Abstract
Vitamin D3 (1) is metabolized by various cytochrome P450 (CYP) enzymes, resulting in the formation of diverse metabolites. Among them, 4α,25-dihydroxyvitamin D3 (6a) and 4β,25-dihydroxyvitamin D3 (6b) are both produced from 25-hydroxyvitamin D3 (2) by CYP3A4. However, 6b is detectable in serum, whereas 6a is not. We hypothesized that the reason for this is a difference in the susceptibility of 6a and 6b to CYP24A1-mediated metabolism. Here, we synthesized 6a and 6b, and confirmed that 6b has greater metabolic stability than 6a. We also identified 4α,24R,25- and 4β,24R,25-trihydroxyvitamin D3 (16a and 16b) as metabolites of 6a and 6b, respectively, by HPLC comparison with synthesized authentic samples. Docking studies suggest that the β-hydroxy group at C4 contributes to the greater metabolic stability of 6b by blocking a crucial hydrogen-bonding interaction between the C25 hydroxy group and Leu325 of CYP24A1.
1. Introduction
Vitamin D3 (1) is a steroid hormone that is synthesized in the skin upon exposure to sunlight or can be absorbed from foods. It is transported to the liver, where it undergoes hydroxylation at C25 by cytochrome (CYP) P450 family members CYP2R1 and CYP27A1 to form 25-hydroxyvitamin D3 (2, 25D) (Scheme 1). The resulting 2 is then transported to the kidneys, where it undergoes further hydroxylation at C1α by CYP27B1 to afford 1α,25-dihydroxyvitamin D3 (3, 1,25D) [1], which regulates multiple physiological functions, including calcium homeostasis [2], bone metabolism [3], cell differentiation [4], and immune regulation [5] by binding to the vitamin D receptor (VDR), which is a nuclear receptor. Subsequently, both 2 and 3 undergo metabolism of their side chains. Specifically, CYP24A1 mediates hydroxylation at the C23S or C24R position of 2 and 3, leading to the formation of vitamin D3 lactone (4) and calcitroic acid (5), respectively, as final metabolites [6,7,8,9,10,11].
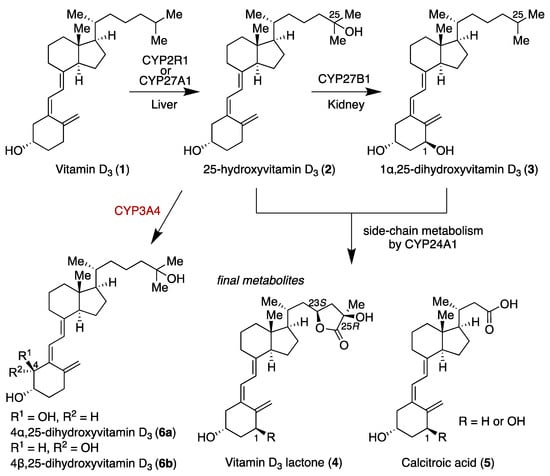
Scheme 1.
Metabolic pathways of vitamin D3 (1).
In 2011, Thummel et al. found a novel vitamin D metabolite, 4β,25-dihydroxyvitamin D3 (6b), which is formed from 2 by hydroxylation at C4β [12]. The conversion of compound 2 to compound 6 may be involved in drug-induced osteomalacia. Notably, in vitro studies revealed that CYP3A4 generated 4α,25-dihydroxyvitamin D3 (6a) and 6b at a ratio of 1:2. Interestingly, however, analysis of human serum showed the presence of only 6b, with no detection of 6a. This intriguing result led us to propose that differential metabolic stability between 6a and 6b might explain this discrepancy. We hypothesized that a difference in metabolic stability between 6a and 6b would account for this finding. Here, we synthesized 6a and 6b, and evaluated their metabolic stability in the presence of CYP24A1, which plays a major role in vitamin D metabolism. We also identified metabolites of 6 generated by CYP24A1 by means of HPLC comparison with synthesized authentic samples.
2. Materials and Methods
2.1. Synthesis of 6a and 6b
All reagents were supplied by commercial sources without further purification. All reactions involving air- or moisture-sensitive reagents were carried out in flame-dried glassware under argon atmosphere. Flash chromatography was performed using silica gel 60 (spherical, particle size 0.040−0.100 mm; Kanto Co., Inc., Tokyo, Japan), and preparative TLC (PLC) was performed using PLC silica gel 60 F254 (0.5 mm, Merck Ltd., Darmstadt, Germany). Optical rotations were measured on a JASCO P-2200 polarimeter (JASCO Co., Inc., Tokyo, Japan). 1H and 13C NMR spectra were recorded on JNM-AL300 (300 MHz, JEOL Ltd., Tokyo, Japan), JNM-ECX 400 (400 MHz, JEOL Ltd., Tokyo, Japan), and JNM-ECA 500 (500 MHz, JEOL Ltd., Tokyo, Japan) spectrometers. Chemical shift in CDCl3 was reported in the scale relative to CDCl3 (7.26 ppm) for 1H NMR. For 13C NMR, the chemical shift was reported in the scale relative to CDCl3 (77.0 ppm) and CD3OD (49.0 ppm) as an internal reference. HRMS (ESI) measurements were performed on a JMS-T100LC spectrometer (JEOL Ltd., Tokyo, Japan).
(S)-3,4-bis((tert-butyldimethylsilyl)oxy)butyl benzoate (S1): To a solution of diol 9 [13] (300 mg, 1.4 mmol) in CH2Cl2 (7 mL) was added 2,6-lutidine (0.44 mL, 3.7 mmol) and tert-butyldimethylsilyl triflate (0.82 mL, 3.6 mmol) at 0 °C. The resulting mixture was allowed to warm to room temperature and stirred for 30 min. The reaction mixture was quenched with H2O, and the aqueous layer was extracted with CH2Cl2 three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 20:1) to give the protected product S1 (627 mg, 99%) as a colorless oil. [α] = –10.6 (c 0.87, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.05 (d, J = 7.9 Hz, 2H), 7.55 (t, J = 7.7 Hz, 1H), 7.44 (t, J = 7.7 Hz, 2H), 4.34–4.51 (m, 2H), 3.86–3.94 (m, 1H), 3.62 (q, J = 4.9 Hz, 1H), 3.48 (q, J = 5.5 Hz, 1H), 2.05–2.15 (m, 1H), 1.81 (td, J = 13.6, 5.6 Hz, 1H), 0.89 (s, 18H), 0.07 (d, J = 4.8 Hz, 12H); 13C NMR (75 MHz, CDCl3) δ 166.5, 132.8, 130.4, 129.5, 128.3, 70.1, 67.5, 61.8, 33.4, 31.1, 25.9, 25.8, 25.7, −3.0, −4.3, −5.0, −5.4; HRMS (ESI) m/z: [M + Na]+ calcd for C23H42O4Si2Na 461.2519, found 461.2493.
