
Using a Quantitative High-Throughput Screening Platform to Identify Molecular Targets and Compounds as Repurposing Candidates for Endometriosis
 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Kits
2.2. Cell Lines and Culture
2.3. Compound Libraries
2.4. High-Throughput Screening (HTS)
2.5. Quality Control (QC) Compound Screen
2.6. Compound Library Screen
2.7. High-Content Imaging (HCI)
2.8. Selection of Compound Hits
2.9. In Silico Analysis of Compound Hits
2.10. Data and Statistical Analysis
3. Results
3.1. Establishing the HTS Quality Control Parameters for Estrogen-Stimulated Cell Survival
3.2. Demonstrating HTS Assay Robustness
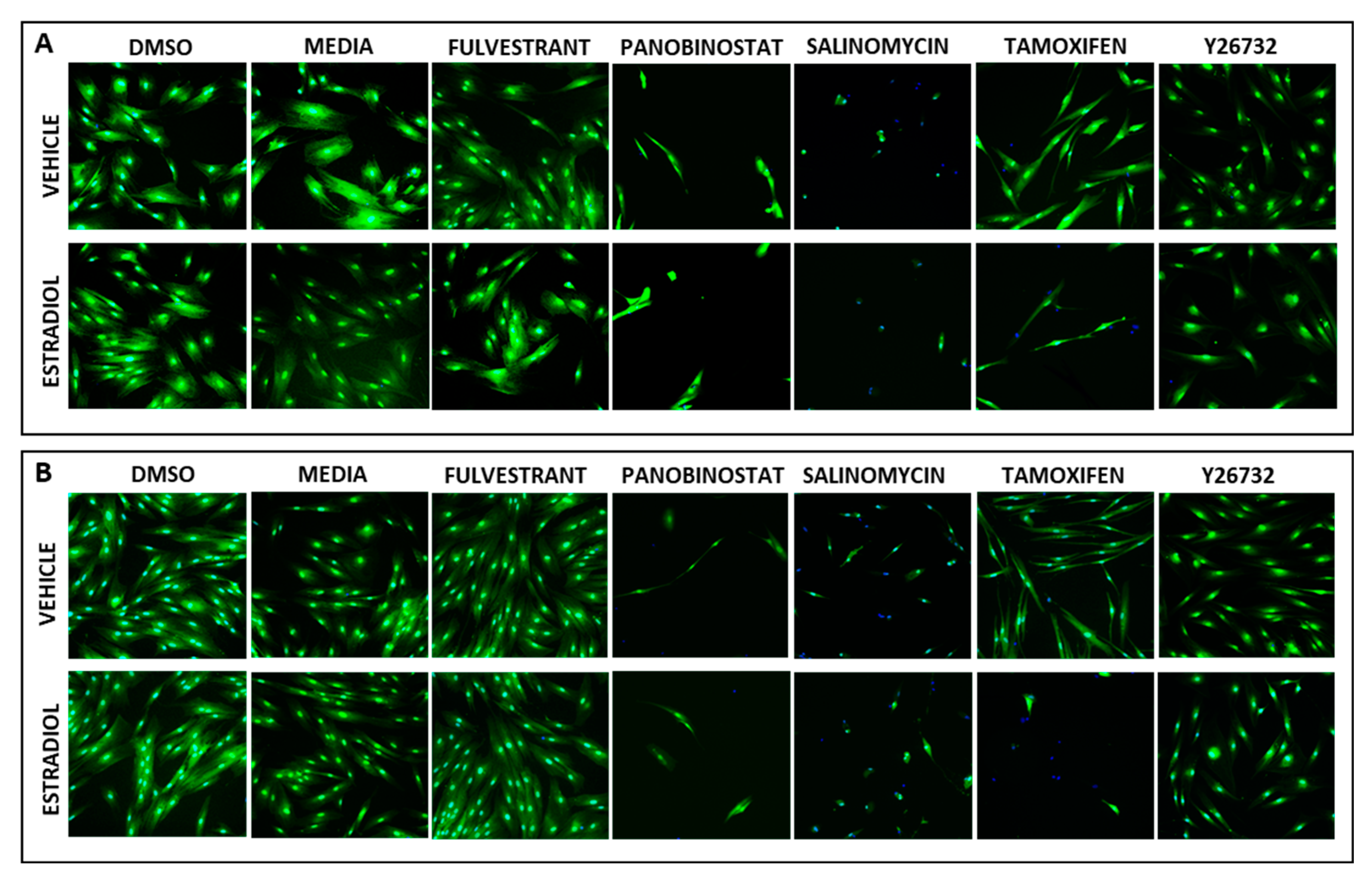
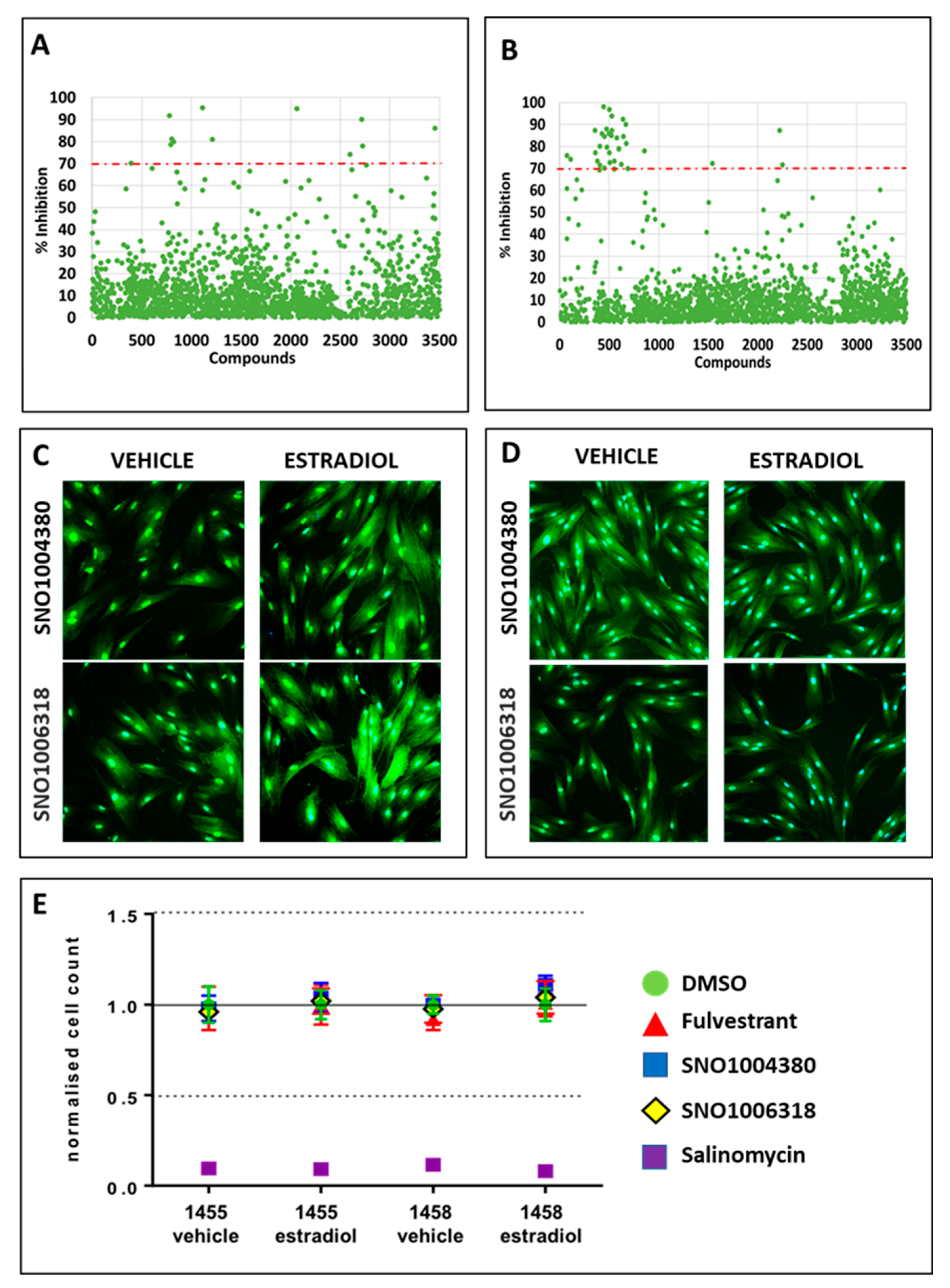
3.3. Identification of Compounds That Specifically Inhibited Estrogen-Stimulated Cell Survival
3.4. Identification of Lead Compounds Using Quantitative HTS (qHTS)
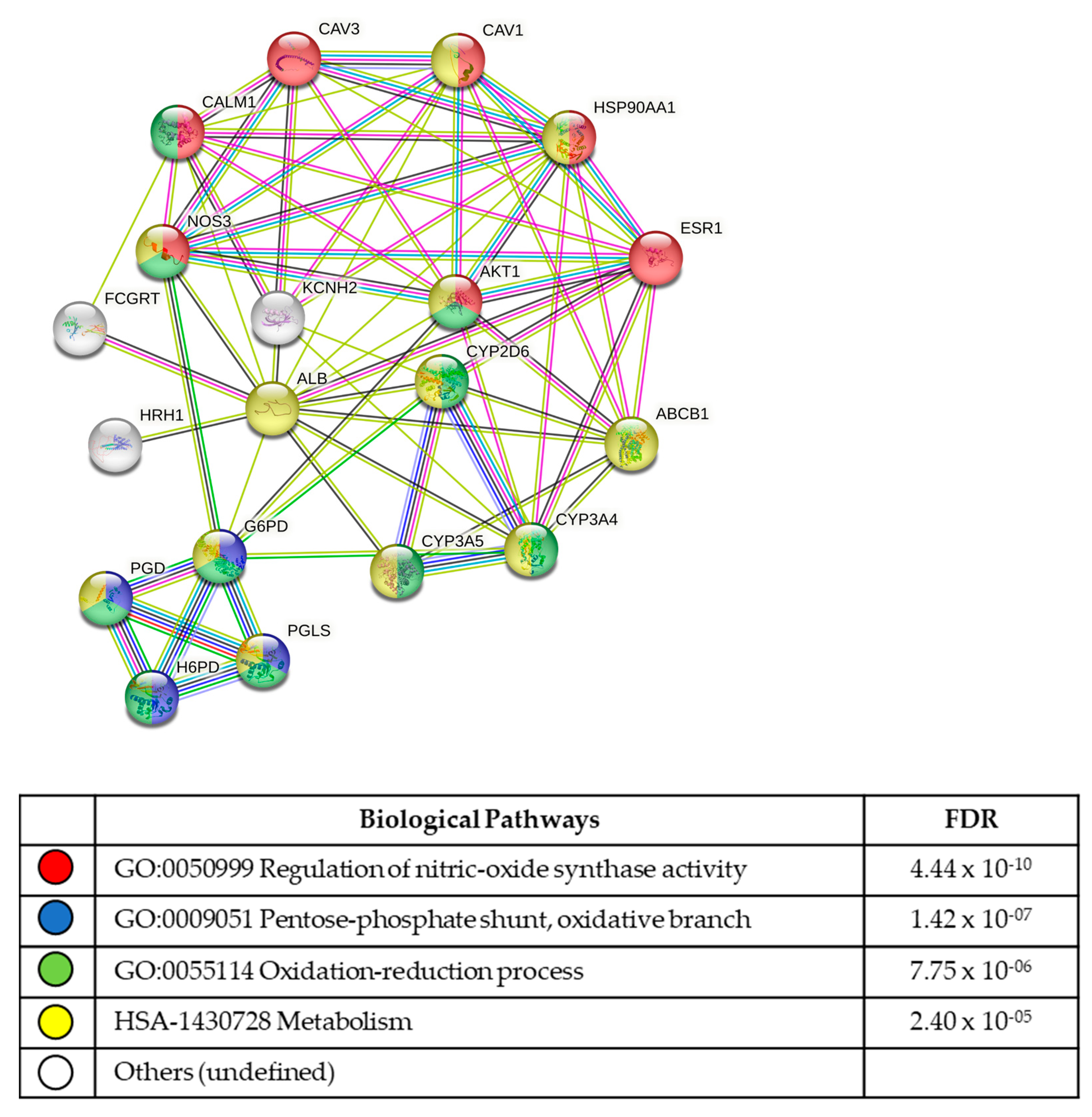
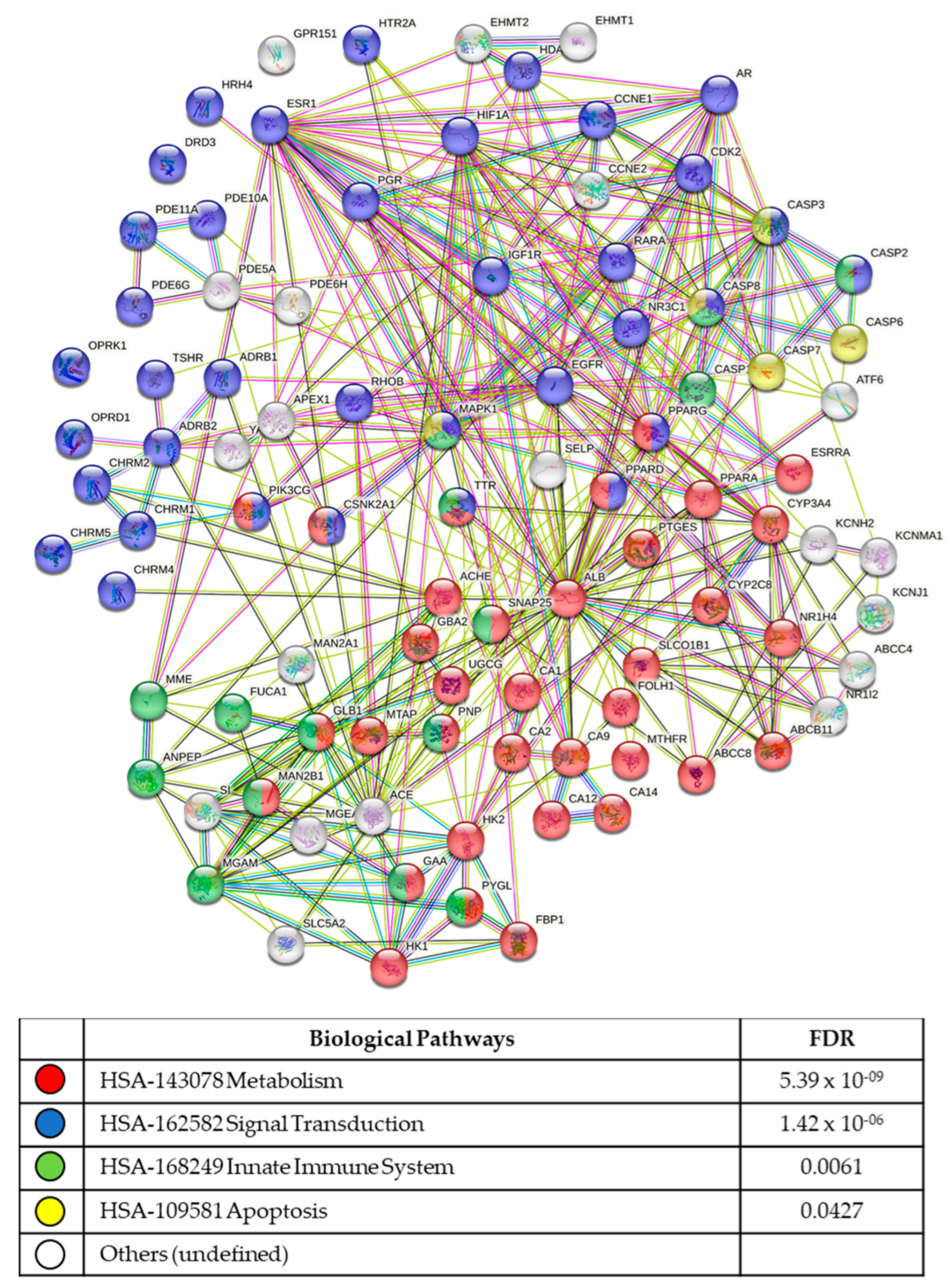
3.5. In Silico Analysis of Compound Molecular Targets and Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Burney, R.O.; Giudice, L.C. Pathogenesis and pathophysiology of endometriosis. Fertil. Steril. 2012, 98, 511–519. [Google Scholar] [CrossRef]
- AIHW. Endometriosis in Australia: Prevalence and Hospitalisations. Available online: https://www.aihw.gov.au/reports/chronic-disease/endometriosis-prevalence-and-hospitalisations/contents/table-of-contents. (accessed on 29 August 2019).
