGFR, respectively. The in vitro cytotoxic activity against the A431 cell line resulted in an IC50 of 300 nM at 72 h, slightly higher than for other nanoITXs described against EGFR [64]. This lower cytotoxic efficacy could be due to a lower internalization rate of the nanoITX, or to different issues regarding the release of the toxic domain once it has been internalized. Inclusion of the furin site in the linker between both domains could be achieved, according to the results described for other ITXs [44]. VHHEGFRαS showed potent inhibition of tumoral growth in A431-cell-xenografted mice, comparable to other immunotoxins whose in vivo antitumor activity has already been characterized [42]. Tumor growth rebound observed once treatment has been completed could be explained by lower tumor retention of the nanoITX, due to its monovalence or to the fast glomerular clearance of VHHEGFRαS, since its molecular weight (35 kDa) is lower than the glomerular filtration threshold [65,66].
The design of new formats of ITXs, thanks to the development of antibody engineering technology, has made it possible to overcome these drawbacks through the construction of multimeric and multispecific formats, with improvements in biodistribution, serum-half life, and tumoral penetration and retention [43,67]. In this sense, we designed a trimeric nanoITX, TriVHHEGFRαS, by the incorporation of a collagen-XVIII-derived trimerization domain (TIEXVIII) between the target and the toxic domain. After its production in P. pastoris, the electrophoretic analysis showed two different bands: a 45 kDa band that was recognized by the anti-α-sarcin antibody, and would consequently correspond to a monomer of TriVHHEGFRαS, and a 30 kDa band, not recognized by the anti-α-sarcin antibody, and that would presumably correspond to a monomer of TriVHHEGFR, without the toxic domain, thus indicating that it was soon cleaved before the whole protein was translated and secreted to the extracellular medium. The structural characterization showed that TriVHHEGFRαS presented a higher content of random structure than its monomeric counterpart, due to the flexible loops that flank the trimerization domain [46]. Size-exclusion chromatography showed that TriVHHEGFRαS eluted in a major peak, with the expected molecular size of 130 kDa, confirming its trimeric format in solution. The functional characterization of TriVHHEGFRαS showed that it reached binding saturation at a concentration of 10 nM, indicating better targeting activity than that of its monomeric counterpart. This improvement in targeting activity was also observed in the evaluation of the cell viability of both nanoITXs, with improved cytotoxic activity observed for the trimeric format, leading to a 30-fold lower IC50 compared to the monomeric format. Both results indicate that the trimeric format represents an advance in the design of therapeutic antibodies, as assessed with other trimeric immunotoxins [43]. It has been postulated that the binding of one monomer of the target domain to its antigen could enhance the binding of the other targeting domains. This increase in avidity is facilitated by the flexible 21 amino acid linker that flanks the trimerization domain [68,69]. Moreover, the enhanced binding activity, together with the release of three toxin molecules to the cytosol, could explain an increase in the overall cytotoxic activity, with a significant decrease in the IC50, and could potentially allow a reduction in the amount of drug administered, with the same therapeutic effect and minimizing the potential adverse effects. Most importantly, the molecular size of TriVHHEGFRαS, far above the renal cut-off, is expected to increase the half-life and decrease the frequency of dosing along with improved tumor retention [48]. In this sense, in vivo experiments with TriVHHEGFRαS are yet to be carried out.
Although anti-EGFR mAb cetuximab and panitumumab have improved the clinical response and overall survival of metastatic colorectal cancer patients [70], several unmet needs remain. Patients that initially responded to cetuximab treatment became refractory to the treatment because of downstream signaling mutations, such as mutations in KRAS or PIK3CA [67,71]. One of the most notable mutations of EGFR, namely EGFRvIII, consists of the deletion of exons 2–7, resulting in a truncated extracellular domain that lacks the ligand binding site, and gains constitutive kinase activity [72]. In addition, intrinsic heterogeneity and tumor evolution allow tumors to escape monospecific therapeutics, due to antigen loss [73,74]. In this sense, antibody technology has allowed the design of multispecific antibodies, that can bind to different epitopes in the same antigen [75], to different antigens on the same cell type, or to different antigens on different cell types, allowing the recruitment of immune cells to the tumoral environment [37,75].