(S)-3-((tert-butyldimethylsilyl)oxy)-4-hydroxybutyl benzoate (10): To a solution of diol S1 (627 mg, 1.4 mmol) in MeOH/THF = 1:1 (17 mL) was added (±)-CSA (40 mg, 0.17 mmol) at room temperature. The resulting mixture was stirred at the same temperature for 1 h. The reaction mixture was quenched with sat. NaHCO3 aq, and the aqueous layer was extracted with CH2Cl2 three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 10:1) to give 10 (77 mg, 54%) as a colorless oil. [α] = –9.8 (c 0.53, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.03 (d, J = 7.2 Hz, 2H), 7.56 (t, J = 7.4 Hz, 1H), 7.44 (t, J = 7.7 Hz, 2H), 4.31–4.49 (m, 2H), 3.96–4.03 (m, 1H), 3.66 (q, J = 4.9 Hz, 1H), 3.55 (q, J = 5.3 Hz, 1H), 1.99 (q, J = 6.3 Hz, 2H), 0.91 (s, 9H), 0.10 (d, J = 2.1 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 166.5, 132.9, 130.2, 129.5, 128.4, 69.7, 66.4, 61.6, 32.9, 25.8, 18.0, −4.6, −4.8; HRMS (ESI) m/z: [M + Na]+ calcd for C17H28O4SiNa 347.1655, found 347.1670.
(3S,4R)-3-((tert-butyldimethylsilyl)oxy)-4-hydroxy-6-(trimethylsilyl)hex-5-yn-1-yl benzoate (11b): To a solution of oxalic acid dichloride (0.48 mL, 5.6 mmol) in CH2Cl2 (7 mL) was added dimethyl sulfoxide (1.0 mL, 14.0 mmol) at –78 °C. The resulting mixture was stirred at the same temperature for 10 min, then a solution of 10 in CH2Cl2 (0.4 M, 7 mL, 2.78 mmol) and triethylamine (3.9 mL, 27.8 mmol) were added dropwise at the same temperature. The resulting mixture was allowed to warm to room temperature and stirred for 1 h. The reaction mixture was quenched with H2O, and the aqueous layer was extracted with CH2Cl2 three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 1:1) to give the aldehyde as a yellow oil. The crude residue was used in the subsequent step without purification. To a solution of trimethylsilylacetylene (1.0 mL, 7.2 mmol) in THF (12 mL) was added n-butyllithium (2.6 M in hexane; 2.3 mL, 6.0 mmol) at –78 °C. The resulting mixture was stirred at the same temperature for 30 min, then a solution of the aldehyde in THF (0.3 M, 9.3 mL, 2.78 mmol) was added dropwise at the same temperature. The resulting mixture was stirred at the same temperature for 15 min. The reaction mixture was quenched with sat. NH4Cl aq, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 30:1) to give 11b (910 mg, 90%) as a colorless oil. [α] = 1.5 (c 5.36, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.04 (d, J = 6.9 Hz, 2H), 7.56 (t, J = 7.4 Hz, 1H), 7.44 (t, J = 7.6 Hz, 2H), 4.48–4.56 (m, 1H), 4.30–4.42 (m, 2H), 3.99–4.04 (m, 1H), 2.16–2.27 (m, 1H), 1.97–2.08 (m, 1H), 0.91 (s, 9H), 0.16 (s, 9H), 0.10 (d, J = 5.2 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 166.4, 132.9, 130.2, 129.5, 128.3, 103.0, 91.7, 71.9, 66.5, 61.6, 31.1, 25.7, 18.0, −0.3, −4.5, −4.7; HRMS (ESI) m/z: [M + Na]+ calcd for C22H36O4Si2Na 443.2050, found 443.2017.
(S)-3-((tert-butyldimethylsilyl)oxy)-4-oxo-6-(trimethylsilyl)hex-5-yn-1-yl benzoate (S2): To a solution of 11b (100 mg, 0.24 mmol) and pyridine (0.38 mL) in CH2Cl2 (19 mL) was added Dess–Martin periodinane (404 mg, 0.95 mmol) at 0 °C. The resulting mixture was allowed to warm to room temperature and stirred for 30 min. The reaction mixture was quenched with sat. Na2S2O3 aq, and the aqueous layer was extracted with CH2Cl2 three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 10:1) to give S2 (94 mg, 94%) as a colorless oil. [α] = +0.7 (c 1.37, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.02 (d, J = 6.9 Hz, 2H), 7.54 (t, J = 7.4 Hz, 1H), 7.43 (t, J = 7.4 Hz, 2H), 4.46–4.56 (m, 1H), 4.32–4.43 (m, 2H), 2.18–2.26 (m, 2H), 0.93 (s, 9H), 0.05–0.22 (m, 15H); 13C NMR (75 MHz, CDCl3) δ 189.5, 166.3, 132.9, 130.0, 129.6, 128.3, 102.4, 100.4, 75.5, 60.1, 33.7, 31.1, 25.7, 18.1, −1.0, −4.7, −5.4; HRMS (ESI) m/z: [M + Na]+ calcd for C22H34O4Si2Na 441.1893, found 441.1877.
(3S,4S)-3-((tert-butyldimethylsilyl)oxy)-4-hydroxy-6-(trimethylsilyl)hex-5-yn-1-yl benzoate (11a): To a solution of S2 (94 mg, 0.22 mmol) in THF (11 mL) was added L-selectride (1.0 M in THF, 0.40 mL, 0.40 mmol) at –78 °C. The resulting mixture was stirred at the same temperature for 1 h. The reaction mixture was quenched with sat. NH4Cl aq, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 10:1) to give 11a (94 mg, 99%) as a colorless oil. [α] = –9.1 (c 0.35, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.05 (d, J = 6.9 Hz, 2H), 7.56 (t, J = 7.4 Hz, 1H), 7.44 (t, J = 7.6 Hz, 2H), 4.32–4.51 (m, 3H), 3.99 (q, J = 5.4 Hz, 1H), 1.99–2.19 (m, 2H), 0.92 (s, 9H), 0.12–0.16 (m, 15H); 13C NMR (75 MHz, CDCl3) δ 166.4, 132.9, 130.1, 129.5, 128.3, 104.6, 90.6, 72.5, 65.8, 61.2, 32.7, 25.9, 18.1, −0.3, −4.3, −4.6; HRMS (ESI) m/z: [M + Na]+ calcd for C22H36O4Si2Na 443.2050, found 443.2054.
(3S,4S)-3,4-bis((tert-butyldimethylsilyl)oxy)-6-(trimethylsilyl)hex-5-yn-1-yl benzoate (S3a): To a solution of 11a (115 mg, 0.27 mmol) in CH2Cl2 (3 mL) was added 2,6-lutidine (98 μL, 0.82 mmol) and tert-butyldimethylsilyl triflate (94 μL, 0.41 mmol) at 0 °C. The resulting mixture was allowed to warm to room temperature and stirred for 30 min. The reaction mixture was quenched with H2O, and the aqueous layer was extracted with CH2Cl2 three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 40:1) to give S3a (143 mg, 98%) as a colorless oil. [α] = –5.6 (c 0.25, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.05 (d, J = 7.2 Hz, 2H), 7.55 (t, J = 7.2 Hz, 1H), 7.43 (t, J = 7.6 Hz, 2H), 4.35–4.58 (m, 3H), 3.81–3.87 (m, 1H), 2.14–2.25 (m, 1H), 1.95–2.09 (m, 1H), 0.90 (d, J = 1.4 Hz, 18H), 0.05–0.16 (m, 21H); 13C NMR (75 MHz, CDCl3) δ 166.5, 132.8, 130.5, 129.5, 128.3, 104.8, 90.5, 71.5, 67.3, 61.8, 31.2, 25.8, 25.8, 18.2, 18.0, −0.3, −4.5, −4.6, −4.8, −4.9; HRMS (ESI) m/z: [M + Na]+ calcd for C28H50O4Si3Na 557.2915, found 557.2909.
