
In Silico Identification of Potential Inhibitors of the SARS-CoV-2 Main Protease among a PubChem Database of Avian Infectious Bronchitis Virus 3CLPro Inhibitors


Abstract
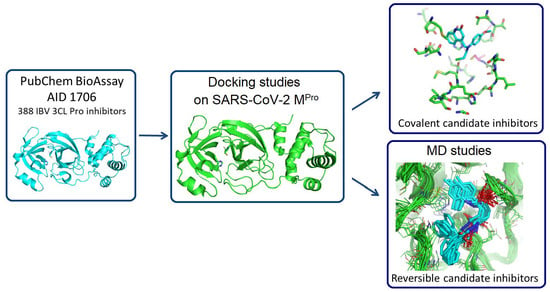
1. Introduction
2. Materials and Methods
2.1. Homology Studies
2.2. Docking Studies
2.2.1. Docking Studies for Covalent Inhibition
2.2.2. Docking Studies for Non-Covalent Inhibition
2.2.3. Molecular Dynamics Studies
3. Results and Discussion
3.1. Homology Studies
3.2. Docking Studies of the 388 Active Compounds from the PubChem BioAssay AID 1706 within the SARS-CoV-2 Protease Active Site
3.2.1. Docking Studies Looking for Covalent Inhibitors
3.2.2. Docking Studies Looking for Non-Covalent Inhibitors
3.3. Molecular Dynamics Studies (Non-Covalent Inhibitors)
3.4. ADMET Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pillaiyar, T.; Meenakshisundaram, S.; Manickam, M. Recent discovery and development of inhibitors targeting coronaviruses. Drug Discov. Today 2020, 25, 668–688. [Google Scholar] [CrossRef]
- Menéndez, J.C. Approaches to the Potential Therapy of COVID-19: A General Overview from the Medicinal Chemistry Perspective. Molecules 2022, 27, 658. [Google Scholar] [CrossRef]
- Castillo-Garit, J.A.; Cañizares-Carmenate, Y.; Pham-The, H.; Pérez-Doñate, V.; Torrens, F.; Pérez-Giménez, F. A Review of Computational Approaches Targeting SARS-CoV-2 Main Protease to the Discovery of New Potential Antiviral Compounds. Curr. Top. Med. Chem. 2023, 23, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Suryavanshi, H.; Chaudhari, R.D.; Patil, V.; Majumdar, S.; Debnath, S.; Biswas, G. Design, synthesis and docking study of Vortioxetine derivatives as a SARS-CoV-2 main protease inhibitor. DARU J. Pharm. Sci. 2022, 30, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Mslati, H.; Gentile, F.; Perez, C.; Cherkasov, A. Comprehensive Consensus Analysis of SARS-CoV-2 Drug Repurposing Campaigns. J. Chem. Inf. Model. 2021, 61, 3771–3788. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Zhang, S.; Amahong, K.; Sun, X.; Lian, X.; Liu, J.; Sun, H.; Lou, Y.; Zhu, F.; Qiu, Y. The miRNA: A small but powerful RNA for COVID-19. Brief. Bioinform. 2021, 22, 1137–1149. [Google Scholar] [CrossRef]
- Zhang, S.; Amahong, K.; Zhang, C.; Li, F.; Gao, J.; Qiu, Y.; Zhu, F. RNA–RNA interactions between SARS-CoV-2 and host benefit viral development and evolution during COVID-19 infection. Brief. Bioinform. 2022, 23, bbab397. [Google Scholar] [CrossRef]
- Gimeno, A.; Mestres-Truyol, J.; Ojeda-Montes, M.J.; Macip, G.; Saldivar-Espinoza, B.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallvé, S. Prediction of Novel Inhibitors of the Main Protease (M-pro) of SARS-CoV-2 through Consensus Docking and Drug Reposition. Int. J. Mol. Sci. 2020, 21, 3793. [Google Scholar] [CrossRef]
- Yamamoto, K.Z.; Yasuo, N.; Sekijima, M. Screening for Inhibitors of Main Protease in SARS-CoV-2: In Silico and In Vitro Approach Avoiding Peptidyl Secondary Amides. J. Chem. Inf. Model 2022, 62, 350–358. [Google Scholar] [CrossRef]