In this sense, we designed and produced a bispecific immunotoxin (BsITXαSDI) that presents VHH7D12 against EGFR and the scFv against GPA33 as the target domains, and a non-immunogenic variant of α-sarcin (αSDI) as the toxic domain [51]. BsITXαSDI was successfully produced and purified to homogeneity in P. pastoris, with a final yield of 3 mg/L of induction, indicating that, as in the case of VHHEGFRαS, the presence of the VHH 7D12 in the N-terminus leads to a higher rate of production.
The structural characterization showed that BsITXαSDI presents a correct folding as a water-soluble globular protein, with a high content of β-sheet structure, as expected given the structure of its two target domains and the contribution of the αSDI. BsITXαSDI exhibited structural stability at temperatures above the physiological threshold of 37 ºC.
The toxic domain of BsITXαSDI, the non-immunogenic α-sarcin variant, kept its ribonucleolytic activity, as shown by the release of the α-fragment in the reticulocyte assay. The results obtained in the binding assays by flow cytometry confirmed that both target domains were functional. Thus, BsITXαSDI, under saturated concentration conditions was able to bind to both the EGFR+ A431 and the GPA33+ SW1222 cell lines, in a similar manner to its monospecific counterparts, VHHEGFRαS and the previously described scFvA33αS [50]. The IC50 values of BsITXαSDI for both cell lines were higher than those obtained with the monospecific variants, suggesting a lower internalization rate, possibly due to the higher molecular weight of the bispecific immunotoxin. However, several studies have shown that bispecific antibodies showing lower affinities for their TAAs are more likely to have better safety profiles since the binding of both targeting domains is required for an efficient intracellular uptake, whereas high-affinity targeting domains could bind to non-malignant cells expressing only a single TAA in their membrane [36,55]. In this sense, the dual targeting of two different TAAs could be an effective tool for overcoming primary tumor heterogeneity, and subsequent tumor escape due to antigen loss, which makes tumors refractory to conventional treatments. In addition, the inclusion of αSDI allows a safer and more effective therapeutic tool since it has proven to be effective both in vitro and in vivo [76] and may prevent the formation of potentially neutralizing anti-drug antibodies (ADAs) by the patient´s immune system.
5. Conclusions
The results obtained from the characterization of the three nanobody-based immunotoxins confirm and support their huge potential in the diagnosis and treatment of cancer. The special features of nanobodies, including high binding affinity, thermostability, and low immunogenicity, are of special interest in the diagnosis and treatment of several cancer types, including hematological tumors [77], glioblastoma [78], and colorectal cancer [79,80,81]. In addition, due to their modular arrangement, it is possible to design new immunotoxins, incorporating different functional domains with multimeric formats or different specificities, that endow these immunotoxins with optimized features against cancer.
The work herein described constitutes an effort to overcome the challenges of treating a heterogeneous disease such as colorectal cancer. The nanoITXs here described benefit from the smaller size of nanobodies and their excellent binding properties allowing deeper tumor penetration. The trimeric ITX improves the overall antitumor efficacy due to its increased avidity and toxic payload. Finally, the combination of multiple target domains directed against different tumoral antigens, combined with the absence of immunogenicity derived from the αSDI, represents a step forward in the treatment of this disease. The next steps will involve the study of the in vivo antitumor efficacy of the trimeric nanoITX, as well as the effect of combined treatments, and also the design of new optimized bispecific formats, using fungal ribotoxin-based nanoITXs as a platform for the design of new therapeutic constructs.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biom13071042/s1, Figure S1: Original SDS-PAGE of purified nanoITXs; Figure S2: SDS-PAGE and Western blot analysis of all purified nanoITXs; Figure S3: In vitro functional characterization.
Author Contributions
Conceptualization, J.N., L.H.-B., R.G.G., L.S. and J.L.; methodology, J.N., L.H.-B. and R.G.G.; formal analysis and investigation, J.N., L.H.-B., R.G.G. and J.L.; writing—original draft preparation, J.N.; writing—review and editing, J.N., L.H.-B., R.G.G., L.S. and J.L.; supervision, J.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Complutense University of Madrid (grant numbers PR75/18-21563 and PR87/19-22627), the PID2020-116692RB-I00 grant from the I+D+I Retos Investigación of the Spanish Ministry of Science and Innovation, and by a REACT-EU grant from the Comunidad de Madrid to the ANTICIPA-REACT project of Complutense University of Madrid. J.N. was supported by a predoctoral fellowship from the Spanish Ministry of Science and Innovation (FPU15/04121).
