
Pharmacological Inhibition of Gasdermin D Suppresses Angiotensin II-Induced Experimental Abdominal Aortic Aneurysms

 , and
, and 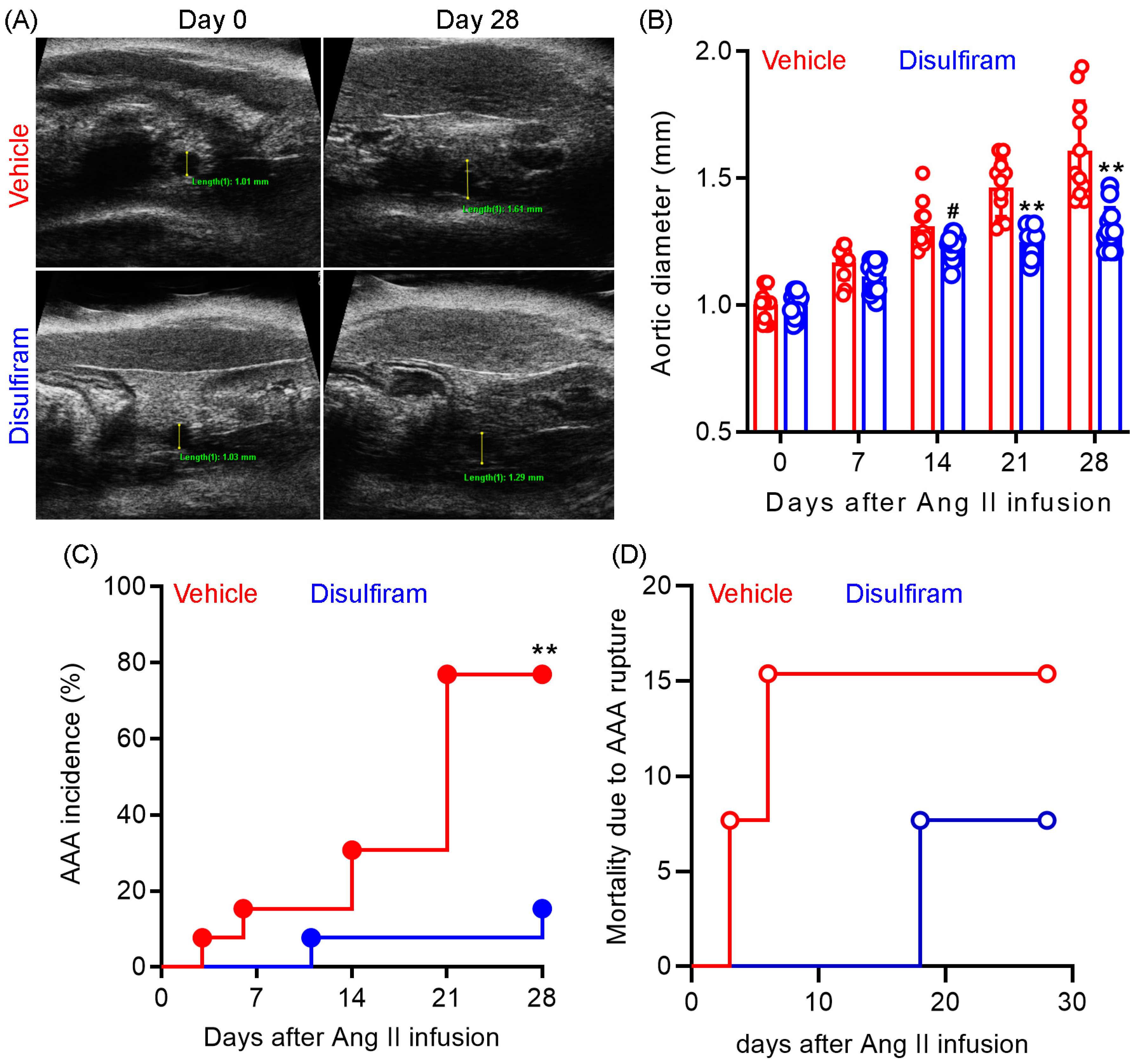{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Induction and Treatment of Experimental AAAs
2.2. Ultrasound Imaging of Experimental AAAs In Vivo
2.3. Measurement of Blood Pressure
2.4. AAA Severity Grading
2.5. Determination of Serum Triglycerides and Total Cholesterol Levels
2.6. Aortic Histological Analyses
2.7. Culture and Treatment of Macrophages In Vitro
2.8. Enzyme-Linked Immunosorbent Assays (ELISA)
2.9. Statistical Analysis
3. Results
3.1. Disulfiram Treatment Suppresses the Formation and Progression of Experimental AAAs
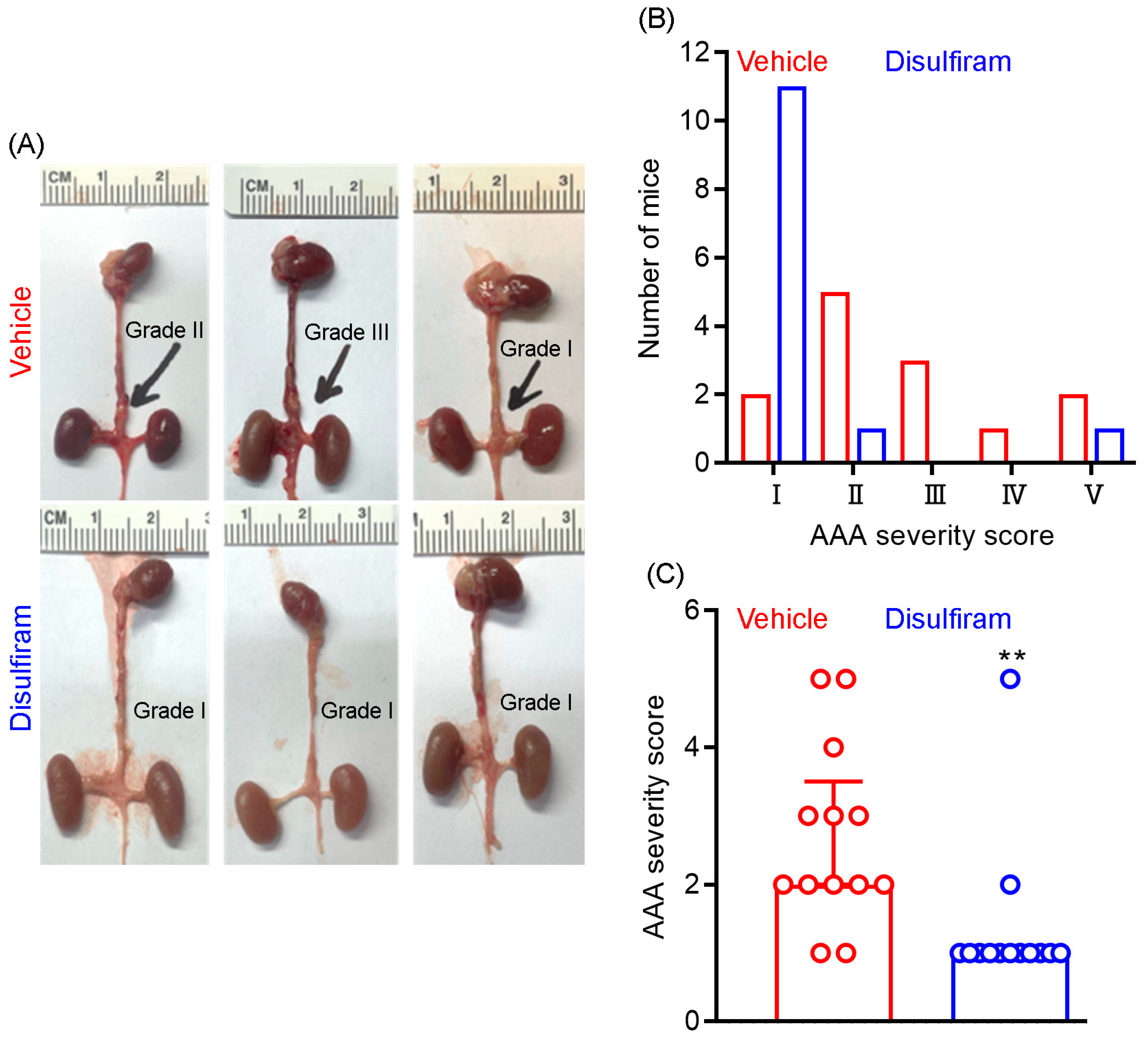
3.2. Disulfiram Treatment Ameliorates Experimental AAA Severity
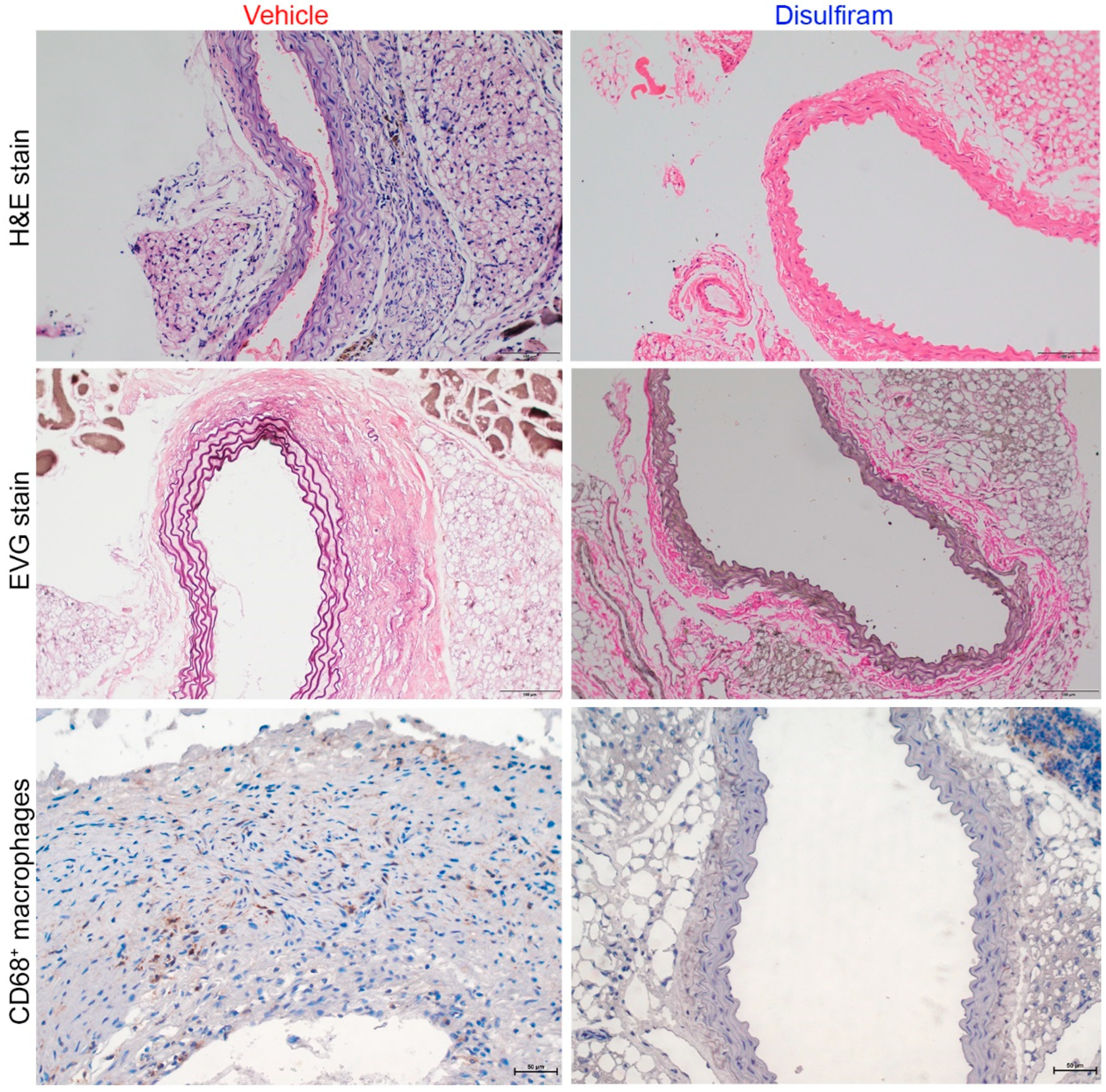
3.3. Disulfiram Treatment Attenuates Aortic Inflammation in Experimental AAAs
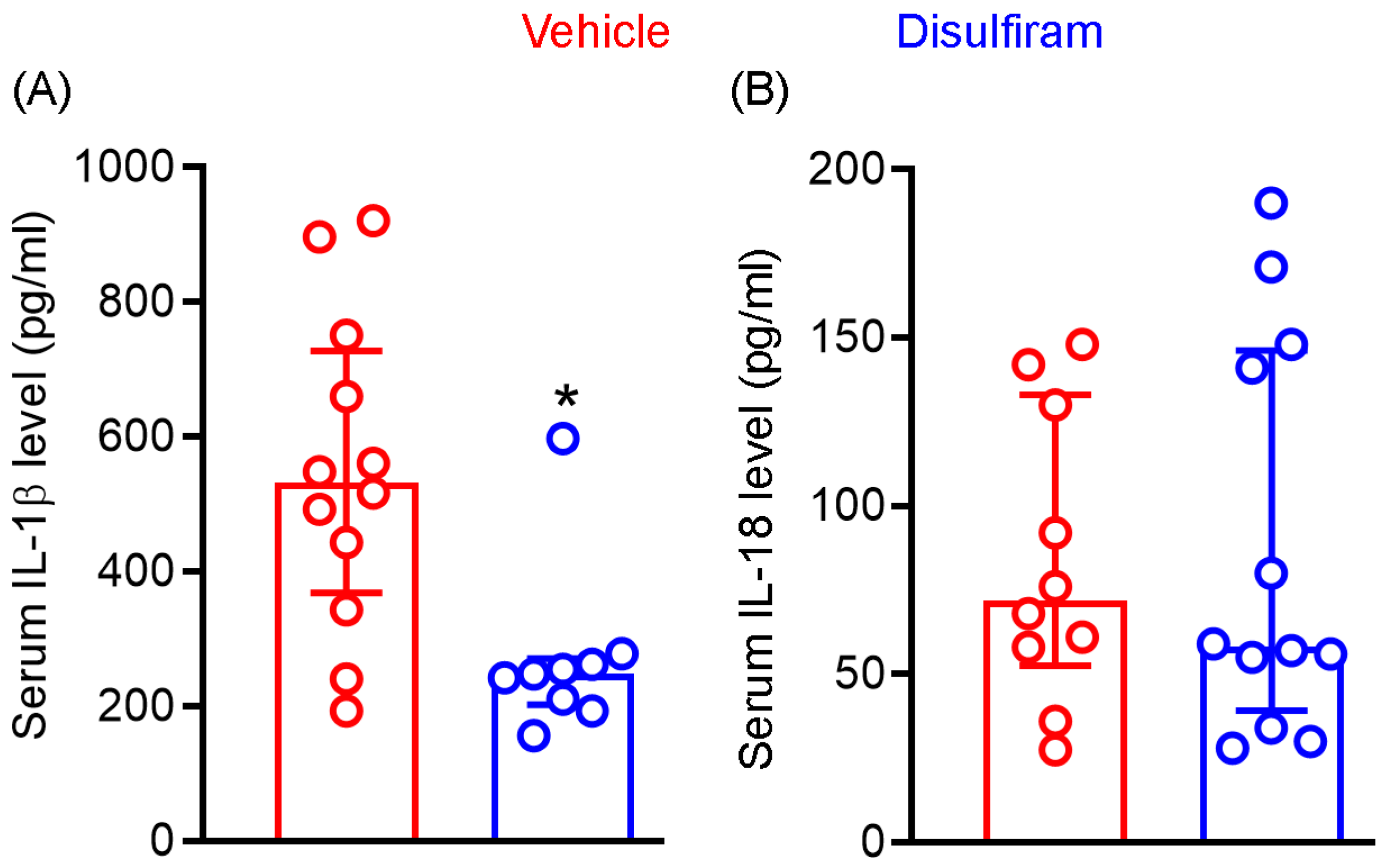
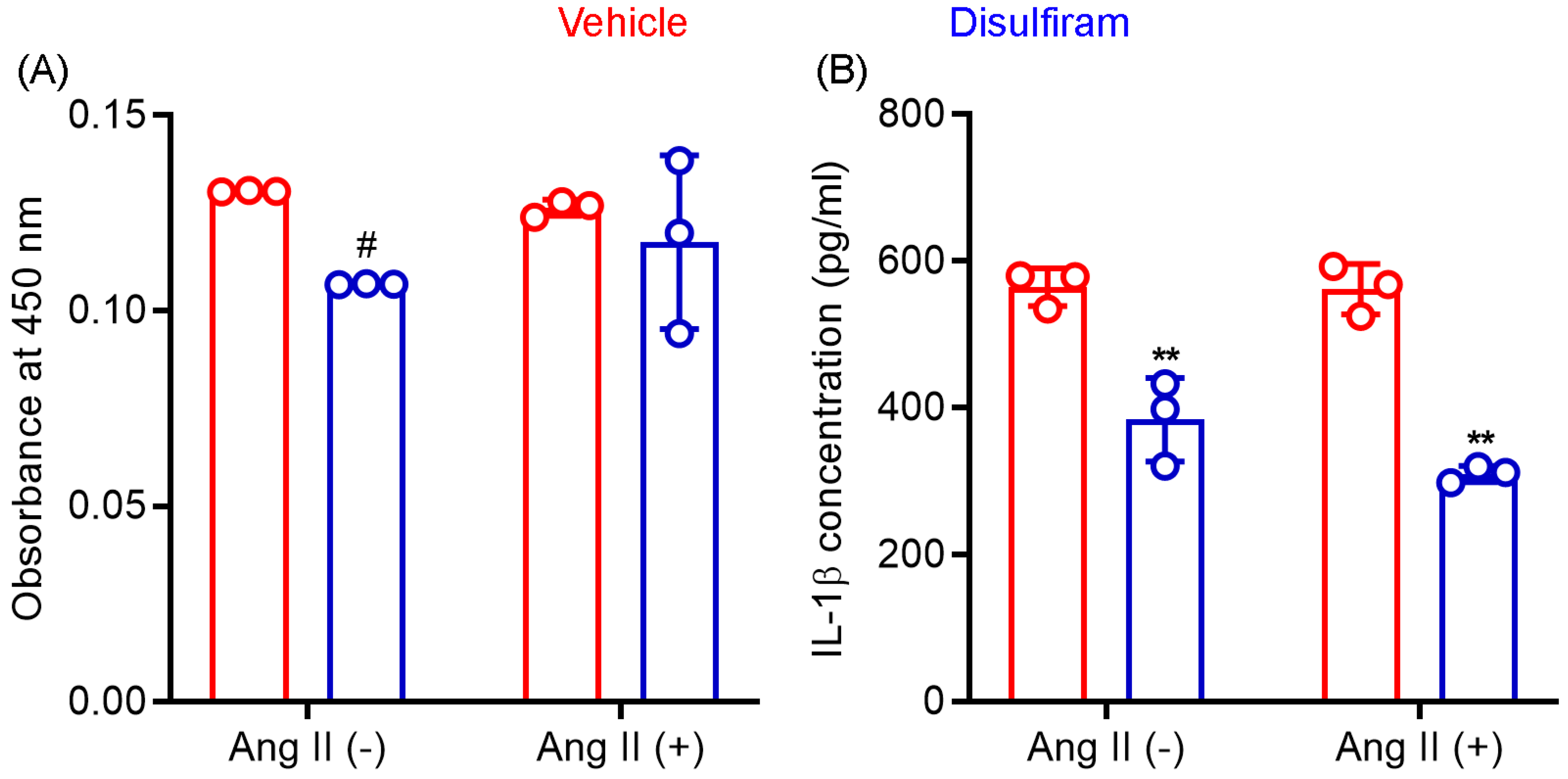
3.4. Disulfiram Treatment Reduces the Systemic Levels of IL-1β Not IL-18 in Experimental AAAs
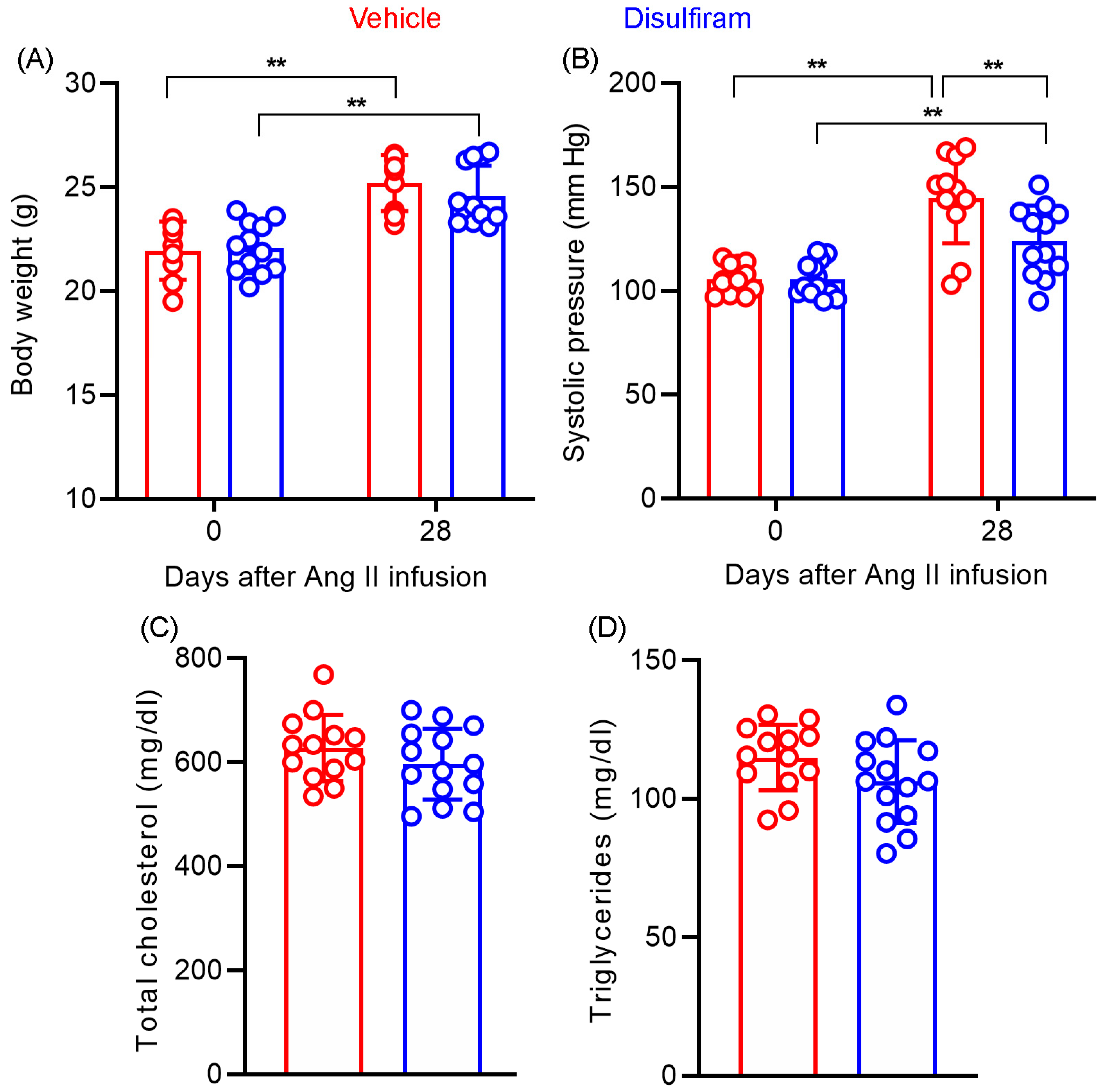
3.5. Disulfiram Treatment Slightly Lowers Angiotensin II-Induced Increase in Systolic Blood Pressure in Experimental AAAs
3.6. Disulfiram Treatment Affects Neither Body Weight Gain Nor Lipid Levels in Experimental AAAs
3.7. Disulfiram Treatment Suppresses IL-1β Release by Macrophages In Vitro
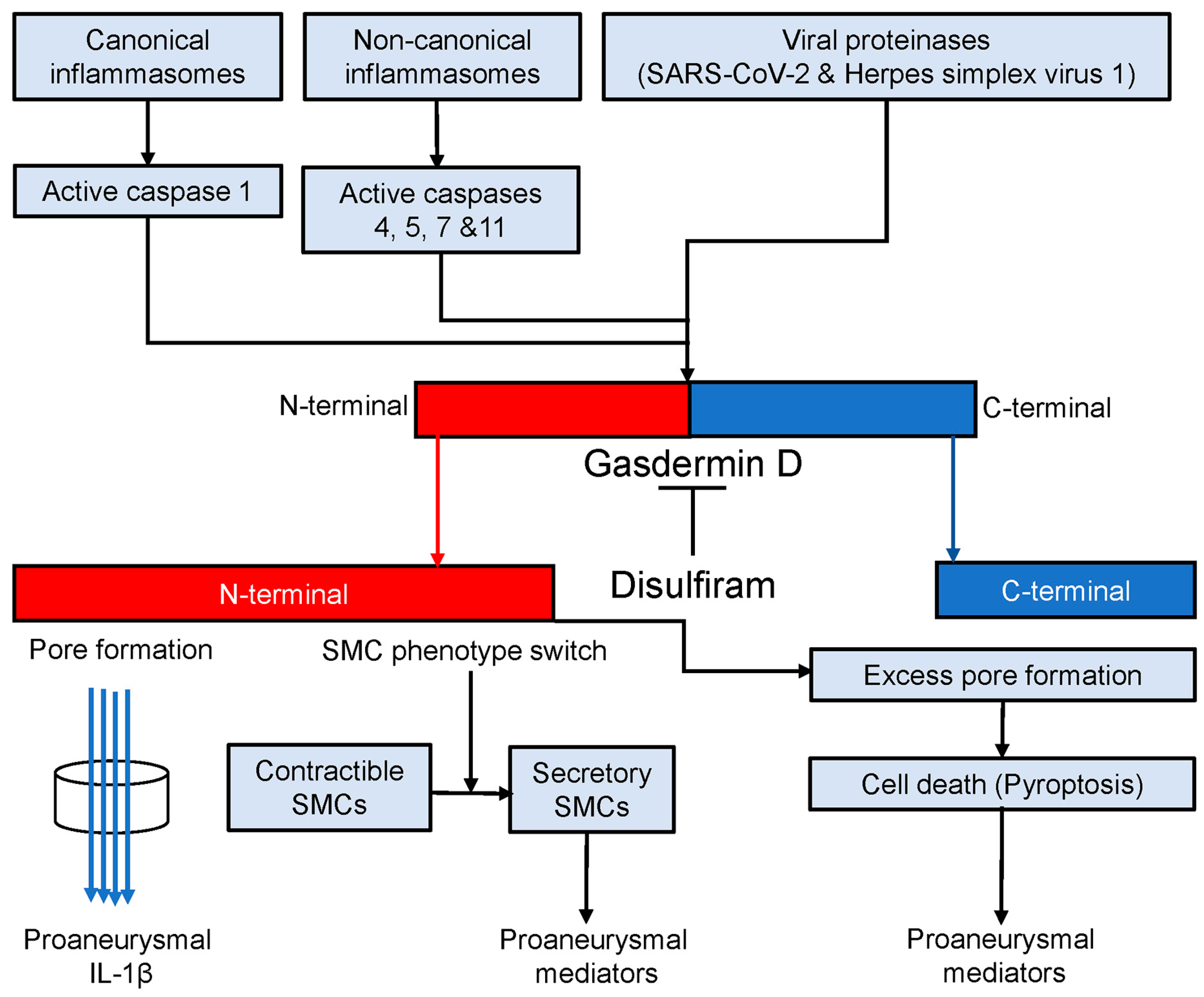
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
References
- Calgi, M.P.; McNeil, J.S. Abdominal Aortic Aneurysms (Etiology, Epidemiology, and Natural History). Anesthesiol. Clin. 2022, 40, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Lederle, F.A.; Johnson, G.R.; Wilson, S.E.; Chute, E.P.; Hye, R.J.; Makaroun, M.S.; Barone, G.W.; Bandyk, D.; Moneta, G.L.; Makhoul, R.G. The Aneurysm Detection and Management Study Screening Program. Arch. Intern. Med. 2000, 160, 1425–1430. [Google Scholar] [CrossRef]
- Bakewell, R.; Krokidis, M.; Winterbottom, A. Endovascular Abdominal Aortic Aneurysm Repair: Overview of Current Guidance, Strategies, and New Technologies, Perspectives from the United Kingdom. J. Clin. Med. 2022, 11, 5415. [Google Scholar] [CrossRef] [PubMed]
- Chaikof, E.L.; Dalman, R.L.; Eskandari, M.K.; Jackson, B.M.; Lee, W.A.; Mansour, M.A.; Mastracci, T.M.; Mell, M.; Murad, M.H.; Nguyen, L.L.; et al. The Society for Vascular Surgery practice guidelines on the care of patients with an abdominal aortic aneurysm. J. Vasc. Surg. 2018, 67, 2–77.e2. [Google Scholar] [CrossRef]
- Golledge, J.; Norman, P.E.; Murphy, M.P.; Dalman, R.L. Challenges and opportunities in limiting abdominal aortic aneurysm growth. J. Vasc. Surg. 2017, 65, 225–233. [Google Scholar] [CrossRef]
- Quintana, R.A.; Taylor, W.R. Cellular Mechanisms of Aortic Aneurysm Formation. Circ. Res. 2019, 124, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Shoji, T.; Ge, Y.; Zheng, X.; Li, Y.; Zhao, S.; Ikezoe, T.; Liu, S.; Huang, J.; Wang, W.; et al. Treatment with the Prolyl Hydroxylase Inhibitor JNJ Promotes Abdominal Aortic Aneurysm Progression in Diabetic Mice. Eur. J. Vasc. Endovasc. Surg. 2022, 63, 484–494. [Google Scholar] [CrossRef]