- Konrad, L.; Dietze, R.; Kudipudi, P.K.; Horne, F.; Meinhold-Heerlein, I. Endometriosis in MRKH cases as a proof for the coelomic metaplasia hypothesis? Reproduction 2019, 158, R41–R47. [Google Scholar] [CrossRef]
- Rei, C.; Williams, T.; Feloney, M. Endometriosis in a Man as a Rare Source of Abdominal Pain: A Case Report and Review of the Literature. Case Rep. Obstet. Gynecol. 2018, 2018, 2083121. [Google Scholar] [CrossRef]
- Zamecnik, M.; Hostakova, D. Endometriosis in a mesothelial cyst of tunica vaginalis of the testis. Report of a case. Ceskoslov. Patol. 2013, 49, 134–136. [Google Scholar]
- Bulun, S.E.; Zeitoun, K.; Takayama, K.; Noble, L.; Michael, D.; Simpson, E.; Johns, A.; Putman, M.; Sasano, H. Estrogen production in endometriosis and use of aromatase inhibitors to treat endometriosis. Endocr. Relat. Cancer 1999, 6, 293–301. [Google Scholar] [CrossRef]
- Kitawaki, J.; Noguchi, T.; Amatsu, T.; Maeda, K.; Tsukamoto, K.; Yamamoto, T.; Fushiki, S.; Osawa, Y.; Honjo, H. Expression of aromatase cytochrome P450 protein and messenger ribonucleic acid in human endometriotic and adenomyotic tissues but not in normal endometrium. Biol. Reprod. 1997, 57, 514–519. [Google Scholar] [CrossRef]
- Guo, S.W. Recurrence of endometriosis and its control. Hum. Reprod. Update 2009, 15, 441–461. [Google Scholar] [CrossRef]
- Schliep, K.C.; Mumford, S.L.; Peterson, C.M.; Chen, Z.; Johnstone, E.B.; Sharp, H.T.; Stanford, J.B.; Hammoud, A.O.; Sun, L.; Buck Louis, G.M. Pain typology and incident endometriosis. Hum. Reprod. 2015, 30, 2427–2438. [Google Scholar] [CrossRef]
- Zondervan, K.T.; Becker, C.M.; Koga, K.; Missmer, S.A.; Taylor, R.N.; Vigano, P. Endometriosis. Nat. Rev. Dis. Prim. 2018, 4, 9. [Google Scholar] [CrossRef]
- Giudice, L.C. Clinical practice. Endometriosis. N. Engl. J. Med. 2010, 362, 2389–2398. [Google Scholar] [CrossRef]
- Enmark, E.; Gustafsson, J.A. Estrogen receptors—An overview. J. Intern. Med. 1999, 246, 133–138. [Google Scholar] [CrossRef]
- Bjornstrom, L.; Sjoberg, M. Mechanisms of estrogen receptor signaling: Convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. 2005, 19, 833–842. [Google Scholar] [CrossRef]
- Titolo, D.; Mayer, C.M.; Dhillon, S.S.; Cai, F.; Belsham, D.D. Estrogen facilitates both phosphatidylinositol 3-kinase/Akt and ERK1/2 mitogen-activated protein kinase membrane signaling required for long-term neuropeptide Y transcriptional regulation in clonal, immortalized neurons. J. Neurosci. 2008, 28, 6473–6482. [Google Scholar] [CrossRef]
- Kelly, M.J.; Levin, E.R. Rapid actions of plasma membrane estrogen receptors. Trends Endocrinol. Metab. 2001, 12, 152–156. [Google Scholar] [CrossRef]
- Vasudevan, N.; Pfaff, D.W. Membrane-initiated actions of estrogens in neuroendocrinology: Emerging principles. Endocr. Rev. 2007, 28, 1–19. [Google Scholar] [CrossRef]
- Maggiolini, M.; Vivacqua, A.; Fasanella, G.; Recchia, A.G.; Sisci, D.; Pezzi, V.; Montanaro, D.; Musti, A.M.; Picard, D.; Ando, S. The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17beta-estradiol and phytoestrogens in breast cancer cells. J. Biol. Chem. 2004, 279, 27008–27016. [Google Scholar] [CrossRef]
- Edwards, D.P. Regulation of signal transduction pathways by estrogen and progesterone. Annu. Rev. Physiol. 2005, 67, 335–376. [Google Scholar] [CrossRef]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Hernandez, J.J.; Pryszlak, M.; Smith, L.; Yanchus, C.; Kurji, N.; Shahani, V.M.; Molinski, S.V. Giving Drugs a Second Chance: Overcoming Regulatory and Financial Hurdles in Repurposing Approved Drugs as Cancer Therapeutics. Front. Oncol. 2017, 7, 273. [Google Scholar] [CrossRef]