Institutional Review Board Statement
The animal study protocol was approved by the Ethics Committee of the Complutense University Animal Experimentation Committee and the Community of Madrid (PROEX298/15) for studies involving animals.
Informed Consent Statement
Not applicable.
Data Availability Statement
All experimental data generated or analyzed during this study are included in the article.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Glynne-Jones, R.; Wyrwicz, L.; Tiret, E.; Brown, G.; Rödel, C.; Cervantes, A.; Arnold, D. Rectal cancer: ESMO clinical practice guidelines for diagnosis, treatment, and follow-up. Ann. Oncol. 2018, 29 (Suppl. S4), 22–40. [Google Scholar] [CrossRef]
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.H.; Watanabe, T. Colorectal Cancer. Nat. Rev. Dis. Prim. 2016, 1, 15065. [Google Scholar] [CrossRef] [PubMed]
- Tintelnot, J.; Stein, A. Immunotherapy in colorectal cancer: Available clinical evidence, challenges, and novel approaches. World J. Gastroenterol. 2019, 25, 3920–3928. [Google Scholar] [CrossRef]
- Sadraeian, M.; Guimaraes, F.E.G.; Araujo, A.P.U.; Worthylake, D.K.; LeCour, L.J.; Pincus, S.H. Selective cytotoxicity of a novel immunotoxin based on pulchellin A chain for cells expressing HIV envelope. Sci. Rep. 2017, 7, 7579. [Google Scholar] [CrossRef] [PubMed]
- Fercher, C.; Keshvari, S.; AMcGuckin, M.; Barnard, R.T. Evolution of the magic bullet: Single chain antibody fragments for the targeted delivery of immunomodulatory proteins. Exp. Biol. Med. 2018, 243, 166–183. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Jun, S.J.; Kim, Y.S. Critical Issues in the development of immunotoxins for anticancer therapy. J. Pharm. Sci. 2020, 109, 104–115. [Google Scholar] [CrossRef]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- Muruganandam, A.; Tanha, J.; Narang, S.; Stanimirovic, D. Selection of phage-displayed llama single-domain antibodies that transmigrate across human blood-brain barrier endothelium. FASEB J. 2002, 16, 240. [Google Scholar] [CrossRef]
- Van der Linden, R.H.; Frenken, L.G.; de Geus, B.; Harmsen, M.M.; Ruuls, R.C.; Stok, W.; de Ron, L.; Wilson, S.; Davis, P.; Verrips, C.T. Comparison of physical chemicals properties of llama VHH antibody fragments and mouse monoclonal antibodies. Biochim. Biophys. Acta 1999, 1431, 37–46. [Google Scholar] [CrossRef]
- De Vos, J.; Devoogdt, N.; Lahoutte, T.; Muyldermans, S. Camelid single-domain antibody-fragment engineering for (pre)clinical in vivo molecular imaging applications: Adjusting the bullet to its target. Expert. Opin. Biol. 2013, 13, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- De Genst, E.; Silence, K.; Decanniere, K.; Conrath, K.; Loris, R.; Kinne, J.; Muyldemars, S.; Wyns, L. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. USA 2006, 103, 4586–4591. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.Y.; Shah, K. Nanobodies: Next generation of cancer diagnostics and therapeutics. Front. Oncol. 2020, 10, 1182. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, K.; Dreier, T.; Silence, K.; de Haard, H.; Lauwereys, M.; Casteels, P.; Beirnaert, E.; Jonckheere, H.; Van de Wiele, C.; Staelens, L.; et al. Formatted anti-tumor necrosis factor alpha VHH proteins derived from camelids show superior potency and targeting to inflamed joints in a murine model of collagen-induced arthritis. Arthritis Rheum. 2006, 54, 1856–1866. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.A.; Verrips, C.T. Nanobodies that neutralize HIV. Vaccines 2019, 7, 77. [Google Scholar] [CrossRef]