- Iida, Y.; Xu, B.; Xuan, H.; Glover, K.J.; Tanaka, H.; Hu, X.; Fujimura, N.; Wang, W.; Schultz, J.R.; Turner, C.R.; et al. Peptide Inhibitor of CXCL4–CCL5 Heterodimer Formation, MKEY, Inhibits Experimental Aortic Aneurysm Initiation and Progression. Arter. Thromb. Vasc. Biol. 2013, 33, 718–726. [Google Scholar] [CrossRef]
- Xu, B.; Xuan, H.; Iida, Y.; Miyata, M.; Dalman, R.L. Pathogenic and Therapeutic Significance of Angiotensin II Type I Receptor in Abdominal Aortic Aneurysms. Curr. Drug Targets 2018, 19, 1318–1326. [Google Scholar] [CrossRef]
- Xu, B.; Iida, Y.; Glover, K.J.; Ge, Y.; Wang, Y.; Xuan, H.; Hu, X.; Tanaka, H.; Wang, W.; Fujimura, N.; et al. Inhibition of VEGF (Vascular Endothelial Growth Factor)-A or its Receptor Activity Suppresses Experimental Aneurysm Progression in the Aortic Elastase Infusion Model. Arter. Thromb. Vasc. Biol. 2019, 39, 1652–1666. [Google Scholar] [CrossRef]
- Tanaka, H.; Xu, B.; Xuan, H.; Ge, Y.; Wang, Y.; Li, Y.; Wang, W.; Guo, J.; Zhao, S.; Glover, K.J.; et al. Recombinant Interleukin-19 Suppresses the Formation and Progression of Experimental Abdominal Aortic Aneurysms. J. Am. Heart Assoc. 2021, 10, e022207. [Google Scholar] [CrossRef] [PubMed]
- Johnston, W.F.; Salmon, M.; Su, G.; Lu, G.; Stone, M.L.; Zhao, Y.; Owens, G.K.; UpchurchJr, G.R.; Ailawadi, G. Genetic and Pharmacologic Disruption of Interleukin-1β Signaling Inhibits Experimental Aortic Aneurysm Formation. Arter. Thromb. Vasc. Biol. 2013, 33, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Wakita, D.; Kurashima, Y.; Crother, T.R.; Rivas, M.N.; Lee, Y.; Chen, S.; Fury, W.; Bai, Y.; Wagner, S.; Li, D.; et al. Role of Interleukin-1 Signaling in a Mouse Model of Kawasaki Disease–Associated Abdominal Aortic Aneurysm. Arter. Thromb. Vasc. Biol. 2016, 36, 886–897. [Google Scholar] [CrossRef]
- Batra, R.; Suh, M.K.; Carson, J.S.; Dale, M.A.; Meisinger, T.M.; Fitzgerald, M.; Opperman, P.J.; Luo, J.; Pipinos, I.I.; Xiong, W.; et al. IL-1β (Interleukin-1β) and TNF-α (Tumor Necrosis Factor-α) Impact Abdominal Aortic Aneurysm Formation by Differential Effects on Macrophage Polarization. Arter. Thromb. Vasc. Biol. 2018, 38, 457–463. [Google Scholar] [CrossRef]
- Meher, A.K.; Spinosa, M.; Davis, J.P.; Pope, N.; Laubach, V.E.; Su, G.; Serbulea, V.; Leitinger, N.; Ailawadi, G.; Upchurch, G.R. Novel Role of IL (Interleukin)-1β in Neutrophil Extracellular Trap Formation and Abdominal Aortic Aneurysms. Arter. Thromb. Vasc. Biol. 2018, 38, 843–853. [Google Scholar] [CrossRef]
- Shi, J.; Guo, J.; Li, Z.; Xu, B.; Miyata, M. Importance of NLRP3 Inflammasome in Abdominal Aortic Aneurysms. J. Atheroscler. Thromb. 2021, 28, 454–466. [Google Scholar] [CrossRef]
- Phie, J.; Thanigaimani, S.; Huynh, P.; Anbalagan, R.; Moran, C.S.; Kinobe, R.; Moxon, J.V.; Field, M.A.; Krishna, S.M.; Golledge, J. Colchicine Does Not Reduce Abdominal Aortic Aneurysm Growth in a Mouse Model. Cardiovasc. Ther. 2022, 2022, 5299370. [Google Scholar] [CrossRef]
- Suehiro, C.; Suzuki, J.; Hamaguchi, M.; Takahashi, K.; Nagao, T.; Sakaue, T.; Uetani, T.; Aono, J.; Ikeda, S.; Okura, T.; et al. Deletion of interleukin-18 attenuates abdominal aortic aneurysm formation. Atherosclerosis 2019, 289, 14–20. [Google Scholar] [CrossRef]