(3S,4R)-3,4-bis((tert-butyldimethylsilyl)oxy)-6-(trimethylsilyl)hex-5-yn-1-yl benzoate (S3b): To a solution of 11b (264 mg, 0.63 mmol) in CH2Cl2 (6 mL) was added 2,6-lutidine (0.22 mL, 1.9 mmol) and tert-butyldimethylsilyl triflate (0.22 mL, 0.94 mmol) at 0 °C. The resulting mixture was allowed to warm to room temperature and stirred for 30 min. The reaction mixture was quenched with H2O, and the aqueous layer was extracted with CH2Cl2 three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 40:1) to give S3b (334 mg, 99%) as a colorless oil. [α] = –26.2 (c 1.38, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.05 (d, J = 7.2 Hz, 2H), 7.56 (t, J = 7.4 Hz, 1H), 7.44 (t, J = 7.4 Hz, 2H), 4.46–4.54 (m, 1H), 4.36–4.42 (m, 1H), 4.28 (d, J = 4.5 Hz, 1H), 3.89–3.94 (m, 1H), 2.08–2.19 (m, 1H), 1.92–2.05 (m, 1H), 0.91 (d, J = 1.4 Hz, 18H), 0.05–0.15 (m, 21H); 13C NMR (75 MHz, CDCl3) δ 166.5, 132.8, 130.5, 129.5, 128.3, 105.8, 90.3, 72.9, 67.8, 61.8, 32.0, 25.9, 25.8, 18.1, −0.3, −4.1, −4.4, −4.8, −5.0; HRMS (ESI) m/z: [M + Na]+ calcd for C28H50O4Si3Na 557.2915, found 557.2872.
(3S,4S)-3,4-bis((tert-butyldimethylsilyl)oxy)hex-5-yn-1-ol (12a): To a solution of S3a (143 mg, 0.27 mmol) in MeOH (0.9 mL) was added K2CO3 (148 mg, 1.1 mmol) at room temperature. The resulting mixture was stirred at the same temperature for 1 h. The reaction mixture was quenched with H2O, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 30:1) to give 12a (68 mg, 71%) as a colorless oil. [α] = –14.5 (c 0.29, CHCl3); 1H NMR (300 MHz, CDCl3) δ 4.38 (q, J = 2.3 Hz, 1H), 3.79 (tt, J = 8.1, 2.9 Hz, 3H), 2.38 (d, J = 2.1 Hz, 1H), 1.84–2.09 (m, 2H), 0.90 (d, J = 1.4 Hz, 18H), 0.09–0.15 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 82.4, 74.0, 73.2, 66.9, 60.1, 34.8, 25.7, 18.1, 18.0, −4.6, −4.7, −4.8, −5.1; HRMS (ESI) m/z: [M + Na]+ calcd for C18H38O3Si2Na 381.2257, found 338.2297.
(3S,4R)-3,4-bis((tert-butyldimethylsilyl)oxy)hex-5-yn-1-ol (12b): To a solution of S3b (206 mg, 0.39 mmol) in MeOH (1.3 mL) was added K2CO3 (213 mg, 1.5 mmol) at room temperature. The resulting mixture was stirred at the same temperature for 1 h. The reaction mixture was quenched with H2O, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 30:1) to give 12b (104 mg, 76%) as a colorless oil. [α] = –22.3 (c 1.60, CHCl3); 1H NMR (300 MHz, CDCl3) δ 4.36 (q, J = 2.2 Hz, 1H), 3.94 (q, J = 4.8 Hz, 1H), 3.78 (td, J = 5.8, 2.3 Hz, 2H), 2.41 (d, J = 2.1 Hz, 1H), 1.82–2.06 (m, 2H), 0.90 (d, J = 1.7 Hz, 18H), 0.09–0.17 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 83.5, 74.1, 66.9, 59.0, 34.6, 25.9, 25.7, 18.2, 18.1, −4.3, −4.6, −4.8, −5.1; HRMS (ESI) m/z: [M + Na]+ calcd for C18H38O3Si2Na 381.2257, found 338.2263.
(5S,6S)-5-ethynyl-6-(2-iodoethyl)-2,2,3,3,8,8,9,9-octamethyl-4,7-dioxa-3,8-disiladecane (S4a): To a solution of 12a (49 mg, 0.38 mmol) in THF (0.6 mL) was added triphenylphosphine (70 mg, 0.33 mmol), imidazole (28 mg, 0.41 mmol), and iodine (112 mg, 0.44 mmol) at –20 °C. The resulting mixture was stirred at the same temperature for 30 min. The resulting mixture was allowed to warm to room temperature and stirred for 20 min. The reaction mixture was quenched with sat. Na2S2O3 aq, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane) to give S4a (54 mg, 83%) as a yellow oil. [α] = –35.2 (c 0.25, CHCl3); 1H NMR (300 MHz, CDCl3) δ 4.37 (q, J = 2.3 Hz, 1H), 3.68–3.74 (m, 1H), 3.38 (qd, J = 5.4, 3.8 Hz, 1H), 3.22 (td, J = 9.5, 6.3 Hz, 1H), 2.33 (d, J = 2.1 Hz, 1H), 2.21–2.30 (m, 1H), 2.04–2.15 (m, 1H), 0.90 (d, J = 0.7 Hz, 18H), 0.11–0.14 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 82.6, 73.9, 73.5, 66.3, 35.4, 31.1, 25.7, 18.1, 18.0, 4.2, −4.3, −4.5, −4.7, −5.1; HRMS (ESI) m/z: [M + Na]+ calcd for C18H37IO2Si2Na 491.1274, found 491.1255.
(5R,6S)-5-ethynyl-6-(2-iodoethyl)-2,2,3,3,8,8,9,9-octamethyl-4,7-dioxa-3,8-disiladecane (S4b): To a solution of 12b (136 mg, 0.38 mmol) in THF (1.6 mL) was added triphenylphosphine (193 mg, 0.91 mmol), imidazole (77.2 mg, 1.1 mmol), and iodine (308 mg, 1.2 mmol) at –20 °C. The resulting mixture was stirred at the same temperature for 30 min. The resulting mixture was allowed to warm to room temperature and stirred for 20 min. The reaction mixture was quenched with sat. Na2S2O3 aq, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane) to give S4b (149 mg, 84%) as a yellow oil. [α] = –28.3 (c 1.54, CHCl3); 1H NMR (300 MHz, CDCl3) δ 4.26 (q, J = 2.1 Hz, 1H), 3.77 (td, J = 5.3, 3.3 Hz, 1H), 3.19–3.35 (m, 2H), 2.39 (d, J = 2.4 Hz, 1H), 2.11–2.20 (m, 2H), 0.90 (d, J = 2.1 Hz, 18H), 0.12–0.15 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 83.3, 75.9, 74.0, 66.8, 37.1, 25.9, 25.8, 3.1, −4.1, −4.5, −5.2; HRMS (ESI) m/z: [M + Na]+ calcd for C18H37IO2Si2Na 491.1274, found 491.1243.
