




Reactivity of Coproheme Decarboxylase with Monovinyl, Monopropionate Deuteroheme



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
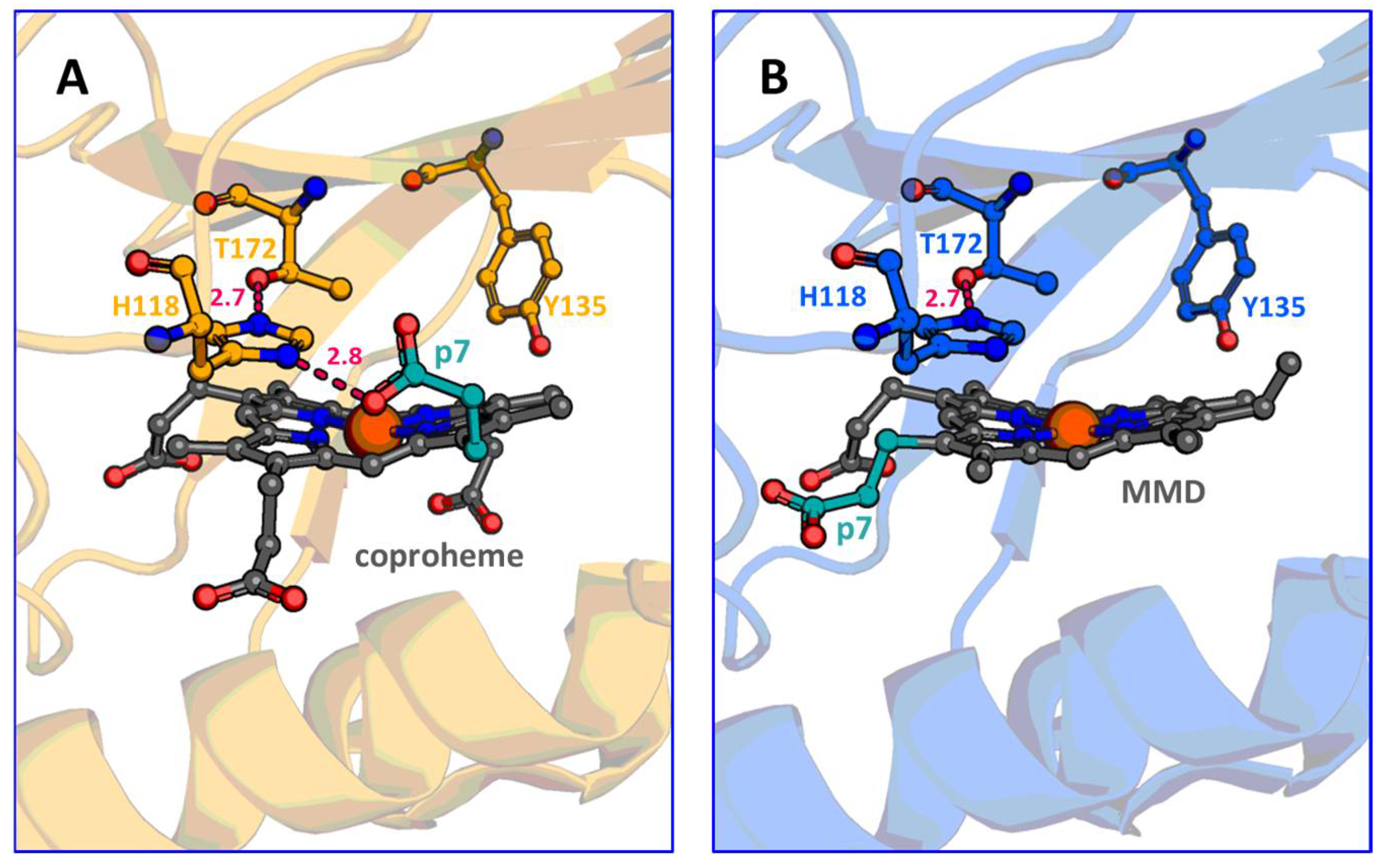{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Expression and Purification of CdChdC Wild-Type and Variants
2.2. Coproheme Decarboxylase Activity
2.3. Production and Purification of Reaction Intermediate MMD
2.4. Stopped-Flow Spectroscopy
3. Results
3.1. MMD Production and Purification
3.2. Reactivity of MMD in CdChdC Wild-Type and the Y135A Variant
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Layer, G. Heme biosynthesis in prokaryotes. Biochim Biophys Acta Mol. Cell Res. 2021, 1868, 118861. [Google Scholar] [CrossRef]
- Dailey, H.A.; Medlock, A.E. A primer on heme biosynthesis. Biol. Chem. 2022, 403, 985–1003. [Google Scholar] [CrossRef]
- Dailey, H.A.; Gerdes, S.; Dailey, T.A.; Burch, J.S.; Phillips, J.D. Noncanonical coproporphyrin-dependent bacterial heme biosynthesis pathway that does not use protoporphyrin. Proc. Natl. Acad. Sci. USA 2015, 112, 2210–2215. [Google Scholar] [CrossRef]
- Dailey, H.A.; Dailey, T.A.; Gerdes, S.; Jahn, D.; Jahn, M.; O’Brian, M.R.; Warren, M.J. Prokaryotic Heme Biosynthesis: Multiple Pathways to a Common Essential Product. Microbiol. Mol. Biol. Rev. 2017, 81, e00048-16. [Google Scholar] [CrossRef]
- Pfanzagl, V.; Holcik, L.; Maresch, D.; Gorgone, G.; Michlits, H.; Furtmüller, P.G.; Hofbauer, S. Coproheme decarboxylases—Phylogenetic prediction versus biochemical experiments. Arch. Biochem. Biophys. 2018, 640, 27–36. [Google Scholar] [CrossRef]
- Celis, A.I.; Streit, B.R.; Moraski, G.C.; Kant, R.; Lash, T.D.; Lukat-Rodgers, G.S.; Rodgers, K.R.; DuBois, J.L. Unusual Peroxide-Dependent, Heme-Transforming Reaction Catalyzed by HemQ. Biochemistry 2015, 54, 4022–4032. [Google Scholar] [CrossRef]
- Hofbauer, S.; Mlynek, G.; Milazzo, L.; Pühringer, D.; Maresch, D.; Schaffner, I.; Furtmüller, P.G.; Smulevich, G.; Djinović-Carugo, K.; Obinger, C. Hydrogen peroxide-mediated conversion of coproheme to heme b by HemQ-lessons from the first crystal structure and kinetic studies. FEBS J. 2016, 283, 4386–4401. [Google Scholar] [CrossRef]
- Milazzo, L.; Gabler, T.; Pühringer, D.; Jandova, Z.; Maresch, D.; Michlits, H.; Pfanzagl, V.; Djinović-Carugo, K.; Oostenbrink, C.; Furtmüller, P.G.; et al. Redox Cofactor Rotates during Its Stepwise Decarboxylation: Molecular Mechanism of Conversion of Coproheme to Heme. ACS Catal. 2019, 9, 6766–6782. [Google Scholar] [CrossRef]