- Mengist, H.M.; Dilnessa, T.; Jin, T. Structural Basis of Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Front. Chem. 2021, 9, 622898. [Google Scholar] [CrossRef] [PubMed]
- Citarella, A.; Scala, A.; Piperno, A.; Micale, N. SARS-CoV-2 Mpro: A Potential Target for Peptidomimetics and Small-Molecule Inhibitors. Biomolecules 2021, 11, 607. [Google Scholar] [CrossRef] [PubMed]
- Agost-Beltrán, L.; de la Hoz-Rodríguez, S.; Bou-Iserte, L.; Rodríguez, S.; Fernández-de-la-Pradilla, A.; González, F.V. Advances in the Development of SARS-CoV-2 Mpro Inhibitors. Molecules 2022, 27, 2523. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulou, I.; Sapountzaki, E.; Rova, U.; Christakopoulos, P. Inhibition of the main protease of SARS-CoV-2 (M(pro)) by repurposing/designing drug-like substances and utilizing nature’s toolbox of bioactive compounds. Comput. Struct. Biotechnol. J. 2022, 20, 1306–1344. [Google Scholar] [CrossRef]
- Liu, S.; Zheng, Q.; Wang, Z. Potential covalent drugs targeting the main protease of the SARS-CoV-2 coronavirus. Bioinformatics 2020, 36, 3295–3298. [Google Scholar] [CrossRef]
- Soulère, L.; Barbier, T.; Queneau, Y. Docking-based virtual screening studies aiming at the covalent inhibition of SARS-CoV-2 M(Pro) by targeting the cysteine 145. Comput. Biol. Chem. 2021, 92, 107463. [Google Scholar] [CrossRef]
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020, 39, e2000028. [Google Scholar] [CrossRef]
- Wang, J. Fast Identification of Possible Drug Treatment of Coronavirus Disease-19 (COVID-19) through Computational Drug Repurposing Study. J. Chem. Inf. Model. 2020, 60, 3277–3286. [Google Scholar] [CrossRef]
- Mishra, B.; Ballaney, P.; Saha, G.; Shinde, A.; Banerjee, S.; Thimmakondu, V.S.; Aduri, R. An in silico discovery of potential 3CL protease inhibitors of SARS-CoV-2 based upon inactivation of the cysteine 145-Histidine 41 catalytic dyad. J. Biomol. Struct. Dyn. 2022, 41, 3167–3186. [Google Scholar] [CrossRef]
- Awoonor-Williams, E.; Abu-Saleh, A.A.A. Covalent and non-covalent binding free energy calculations for peptidomimetic inhibitors of SARS-CoV-2 main protease. Phys. Chem. 2021, 23, 6746–6757. [Google Scholar] [CrossRef]
- Xie, X.Q. Exploiting PubChem for Virtual Screening. Expert Opin. Drug Discov. 2010, 5, 1205–1220. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Zhou, Z.; Han, L.; Karapetyan, K.; Dracheva, S.; Shoemaker, B.A.; et al. PubChem’s BioAssay Database. Nucleic Acids Res. 2012, 40, D400–D412. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, S.; Nath, P.; Chatterjee, M.; Das, N.; Kalita, D.; Roy, P.; Satapathi, S. Repurposing therapeutics for COVID-19: Rapid prediction of commercially available drugs through machine learning and docking. PLoS ONE 2020, 15, e0241543. [Google Scholar] [CrossRef]
- Alves, V.M.; Bobrowski, T.; Melo-Filho, C.C.; Korn, D.; Auerbach, S.; Schmitt, C.; Muratov, E.N.; Tropsha, A. QSAR Modeling of SARS-CoV M(pro) Inhibitors Identifies Sufugolix, Cenicriviroc, Proglumetacin, and other Drugs as Candidates for Repurposing against SARS-CoV-2. Mol. Inform. 2021, 40, e2000113. [Google Scholar] [CrossRef] [PubMed]