- Mei, Y.; Chen, Y.; Sivaccumar, J.P.; An, Z.; Xia, N.; Luo, W. Research progress and applications of nanobody in human infectious diseases. Front. Pharmacol. 2022, 13, 963978. [Google Scholar] [CrossRef]
- Roovers, R.C.; Laeremans, T.; Huang, L.; de Taeye, S.; Verkleij, A.J.; Revets, H.; de Haard, H.J.; van Bergen en Henegouwen, P.M.P. Efficient inhibition of EGFR signalling and of tumor growth by antagonistic anti EGFR nanobodies. Cancer Immunol. Immunother. 2007, 56, 303–317. [Google Scholar] [CrossRef]
- Moradi, A.; Pourseif, M.M.; Jafari, B.; Parvizpour, S. Nanobody-based therapeutics against colorectal cancer: Precision therapies based on the personal mutanome profile and tumor neoantigens. Pharmacol. Res. 2020, 156, 104790. [Google Scholar] [CrossRef]
- Li, M.; Liu, Z.S.; Liu, X.L.; Hui, Q.; Lu, S.Y.; Qu, L.L.; Li, Y.S.; Zhou, Y.; Ren, H.L.; Hu, P. Clinical targeting recombinant immunotoxins for cancer therapy. Oncotargets 2017, 10, 3645–3665. [Google Scholar] [CrossRef]
- Recht, L.D.; Raso, V.; Davis, R.; Salmonsen, R. Immunotoxin sensitivity of Chinese hamster ovary cells expressing human transferrin receptors with differing internalization rates. Cancer Immunl. Immunother. 1996, 42, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Alewine, C.; Hassan, R.; Pastan, I. Advances in anticancer immunotoxin therapy. Oncologist 2015, 20, 176–185. [Google Scholar] [CrossRef]
- Waltzman, R.J.; Sarkar, A.; Williams, E.T.; Iberg, A.T.; Higgins, J.T.; Willert, E.K. MT-5111: A novel HER2 targeting engineered toxin body in clinical development. J. Clin. Oncol. 2020, 38, 433. [Google Scholar] [CrossRef]
- Weaver, M.; Laske, D.W. Transferrin receptor ligand-targeted toxin conjugate (Tf-CRM107) for therapy of malignant gliomas. J. Neurooncol. 2003, 65, 3–13. [Google Scholar] [CrossRef]
- Powell, D.J.; Attia, P.; Ghetie, V.; Schindler, J.; Vitetta, E.S.; Rosenberg, S.A. Partial reduction of human FOXP3+ CD4 T cells in vivo after CD25-directed recombinant immunotoxin administration. J. Immunother. 2008, 31, 189–198. [Google Scholar] [CrossRef]
- Yeo, D.; Castelletti, L.; van Zandwijk, N.; Rasko, E.J.J. Hitting the bull´s eye: Mesothelin´s role as a biomarker and therapeutic target for malignant plural mesothelioma. Cancers 2021, 13, 3932. [Google Scholar] [CrossRef]
- Catimel, B.; Ritter, G.; Welt, S.; Old, L.J.; Cohen, L.; Nerrie, M.A.; White, S.J.; Heath, J.K.; Demediuk, B.; Domagala, T.; et al. Purification and characterization of a novel restricted antigen expressed by normal and transformed human colonic epithelium. J. Biol. Chem. 1996, 271, 25664–25670. [Google Scholar] [CrossRef]
- Heath, J.K.; White, S.J.; Johnstone, C.N.; Catimel, B.; Simpson, R.J.; Moritz, R.L.; Tu, G.F.; Ji, H.; Whitehead, R.H.; Groenen, L.C.; et al. The human A33 antigen is a transmembrane glycoprotein and a novel member of the immunoglobulin superfamily. Biochemistry 1997, 94, 469–474. [Google Scholar] [CrossRef]