- Fox, S.; Farr-Jones, S.; Sopchak, L.; Boggs, A.; Nicely, H.W.; Khoury, R.; Biros, M. High-throughput screening: Update on practices and success. J. Biomol. Screen. 2006, 11, 864–869. [Google Scholar] [CrossRef] [PubMed]
- Badr, C.E.; Wurdinger, T.; Tannous, B.A. Functional drug screening assay reveals potential glioma therapeutics. Assay Drug Dev. Technol. 2011, 9, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.P.; Tesdorpf, U.J. Phenotypic Drug Discovery with High Content Screening; Perkin Elmer: Waltham, MA, USA, 2015. [Google Scholar]
- Holdsworth-Carson, S.J.; Colgrave, E.M.; Donoghue, J.F.; Fung, J.N.; Churchill, M.L.; Mortlock, S.; Paiva, P.; Healey, M.; Montgomery, G.W.; Girling, J.E.; et al. Generation of immortalized human endometrial stromal cell lines with different endometriosis risk genotypes. Mol. Hum. Reprod. 2019, 25, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Saunders, D.N.; Falkenberg, K.J.; Simpson, K.J. High-throughput approaches to measuring cell death. Cold Spring Harb. Protoc. 2014, 591–601. [Google Scholar] [CrossRef]
- Dragiev, P.; Nadon, R.; Makarenkov, V. Two effective methods for correcting experimental high-throughput screening data. Bioinformatics 2012, 28, 1775–1782. [Google Scholar] [CrossRef][Green Version]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Mitani, M.; Yamanishi, T.; Miyazaki, Y.; Otake, N. Salinomycin effects on mitochondrial ion translocation and respiration. Antimicrob. Agents Chemother. 1976, 9, 655–660. [Google Scholar] [CrossRef]
- Iversen, P.W.; Beck, B.; Chen, Y.F.; Dere, W.; Devanarayan, V.; Eastwood, B.J.; Farmen, M.W.; Iturria, S.J.; Montrose, C.; Moore, R.A.; et al. HTS Assay Validation. In Assay Guidance Manual; Sittampalam, G.S., Grossman, A., Brimacombe, K., Arkin, M., Auld, D., Austin, C., Baell, J., Bejcek, B., Caaveiro, J.M.M., Chung, T.D.Y., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Sittampalam, G.S.; Kahl, S.D.; Janzen, W.P. High-throughput screening: Advances in assay technologies. Curr. Opin. Chem. Biol. 1997, 1, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Kuohung, W.; Burnett, M.; Mukhtyar, D.; Schuman, E.; Ni, J.; Crowley, W.F.; Glicksman, M.A.; Kaiser, U.B. A high-throughput small-molecule ligand screen targeted to agonists and antagonists of the G-protein-coupled receptor GPR54. J. Biomol. Screen. 2010, 15, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Baurle, S.; Nagel, J.; Peters, O.; Brauer, N.; Ter Laak, A.; Preusse, C.; Rottmann, A.; Heldmann, D.; Bothe, U.; Blume, T.; et al. Identification of a Benzimidazolecarboxylic Acid Derivative (BAY 1316957) as a Potent and Selective Human Prostaglandin E2 Receptor Subtype 4 (hEP4-R) Antagonist for the Treatment of Endometriosis. J. Med. Chem. 2019, 62, 2541–2563. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yang, Y.; Li, W.; Tian, X.; Cui, H.; Zhang, Q. Identification of small-molecule ligands that bind to MiR-21 as potential therapeutics for endometriosis by screening ZINC database and in-vitro assays. Gene 2018, 662, 46–53. [Google Scholar] [CrossRef]