(4S,5R)-4,5-bis((tert-butyldimethylsilyl)oxy)hept-6-ynenitrile (13a). To a solution of S4a (54 mg, 0.11 mmol) in DMF (0.3 mL) was added sodium cyanide (8.4 mg, 0.17 mmol) at room temperature. The resulting mixture was allowed to warm to 90 °C and stirred for 20 min. The reaction mixture was quenched with sat. NaHCO3 aq, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 80:1) to give 13a (42 mg, 99%) as a yellow oil. [α] = –27.6 (c 0.29, CHCl3); 1H NMR (300 MHz, CDCl3) δ 4.38 (q, J = 2.3 Hz, 1H), 3.67–3.73 (m, 1H), 2.39–2.58 (m, 2H), 2.36 (d, J = 2.1 Hz, 1H), 2.04–2.16 (m, 1H), 1.90–2.02 (m, 1H), 0.90 (s, 18H), 0.11–0.14 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 119.9, 82.0, 74.0, 72.3, 66.4, 31.1, 27.6, 25.7, 18.1, 18.0, −4.6, −4.8, −5.1; HRMS (ESI) m/z: [M + Na]+ calcd for C19H37NO2Si2Na 390.2261, found 390.2290.
(4S,5R)-4,5-bis((tert-butyldimethylsilyl)oxy)hept-6-ynenitrile (13b): To a solution of S4b (133 mg, 0.28 mmol) in DMF (0.7 mL) was added sodium cyanide (21.0 mg, 0.43 mmol) at room temperature. The resulting mixture was allowed to warm to 90 °C and stirred for 20 min. The reaction mixture was quenched with sat. NaHCO3 aq, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 80:1) to give 13b (105 mg, 99%) as a yellow oil. [α] = –32.0 (c 2.14, CHCl3); 1H NMR (300 MHz, CDCl3) δ 4.27 (q, J = 2.1 Hz, 1H), 3.82 (td, J = 5.3, 3.2 Hz, 1H), 2.45–2.51 (m, 2H), 2.42 (d, J = 2.1 Hz, 1H), 1.89–2.12 (m, 2H), 0.90 (s, 18H), 0.11–0.15 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 120.3, 83.0, 74.4, 73.7, 66.7, 28.2, 25.8, 25.7, 18.1, 13.0, −4.2, −4.6, −4.8, −5.2; HRMS (ESI) m/z: [M + Na]+ calcd for C19H37NO2Si2Na 390.2261, found 390.2249.
(5S,6S)-5-(but-3-en-1-yl)-6-ethynyl-2,2,3,3,8,8,9,9-octamethyl-4,7-dioxa-3,8-disiladecane (8a): To a solution of 13a (42 mg, 0.11 mmol) in CH2Cl2 (0.6 mL) was added diisobutylaluminium hydride (1.0 M in hexane; 0.14 mL, 0.14 mmol) at 0 °C. The resulting mixture was stirred at the same temperature for 30 min. To the reaction mixture was added 2-propanol (0.098 mL), silica gel (200 mg), and water (1 mL) at room temperature. The resulting mixture was stirred at the same temperature for 30 min. The reaction mixture was filtered through a pad of Celite and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 10:1) to give the aldehyde as a yellow oil. The crude residue was used in the subsequent step without purification. To a solution of methyltriphenylphosphonium iodide (240 mg, 0.39 mmol) in THF (0.6 mL) was added sodium bis(trimethylsilyl)amide (1.9 M in THF, 0.29 mL, 0.56 mmol) at 0 °C. The resulting mixture was stirred at the same temperature for 30 min, then a solution of the aldehyde in THF (0.2 M, 0.55 mL, 0.11 mmol) was added dropwise at the same temperature. The resulting mixture was allowed to warm to room temperature and stirred for 1 h. The reaction mixture was quenched with sat. NH4Cl aq, and the aqueous layer was extracted with n-hexane three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane) to give 8a (27 mg, 64%) as a colorless oil. [α] = –18.0 (c 0.22, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.78–5.88 (m, 1H), 4.94–5.06 (m, 2H), 4.34 (q, J = 2.3 Hz, 1H), 3.57–3.61 (m, 1H), 2.33 (d, J = 2.3 Hz, 1H), 2.18–2.27 (m, 1H), 2.02–2.11 (m, 1H), 1.78–1.86 (m, 1H), 1.67–1.75 (m, 1H), 0.90 (s, 18H), 0.07–0.14 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 138.9, 114.3, 83.0, 74.0, 73.2, 66.8, 30.8, 29.6, 25.8, 25.8, 18.2, 18.1, −4.4, −4.5, −4.7, −5.0; HRMS (ESI) m/z: [M + Na]+ calcd for C20H40O2Si2Na 391.2465, found 391.2465.
(5S,6R)-5-(but-3-en-1-yl)-6-ethynyl-2,2,3,3,8,8,9,9-octamethyl-4,7-dioxa-3,8-disiladecane (8b): To a solution of 13b (32.5 mg, 0.089 mmol) in CH2Cl2 (0.4 mL) was added diisobutylaluminium hydride (1.0 M in hexane; 0.11 mL, 0.11 mmol) at 0 °C. The resulting mixture was stirred at the same temperature for 30 min. To the reaction mixture was added 2-propanol (0.074 mL), silica gel (200 mg), and water (1 mL) at room temperature. The resulting mixture was stirred at the same temperature for 30 min. The reaction mixture was filtered through a pad of Celite and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 10:1) to give the aldehyde as a yellow oil. The crude residue was used in the subsequent step without purification. To a solution of methyltriphenylphosphonium iodide (189 mg, 0.47 mmol) in THF (0.5 mL) was added sodium bis(trimethylsilyl)amide (1.9 M in THF, 0.23 mL, 0.44 mmol) at 0 °C. The resulting mixture was stirred at the same temperature for 30 min, then a solution of the aldehyde in THF (0.2 M, 0.45 mL, 0.089 mmol) was added dropwise at the same temperature. The resulting mixture was allowed to warm to room temperature and stirred for 1 h. The reaction mixture was quenched with sat. NH4Cl aq, and the aqueous layer was extracted with n-hexane three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane) to give 8b (23 mg, 70%) as a colorless oil. [α] = –29.1 (c 3.30, CHCl3); 1H NMR (300 MHz, CDCl3) δ 5.76–5.89 (m, 1H), 4.93–5.05 (m, 2H), 4.23 (q, J = 2.3 Hz, 1H), 3.73 (q, J = 5.2 Hz, 1H), 2.35 (d, J = 2.4 Hz, 1H), 2.13 (q, J = 7.3 Hz, 2H), 1.59–1.82 (m, 2H), 0.91 (s, 18H), 0.08–0.15 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 138.8, 114.3, 84.2, 75.2, 73.3, 66.7, 32.0, 28.9, 26.0, 25.8, 18.2, −4.1, −4.5, −4.5, −5.1; HRMS (ESI) m/z: [M + Na]+ calcd for C20H40O2Si2Na 391.2465, found 391.2442.