- Pereira-Fantini, P.M.; Judd, L.M.; Kalantzis, A.; Peterson, A.; Ernst, M.; Heath, J.K.; Giraud, A.S. A33 antigen-deficient mice have defective colonic mucosal repair. Inflamm. Bowel Dis. 2010, 16, 604–612. [Google Scholar] [CrossRef]
- Ackerman, M.E.; Chalouni, C.; Schmidt, M.M.; Raman, V.V.; Ritter, G.; Old, L.J.; Mellman, I.; Wittrup, K.D. A33 antigen displays persistent surface expression. Cancer Immunol. Immunother. 2008, 57, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Simon, N.M.; FitzGerald, D. Immunotoxin therapies for the treatment of epidermal growth factor receptor-dependent cancers. Toxins 2016, 8, 137. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal. Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Roskoski, R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharm. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Polito, L.; Bortolotti, M.; Battelli, M.G.; Calafato, G.; Bolognesi, A. Ricin: An ancient story for a timeless plant toxin. Toxins 2019, 11, 324. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Xu, K.; Li, S.; Cao, L.; Nan, Y.; Li, Q.; Li, W.; Hong, Z. A single-domain antibody-based anti-PSMA recombinant immunotoxin exhibits specificity and efficacy for prostate cancer therapy. Int. J. Mol. Sci. 2021, 22, 5501. [Google Scholar] [CrossRef]
- Mazor, Y.; Sachsenmeier, K.F.; Yang, C.; Hansen, A.; Filderman, J.; Mulgrew, K.; Wu, H.; Dall’Acqua, W.F. Enhanced tumor-targeting selectivity by modulating bispecific antibody binding affinity and format valence. Sci. Rep. 2017, 7, 40098. [Google Scholar] [CrossRef]
- Mazor, R.; King, E.; Pastan, I. Anti-drug antibodies to LMB-100 are enhanced by mAbs targeting OX40 and CTLA4 but not by mAbs targeting PD1 or PDL-1. Cell Immunol. 2018, 334, 38–41. [Google Scholar] [CrossRef]
- Lacadena, J.; Álvarez-García, E.; Carreras-Sangrà, N.; Herrero-Galán, E.; Alegre-Cebollada, J.; García-Ortega, L.; Oñaderra, M.; Gavilanes, J.G.; Martínez-del-Pozo, A. Fungal ribotoxins: Molecular dissection of a family of natural killers. FEMS Microbiol. Rev. 2007, 31, 212–237. [Google Scholar] [CrossRef]
- Olombrada, M.; Lázaro-Gorines, R.; López-Rodríguez, J.C.; Martínez-del-Pozo, A.; Oñaderra, M.; Maestro-López, M.; Lacadena, J.; Gavilanes, J.G.; García-Ortega, L. Fungal ribotoxins: A review of potential biotechnological applications. Toxins 2017, 9, 71. [Google Scholar] [CrossRef]
- Lázaro-Gorines, R.; López-Rodríguez, J.C.; Benedé, S.; González, M.; Mayorga, C.; Vogel, L.; Martínez-del-Pozo, A.; Lacadena, J.; Villalba, M. Derp 1-based immunotoxin as potential tool for the treatment of dust mite respiratory allergy. Sci. Rep. 2020, 10, 12255. [Google Scholar] [CrossRef]
- Carreras-Sangrà, N.; Tomé-Amat, J.; García-Ortega, L.; Batt, C.A.; Oñaderra, M.; Martínez-del-Pozo, A.; Gavilanes, J.G.; Lacadena, J. Production and characterization of a colon cancer-specific immunotoxin based on the fungal ribotoxin α-sarcin. Protein Eng. Des. Sel. 2012, 25, 425–435. [Google Scholar] [CrossRef]
- Tomé-Amat, J.; Olombrada, M.; Ruiz-de-la-Herrán, J.; Pérez-Gómez, E.; Andradas, C.; Sánchez, C.; Martínez, L.; Martínez-del-Pozo, A.; Gavilanes, J.G.; Lacadena, J. Efficient in vivo antitumor effect of an immunotoxin based on ribotoxin α-sarcin in nude mice bearing human colorectal cancer xenografts. Springer Plus 2015, 4, 168. [Google Scholar] [CrossRef] [PubMed]