- Rekker, K.; Saare, M.; Eriste, E.; Tasa, T.; Kukuskina, V.; Roost, A.M.; Anderson, K.; Samuel, K.; Karro, H.; Salumets, A.; et al. High-throughput mRNA sequencing of stromal cells from endometriomas and endometrium. Reproduction 2017, 154, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Gu, C.; Ye, M.; Zhang, Z.; Li, L.; Fan, W.; Meng, Y. Integration analysis of microRNA and mRNA paired expression profiling identifies deregulated microRNA-transcription factor-gene regulatory networks in ovarian endometriosis. Reprod. Biol. Endocrinol. 2018, 16, 4. [Google Scholar] [CrossRef]
- An, W.F.; Tolliday, N. Cell-based assays for high-throughput screening. Mol. Biotechnol. 2010, 45, 180–186. [Google Scholar] [CrossRef]
- Ren, C.E.; Zhu, X.; Li, J.; Lyle, C.; Dowdy, S.; Podratz, K.C.; Byck, D.; Chen, H.B.; Jiang, S.W. Microarray analysis on gene regulation by estrogen, progesterone and tamoxifen in human endometrial stromal cells. Int. J. Mol. Sci. 2015, 16, 5864–5885. [Google Scholar] [CrossRef]
- Huhtinen, K.; Desai, R.; Stahle, M.; Salminen, A.; Handelsman, D.J.; Perheentupa, A.; Poutanen, M. Endometrial and endometriotic concentrations of estrone and estradiol are determined by local metabolism rather than circulating levels. J. Clin. Endocrinol. Metab. 2012, 97, 4228–4235. [Google Scholar] [CrossRef]
- Malutan, A.M.; Drugan, T.; Costin, N.; Ciortea, R.; Bucuri, C.; Rada, M.P.; Mihu, D. Pro-inflammatory cytokines for evaluation of inflammatory status in endometriosis. Cent. Eur. J. Immunol. 2015, 40, 96–102. [Google Scholar] [CrossRef]
- Novella-Maestre, E.; Carda, C.; Ruiz-Sauri, A.; Garcia-Velasco, J.A.; Simon, C.; Pellicer, A. Identification and quantification of dopamine receptor 2 in human eutopic and ectopic endometrium: A novel molecular target for endometriosis therapy. Biol. Reprod. 2010, 83, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Cordeaux, Y.; Pasupathy, D.; Bacon, J.; Charnock-Jones, D.S.; Smith, G.C. Characterization of serotonin receptors in pregnant human myometrium. J. Pharmacol. Exp. Ther. 2009, 328, 682–691. [Google Scholar] [CrossRef] [PubMed]
- Willets, J.M.; Taylor, A.H.; Shaw, H.; Konje, J.C.; Challiss, R.A. Selective regulation of H1 histamine receptor signalling by G protein-coupled receptor kinase 2 in uterine smooth muscle cells. Mol. Endocrinol. 2008, 22, 1893–1907. [Google Scholar] [CrossRef] [PubMed]
- Heltianu, C.; Simionescu, M.; Simionescu, N. Histamine receptors of the microvascular endothelium revealed in situ with a histamine-ferritin conjugate: Characteristic high-affinity binding sites in venules. J. Cell Biol. 1982, 93, 357–364. [Google Scholar] [CrossRef]
- Dey, S.K.; Villanueva, C.; Abdou, N.I. Histamine receptors on rabbit blastocyst and endometrial cell membranes. Nature 1979, 278, 648–649. [Google Scholar] [CrossRef] [PubMed]
- Bodis, J.; Tinneberg, H.R.; Schwarz, H.; Papenfuss, F.; Torok, A.; Hanf, V. The effect of histamine on progesterone and estradiol secretion of human granulosa cells in serum-free culture. Gynecol. Endocrinol. 1993, 7, 235–239. [Google Scholar] [CrossRef]
- Cocchiara, R.; Albeggiani, G.; Di Trapani, G.; Azzolina, A.; Lampiasi, N.; Rizzo, F.; Diotallevi, L.; Gianaroli, L.; Geraci, D. Estradiol enhances in vitro the histamine release induced by embryonic histamine-releasing factor (EHRF) from uterine mast cells. Hum. Reprod. 1992, 7, 1036–1041. [Google Scholar] [CrossRef][Green Version]
- Zaitsu, M.; Narita, S.; Lambert, K.C.; Grady, J.J.; Estes, D.M.; Curran, E.M.; Brooks, E.G.; Watson, C.S.; Goldblum, R.M.; Midoro-Horiuti, T. Estradiol activates mast cells via a non-genomic estrogen receptor-alpha and calcium influx. Mol. Immunol. 2007, 44, 1977–1985. [Google Scholar] [CrossRef]