- Delijewski, M.; Haneczok, J. AI drug discovery screening for COVID-19 reveals zafirlukast as a repurposing candidate. Med. Drug Discov. 2021, 9, 100077. [Google Scholar] [CrossRef] [PubMed]
- Nand, M.; Maiti, P.; Joshi, T.; Chandra, S.; Pande, V.; Kuniyal, J.C.; Ramakrishnan, M.A. Virtual screening of anti-HIV1 compounds against SARS-CoV-2: Machine learning modeling, chemoinformatics and molecular dynamics simulation based analysis. Sci. Rep. 2020, 10, 20397. [Google Scholar] [CrossRef] [PubMed]
- Santana, M.V.S.; Silva-Jr, F.P. De novo design and bioactivity prediction of SARS-CoV-2 main protease inhibitors using recurrent neural network-based transfer learning. BMC Chem. 2021, 15, 8. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Xue, X.; Yu, H.; Yang, H.; Xue, F.; Wu, Z.; Shen, W.; Li, J.; Zhou, Z.; Ding, Y.; Zhao, Q.; et al. Structures of two coronavirus main proteases: Implications for substrate binding and antiviral drug design. J. Virol. 2008, 82, 2515–2527. [Google Scholar] [CrossRef]
- Dalton, S.E.; Campos, S. Covalent Small Molecules as Enabling Platforms for Drug Discovery. Chembiochem 2020, 21, 1080–1100. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Samanta, I.; Mondal, A.; Liu, W.R. Covalent Inhibition in Drug Discovery. Chemmedchem 2019, 14, 889–906. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Thompson, M.A. ArgusLaB 4.0.1; WA planetaria Software LLC: Seattle, WA, USA, 2004. [Google Scholar]
- Oda, A.; Okayasu, M.; Kamiyama, Y.; Yoshida, T.; Takahashi, O.; Matsuzaki, H. Evaluation of docking accuracy and investigations of roles of parameters and each term in scoring functions for protein–ligand docking using ArgusLab software. Bull. Chem. Soc. Jpn. 2007, 80, 1920–1925. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Case, D.A.; Darden, T.A.; Cheatham, T.E.; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Crowley, M.; Walker, R.C.; Zhang, W. Amber 10; University of California: San Francisco, CA, USA, 2008. [Google Scholar]
- Gerber, P.R.; Müller, K. MAB, a generally applicable molecular force field for structure modelling in medicinal chemistry. J. Comput. Aided Mol. Des. 1995, 9, 251–268. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R. An extended Hückel theory. I. hydrocarbons. J. Chem. Phys. 1963, 39, 1397–1412. [Google Scholar] [CrossRef]
- Sturgeon, J.B.; Laird, B.B. Symplectic algorithm for constant-pressure molecular dynamics using a Nosé–Poincaré thermostat. J. Chem. Phys. 2000, 112, 3474–3482. [Google Scholar] [CrossRef]
- Khelfaoui, H.; Harkati, D.; Saleh, B.A. Molecular docking, molecular dynamics simulations and reactivity, studies on approved drugs library targeting ACE2 and SARS-CoV-2 binding with ACE2. J. Biomol. Struct. Dyn. 2021, 39, 7246–7262. [Google Scholar] [CrossRef]
- Kurkcuoglu, Z.; Koukos, P.I.; Citro, N.; Trellet, M.E.; Rodrigues, J.; Moreira, I.S.; Roel-Touris, J.; Melquiond, A.S.J.; Geng, C.; Schaarschmidt, J.; et al. Performance of HADDOCK and a simple contact-based protein-ligand binding affinity predictor in the D3R Grand Challenge 2. J. Comput. Aided Mol. Des. 2018, 32, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Vangone, A.; Schaarschmidt, J.; Koukos, P.; Geng, C.; Citro, N.; Trellet, M.E.; Xue, L.C.; Bonvin, A. Large-scale prediction of binding affinity in protein-small ligand complexes: The PRODIGY-LIG web server. Bioinformatics 2019, 35, 1585–1587. [Google Scholar] [CrossRef]
