
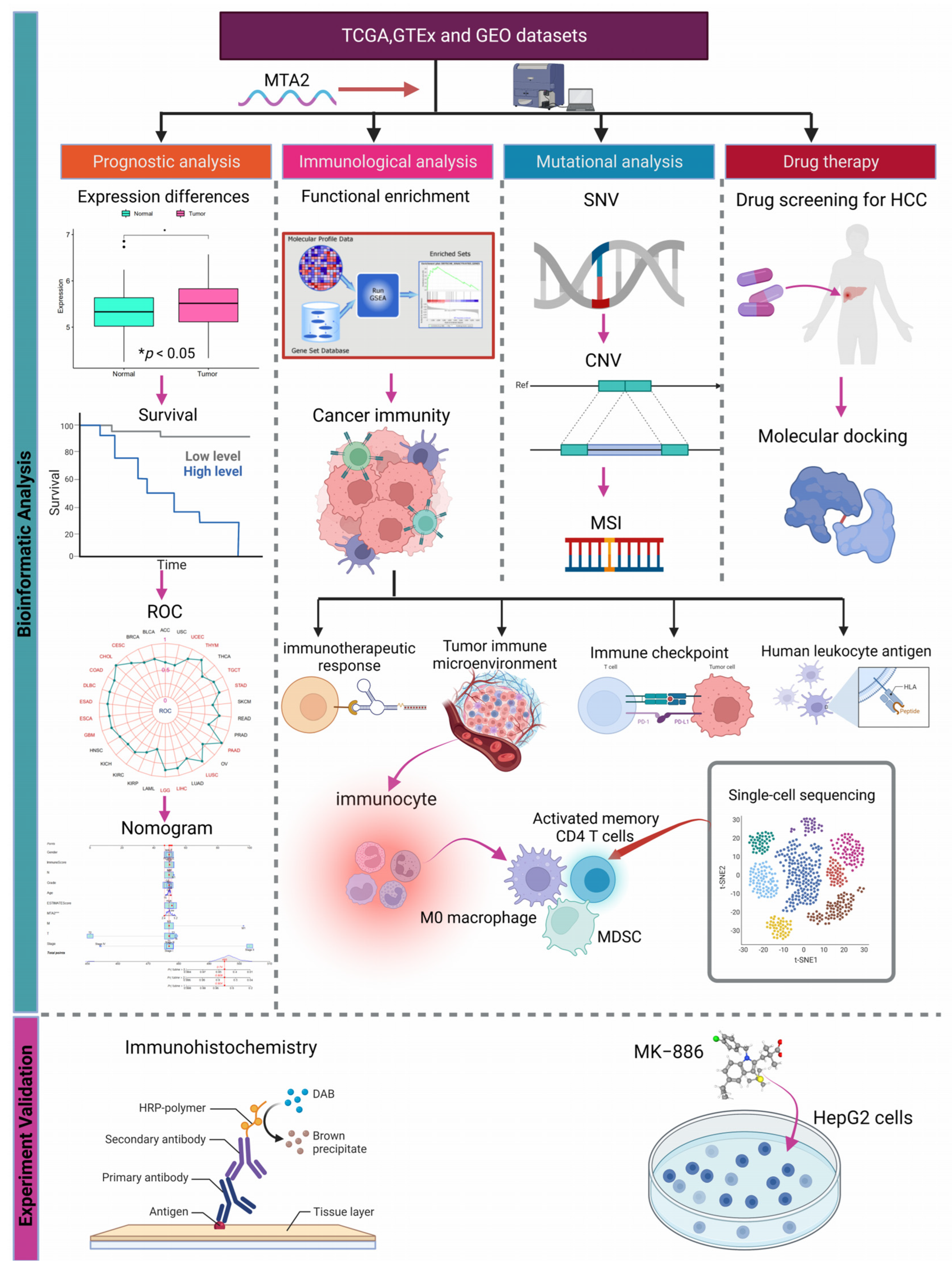
Prognostic, Immunological, and Mutational Analysis of MTA2 in Pan-Cancer and Drug Screening for Hepatocellular Carcinoma