- Michlits, H.; Lier, B.; Pfanzagl, V.; Djinović-Carugo, K.; Furtmüller, P.G.; Oostenbrink, C.; Obinger, C.; Hofbauer, S. Actinobacterial coproheme decarboxylases use histidine as distal base to promote Compound I formation. ACS Catal. 2020, 10, 5405–5413. [Google Scholar] [CrossRef]
- Streit, B.R.; Celis, A.I.; Moraski, G.C.; Shisler, K.A.; Shepard, E.M.; Rodgers, K.R.; Lukat-Rodgers, G.S.; DuBois, J.L. Decarboxylation involving a ferryl, propionate, and a tyrosyl group in a radical relay yields heme. J. Biol. Chem. 2018, 293, 3989–3999. [Google Scholar] [CrossRef]
- Celis, A.I.; Gauss, G.H.; Streit, B.R.; Shisler, K.; Moraski, G.C.; Rodgers, K.R.; Lukat-Rodgers, G.S.; Peters, J.W.; DuBois, J.L. Structure-Based Mechanism for Oxidative Decarboxylation Reactions Mediated by Amino Acids and Heme Propionates in Coproheme Decarboxylase (HemQ). J. Am. Chem. Soc. 2017, 139, 1900–1911. [Google Scholar] [CrossRef]
- Liu, W.; Pang, Y.; Song, Y.; Li, X.; Tan, H.; Chen, G. Reorienting Mechanism of Harderoheme in Coproheme Decarboxylase-A Computational Study. Int. J. Mol. Sci. 2022, 23, 2564. [Google Scholar] [CrossRef]
- Sebastiani, F.; Michlits, H.; Lier, B.; Becucci, M.; Furtmüller, P.G.; Oostenbrink, C.; Obinger, C.; Hofbauer, S.; Smulevich, G. Reaction intermediate rotation during the decarboxylation of coproheme to heme b in C. diphtheriae. Biophys. J. 2021, 120, 3600–3614. [Google Scholar] [CrossRef] [PubMed]
- Milazzo, L.; Hofbauer, S.; Howes, B.D.; Gabler, T.; Furtmüller, P.G.; Obinger, C.; Smulevich, G. Insights into the Active Site of Coproheme Decarboxylase from Listeria monocytogenes. Biochemistry 2018, 57, 2044–2057. [Google Scholar] [CrossRef] [PubMed]
- Milazzo, L.; Gabler, T.; Pfanzagl, V.; Michlits, H.; Furtmüller, P.G.; Obinger, C.; Hofbauer, S.; Smulevich, G. The hydrogen bonding network of coproheme in coproheme decarboxylase from Listeria monocytogenes: Effect on structure and catalysis. J. Inorg. Biochem. 2019, 195, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, F.; Baroni, C.; Patil, G.; Dali, A.; Becucci, M.; Hofbauer, S.; Smulevich, G. The Role of the Hydrogen Bond Network in Maintaining Heme Pocket Stability and Protein Function Specificity of C. diphtheriae Coproheme Decarboxylase. Biomolecules 2023, 13, 235. [Google Scholar] [CrossRef]
- Hofbauer, S.; Howes, B.D.; Flego, N.; Pirker, K.F.; Schaffner, I.; Mlynek, G.; Djinović-Carugo, K.; Furtmüller, P.G.; Smulevich, G.; Obinger, C. From chlorite dismutase towards HemQ—The role of the proximal H-bonding network in heme binding. Biosci. Rep. 2016, 36, e00312. [Google Scholar] [CrossRef]