4α,25-dihydroxyvitamin D3 (6a): To a solution of CD-ring 7 [14] (19 mg, 0.040 mmol), 8a (18 mg, 0.048 mmol), and triethylamine (0.4 mL) in toluene (0.4 mL) was added tetrakis(triphenylphosphine)palladium(0) (5 mg, 0.0040 mmol) at room temperature. The resulting mixture was allowed to warm to 90 °C and stirred for 2 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane) to give the coupling product (20 mg, 66%) as a yellow oil. The crude residue was used in the subsequent step without purification. To a solution of the coupling product (20 mg, 0.026 mmol) in THF (0.3 mL) was added tetra-n-butylammonium fluoride (1.0 M in THF, 0.26 mL, 0.26 mmol) at room temperature. The resulting mixture was stirred at the same temperature for 12 h. The reaction mixture was quenched with sat. NaHCO3 aq, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (methanol/chloroform 1:20) to give 6a (8 mg, 77%) as a yellow oil. [α] = +162.5 (c 0.08, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.58 (d, J = 10.1 Hz, 1H), 6.04 (d, J = 11.4 Hz, 1H), 5.12 (s, 1H), 4.87 (s, 1H), 3.96 (d, J = 8.2 Hz, 1H), 3.57 (td, J = 8.9, 3.7 Hz, 1H), 2.90 (d, J = 12.8 Hz, 1H), 2.36 (td, J = 8.8, 4.4 Hz, 1H), 2.16–2.21 (m, 1H), 2.07–2.13 (m, 1H), 2.00 (t, J = 9.2 Hz, 2H), 1.88 (t, J = 8.2 Hz, 1H), 1.21–1.82 (m, 22H), 0.93 (d, J = 6.4 Hz, 3H), 0.53 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 144.1, 143.5, 137.3, 119.1, 117.1, 114.6, 78.4, 74.6, 71.1, 56.5, 56.4, 52.1, 45.9, 44.4, 40.5, 36.4, 36.1, 32.2, 29.7, 29.4, 29.2, 27.7, 23.5, 22.2, 20.8, 18.8, 12.0; HRMS (ESI) m/z: [M + Na]+ calcd for C27H44O3Na 439.3188, found 439.3177.
4β,25-dihydroxyvitamin D3 (6b): To a solution of CD-ring 7 (38 mg, 0.080 mmol), 8b (35 mg, 0.096 mmol), and triethylamine (0.8 mL) in toluene (0.8 mL) was added tetrakis(triphenylphosphine)palladium(0) (9 mg, 0.0080 mmol) at room temperature. The resulting mixture was allowed to warm to 90 °C and stirred for 2 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane) to give the coupling product (39 mg, 65%) as a yellow oil. The crude residue was used in the subsequent step without purification. To a solution of the coupling product (9 mg, 0.012 mmol) in THF (0.6 mL) was added tetra-n-butylammonium fluoride (1.0 M in THF, 0.12 mL, 0.12 mmol) at room temperature. The resulting mixture was stirred at the same temperature for 12 h. The reaction mixture was quenched with sat. NaHCO3 aq, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (methanol/chloroform 1:20) to give 6b (5 mg, 100%) as a yellow oil. [α] = +39.0 (c 0.21, CHCl3); 1H NMR (500 MHz, CDCl3) δ 6.51 (d, J = 10.9 Hz, 1H), 6.04 (d, J = 11.5 Hz, 1H), 5.17 (s, 1H), 4.92 (s, 1H), 4.22 (s, 1H), 3.86 (s, 1H), 2.86 (d, J = 13.2 Hz, 1H), 2.35–2.38 (m, 1H), 2.13–2.18 (m, 1H), 2.00–2.02 (m, 2H), 1.84–1.85 (m, 3H), 1.21–1.71 (m, 21H), 0.93 (d, J = 6.3 Hz, 3H), 0.54 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 145.4, 142.6, 137.2, 123.5, 117.0, 115.4, 71.9, 71.1, 56.5, 56.4, 46.0, 44.4, 40.5, 36.4, 36.1, 32.0, 30.1, 29.7, 29.4, 29.2, 29.2, 27.7, 23.6, 22.2, 20.8, 18.8, 12.0; HRMS (ESI) m/z: [M + Na]+ calcd for C27H44O3Na 439.3188, found 439.3181.
2.2. Metabolism of 6a and 6b by CYP24A1
The metabolism of 6a and 6b by CYP24A1 was examined using recombinant human CYP24A1 as described in previous studies [7,15]. Briefly, the reaction mixture containing 20 nM CYP24A1, 2.0 μM bovine adrenodoxin, 0.2 μM bovine adrenodoxin reductase, 5 μM each substrate, 1 mM NADPH, and 1 mM ethylenediaminetetraacetic acid (EDTA) in 100 mM Tris-HCl (pH 7.4) was incubated at 37 °C for 15 or 30 min. The metabolites were extracted with 4 volumes of chloroform/methanol (3:1) and analyzed by reversed-phase HPLC under the same conditions followed in our previous study [16].
2.3. Synthesis of 16a and 16b and Identification of New Metabolites
4α,24R,25-trihydroxyvitamin D3 (16a): To a solution of CD-ring 15 [17] (9.6 mg, 0.016 mmol), 8a (5 mg, 0.014 mmol), and triethylamine (0.11 mL) in toluene (0.11 mL) was added tetrakis(triphenylphosphine)palladium(0) (3.2 mg, 0.0016 mmol) at room temperature. The resulting mixture was allowed to warm to 90 °C and stirred for 2 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane) to give the coupling product (11 mg, 88%) as a yellow oil. The crude residue was used in the subsequent step without purification. To a solution of the coupling product (11 mg, 0.012 mmol) in THF (0.1 mL) was added tetra-n-butylammonium fluoride (1.0 M in THF, 144 μL, 0.144 mmol) at room temperature. The resulting mixture was stirred at the same temperature for 12 h. The reaction mixture was quenched with sat. NaHCO3 aq, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (methanol/chloroform 1:15) to give 16a (4.9 mg, 94%) as a yellow oil. [α] = +36.6 (c 0.29, CHCl3); 1H NMR (300 MHz, CDCl3) δ 6.59 (d, J = 11.4 Hz, 1H), 6.04 (d, J = 11.4 Hz, 1H), 5.12 (s, 1H), 4.88 (s, 1H), 3.96 (d, J = 8.6 Hz, 1H), 3.54–3.61 (m, 1H), 3.34 (t, J = 5.8 Hz, 1H), 2.90 (d, J = 12.4 Hz, 1H), 2.32–2.40 (m, 1H), 1.86–2.27 (m, 9H), 1.25–1.71 (m, 10H), 1.22 (s, 3H), 1.17 (s, 3H), 0.94 (d, J = 6.2 Hz, 3H), 0.54 (s, 3H); 13C NMR (125 MHz, CD3OD) δ 146.0, 144.1, 140.0, 120.7, 118.7, 114.2, 79.7, 79.2, 75.0, 73.9, 58.1, 57.6, 47.0, 41.9, 37.2, 34.2, 33.2, 32.7, 30.1, 28.8, 28.7, 25.7, 24.9, 24.6, 23.3, 19.3, 12.4; HRMS (ESI) m/z: [M + Na]+ calcd for C27H44O4Na 455.3137, found 455.3120.