- Zierau, O.; Zenclussen, A.C.; Jensen, F. Role of female sex hormones, estradiol and progesterone, in mast cell behavior. Front. Immunol. 2012, 3, 169. [Google Scholar] [CrossRef]
- Needham, K.; Fadia, M.; Dahlstrom, J.E.; Harrington, K.; Shadbolt, B.; Robson, S.J. Significance of mast cell distribution in placental tissue and membranes in spontaneous preterm birth. J. Inflamm. Res. 2016, 9, 141–145. [Google Scholar] [CrossRef]
- Cote, F.; Fligny, C.; Bayard, E.; Launay, J.M.; Gershon, M.D.; Mallet, J.; Vodjdani, G. Maternal serotonin is crucial for murine embryonic development. Proc. Natl. Acad. Sci. USA 2007, 104, 329–334. [Google Scholar] [CrossRef]
- Hayashi, T.; Sumi, D.; Matsui-Hirai, H.; Fukatsu, A.; Arockia Rani, P.J.; Kano, H.; Tsunekawa, T.; Iguchi, A. Sarpogrelate HCl, a selective 5-HT2A antagonist, retards the progression of atherosclerosis through a novel mechanism. Atherosclerosis 2003, 168, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, C.; Bodelsson, G.; Bodelsson, M.; Stjernquist, M. 5-Hydroxytryptamine contracts human uterine artery smooth muscle predominantly via 5-HT2 receptors. Hum. Reprod. 1997, 12, 361–367. [Google Scholar] [CrossRef] [PubMed][Green Version]
- John Jayakumar, J.A.K.; Panicker, M.M.; Basu, B. Serotonin 2A (5-HT(2A)) receptor affects cell-matrix adhesion and the formation and maintenance of stress fibers in HEK293 cells. Sci. Rep. 2020, 10, 21675. [Google Scholar] [CrossRef] [PubMed]
- Duerschmied, D.; Suidan, G.L.; Demers, M.; Herr, N.; Carbo, C.; Brill, A.; Cifuni, S.M.; Mauler, M.; Cicko, S.; Bader, M.; et al. Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood 2013, 121, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Courteix, C.; Dupuis, A.; Martin, P.Y.; Sion, B. 5-HT2A Receptors and Pain. In 5-HT2A Receptors in the Central Nervous System; The Receptors; Guiard, B., Di Giovanni, G., Eds.; Humana Press: Cham, Switzerland, 2018; Volume 32, pp. 339–352. [Google Scholar] [CrossRef]
- Pierce, S.R.; Fang, Z.; Yin, Y.; West, L.; Asher, M.; Hao, T.; Zhang, X.; Tucker, K.; Staley, A.; Fan, Y.; et al. Targeting dopamine receptor D2 as a novel therapeutic strategy in endometrial cancer. J. Exp. Clin. Cancer Res. 2021, 40, 61. [Google Scholar] [CrossRef]
- Ishizawa, J.; Kojima, K.; Chachad, D.; Ruvolo, P.; Ruvolo, V.; Jacamo, R.O.; Borthakur, G.; Mu, H.; Zeng, Z.; Tabe, Y.; et al. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Sci. Signal. 2016, 9, ra17. [Google Scholar] [CrossRef]
- Chi, A.S.; Tarapore, R.S.; Hall, M.D.; Shonka, N.; Gardner, S.; Umemura, Y.; Sumrall, A.; Khatib, Z.; Mueller, S.; Kline, C.; et al. Pediatric and adult H3 K27M-mutant diffuse midline glioma treated with the selective DRD2 antagonist ONC201. J. Neurooncol. 2019, 145, 97–105. [Google Scholar] [CrossRef]
- Bilibio, J.P.; Matte, U.; de Conto, E.; Genro, V.K.; Souza, C.A.; Cunha-Filho, J.S. Dopamine receptor D2 genotype (3438) is associated with moderate/severe endometriosis in infertile women in Brazil. Fertil. Steril. 2013, 99, 1340–1345. [Google Scholar] [CrossRef]
- Delgado-Rosas, F.; Gomez, R.; Ferrero, H.; Gaytan, F.; Garcia-Velasco, J.; Simon, C.; Pellicer, A. The effects of ergot and non-ergot-derived dopamine agonists in an experimental mouse model of endometriosis. Reproduction 2011, 142, 745–755. [Google Scholar] [CrossRef]