- Sebastiani, F.; Risorti, R.; Niccoli, C.; Michlits, H.; Becucci, M.; Hofbauer, S.; Smulevich, G. An active site at work—The role of key residues in C. diphteriae coproheme decarboxylase. J. Inorg. Biochem. 2022, 229, 111718. [Google Scholar] [CrossRef]
- Sebastiani, F.; Niccoli, C.; Michlits, H.; Risorti, R.; Becucci, M.; Hofbauer, S.; Smulevich, G. Spectroscopic evidence of the effect of hydrogen peroxide excess on the coproheme decarboxylase from actinobacterial Corynebacterium diphtheriae. J. Raman Spectrosc. 2022, 53, 890–901. [Google Scholar] [CrossRef]
- Streit, B.R.; Celis, A.I.; Shisler, K.; Rodgers, K.R.; Lukat-Rodgers, G.S.; DuBois, J.L. Reactions of Ferrous Coproheme Decarboxylase (HemQ) with O2 and H2O2 Yield Ferric Heme b. Biochemistry 2017, 56, 189–201. [Google Scholar] [CrossRef]
- Hofbauer, S.; Dalla Sega, M.; Scheiblbrandner, S.; Jandova, Z.; Schaffner, I.; Mlynek, G.; Djinović-Carugo, K.; Battistuzzi, G.; Furtmüller, P.G.; Oostenbrink, C.; et al. Chemistry and Molecular Dynamics Simulations of Heme b-HemQ and Coproheme-HemQ. Biochemistry 2016, 55, 5398–5412. [Google Scholar] [CrossRef] [PubMed]
- Celis, A.I.; Choby, J.E.; Kentro, J.; Skaar, E.P.; DuBois, J.L. Control of Metabolite Flux during the Final Steps of Heme b Biosynthesis in Gram-Positive Bacteria. Biochemistry 2019, 58, 5259–5270. [Google Scholar] [CrossRef]
- Hofbauer, S.; Hagmüller, A.; Schaffner, I.; Mlynek, G.; Krutzler, M.; Stadlmayr, G.; Pirker, K.F.; Obinger, C.; Daims, H.; Djinović-Carugo, K.; et al. Structure and heme-binding properties of HemQ (chlorite dismutase-like protein) from Listeria monocytogenes. Arch. Biochem. Biophys. 2015, 574, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Hofbauer, S.; Pfanzagl, V.; Michlits, H.; Schmidt, D.; Obinger, C.; Furtmüller, P.G. Understanding molecular enzymology of porphyrin-binding alpha + beta barrel proteins—One fold, multiple functions. Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140536. [Google Scholar] [CrossRef] [PubMed]
- Michlits, H.; Valente, N.; Mlynek, G.; Hofbauer, S. Initial Steps to Engineer Coproheme Decarboxylase to Obtain Stereospecific Monovinyl, Monopropionyl Deuterohemes. Front. Bioeng. Biotechnol. 2022, 9, 807678. [Google Scholar] [CrossRef] [PubMed]
- Teale, F.W. Cleavage of the haem-protein link by acid methylethylketone. Biochim. Biophys. Acta 1959, 35, 543. [Google Scholar] [CrossRef]
- Furtmüller, P.G.; Burner, U.; Jantschko, W.; Regelsberger, G.; Obinger, C. The reactivity of myeloperoxidase compound I formed with hypochlorous acid. Redox Rep. 2000, 5, 173–178. [Google Scholar] [CrossRef]