4β,24R,25-trihydroxyvitamin D3 (16b): To a solution of CD-ring 15 (9.6 mg, 0.016 mmol), 8b (5 mg, 0.014 mmol), and triethylamine (0.11 mL) in toluene (0.11 mL) was added tetrakis(triphenylphosphine)palladium(0) (3.2 mg, 0.0016 mmol) at room temperature. The resulting mixture was allowed to warm to 90 °C and stirred for 2 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane) to give the coupling product (8.5 mg, 68%) as a yellow oil. The crude residue was used in the subsequent step without purification. To a solution of the coupling product (8.5 mg, 0.0096 mmol) in THF (0.1 mL) was added tetra-n-butylammonium fluoride (1.0 M in THF, 115 μL, 0.115 mmol) at room temperature. The resulting mixture was stirred at the same temperature for 12 h. The reaction mixture was quenched with sat. NaHCO3 aq, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (methanol/chloroform 1:15) to give 16b (3.5 mg, 84%) as a yellow oil. [α] = +47.7 (c 0.13, CHCl3); 1H NMR (300 MHz, CDCl3) δ 6.51 (d, J = 11.4 Hz, 1H), 6.04 (d, J = 11.4 Hz, 1H), 5.17 (s, 1H), 4.92 (s, 1H), 4.22 (d, J = 2.8 Hz, 1H), 3.84–3.88 (m, 1H), 3.65 (s, 1H), 3.34 (t, J = 5.8 Hz, 1H), 2.86 (d, J = 12.4 Hz, 1H), 2.33–2.42 (m, 1H), 2.11–2.20 (m, 1H), 1.17–2.04 (m, 23H), 0.94 (d, J = 6.2 Hz, 3H), 0.55 (s, 3H); 13C NMR (125 MHz, CD3OD) δ 144.9, 139.9, 123.4, 118.6, 115.0, 79.8, 78.1, 73.9, 73.1, 58.1, 57.6, 47.1, 41.9, 37.2, 34.2, 33.2, 30.5, 30.1, 28.8, 28.7, 25.7, 24.9, 24.6, 23.3, 19.3, 12.4; HRMS (ESI) m/z: [M + Na]+ calcd for C27H44O4Na 455.3137, found 455.3117.
2.4. Docking Study
The initial structure of the human CYP24A1 was obtained from the AlphaFold protein structure database [18] (AF-Q07973-F1-model_v2.pdb) and refined for docking simulations using the Protein Preparation Wizard script within Maestro (Schrödinger, LLC, New York, NY, USA). For all compound molecules, ionization and energy minimization were performed using the OPLS3e force field in the LigPrep script in Maestro. These minimized structures were used as input structures for the docking simulations using the Glide SP docking [19,20] program (Schrödinger, LLC, New York, NY, USA), with a grid box defined by a potential binding site position from SiteMap [21,22] (Schrödinger, LLC, New York, NY, USA). In docking simulations, we also introduced hydrogen-bonding constraints between the sidechain of The395 and any polar atoms in the compound molecules, because this hydrogen-bonding formation is a key interaction in known complexes of CYP24A1 bound to the vitamin D3 analogs [23]. After the docking simulations were completed, the lowest distances between the Fe atom of the HEM molecule and the C24 of the compounds from 100 poses on a binding site were selected.
3. Results and Discussion
3.1. Synthesis of 4α,25-Dihydroxyvitamin D3 (6a) and 4β,25-Dihydroxyvitamin D3 (6b)
The retrosynthetic plan for the synthesis of 4α,25-dihydroxyvitamin D3 (6a) and 4β,25-dihydroxyvitamin D3 (6b) is depicted in Scheme 2. We planned to construct the triene system in 6 using a palladium-catalyzed coupling reaction [24] of the bromoolefin 7 [24] (CD-ring) and the A-ring precursor enyne 8. The synthesis of enyne 8 was planned starting from diol 9, which can be derived from L-(–)-malic acid [13].
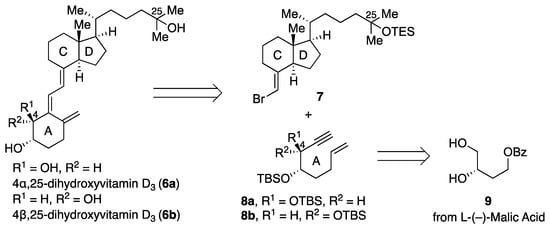
Scheme 2.
Retrosynthetic analysis for 4α,25-dihydroxyvitamin D3 (6a) and 4β,25-dihydroxyvitamin D3 (6b).
The syntheses of 4α,25-dihydroxyvitamin D3 (6a) and 4β,25-dihydroxyvitamin D3 (6b) are illustrated in Scheme 3A. The primary hydroxy alcohol 10 was synthesized in 54% yield by the reaction of 9, derived from L-(–)-malic acid, with TBSOTf followed by selective deprotection of the primary TBS ether in the presence of camphorsulfonic acid (CSA). After oxidation of the primary hydroxy group in 10 under Swern conditions, the resulting aldehyde was reacted with TMS acetylene in the presence of n-butyllithium to stereoselectively give 4β-11b in 90% yield (2 steps), where the stereochemistry at C4 is controlled according to the Felkin–Anh model (Scheme 3B). The 4α-isomer 11a was stereoselectively obtained by oxidation of 11b with Dess–Martin periodinane followed by reduction of the resulting ketone with a bulky reducing agent, L-selectride. The alcohols 11a and 11b were then converted to 6a and 6b, respectively, as follows. The secondary hydroxy groups in 11a and 11b were protected as TBS ethers in the presence of TBSOTf and 2,6-lutidine, and the benzoyl and TMS groups were removed with potassium carbonate to give alcohols 12a and 12b in 70% and 76% yields, respectively. Treatment of 12a and 12b with iodine and PPh3 followed by reaction with sodium cyanide gave the corresponding nitriles 13a and 13b. Reduction of the nitriles 13 with DIBAL-H afforded the corresponding aldehydes, which were then reacted with Wittig reagent 14 to give the A-ring synthons, enynes 8a and 8b, in 63% and 70% yield, respectively. Coupling of 8a and 8b with the CD-ring synthon, bromoolefin 7, was performed in the presence of a Pd catalyst, and the coupling products were deprotected with TBAF to remove the silyl ether groups, affording vitamin D metabolites 6a and 6b in 50% yield and 65% yield, respectively.