 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Pan-Cancer Data Acquisition
2.2. Construction of PPI Networks of MTA2
2.3. The Mutational Landscape of MTA2 in Pan-Cancer
2.4. Association of MTA2 with Prognosis in Pan-Cancer
2.5. Correlation of MTA2 Expression with Tumor Mutation Burden (TMB) and Microsatellite Instability (MSI)
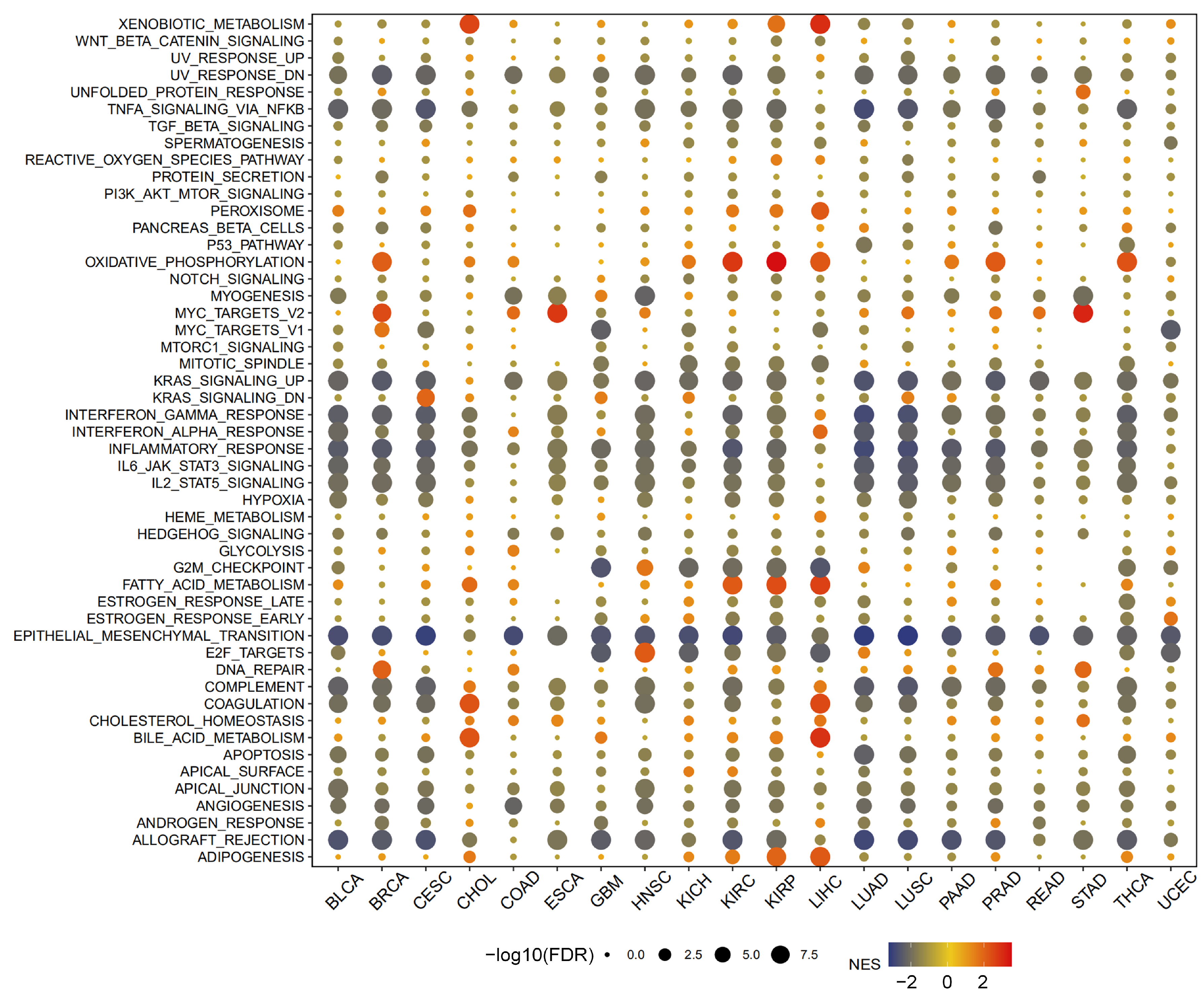