- Douangamath, A.; Fearon, D.; Gehrtz, P.; Krojer, T.; Lukacik, P.; Owen, C.D.; Resnick, E.; Strain-Damerell, C.; Aimon, A.; Ábrányi-Balogh, P.; et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 2020, 11, 5047. [Google Scholar] [CrossRef]
- Grygorenko, O.O.; Volochnyuk, D.M.; Vashchenko, B.V. Emerging Building Blocks for Medicinal Chemistry: Recent Synthetic Advances. Eur. J. Org. Chem. 2021, 2021, 6478–6510. [Google Scholar] [CrossRef]
- Yoshino, R.; Yasuo, N.; Sekijima, M. Identification of key interactions between SARS-CoV-2 main protease and inhibitor drug candidates. Sci. Rep. 2020, 10, 12493. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions, and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions, or products referred to in the content. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound CID— H-Bonds and Interactions with Amino Acids | Binding Modes |
|---|---|
 |  |
 |  |
 |  |
 |  |
| Compound CID— H-Bonds and Interactions with Amino Acids | Binding Modes and Docking Score (kcal/mol) |
|---|---|
 |  |
 |  |
 |  |
 |  |
 |  |
| Compounds CID | Ligand RMSD (Å) | Residues Interacting via H-Bonds during the MD Simulations |
|---|---|---|
| 1632360 | 1.602 | Asn142, Gly143, Glu166 |
| 645492 | 1.087 | Met49, Ser144, Gln189 |
| 2193552 | 1.851 | His41, Met49, Asn51, Glu166, Asp187, Thr190, Gln192, Gln189 |
| 4586109 | 3.344 | Gly143, Ser144, His163, Glu166, Gln189, |
| 654498 | 1.717 | Met49, Glu166, Gln189, |
| Compounds CID | 1632360 | 645492 | 2193552 | 4586109 | 654498 |
|---|---|---|---|---|---|
| ΔGBinding t = 0 | −9.16 (Gly143) | −9.21 (Gln189, Met49, Cys145) | −10.23 (Glu166, Thr190, Gln192) | −9.42 (Gly143, Ser144, His163, Glu166) | −8.52 (Met49, Gln189) |
| ΔGBinding t = 1000 | −9.34 (Gly143, Glu166) | −9.24 (Gln189) | −9.95 (His41, Glu166, Asp187, Gln189, Thr190, Gln192) | −9.91 (Gly143, Ser144, His163, Gln189) | −9.08 (Met49, Glu166, Gln189) |
| ΔGBinding t = 2000 | −9.47 (Gly143) | −9.08 (Gln189) | −10.28 (His41, Glu166, Asp187, Gln189, Thr190, Gln192) | −9.89 (Gly143, Ser144, His163, Gln189) | −9.17 (Cys145) |
| ΔGBinding t = 3000 | −9.37 (Asn142) | −9.28 (Met49) | −10.33 (His41, Glu166, Gln189, Thr190, Gln192) | −10.20 (Gly143, Ser144, His163, Gln189) | −8.70 (Glu166, Gln189) |
| ΔGBinding t = 4000 | −9.16 (Cys145) | −9.09 (Cys145) | −10.63 (Met49 Glu166, Thr190, Gln192) | −9.79 (Gly143 Ser144, Cys145, His163) | −9.05 (Met49, Glu166, Gln189) |
| ΔGBinding t = 5000 | −9.12 (Glu166) | −9.22 (Ser144) | −10.52 (Met49, Asn51, Glu166, Thr190, Gln192) | −9.82 (Gly143 Ser144, His163, Gln189) | −9.36 (Glu166, Gln189) |
| Property | Unit | CID 843322 | CID 1154427 | CID 4868361 | CID 4961646 | CID 1632360 | CID 645492 | CID 2193552 | CID 4586109 | CID 654498 |
|---|---|---|---|---|---|---|---|---|---|---|
| Molecular weight | g/mol | 255.10 | 356.80 | 303.74 | 292.72 | 429.51 | 499.58 | 476.52 | 474.59 | 485.56 |