- Liu, C.-L.; Ren, J.; Wang, Y.; Zhang, X.; Sukhova, G.K.; Liao, M.; Santos, M.; Luo, S.; Yang, D.; Xia, M.; et al. Adipocytes promote interleukin-18 binding to its receptors during abdominal aortic aneurysm formation in mice. Eur. Heart J. 2020, 41, 2456–2468. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.H.; Schroder, K. Inflammasome signaling and regulation of interleukin-1 family cytokines. J. Exp. Med. 2020, 217, e20190314. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, W.; Zhou, R. NLRP3 inflammasome activation and cell death. Cell. Mol. Immunol. 2021, 18, 2114–2127. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Wu, H. Structural Mechanisms of NLRP3 Inflammasome Assembly and Activation. Annu. Rev. Immunol. 2022, 41, 301–316. [Google Scholar] [CrossRef]
- Ren, P.; Wu, D.; Appel, R.; Zhang, L.; Zhang, C.; Luo, W.; Robertson, A.A.B.; Cooper, M.A.; Coselli, J.S.; Milewicz, D.M.; et al. Targeting the NLRP3 Inflammasome with Inhibitor MCC950 Prevents Aortic Aneurysms and Dissections in Mice. J. Am. Heart Assoc. 2020, 9, e014044. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Zhang, L.; Liu, Z.; Mao, C.; Ma, Z.; Li, W.; Yu, F.; Wang, Y.; Huang, Y.; Zhang, W.; et al. Targeting macrophage TFEB-14-3-3 epsilon Interface by naringenin inhibits abdominal aortic aneurysm. Cell Discov. 2022, 8, 1–21. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef]
- Lieberman, J.; Wu, H.; Kagan, J.C. Gasdermin D activity in inflammation and host defense. Sci. Immunol. 2019, 4, eaav1447. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.-D. Gasdermin D: The long-awaited executioner of pyroptosis. Cell Res. 2015, 25, 1183–1184. [Google Scholar] [CrossRef]
- Xia, S.; Zhang, Z.; Magupalli, V.G.; Pablo, J.L.; Dong, Y.; Vora, S.M.; Wang, L.; Fu, T.-M.; Jacobson, M.P.; Greka, A.; et al. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature 2021, 593, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Mulvihill, E.; Sborgi, L.; A Mari, S.; Pfreundschuh, M.; Hiller, S.; Müller, D.J. Mechanism of membrane pore formation by human gasdermin-D. EMBO J. 2018, 37, e98321. [Google Scholar] [CrossRef]
- He, W.-T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Evavold, C.L.; Ruan, J.; Tan, Y.; Xia, S.; Wu, H.; Kagan, J.C. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 2017, 48, 35–44.e6. [Google Scholar] [CrossRef] [PubMed]
- Puylaert, P.; Van Praet, M.; Vaes, F.; Neutel, C.H.G.; Roth, L.; Guns, P.-J.; De Meyer, G.R.Y.; Martinet, W. Gasdermin D Deficiency Limits the Transition of Atherosclerotic Plaques to an Inflammatory Phenotype in ApoE Knock-Out Mice. Biomedicines 2022, 10, 1171. [Google Scholar] [CrossRef] [PubMed]
- Opoku, E.; Traughber, C.A.; Zhang, D.; Iacano, A.J.; Khan, M.; Han, J.; Smith, J.D.; Gulshan, K. Gasdermin D Mediates Inflammation-Induced Defects in Reverse Cholesterol Transport and Promotes Atherosclerosis. Front. Cell Dev. Biol. 2021, 9, 715221. [Google Scholar] [CrossRef]