- Gasselhuber, B.; Carpena, X.; Graf, M.M.H.; Pirker, K.F.; Nicolussi, A.; Sündermann, A.; Hofbauer, S.; Zamocky, M.; Furtmüller, P.G.; Jakopitsch, C.; et al. Eukaryotic Catalase-Peroxidase: The Role of the Trp-Tyr-Met Adduct in Protein Stability, Substrate Accessibility, and Catalysis of Hydrogen Peroxide Dismutation. Biochemistry 2015, 54, 5425–5438. [Google Scholar] [CrossRef]
- Pfanzagl, V.; Nys, K.; Bellei, M.; Michlits, H.; Mlynek, G.; Battistuzzi, G.; Djinović-Carugo, K.; Van Doorslaer, S.; Furtmüller, P.G.; Hofbauer, S.; et al. Roles of distal aspartate and arginine of B-class dye-decolorizing peroxidase in heterolytic hydrogen peroxide cleavage. J. Biol. Chem. 2018, 293, 14823–14838. [Google Scholar] [CrossRef]
- Marzocchi, M.; Smulevich, G. Relationship between heme vinyl conformation and the protein matrix in peroxidases. J. Raman Spectrosc. 2003, 34, 725–736. [Google Scholar] [CrossRef]
- Sugiyama, K.; Highet, R.J.; Woods, A.; Cotter, R.J.; Osawa, Y. Hydrogen peroxide-mediated alteration of the heme prosthetic group of metmyoglobin to an iron chlorin product: Evidence for a novel oxidative pathway. Proc. Natl. Acad. Sci. USA 1997, 94, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Poulos, T.L.; Kraut, J. The stereochemistry of peroxidase catalysis. J. Biol. Chem. 1980, 255, 8199–8205. [Google Scholar] [CrossRef] [PubMed]
- Poulos, T.L.; Freer, S.T.; Alden, R.A.; Edwards, S.L.; Skogland, U.; Takio, K.; Eriksson, B.; Xuong, N.; Yonetani, T.; Kraut, J. The crystal structure of cytochrome c peroxidase. J. Biol. Chem. 1980, 255, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Poulos, T.L. Heme enzyme structure and function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, G.; Michlits, H.; Furtmüller, P.G.; Hofbauer, S. Reactivity of Coproheme Decarboxylase with Monovinyl, Monopropionate Deuteroheme. Biomolecules 2023, 13, 946. https://doi.org/10.3390/biom13060946
Patil G, Michlits H, Furtmüller PG, Hofbauer S. Reactivity of Coproheme Decarboxylase with Monovinyl, Monopropionate Deuteroheme. Biomolecules. 2023; 13(6):946. https://doi.org/10.3390/biom13060946
Chicago/Turabian StylePatil, Gaurav, Hanna Michlits, Paul G. Furtmüller, and Stefan Hofbauer. 2023. "Reactivity of Coproheme Decarboxylase with Monovinyl, Monopropionate Deuteroheme" Biomolecules 13, no. 6: 946. https://doi.org/10.3390/biom13060946
APA StylePatil, G., Michlits, H., Furtmüller, P. G., & Hofbauer, S. (2023). Reactivity of Coproheme Decarboxylase with Monovinyl, Monopropionate Deuteroheme. Biomolecules, 13(6), 946. https://doi.org/10.3390/biom13060946