| LogP | 2.55 | 2.92 | 2.32 | 2.27 | 3.70 | 3.60 | 3.92 | 4.89 | 3.22 | |
| H-bond donors | 1 | 1 | 2 | 0 | 3 | 1 | 3 | 1 | 1 | |
| H-bond acceptors | 2 | 3 | 2 | 4 | 3 | 5 | 6 | 4 | 5 | |
| Rotatable bonds | 3 | 6 | 7 | 4 | 11 | 11 | 11 | 10 | 11 | |
| PSA | Å2 | 52.89 | 62.40 | 61.44 | 58.95 | 87.30 | 121.50 | 113.96 | 124.04 | 121.50 |
| Caco2 permeability | log Papp in 10–6 cm/s | 1.324 | 1.133 | 1.307 | 1.315 | 0.793 | 0.739 | 0.626 | 1.04 | 0.83 |
| Intestinal absorption (human) | % Absorbed | 91.937 | 96.64 | 91.279 | 96.937 | 93.663 | 94.393 | 70.344 | 93.421 | 93.919 |
| VDss (human) | log L/kg | 0.011 | −0.076 | −0.021 | −0.21 | −0.24 | 0.166 | −1.842 | −0.714 | 0.117 |
| Fraction unbound (human) | Fu | 0.319 | 0.038 | 0 | 0.34 | 0.024 | 0.086 | 0 | 0.278 | 0.072 |
| BBB permeability | log BB | 0.079 | −0.077 | 0.244 | 0.122 | −0.662 | −0.429 | −1.359 | −0.996 | −0.435 |
| CNS permeability | log PS | −2.781 | −2.35 | −2.088 | −2.877 | −2.191 | −2.239 | −2.896 | −2.257 | −2.308 |
| CYP2D6 inhibitor | Yes/No | No | No | No | No | No | No | No | No | No |
| CYP3A4 inhibitor | Yes/No | No | No | No | No | Yes | Yes | No | No | Yes |
| Total Clearance | log ml/min/kg | −0.054 | 0.086 | −0.069 | 0.189 | 0.241 | 0.035 | −0.045 | 0.624 | 0.091 |
| Hepatotoxicity | Yes/No (probability %) | No (61) | No (71) | No (54) | Yes (51) | No (59) | No (58) | No (75) | No (57) | No (58) |
| Carcinogenicity | Yes/No (probability %) | No (59) | Yes (52) | Yes (66) | Yes (66) | Yes (56) | Yes (52) | No (69) | No (53) | Yes (52) |
| Immunotoxicity | Yes/No (probability %) | No (99) | Yes (87) | No (99) | No (98) | No (99) | No (99) | No (99) | No (99) | No (99) |
| Mutagenicity | Yes/No (probability %) | Yes (55) | Yes (52) | No (55) | No (57) | No (52) | Yes (55) | No (79) | No (65) | Yes (56) |
| Cytotoxicity | Yes/No (probability %) | No (76) | Yes (57) | No (72) | No (68) | No (77) | No (61) | No (60) | No (66) | No (63) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soulère, L.; Barbier, T.; Queneau, Y. In Silico Identification of Potential Inhibitors of the SARS-CoV-2 Main Protease among a PubChem Database of Avian Infectious Bronchitis Virus 3CLPro Inhibitors. Biomolecules 2023, 13, 956. https://doi.org/10.3390/biom13060956
Soulère L, Barbier T, Queneau Y. In Silico Identification of Potential Inhibitors of the SARS-CoV-2 Main Protease among a PubChem Database of Avian Infectious Bronchitis Virus 3CLPro Inhibitors. Biomolecules. 2023; 13(6):956. https://doi.org/10.3390/biom13060956
Chicago/Turabian StyleSoulère, Laurent, Thibaut Barbier, and Yves Queneau. 2023. "In Silico Identification of Potential Inhibitors of the SARS-CoV-2 Main Protease among a PubChem Database of Avian Infectious Bronchitis Virus 3CLPro Inhibitors" Biomolecules 13, no. 6: 956. https://doi.org/10.3390/biom13060956
APA StyleSoulère, L., Barbier, T., & Queneau, Y. (2023). In Silico Identification of Potential Inhibitors of the SARS-CoV-2 Main Protease among a PubChem Database of Avian Infectious Bronchitis Virus 3CLPro Inhibitors. Biomolecules, 13(6), 956. https://doi.org/10.3390/biom13060956