- Lázaro-Gorines, R.; Ruiz-de-la-Herrán, J.; Navarro, R.; Sanz, L.; Álvarez-Vallina, L.; Martínez-del-Pozo, A.; Gavilanes, J.G.; Lacadena, J. A novel carcinoembryonic antigen (CEA)-targeted trimeric immunotoxin shows significantly enhanced antitumor activity in human colorectal cancer xenografts. Sci. Rep. 2019, 9, 11680. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-de-la-Herrán, J.; Tomé-Amat, J.; Lázaro-Gorines, R.; Gavilanes, J.G.; Lacadena, J. Inclusion of a furin cleavage site enhances antitumor efficacy against colorectal cancer cells of ribotoxin α-sarcin-or RNase T1-based immunotoxins. Toxins 2019, 11, 593. [Google Scholar] [CrossRef]
- Gainkam, L.O.; Huang, L.; Caveliers, V.; Keyartes, M.; Hernot, S.; Vaneycken, I.; Vanhove, C.; Revets, H.; De Baetselier, P.; Lahoutte, P. Comparison of the biodistribution and tumor targeting of two 99mtc-labeled anti-EGFR nanobodies in mice, using pinhole SPECT/micro-CT. J. Nucl. Med. 2008, 49, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Toribio, A.; Lacadena, J.; Nuñez-Prado, N.; Álvarez-Cienfuegos, A.; Villate, M.; Compte, M.; Sanz, L.; Blanco, F.J. Álvarez-Vallina, L. Efficient production of single-chain fragment variable-based N-terminal trimerbodies in Pichia pastoris. Microb. Cell. Factories 2014, 13, 116. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, K.; Lykke-Harwood, S.; Compte, M.; Merino, N.; Molgaard, K.; Lykkemark, S.; Alvarez-Mendez, A.; Blanco, F.J. Álvarez-Vallina, L. Carcynoembryonic antigen (CEA)-specific 4-1BB.Costimulation induced by CEA-targeted 4-1BB-Agonistic trimerbodies. Front. Immunol. 2019, 10, 1791. [Google Scholar] [CrossRef]
- Rios, X.; Compte, M.; Gómez-Vallejo, V.; Cossío, U.; Baz, Z.; Morcillo, M.A.; Ramos-Cabrer, P.; Alvarez-Vallina, L.; Llop, J. Immuno-PET imaging and pharmacokinetics of an anti-CEA scFv-based trimerbody and its monomeric counterpart in human gastric carcinoma-bearing mice. Mol. Pharm. 2019, 16, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Damasceno, L.; Lee, F.; Ritter, G.; Old, L.; Batt, C. High-level expression of a phage display-derived scFv in Pichia pastoris. Methods Mol. Biol. 2009, 562, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Tomé-Amat, J.; Ruiz-de-la-Herrán, J.; Martínez-del-Pozo, A.; Gavilanes, J.G.; Lacadena, J. α-sarcin and RNase T1 based immunoconjugates: The role of intracelular trafficking in cytotoxic efficiency. FEBS J. 2015, 282, 673–684. [Google Scholar] [CrossRef]
- Jones, T.D.; Hearn, A.R.; Holgate, R.; Kozub, D.; Fogg, M.H.; Carr, F.J.; Baker, M.P.; Lacadena, J.; Gehlsen, K.R. A deimmunized form of the ribotoxin α-sarcin, lacking CD4+ T cell epitopes and its use as an immunotoxin warhead. Protein Eng. Des. Sel. 2016, 29, 531–540. [Google Scholar] [CrossRef]
- Kao, R.; Martínez-Ruiz, A.; Martínez-del-Pozo, Á.; Crameri, R.; Davies, J. Mitogillin and related fungal ribotoxins. Methods Enzymol. 2001, 341, 324–335. [Google Scholar] [CrossRef]
- Martínez-Ruiz, A.; García-Ortega, L.; Kao, R.; Lacadena, J.; Oñaderra, M.; Mancheño, J.M.; Davies, J.; Martínez-del-Pozo, Á.; Gavilanes, J.G. RNase U2 and α-sarcin: A study of relantionships. Methods Enzymol. 2001, 341, 335–351. [Google Scholar] [CrossRef]