- Jiang, M.; Sun, X.; Liu, S.; Tang, Y.; Shi, Y.; Bai, Y.; Wang, Y.; Yang, Q.; Yang, Q.; Jiang, W.; et al. Caspase-11-Gasdermin D-Mediated Pyroptosis Is Involved in the Pathogenesis of Atherosclerosis. Front. Pharmacol. 2021, 12, 657486. [Google Scholar] [CrossRef]
- Fidler, T.P.; Xue, C.; Yalcinkaya, M.; Hardaway, B.; Abramowicz, S.; Xiao, T.; Liu, W.; Thomas, D.G.; Hajebrahimi, M.A.; Pircher, J.; et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature 2021, 592, 296–301. [Google Scholar] [CrossRef]
- Hu, R.; Liang, J.; Ding, L.; Zhang, W.; Wang, Y.; Zhang, Y.; Zhang, D.; Pei, L.; Liu, X.; Xia, Z.; et al. Gasdermin D inhibition ameliorates neutrophil mediated brain damage in acute ischemic stroke. Cell Death Discov. 2023, 9, 1–14. [Google Scholar] [CrossRef]
- Ye, X.; Zhang, P.; Zhang, Y.; Luan, J.; Xu, C.; Wu, Z.; Ju, D.; Hu, W. GSDMD contributes to myocardial reperfusion injury by regulating pyroptosis. Front. Immunol. 2022, 13, 893914. [Google Scholar] [CrossRef]
- Wang, K.; Sun, Z.; Ru, J.; Wang, S.; Huang, L.; Ruan, L.; Lin, X.; Jin, K.; Zhuge, Q.; Yang, S. Ablation of GSDMD Improves Outcome of Ischemic Stroke Through Blocking Canonical and Non-canonical Inflammasomes Dependent Pyroptosis in Microglia. Front. Neurol. 2020, 11, 577927. [Google Scholar] [CrossRef]
- Han, J.; Dai, S.; Zhong, L.; Shi, X.; Fan, X.; Zhong, X.; Lin, W.; Su, L.; Lin, S.; Han, B.; et al. GSDMD (Gasdermin D) Mediates Pathological Cardiac Hypertrophy and Generates a Feed-Forward Amplification Cascade via Mitochondria-STING (Stimulator of Interferon Genes) Axis. Hypertension 2022, 79, 2505–2518. [Google Scholar] [CrossRef]
- Lin, S.; Dai, S.; Lin, J.; Liang, X.; Wang, W.; Huang, W.; Ye, B.; Hong, X. Oridonin Relieves Angiotensin II-Induced Cardiac Remodeling via Inhibiting GSDMD-Mediated Inflammation. Cardiovasc. Ther. 2022, 2022, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Iida, Y.; Xu, B.; Schultz, G.M.; Chow, V.; White, J.J.; Sulaimon, S.; Hezi-Yamit, A.; Peterson, S.R.; Dalman, R.L. Efficacy and Mechanism of Angiotensin II Receptor Blocker Treatment in Experimental Abdominal Aortic Aneurysms. PLoS ONE 2012, 7, e49642. [Google Scholar] [CrossRef]
- Daugherty, A.; Manning, M.W.; Cassis, L.A. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E–deficient mice. J. Clin. Investig. 2000, 105, 1605–1612. [Google Scholar] [CrossRef]
- Daugherty, A.; Manning, M.; A Cassis, L. Antagonism of AT2 receptors augments Angiotensin II-induced abdominal aortic aneurysms and atherosclerosis. Br. J. Pharmacol. 2001, 134, 865–870. [Google Scholar] [CrossRef]
- Weber, A.N.R.; Bittner, Z.A.; Shankar, S.; Liu, X.; Chang, T.-H.; Jin, T.; Tapia-Abellán, A. Recent insights into the regulatory networks of NLRP3 inflammasome activation. J. Cell Sci. 2020, 133, jcs248344. [Google Scholar] [CrossRef]
- Wang, W.; Xu, B.; Xuan, H.; Ge, Y.; Wang, Y.; Wang, L.; Huang, J.; Fu, W.; Michie, S.A.; Dalman, R.L. Hypoxia-inducible factor 1 in clinical and experimental aortic aneurysm disease. J. Vasc. Surg. 2018, 68, 1538–1550.e2. [Google Scholar] [CrossRef] [PubMed]
- Shoji, T.; Guo, J.; Ge, Y.; Li, Y.; Li, G.; Ikezoe, T.; Wang, W.; Zheng, X.; Zhao, S.; Fujimura, N.; et al. Type I Interferon Receptor Subunit 1 Deletion Attenuates Experimental Abdominal Aortic Aneurysm Formation. Biomolecules 2022, 12, 1541. [Google Scholar] [CrossRef]