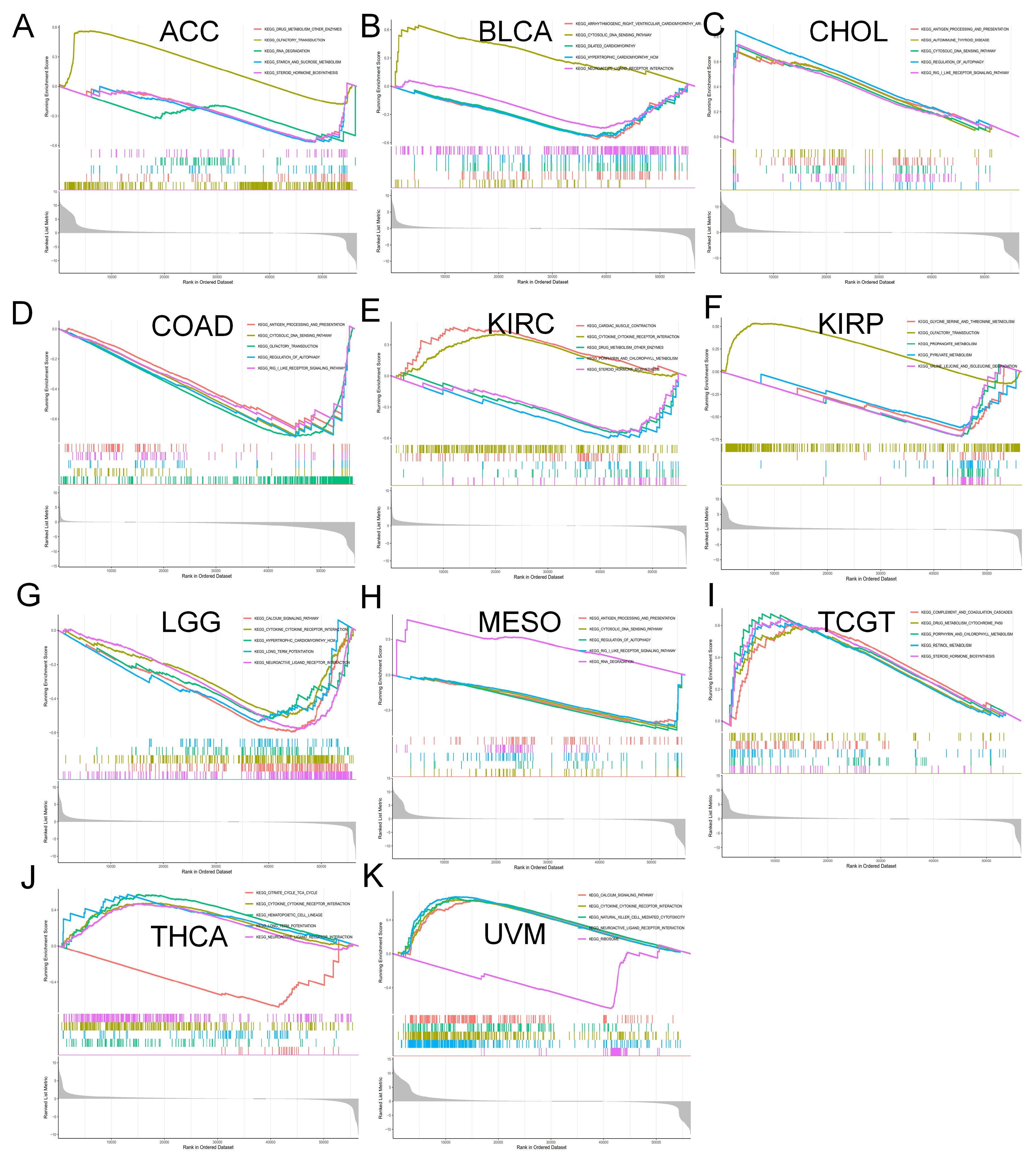
2.6. Enrichment Analysis of MTA2 in Pan-Cancer
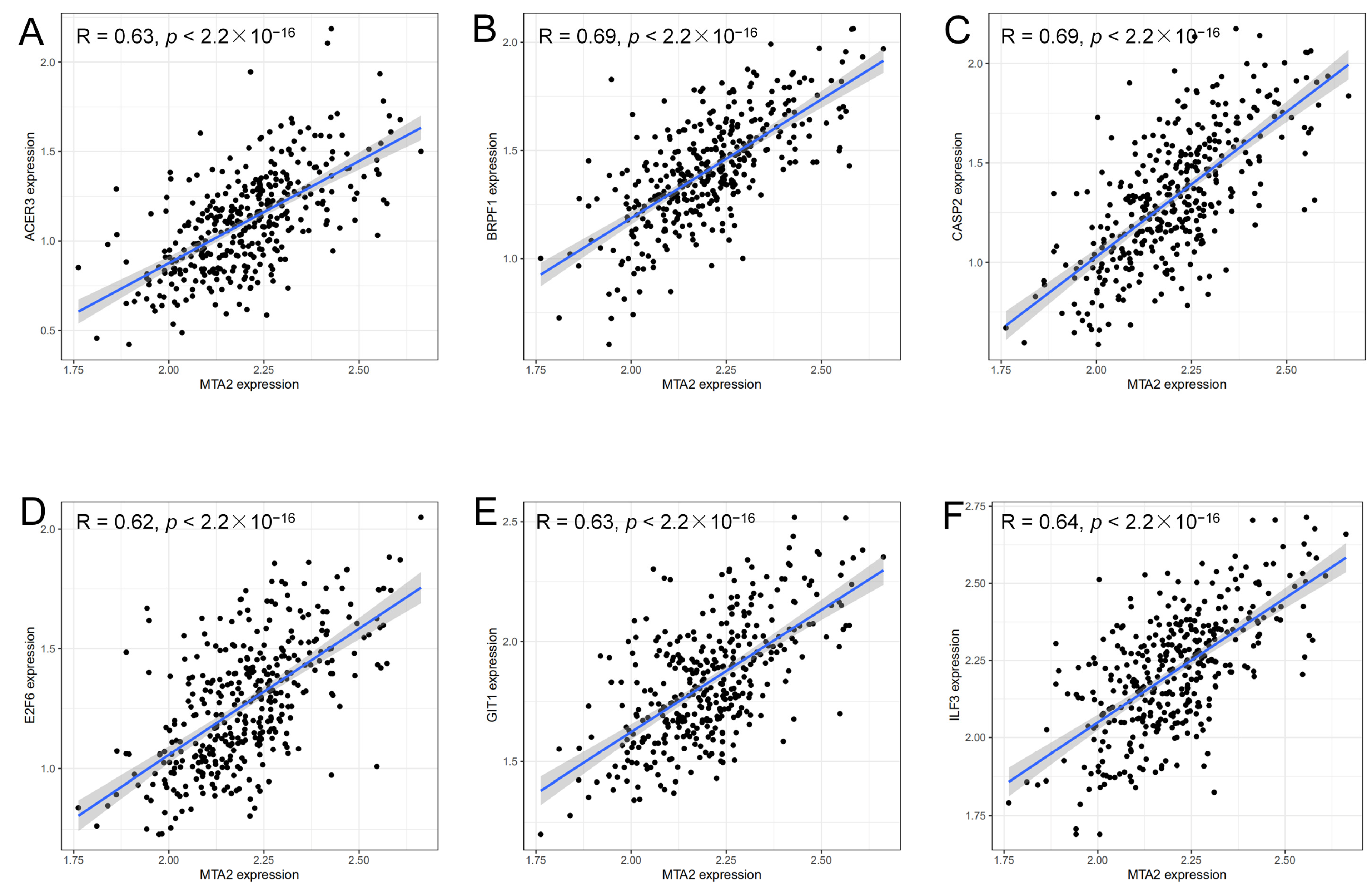
2.7. Analysis of Genes Co-Expressed with MTA2