- Novella-Maestre, E.; Carda, C.; Noguera, I.; Ruiz-Sauri, A.; Garcia-Velasco, J.A.; Simon, C.; Pellicer, A. Dopamine agonist administration causes a reduction in endometrial implants through modulation of angiogenesis in experimentally induced endometriosis. Hum. Reprod. 2009, 24, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Berga, S.L.; Meng, Q.; Xia, M.; Kohout, T.A.; van Duin, M.; Taylor, R.N. Cabergoline Stimulates Human Endometrial Stromal Cell Decidualization and Reverses Effects of Interleukin-1beta In Vitro. J. Clin. Endocrinol. Metab. 2021, 106, 3591–3604. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K. Cell cycle regulatory control for uterine stromal cell decidualization in implantation. Reproduction 2009, 137, 889–899. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
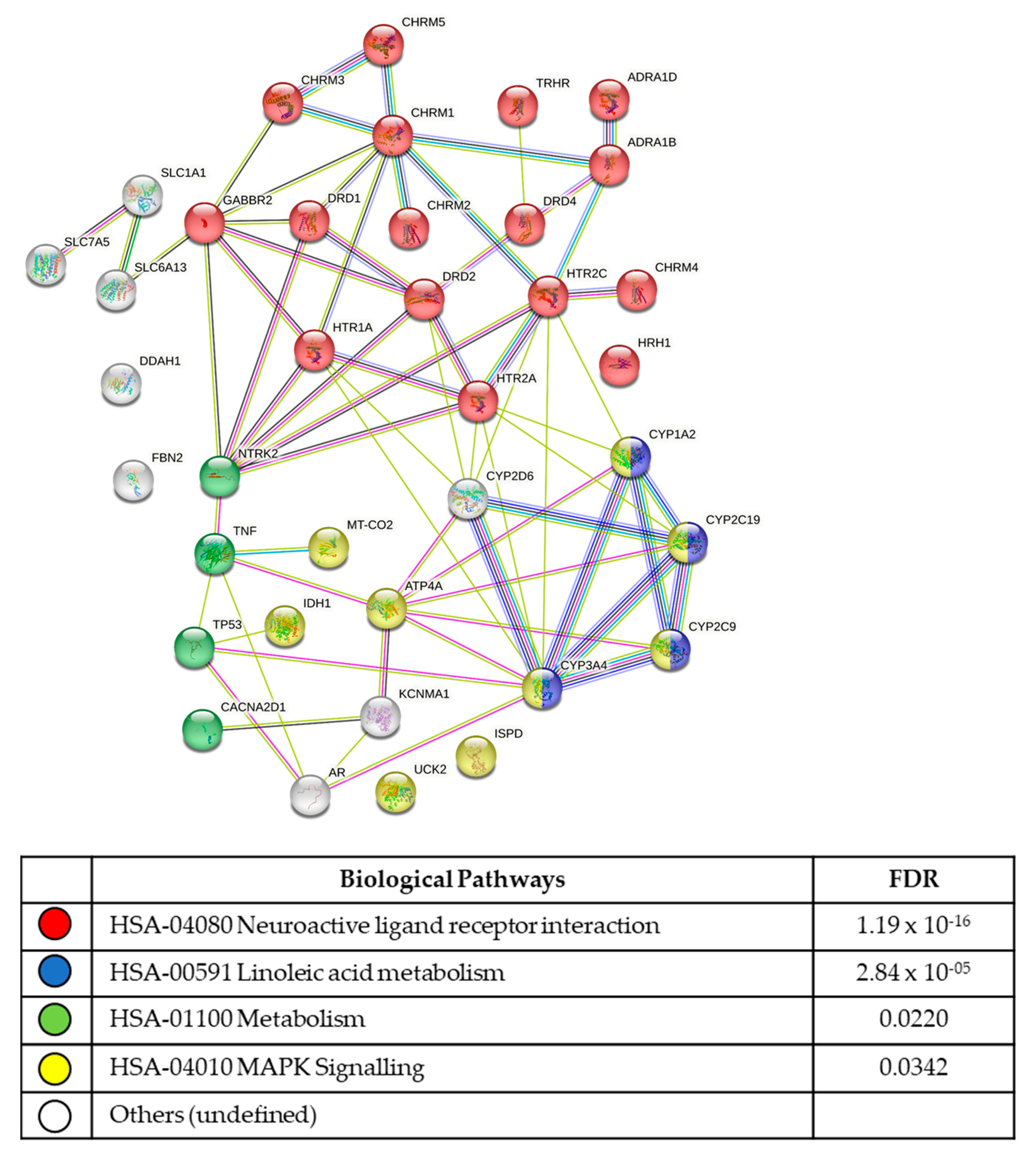{kind=link}
| Compound Name | Generic Name | Compound Group | IC50 [µM] 1455 | IC50 [µM] 1458 |
|---|---|---|---|---|
| SN01006330 | 7,8-Dimethoxyflavone | anti-inflammatory | 0.08 | 0.19 |
| SN01005561 | Aminothiazole | anti-microbial | 0.12 | 2.08 |
| SN01005320 | Benazepril HCl | anti-hypertensive | 9.37 | 0.06 |
| SN01005071 | Ceftibuten | anti-microbial | 2.29 | 0.07 |
| SN00852779 | Chlordiazepoxide | anti-depressant | 0.02 | 0.08 |
| SN01005451 | Chloroquinalol | anti-microbial | >20 | 0.08 |
| SN01006117 | Cytidine triphosphate disodium | anti-inflammatory | 0.02 | 0.08 |
| SN01005419 | Editol | anti-inflammatory | ND | 0.87 |
| SN01004366 | Hydroxyzine Pamoate | anti-histamine | 0.02 | ND |
| SN01004587 | Indoprofen | anti-inflammatory | 0.02 | 0.001 |
| SN01004583 | Ketotifen Fumarate | anti-histamine | 0.22 | ND |
| SN01005061 | Pantoprazole | proton pump inhibitor | 0.19 | 0.37 |
| SN01005391 | Pregabalin | anti-inflammatory | 0.08 | 0.08 |
| SN01004486 | Promazine HCl | anti-psychotic | 0.02 | 0.15 |
| SN01005316 | Repaglinide | anti-diabetic | >20 | 0.07 |
| SN01005445 | Sildenafil Citrate | anti-inflammatory | 1.88 | 0.96 |
| SN01004511 | Spectinomycin HCl | anti-microbial | 0.02 | ND |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Churchill, M.L.; Holdsworth-Carson, S.J.; Cowley, K.J.; Luu, J.; Simpson, K.J.; Healey, M.; Rogers, P.A.W.; Donoghue, J.F. Using a Quantitative High-Throughput Screening Platform to Identify Molecular Targets and Compounds as Repurposing Candidates for Endometriosis. Biomolecules 2023, 13, 965. https://doi.org/10.3390/biom13060965
Churchill ML, Holdsworth-Carson SJ, Cowley KJ, Luu J, Simpson KJ, Healey M, Rogers PAW, Donoghue JF. Using a Quantitative High-Throughput Screening Platform to Identify Molecular Targets and Compounds as Repurposing Candidates for Endometriosis. Biomolecules. 2023; 13(6):965. https://doi.org/10.3390/biom13060965
Chicago/Turabian StyleChurchill, Molly L., Sarah J. Holdsworth-Carson, Karla J. Cowley, Jennii Luu, Kaylene J. Simpson, Martin Healey, Peter A. W. Rogers, and J. F. Donoghue. 2023. "Using a Quantitative High-Throughput Screening Platform to Identify Molecular Targets and Compounds as Repurposing Candidates for Endometriosis" Biomolecules 13, no. 6: 965. https://doi.org/10.3390/biom13060965
APA StyleChurchill, M. L., Holdsworth-Carson, S. J., Cowley, K. J., Luu, J., Simpson, K. J., Healey, M., Rogers, P. A. W., & Donoghue, J. F. (2023). Using a Quantitative High-Throughput Screening Platform to Identify Molecular Targets and Compounds as Repurposing Candidates for Endometriosis. Biomolecules, 13(6), 965. https://doi.org/10.3390/biom13060965