- Usui, F.; Shirasuna, K.; Kimura, H.; Tatsumi, K.; Kawashima, A.; Karasawa, T.; Yoshimura, K.; Aoki, H.; Tsutsui, H.; Noda, T.; et al. Inflammasome Activation by Mitochondrial Oxidative Stress in Macrophages Leads to the Development of Angiotensin II–Induced Aortic Aneurysm. Arter. Thromb. Vasc. Biol. 2015, 35, 127–136. [Google Scholar] [CrossRef]
- Ye, D.; Howatt, D.A.; Li, A.; Daugherty, Z.; Lu, H.S.; Wu, C. Gsdmd deficiency protects against aortic rupture. BioRxiv 2021, 8, 425983. [Google Scholar]
- Gao, J.; Chen, Y.; Wang, H.; Li, X.; Li, K.; Xu, Y.; Xie, X.; Guo, Y.; Yang, N.; Zhang, X.; et al. Gasdermin D Deficiency in Vascular Smooth Muscle Cells Ameliorates Abdominal Aortic Aneurysm Through Reducing Putrescine Synthesis. Adv. Sci. 2022, 10, e2204038. [Google Scholar] [CrossRef]
- Liao, F.; Wang, L.; Wu, Z.; Luo, G.; Qian, Y.; He, X.; Ding, S.; Pu, J. Disulfiram protects against abdominal aortic aneurysm by ameliorating vascular smooth muscle cells pyroptosis. Cardiovasc. Drugs Ther. 2022. [Google Scholar] [CrossRef]
- Matikainen, S.; Nyman, T.A.; Cypryk, W. Function and Regulation of Noncanonical Caspase-4/5/11 Inflammasome. J. Immunol. 2020, 204, 3063–3069. [Google Scholar] [CrossRef]
- Wright, S.S.; Vasudevan, S.O.; Rathinam, V.A. Mechanisms and Consequences of Noncanonical Inflammasome-Mediated Pyroptosis. J. Mol. Biol. 2022, 434, 167245. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Reilly, B.; Tan, C.; Wang, P.; Aziz, M. Extracellular CIRP Induces Macrophage Extracellular Trap Formation Via Gasdermin D Activation. Front. Immunol. 2021, 12, 780210. [Google Scholar] [CrossRef]
- Keshavan, S.; Gupta, G.; Martin, S.; Fadeel, B. Multi-walled carbon nanotubes trigger lysosome-dependent cell death (pyroptosis) in macrophages but not in neutrophils. Nanotoxicology 2021, 15, 1125–1150. [Google Scholar] [CrossRef]
- Jensen, J.C.; Faiman, M.D. Hypothermia as an index of the disulfiram-ethanol reaction in the rat. Alcohol 1984, 1, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.C.; Faiman, M.D. Disulfiram-Ethanol Reaction in the Rat. 1. Blood Alcohol, Acetaldehyde, and Liver Aldehyde Dehydrogenase Relationships. Alcohol. Clin. Exp. Res. 1986, 10, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Johansson, B.; Petersen, E.N.; Arnold, E. Diethylthiocarbamic acid methyl ester: A potent inhibitor of aldehyde dehydrogenase found in rats treated with disulfiram or diethyldithiocarbamic acid methyl ester. Biochem. Pharmacol. 1989, 38, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Petersen, E.N. Pharmacological effects of diethylthiocarbamic acid methyl ester, the active metabolite of disulfiram? Eur. J. Pharmacol. 1989, 166, 419–425. [Google Scholar] [CrossRef]
- Bilska-Wilkosz, A.; Kotańska, M.; Górny, M.; Filipek, B.; Iciek, M. Can Lipoic Acid Attenuate Cardiovascular Disturbances Induced by Ethanol and Disulfiram Administration Separately or Jointly in Rats? Oxidative Med. Cell. Longev. 2019, 2019, 1–10. [Google Scholar] [CrossRef]