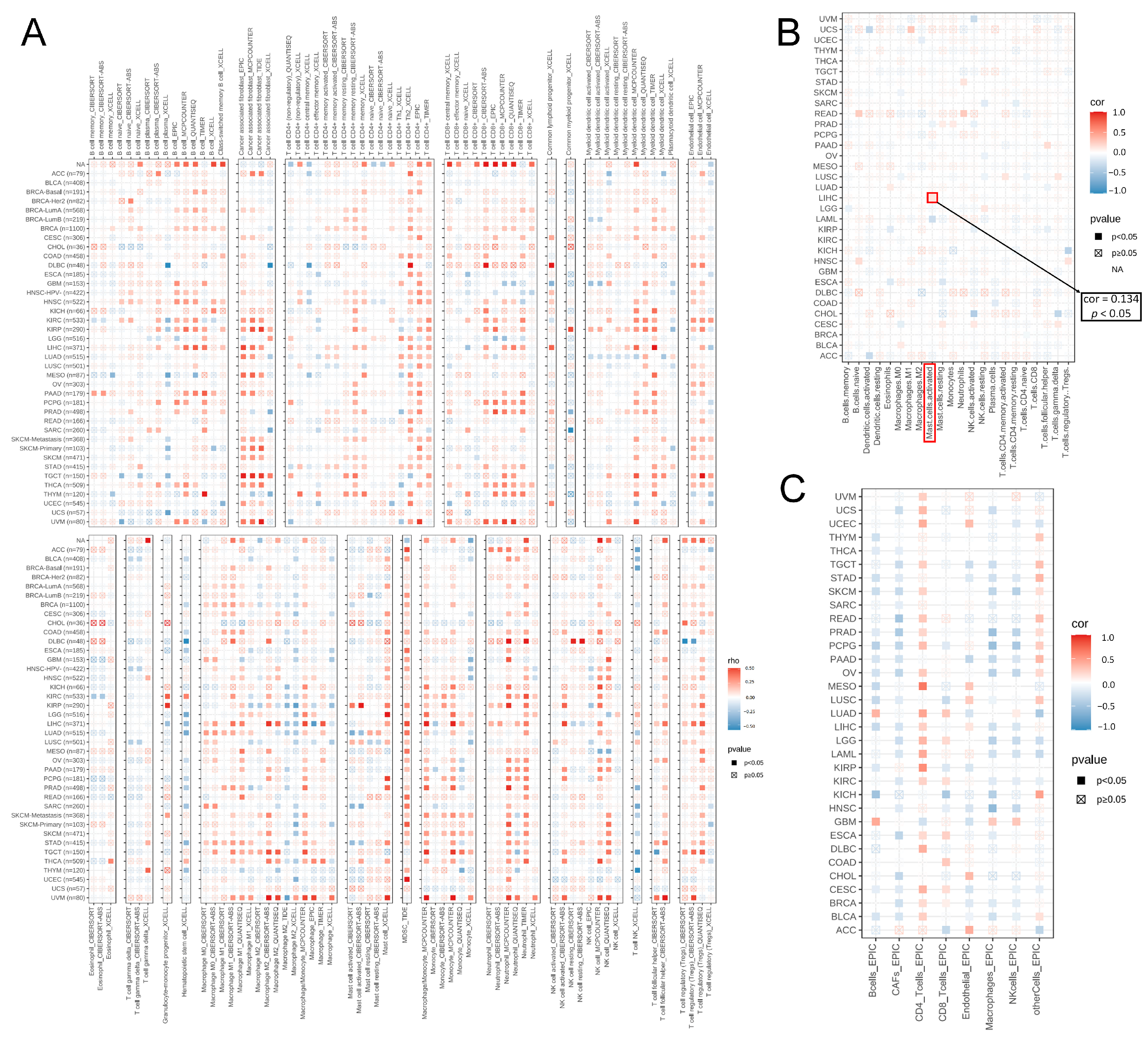
2.8. The Association between MTA2 and Immunity in LIHC
2.9. Single Cell Sequencing Analysis
2.10. Screening of Antitumor Drugs