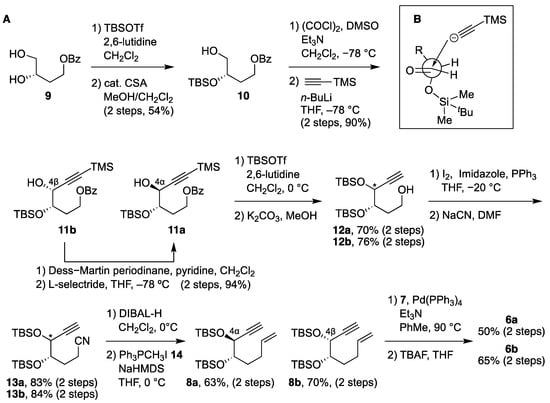
Scheme 3.
(A) Synthesis of 6a and 6b. (B) Felkin–Anh model for the reaction of the aldehyde derived from 10 with TMS acetylide.
3.2. Metabolism of 6a and 6b by CYP24A1
A reconstituted system containing human CYP24A1, adrenodoxin reductase, and adrenodoxin was employed to examine the metabolism of 6a and 6b. The conversion ratios of the substrate to its metabolites were obtained from the peak area ratio in HPLC chromatograms after 15 min incubation of 6 with CYP24A1 (Figure S1). The 6a was sequentially metabolized by CYP24A1 to produce multiple metabolites, as well as 1,25D3 and 25D3 [6,7]. The total conversion ratio of 6a to the multiple metabolites was 38.2%, whereas the conversion ratio of 6b was only 9.8%. These results suggest that the difference in serum concentrations between 6a and 6b can be explained by the difference in their metabolic stability, as we had hypothesized. Specifically, the high resistance of 6b to metabolism by CYP24A1 may explain why 6b is detected in serum, whereas 6a is not.
3.3. Identification of 16a and 16b as Metabolites of 4,25-(OH)2-D3 (6)
Next, we set out to identify the major metabolites of 6a and 6b generated by CYP24A1. Since CYP24A1 is known to hydroxylate C24 of 2 with R-stereochemistry [7], we hypothesized that 6 would undergo 24R-hydroxylation by CYP24A1 to afford 4,24R,25-trihydroxyvitamin D3 (7). To test this hypothesis, we synthesized the 4α compound 16a and the 4β compound 16b and compared their HPLC retention times with the metabolites of 6a and 6b produced by CYP24A1.
Compounds 16a and 16b were synthesized by a similar procedure to that described for the 4-hydroxylated metabolites of 6 (Scheme 4). Specifically, the CD ring 15 [17] bearing an R-hydroxy group at C24 was reacted with the A-ring precursor enyne 8a or 8b in the presence of a palladium catalyst to give the coupling product; deprotection of the silyl ether using TBAF afforded 16a and 16b in 80% and 58% yields, respectively.
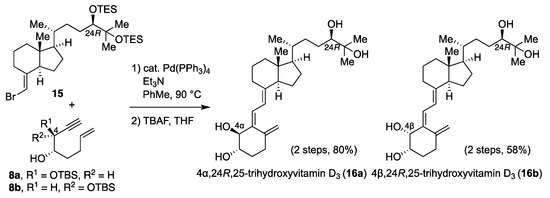
Scheme 4.
Synthesis of 16a and 16b.
Then, we compared the HPLC retention times of the synthesized 16a and 16b with those of the metabolites produced from 6 by CYP24A1. The retention times of 16a and 16b were consistent with those of the main metabolites produced by CYP24A1 from 6a and 6b (peak A and peak B in Figure 1A,B), respectively. In HPLC analyses on a chiral column (SUMICHIRAL OA-7000), the retention times of 16 matched those of the metabolites generated from 6 (Figure S2), indicating that the main metabolites produced by CYP24A1 from 6a and 6b are 16a and 16b, respectively. The conversion ratios to 16a and 16b from 6a and 6b were 25.6 and 7.0 %, respectively (Table 1).
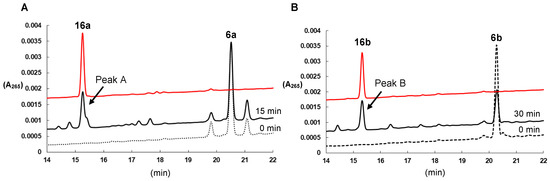
Figure 1.
(A) HPLC profiles of 16a (red line) and metabolites of 6a after incubation with CYP24A1 for 0 min (dotted line) and 15 min (solid line). Peak A, marked with an arrow, indicates a major metabolite. (B) HPLC profiles of 16b (red line) and metabolites of 6b after incubation with CYP24A1 for 0 min (dotted line) and 30 min (solid line) after incubation with CYP24A1. Peak B, marked with an arrow, indicates a major metabolite.

Table 1.
Evaluation of metabolic conversion ratios by human CYP24A1.
3.4. Docking Study
To investigate the reason for the difference in metabolic stability between 6a and 6b, docking studies with CYP24A1 and 6a and 6b were carried out. In the docking of 6a with CYP24A1, hydrogen bonds are formed between the C25 and C4α hydroxy groups of 6a and Leu325 and Thr395 of CYP24A1, resulting in the formation of a stable complex in which the side chain of the CD-ring in 6a is located close to the heme iron (Figure 2A). In contrast, in the docking of 6b with CYP24A1, hydrogen bonds were formed between the C4β and C3 hydroxy groups of 6b and the carboxylic acid of the side chain in heme and Thr395 of CYP24A1 (Figure 2B). In this case, the side chain of the CD ring in 6b is located away from the heme iron, and the hydrogen bond between the C25 hydroxy group and the Leu325, which plays an important role in the metabolism of vitamin D by CYP24A1 [24], is not formed. This may explain why 6b is less susceptible to metabolism by CYP24A1.
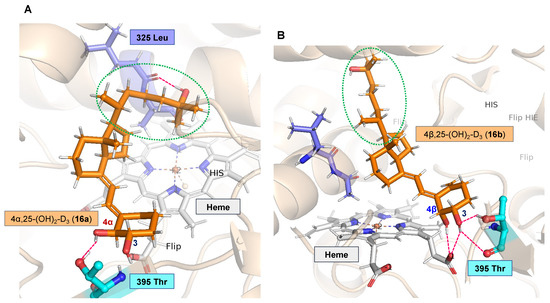
Figure 2.
(A) Docking model of 6a and CYP24A1. (B) Docking model of 6b and CYP24A1.
4. Conclusions
4α,25-Dihydroxyvitamin D3 (6a) and 4β,25-dihydroxyvitamin D3 (6b) are both produced from 25-hydroxyvitamin D3 (2) by CYP3A4, but 6b is detectable in serum, whereas 6a is not. Our findings show that 6a is a better substrate of CYP24A1 than 6b, and thus the greater metabolic stability of 6b can account for its presence in human serum. Furthermore, major metabolites of 6a and 6b generated by CYP24A1 were identified as 4,24R,25-trihydroxyvitamin D3 (16a, 16b) by HPLC, by comparison with synthesized authentic samples. Docking studies indicated that while 6a can form a hydrogen bond between the hydroxy group at C25 and Leu325 in CYP24A1, the presence of the 4β-hydroxy group in 6b prevents the formation of this hydrogen bond, which plays a crucial role in the metabolic activity of CYP24A1.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biom13071036/s1, Figure S1: HPLC profiles of metabolism of 6a and 6b by CYP24A1, Figure S2: HPLC profiles of metabolism of 6a and 6b by CYP24A1 with chiral column, Figure S3: 1H and 13C NMR Spectra.