- Hellström, E.; Tottmar, O. Acute effects of ethanol and acetaldehyde on blood pressure and heart rate in disulfiram-treated and control rats. Pharmacol. Biochem. Behav. 1982, 17, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- Hellström, E.; Tottmar, O.; Fried, R. Implantation of disulfiram in rats. Pharmacol. Biochem. Behav. 1980, 13, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Rapsomaniki, E.; Timmis, A.; George, J.; Pujades-Rodriguez, M.; Shah, A.D.; Denaxas, S.; White, I.R.; Caulfield, M.J.; Deanfield, E.J.; Smeeth, L.; et al. Blood pressure and incidence of twelve cardiovascular diseases: Lifetime risks, healthy life-years lost, and age-specific associations in 1·25 million people. Lancet 2014, 383, 1899–1911. [Google Scholar] [CrossRef]
- Jahangir, E.; Lipworth, L.; Edwards, T.L.; Kabagambe, E.K.; Mumma, M.T.; A Mensah, G.; Fazio, S.; Blot, W.J.; A Sampson, U.K. Smoking, sex, risk factors and abdominal aortic aneurysms: A prospective study of 18 782 persons aged above 65 years in the Southern Community Cohort Study. J. Epidemiol. Community Health 2015, 69, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Umemoto, T. Association of Hypertension with Abdominal Aortic Aneurysm Expansion. Ann. Vasc. Surg. 2016, 39, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Manapurathe, D.T.; Moxon, J.V.; Krishna, S.M.; Quigley, F.; Bourke, M.; Bourke, B.; Jones, R.E.; Golledge, J. Cohort Study Examining the Association of Optimal Blood Pressure Control at Entry with Infrarenal Abdominal Aortic Aneurysm Growth. Front. Cardiovasc. Med. 2022, 9, 868889. [Google Scholar] [CrossRef]
- Golledge, J.; Singh, T.P. Effect of blood pressure lowering drugs and antibiotics on abdominal aortic aneurysm growth: A systematic review and meta-analysis. Heart 2021, 107, 1465–1471. [Google Scholar] [CrossRef]
- Golledge, J.; Pinchbeck, J.; Tomee, S.M.; Rowbotham, S.E.; Singh, T.; Moxon, J.V.; Jenkins, J.S.; Lindeman, J.H.; Dalman, R.L.; McDonnell, L.; et al. Efficacy of Telmisartan to Slow Growth of Small Abdominal Aortic Aneurysms. JAMA Cardiol. 2020, 5, 1374. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Shiraya, S.; Miyake, T.; Yamakawa, S.; Aoki, M.; Makino, H.; Nishimura, M.; Morishita, R. Inhibition of experimental abdominal aortic aneurysm in a rat model by the angiotensin receptor blocker valsartan. Int. J. Mol. Med. 2008, 22, 703–708. [Google Scholar] [PubMed]
- Kaschina, E.; Schrader, F.; Sommerfeld, M.; Kemnitz, U.R.; Grzesiak, A.; Krikov, M.; Unger, T. Telmisartan prevents aneurysm progression in the rat by inhibiting proteolysis, apoptosis and inflammation. J. Hypertens. 2008, 26, 2361–2373. [Google Scholar] [CrossRef]
- Xuan, H.; Xu, B.; Wang, W.; Tanaka, H.; Fujimura, N.; Miyata, M.; Michie, S.A.; Dalman, R.L. Inhibition or deletion of angiotensin II type 1 receptor suppresses elastase-induced experimental abdominal aortic aneurysms. J. Vasc. Surg. 2018, 67, 573–584.e2. [Google Scholar] [CrossRef]
- Ricci, M.A.; Slaiby, J.M.; Gadowski, G.R.; Hendley, E.D.; Nichols, P.; Pilcher, D.B. Effects of Hypertension and Propranolol upon Aneurysm Expansion in the Anidjar/Dobrin Aneurysm Model. Ann. N. Y. Acad. Sci. 1996, 800, 89–96. [Google Scholar] [CrossRef]