2.11. Molecular Docking between MTA2 Protein and MK-886
2.12. Immunohistochemistry
2.13. Cell Culture
2.14. CCK-8 Cell Proliferation Assay
2.15. Cell Viability Assay
2.16. Statistical Methods
3. Results
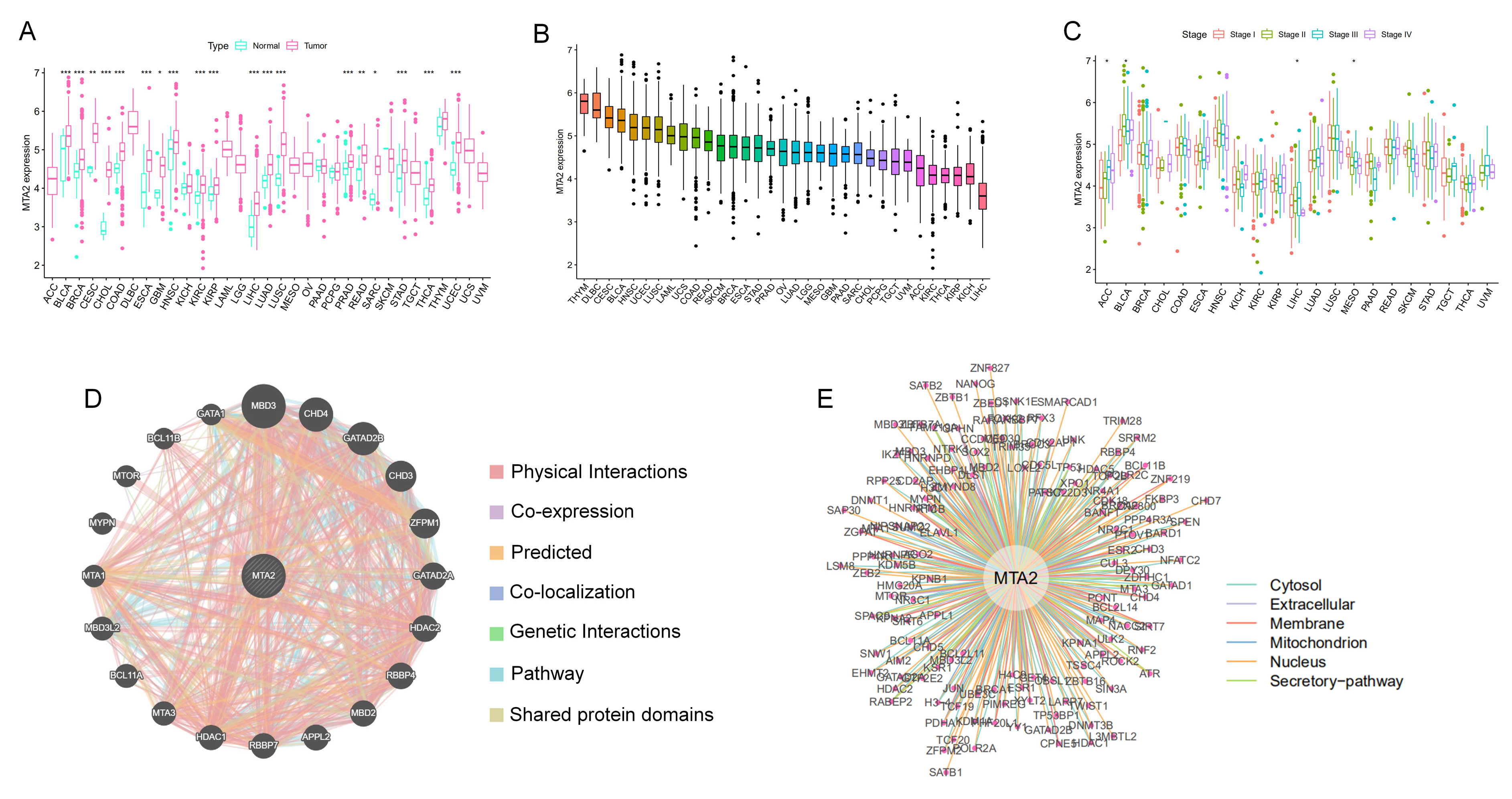
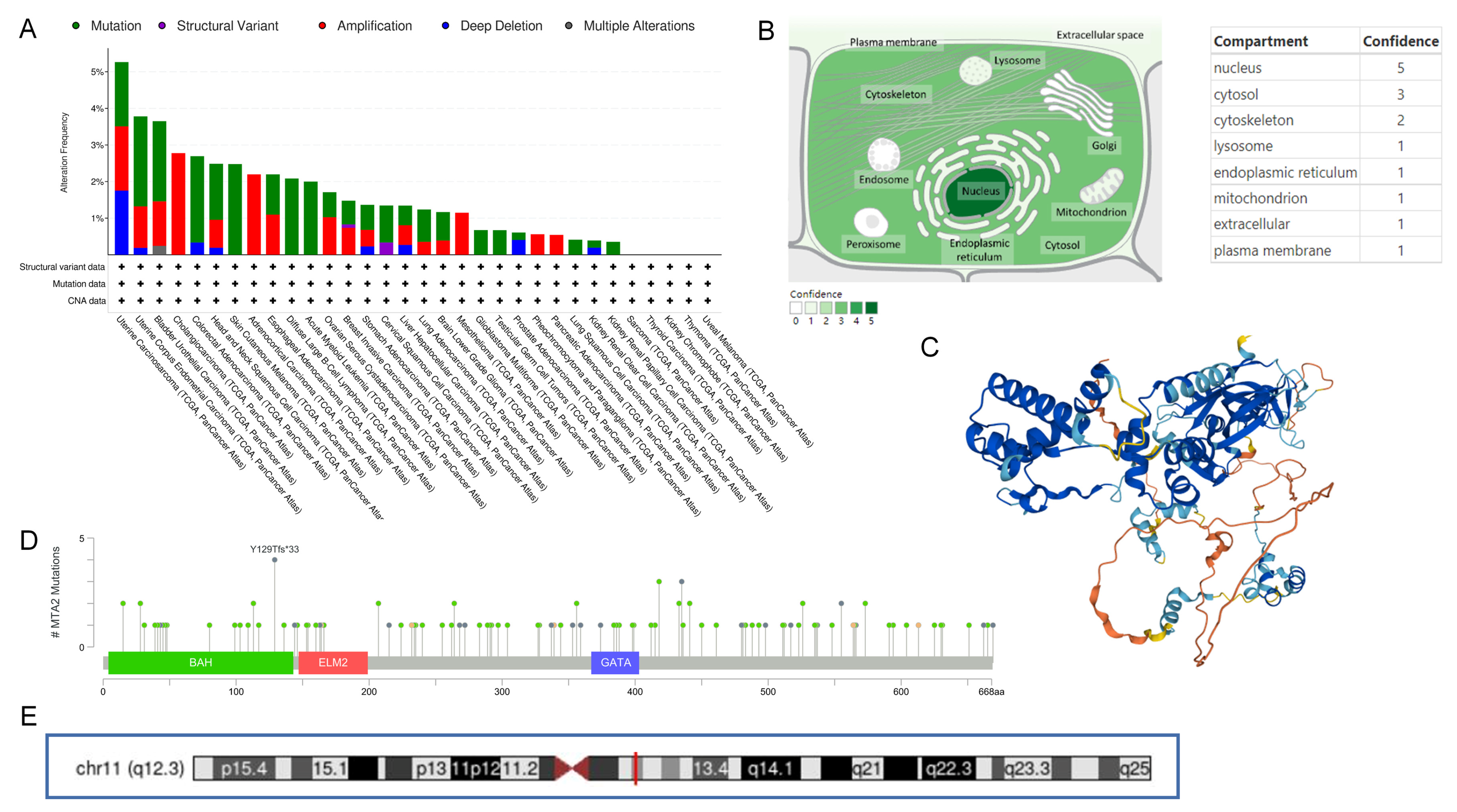
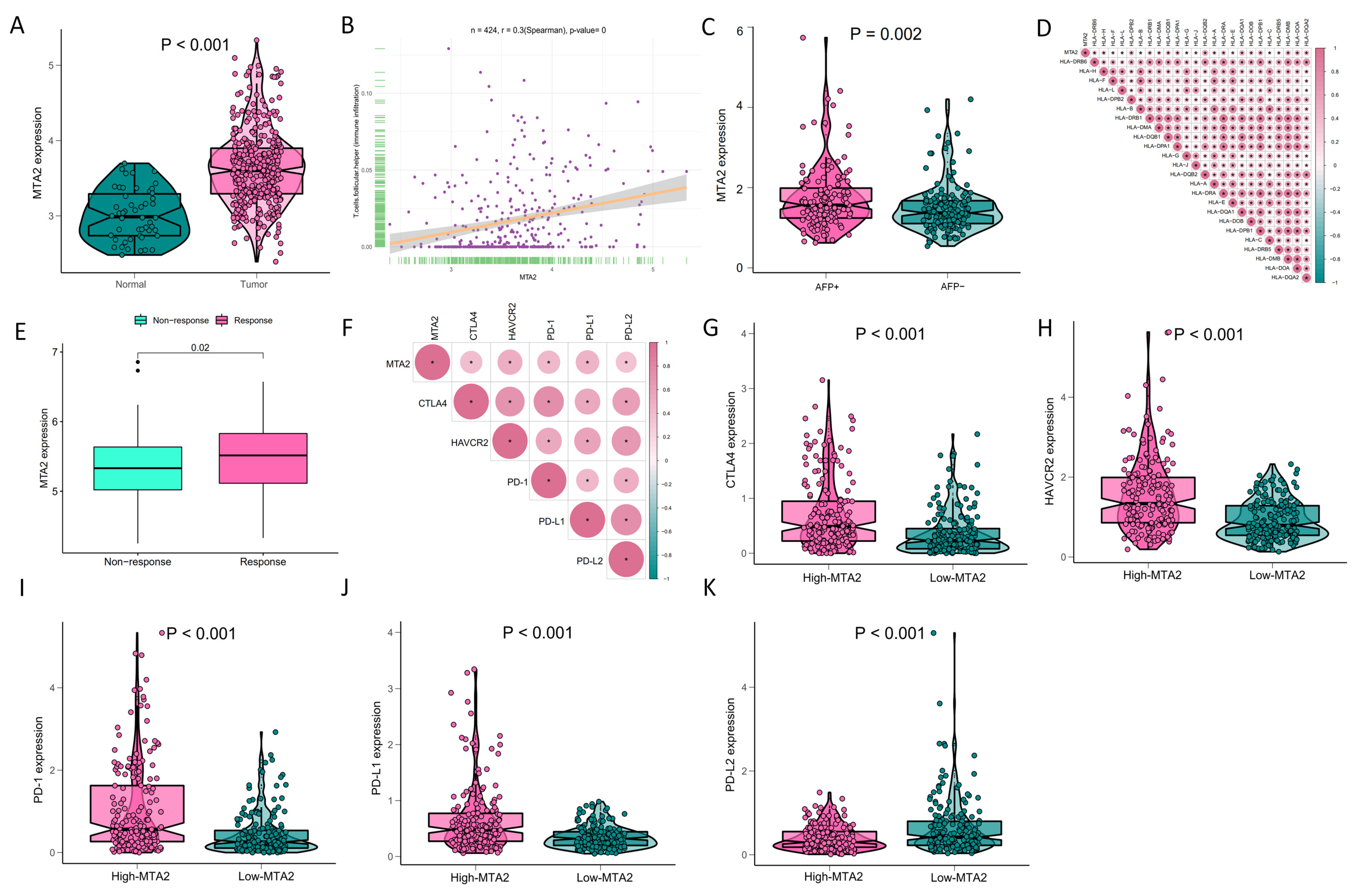