- Lee, F.T.; Mountain, A.J.; Kelly, M.P.; Hall, C.; Rigopoulos, A.; Johns, T.G.; Smyth, F.E.; Brechbiel, M.W.; Nice, E.C.; Burgess, A.W.; et al. Enhanced efficacy of radioimmunotherapy with 90Y-CHX-A”-DTPA-hu3S193 by inhibition of epidermal growth factor receptor (EGFR) signaling with EGFR tyrosine kinase inhibitor AG1478. Clin. Cancer Res. 2005, 11, 7080–7086. [Google Scholar] [CrossRef]
- Sanz, L.; Ibáñez-Pérez, R.; Guerrero-Ochoa, P.; Lacadena, J.; Anel, A. Antibody-based immunotoxins for colorectal cancer therapy. Biomedicines 2021, 9, 1729. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Wang, P.; Dong, S.; Zhou, Z.; Cao, Y.; Yagita, H.; He, X.; Zheng, S.G.; Fisher, S.J.; Fujinami, R.S.; et al. Depletion of PD-1 positive cells ameliorates autoimmune disease. Nat. Biomed. Eng. 2019, 3, 292–305. [Google Scholar] [CrossRef] [PubMed]
- Biteghe, F.A.N.; Mungra, N.; Chalomie, N.E.T.; Ndong, J.; Engohang-Ndong, J.; Vignaux, G.; Padayachee, E.; Naran, K.; Barth, S. Advances in epidermal growth factor specific immunotherapy: Lessons to be learned from armed antibodies. Oncotarget 2021, 11, 3531–3557. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.R.; Bagchi, A.; Roovers, R.C.; van Bergen en Henegouwen, P.M.P.; Ferguson, K.M. Structural evaluation of EGFR inhibition mechanisms for nanobodies/VHH domains. Structure 2013, 21, 1214–1224. [Google Scholar] [CrossRef]
- Herrero-Galán, E.; García-Ortega, L.; Lacadena, J.; Martínez-del-Pozo, A.; Olmo, N.; Gavilanes, J.G.; Oñaderra, M. A non-cytotoxic but ribonucleolytically specific ribotoxin variant: Implication of tryptophan residues in the cytotoxicity of Hirsutellin A. Biol. Chem. 2012, 393, 449–456. [Google Scholar] [CrossRef]
- Harmsen, M.M.; de-Haard, H.J. Properties, production and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol. 2007, 77, 13–22. [Google Scholar] [CrossRef]
- Campos-Olivas, R.; Bruix, M.; Santoro, J.; Martínez-del-Pozo, A.; Lacadena, J.; Gavilanes, J.G.; Rico, M. 1H and 15N nuclear magnetic resonance assignment and secondary structure of the cytotoxic ribonuclease α-sarcin. Protein Sci. 1996, 5, 969–972. [Google Scholar] [CrossRef]
- Pérez-Cañadillas, J.M.; Santoro, J.; Campos-Olivas, R.; Lacadena, J.; Martínez-del-Pozo, A.; Gavilanes, J.G.; Rico, M.; Bruix, M. The highly refined solution structure of the cytotoxic ribonuclease α-sarcin reveals the structural requirements for substrate recognition and ribonucleolytic activity. J. Mol. Biol. 2000, 299, 1061–1073. [Google Scholar] [CrossRef]
- Sun, S.; Ding, Z.; Yang, X.; Zhao, X.; Zhao, M.; Gao, L.; Chen, Q.; Xie, S.; Liu, A.; Yin, S.; et al. Nanobody: A small antibody with big implications for tumor therapeutic strategy. Int. J. Nanomed. 2021, 16, 2337–2356. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Xiong, J.; Gu, X.; Chen, X.; Wu, S.; Wang, Z.; Wang, D.; Tu, J.; Xie, J. Novel recombinant immunotoxin of EGFR specific nanobody fused with cucurmosin, construction and antitumor efficiency in vitro. Oncotarget 2017, 8, 38568–38580. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.M.; Wittrup, K.D. A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Mol. Cancer Ther. 2009, 8, 2861–2871. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Deng, C.; Zhang, C.; Xiong, J.; Gu, X.; Wang, Z.; Tu, J.; Xie, J. Preparation of a novel EGFR specific immunotoxin and its efficacy of anti-colorectal cancer in vitro and in vivo. Clin. Transl. Oncol. 2021, 23, 1549–1560. [Google Scholar] [CrossRef]