Author Contributions
Conceptualization, Y.M., R.S., T.S., K.Y. and K.N.; funding acquisition, K.N.; investigation, Y.M., R.S., K.I., N.N., M.O., M.T. and T.H.; writing—original draft, Y.M., R.S., M.O. and K.N. All authors have read and agreed to the published version of the manuscript.
Funding
Grants-in-aid for Scientific Research 22K19101 (KN).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bouillon, R.; Okamura, W.H.; Norman, A.W. Structure-Function Relationships in the Vitamin D Endocrine System. Endocr. Rev. 1995, 16, 200–257. [Google Scholar] [PubMed]
- Lieben, L.; Masuyama, R.; Torrekens, S.; Looveren, R.V.; Schrooten, J.; Baatsen, P.; Lafage-Proust, M.; Dresselaers, T.; Feng, J.Q.; Bonewald, L.F.; et al. Normocalcemia is maintained in mice under conditions of calcium malabsorption by vitamin D–induced inhibition of bone mineralization. J. Clin. Investig. 2012, 122, 1803–1815. [Google Scholar] [CrossRef]
- Kato, S.; Sekine, K.; Matsumoto, T.; Yoshizawa, T. Molecular genetics of vitamin D receptor acting in bone. J. Bone Miner. Metab. 1998, 16, 65–71. [Google Scholar] [CrossRef]
- Abe, E.; Miyaura, C.; Sakagami, H.; Takada, M.; Konno, K.; Yamazaki, T.; Yoshiki, S.; Suda, T. Differentiation of mouse myeloid leukemia cells induced by 1α,25-dihydroxyvitamin D3. Proc. Natl. Acad. Sci. USA 1981, 78, 4990–4994. [Google Scholar] [CrossRef]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef]
- Sakaki, T.; Sawada, N.; Nonaka, Y.; Ohyama, Y.; Inouye, K. Metabolic studies using recombinant Escherichia coli cells producing rat mitochondrial CYP24. CYP24 can convert 1α,25-dihydroxyvitamin D3 to calcitroic acid. Eur. J. Biol. Chem. 1999, 262, 43–48. [Google Scholar] [CrossRef]
- Sakaki, T.; Sawada, N.; Komai, K.; Shiozawa, S.; Yamada, S.; Yamamoto, K.; Ohyama, Y.; Inouye, K. Dual metabolic pathway of 25-hydroxyvitamin D3 catalyzed by human CYP24. Eur. J. Biochem. 2000, 267, 6158–6165. [Google Scholar] [CrossRef]
- Yamada, S.; Nakayama, K.; Takayama, H.; Shinki, T.; Takasaki, Y.; Suda, T. Isolation, identification, and metabolism of (23S,25R)-25-hydroxyvitamin D3 26,23-lactol. A biosynthetic precursor of (23S,25R)-25-hydroxyvitamin D3 26,23-lactone. J. Biol. Chem. 1984, 259, 884–889. [Google Scholar] [CrossRef]
- Ishizuka, S.; Norman, A.W. Metabolic pathways from 1α,25-dihydroxyvitamin D3 to 1α,25-dihydroxyvitamin D3-26,23-lactone. Stereo-retained and stereo-selective lactonization. J. Biol. Chem. 1987, 262, 7165–7170. [Google Scholar] [CrossRef]
- Yu, O.B.; Arnold, L.A. Calcitroic Acid—A Review. ACS Chem. Biol. 2016, 11, 2665–2672. [Google Scholar] [CrossRef]
- Kaufmann, M.; Martineau, C.; Arabian, A.; Traynor, M.; St-Arnaud, R.; Jones, G. Calcioic acid: In vivo detection and quantification of the terminal C24-oxidation product of 25-hydroxyvitamin D3 and related intermediates in serum of mice treated with 24,25-dihydroxyvitamin D3. J. Steroid Biochem. Mol. Biol. 2019, 188, 23–28. [Google Scholar] [CrossRef]
- Wang, Z.; Lin, Y.S.; Zheng, X.E.; Senn, T.; Hashizume, T.; Scian, M.; Dickmann, L.J.; Nelson, S.D.; Baillie, T.A.; Hebert, M.F.; et al. An Inducible Cytochrome P450 3A4-Dependent Vitamin D Catabolic Pathway. Mol. Pharmacol. 2012, 81, 498–509. [Google Scholar] [CrossRef]
- Hanessian, S.; Ugolini, D.; Dubé, D.; Glamyan, A. Facile access to (S)-1,2,4-butanetriol and its derivatives. Can. J. Chem. 1984, 62, 2146–2147. [Google Scholar] [CrossRef]
- Gogoi, P.; Sigüeiro, R.; Eduardo, S.; Mouriño, A. An Expeditious Route to 1α,25-Dihydroxyvitamin D3 and Its Analogues by an Aqueous Tandem Palladium-Catalyzed A-Ring Closure and Suzuki Coupling to the C/D Unit. Chem. Eur. J. 2010, 16, 1432–1435. [Google Scholar] [CrossRef]
- Urushino, N.; Yasuda, K.; Ikushiro, S.; Kamakura, M.; Ohta, M.; Sakaki, T. Metabolism of 1α,25-dihydroxyvitamin D2 by human CYP24A1. Biochem. Biophys. Res. Commun. 2009, 384, 144–148. [Google Scholar] [CrossRef]
- Kawagoe, F.; Mototani, S.; Yasuda, K.; Mano, H.; Sakaki, T.; Kittaka, A. Stereoselective Synthesis of 24-Fluoro-25-Hydroxyvitamin D3 Analogues and Their Stability to hCYP24A1-Dependent Catabolism. Int. J. Mol. Sci. 2021, 22, 11863. [Google Scholar] [CrossRef]
- Sakamoto, R.; Nagata, A.; Ohshita, H.; Mizumoto, Y.; Iwaki, M.; Yasuda, K.; Sakaki, T.; Nagasawa, K. Chemical Synthesis of Side-Chain-Hydroxylated Vitamin D3 Derivatives and Their Metabolism by CYP27B1. ChemBioChem 2021, 22, 2896–2900. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Halgren, T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Rhieu, S.Y.; Annalora, A.J.; Gathungu, R.M.; Vouros, P.; Uskokovic, M.R.; Schuster, I.; Palmore, G.T.R.; Reddy, G.S. A new insight into the role of rat cytochrome P450 24A1 in metabolism of selective analogs of 1α,25-dihydroxyvitamin D3. Arch. Biochem. Biophys. 2011, 509, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Dumas, J.; Villa, M. New strategies for the synthesis of vitamin D metabolites via palladium-catalyzed reactions. J. Am. Chem. Soc. 1992, 114, 9836–9845. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).