- Mieth, A.; Revermann, M.; Babelova, A.; Weigert, A.; Schermuly, R.T.; Brandes, R.P. l -Type Calcium Channel Inhibitor Diltiazem Prevents Aneurysm Formation by Blood Pressure–Independent Anti-Inflammatory Effects. Hypertension 2013, 62, 1098–1104. [Google Scholar] [CrossRef]
- Park, S.M.; Hong, M.-K.; Kim, S.H.; Jung, S.; Kim, B.K.; Choi, D. Comparison of Efficacy between Ramipril and Carvedilol on Limiting the Expansion of Abdominal Aortic Aneurysm in Mouse Model. J. Cardiovasc. Pharmacol. Ther. 2019, 24, 172–181. [Google Scholar] [CrossRef]
- Kurobe, H.; Matsuoka, Y.; Hirata, Y.; Sugasawa, N.; Maxfield, M.W.; Sata, M.; Kitagawa, T. Azelnidipine suppresses the progression of aortic aneurysm in wild mice model through anti-inflammatory effects. J. Thorac. Cardiovasc. Surg. 2013, 146, 1501–1508. [Google Scholar] [CrossRef]
- Slaiby, J.M.; Ricci, M.A.; Gadowski, G.R.; Hendley, E.D.; Pilcher, D.B. Expansion of aortic aneurysms is reduced by propranolol in a hypertensive rat model. J. Vasc. Surg. 1994, 20, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Matsumoto, Y.; Do.E, Z.; Kanazawa, M.; Satoh, K.; Shimizu, T.; Sato, A.; Fukumoto, Y.; Shimokawa, H. Combination Therapy with Atorvastatin and Amlodipine Suppresses Angiotensin II-Induced Aortic Aneurysm Formation. PLoS ONE 2013, 8, e72558. [Google Scholar] [CrossRef] [PubMed]
- Cassis, L.A.; Gupte, M.; Thayer, S.; Zhang, X.; Charnigo, R.; Howatt, D.A.; Rateri, D.L.; Daugherty, A.; Glenn, D.J.; Cardema, M.C.; et al. ANG II infusion promotes abdominal aortic aneurysms independent of increased blood pressure in hypercholesterolemic mice. Am. J. Physiol. Circ. Physiol. 2009, 296, H1660–H1665. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Yao, L.; Roetker, N.S.; Alonso, A.; Lutsey, P.L.; Steenson, C.C.; Lederle, F.A.; Hunter, D.W.; Bengtson, L.G.; Guan, W.; et al. Lifetime risk and risk factors for abdominal aortic aneurysm in a 24-year prospective study: The aric study (atherosclerosis risk in communities). Arter. Thromb. Vasc. Biol. 2016, 36, 2468–2477. [Google Scholar] [CrossRef]
- Klarin, D.; Damrauer, S.M.; Cho, K.; Sun, Y.V.; Teslovich, T.M.; Honerlaw, J.; Gagnon, D.R.; Duvall, S.L.; Li, J.; Peloso, G.M.; et al. Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nat. Genet. 2018, 50, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Police, S.B.; Thatcher, S.E.; Charnigo, R.; Daugherty, A.; Cassis, L.A. Obesity Promotes Inflammation in Periaortic Adipose Tissue and Angiotensin II-Induced Abdominal Aortic Aneurysm Formation. Arter. Thromb. Vasc. Biol. 2009, 29, 1458–1464. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ait-Oufella, H.; Herbin, O.; Bonnin, P.; Ramkhelawon, B.; Taleb, S.; Huang, J.; Offenstadt, G.; Combadiere, C.; Rénia, L.; et al. TGF-β activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II–infused mice. J. Clin. Investig. 2010, 120, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Ikezoe, T.; Shoji, T.; Guo, J.; Shen, F.; Lu, H.S.; Daugherty, A.; Nunokawa, M.; Kubota, H.; Miyata, M.; Xu, B.; et al. No Effect of Hypercholesterolemia on Elastase-Induced Experimental Abdominal Aortic Aneurysm Progression. Biomolecules 2021, 11, 1434. [Google Scholar] [CrossRef]