- Wu, T.; Liu, M.; Huang, H.; Sheng, Y.; Xiao, H.; Liu, Y. Clustered nanobody-drug conjugates for targeted cancer therapy. Chem. Commun. 2020, 56, 9344–9347. [Google Scholar] [CrossRef]
- Cuesta, A.M.; Sánchez-Martín, D.; Sanz, L.; Bonet, J.; Compte, M.; Kremer, L.; Blanco, F.J.; Oliva, B.; Alvarez-Vallina, L. In vivo tumor targeting and imaging with engineered trivalent antibody fragments containing collagen derivated sequences. PLoS ONE 2009, 4, e5381. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, A.M.; Sainz-Pastor, M.; Bonet, J.; Oliva, B.; Álvarez-Vallina, L. Multivalent antibodies: When design surpasses evolution. Trends Biotechnol. 2010, 28, 355–362. [Google Scholar] [CrossRef]
- Biller, L.H.; Schrag, D. Diagnosis and treatment of metastatic colorectal cancer: A review. JAMA 2021, 325, 669–685. [Google Scholar] [CrossRef]
- Karapetics, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Cvrljevic, A.N.; Johns, T.G. The epidermal growth factor receptor variant III (EGFRvIII). Where wild things are altered. FEBS J. 2013, 280, 5330–5370. [Google Scholar] [CrossRef] [PubMed]
- Vyas, M.; Müller, R.; von Strandmann, E.P. Antigen loss variants: Catching hold of escaping foes. Front. Immunol. 2017, 8, 175. [Google Scholar] [CrossRef]
- El-Sayes, N.; Vito, A.; Mossman, K. Tumor heterogeneity: A great barrier in the age of cancer immunotherapy. Cancers 2021, 13, 806. [Google Scholar] [CrossRef]
- Zhang, C.; Cai, Y.; Dai, X.; Wu, J.; Lan, Y.; Zhang, H.; Lu, M.; Liu, J.; Xie, J. Novel EGFR-bispecific recombinant immunotoxin based on cucurmosin shows potent anti-tumor efficiency in vitro. Oncol. Rep. 2021, 45, 493–500. [Google Scholar] [CrossRef]
- Narbona, J.; Gordo, R.G.; Tomé-Amat, J.; Lacadena, J. A new optimized version of a colorectal cancer targeted immunotoxin based on a non-immunogenic varant of the ribotoxin α-sarcin. Cancers 2023, 15, 1114. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Li, S.; Zhang, W.; Sun, J.; Ren, G.; Dong, Q. A novel VHH antibody targeting the B cell-activating factor for B-cell lymphoma. Int. J. Mol. Sci. 2014, 15, 9481–9496. [Google Scholar] [CrossRef]
- Van de Water, J.A.J.M.; Bagci-Onder, T.; Agarwal, A.S.; Wakimoto, H.; Roovers, R.C.; Zhu, Y.; Kasmieh, R.; Bhere, D.; Van Bergen en Henegouwen, P.M.P.; Shah, K. Therapeutic stem cells expressing variants of EGFR-specific nanobodies have antitumor effects. Proc. Natl. Acad. Sci. USA 2012, 41, 16642–16647. [Google Scholar] [CrossRef]
- Behar, G.; Chames, P.; Teulon, I.; Cornillon, A.; Alshoukr, F.; Roquet, F.; Pugnière, M.; Teillaud, J.L.; Gruaz-Guyon, A.; Pèlegrin, A.; et al. Llama single-domain antibodies directed against nonconventional epitopes of tumor-associated carcinoembryionic antigen absent from non-specific cross-reacting antigen. FEBS J. 2009, 276, 3881–3893. [Google Scholar] [CrossRef]
- Allegra, A.; Immao, V.; Gerace, D.; Vaddinelli, D.; Allegra, A.G.; Musolino, C. Nanobodies and cancer: Current status and new perspectives. Cancer Investig. 2018, 36, 221–237. [Google Scholar] [CrossRef]
- Khirehgesh, M.R.; Sharifi, J.; Safari, F.; Akbari, B. Immunotoxins and nanobody-based immunotoxins: Review and update. J. Drug. Target. 2021, 29, 848–862. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).