- Mulorz, J.; Spin, J.M.; Beck, H.C.; Thi, M.L.T.; Wagenhäuser, M.U.; Rasmussen, L.M.; Lindholt, J.S.; Tsao, P.S.C.; Steffensen, L.B. Hyperlipidemia does not affect development of elastase-induced abdominal aortic aneurysm in mice. Atherosclerosis 2020, 311, 73–83. [Google Scholar] [CrossRef]
- Police, S.B.; Putnam, K.; Thatcher, S.; Batifoulier-Yiannikouris, F.; Daugherty, A.; Cassis, L.A. Weight loss in obese C57BL/6 mice limits adventitial expansion of established angiotensin II-induced abdominal aortic aneurysms. Am. J. Physiol. Circ. Physiol. 2010, 298, H1932–H1938. [Google Scholar] [CrossRef]
- Humphries, F.; Shmuel-Galia, L.; Ketelut-Carneiro, N.; Li, S.; Wang, B.; Nemmara, V.V.; Wilson, R.; Jiang, Z.; Khalighinejad, F.; Muneeruddin, K.; et al. Succination inactivates gasdermin D and blocks pyroptosis. Science 2020, 369, 1633–1637. [Google Scholar] [CrossRef]
- Rathkey, J.K.; Zhao, J.; Liu, Z.; Chen, Y.; Yang, J.; Kondolf, H.C.; Benson, B.L.; Chirieleison, S.M.; Huang, A.Y.; Dubyak, G.R.; et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci. Immunol. 2018, 3, eaat2738. [Google Scholar] [CrossRef]
- Balasubramanian, A.; Ghimire, L.; Hsu, A.Y.; Kambara, H.; Liu, X.; Hasegawa, T.; Xu, R.; Tahir, M.; Yu, H.; Lieberman, J.; et al. Palmitoylation of gasdermin d directs its membrane translocation and pore formation in pyroptosis. BioRxiv 2023, 21, 529402. [Google Scholar] [CrossRef]
- Silva, C.M.S.; Wanderley, C.W.S.; Veras, F.P.; Gonçalves, A.V.; Lima, M.H.F.; Toller-Kawahisa, J.E.; Gomes, G.F.; Nascimento, D.C.; Monteiro, V.V.S.; Paiva, I.M.; et al. Gasdermin-D activation by SARS-CoV-2 triggers NET and mediate COVID-19 immunopathology. Crit. Care 2022, 26, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zeng, Q.; Xiao, J.; Qin, S.; Wang, Y.; Shan, T.; Hu, D.; Zhu, Y.; Liu, K.; Zheng, K.; et al. Herpes Simplex Virus 1 Induces Microglia Gasdermin D-Dependent Pyroptosis Through Activating the NLR Family Pyrin Domain Containing 3 Inflammasome. Front. Microbiol. 2022, 13, 838808. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, J.; Shi, J.; Qin, M.; Wang, Y.; Li, Z.; Shoji, T.; Ikezoe, T.; Ge, Y.; Xu, B. Pharmacological Inhibition of Gasdermin D Suppresses Angiotensin II-Induced Experimental Abdominal Aortic Aneurysms. Biomolecules 2023, 13, 899. https://doi.org/10.3390/biom13060899
Guo J, Shi J, Qin M, Wang Y, Li Z, Shoji T, Ikezoe T, Ge Y, Xu B. Pharmacological Inhibition of Gasdermin D Suppresses Angiotensin II-Induced Experimental Abdominal Aortic Aneurysms. Biomolecules. 2023; 13(6):899. https://doi.org/10.3390/biom13060899
Chicago/Turabian StyleGuo, Jia, Jinyun Shi, Min Qin, Yan Wang, Zhidong Li, Takahiro Shoji, Toru Ikezoe, Yingbin Ge, and Baohui Xu. 2023. "Pharmacological Inhibition of Gasdermin D Suppresses Angiotensin II-Induced Experimental Abdominal Aortic Aneurysms" Biomolecules 13, no. 6: 899. https://doi.org/10.3390/biom13060899
APA StyleGuo, J., Shi, J., Qin, M., Wang, Y., Li, Z., Shoji, T., Ikezoe, T., Ge, Y., & Xu, B. (2023). Pharmacological Inhibition of Gasdermin D Suppresses Angiotensin II-Induced Experimental Abdominal Aortic Aneurysms. Biomolecules, 13(6), 899. https://doi.org/10.3390/biom13060899