3.1. Differential Expression of MTA2 between Tumor and Normal Tissues in Pan-Cancer and Mutation of MTA2
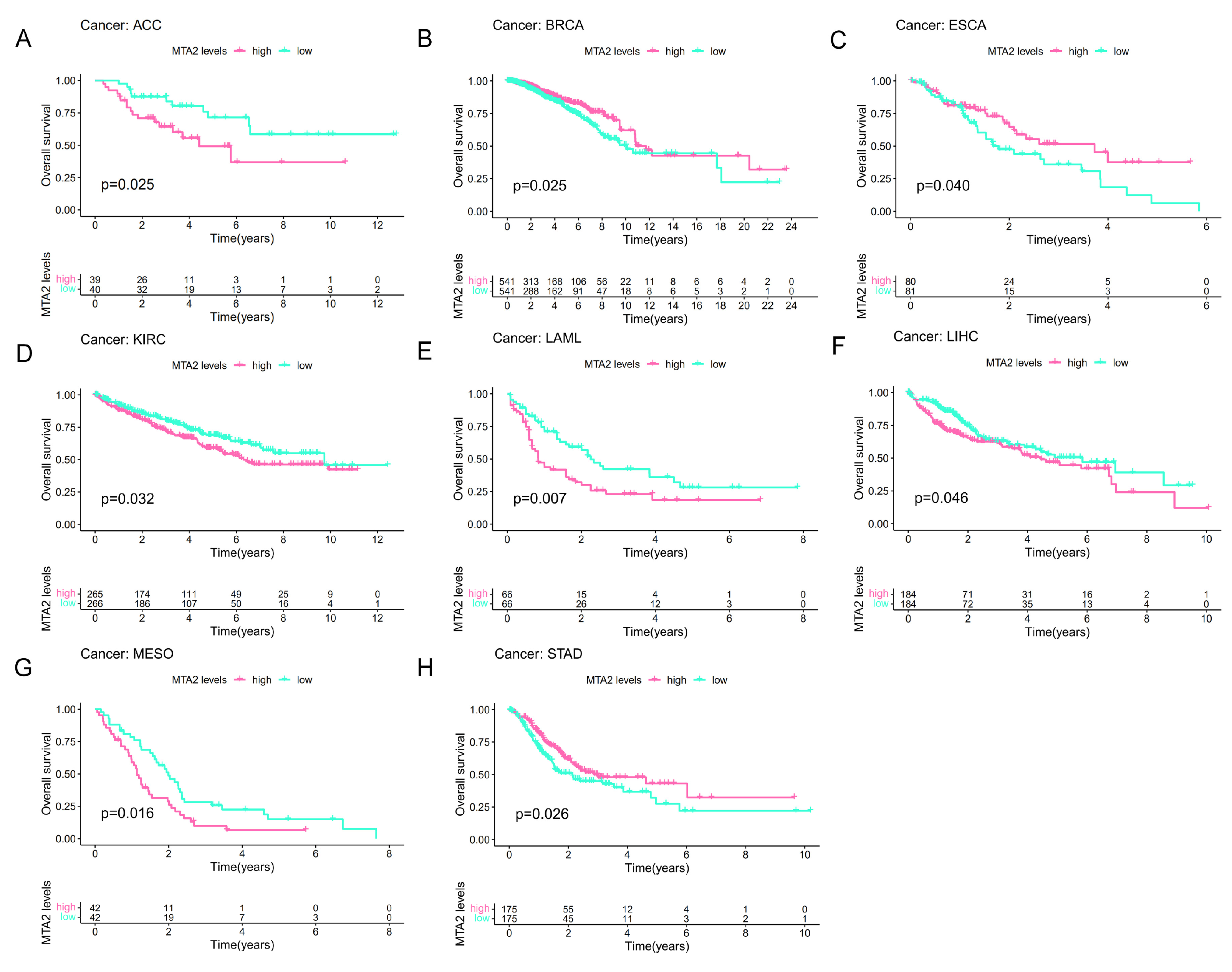
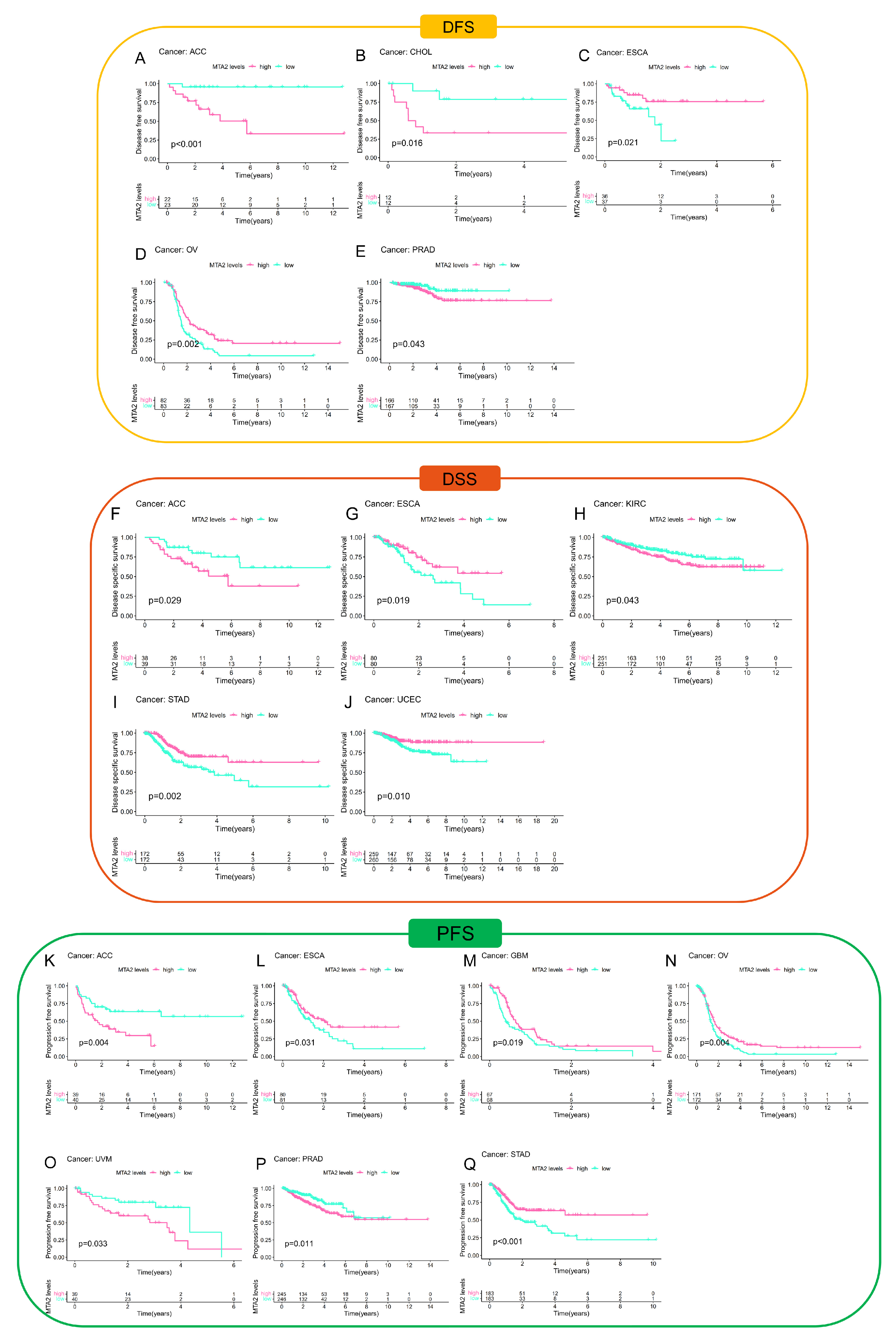
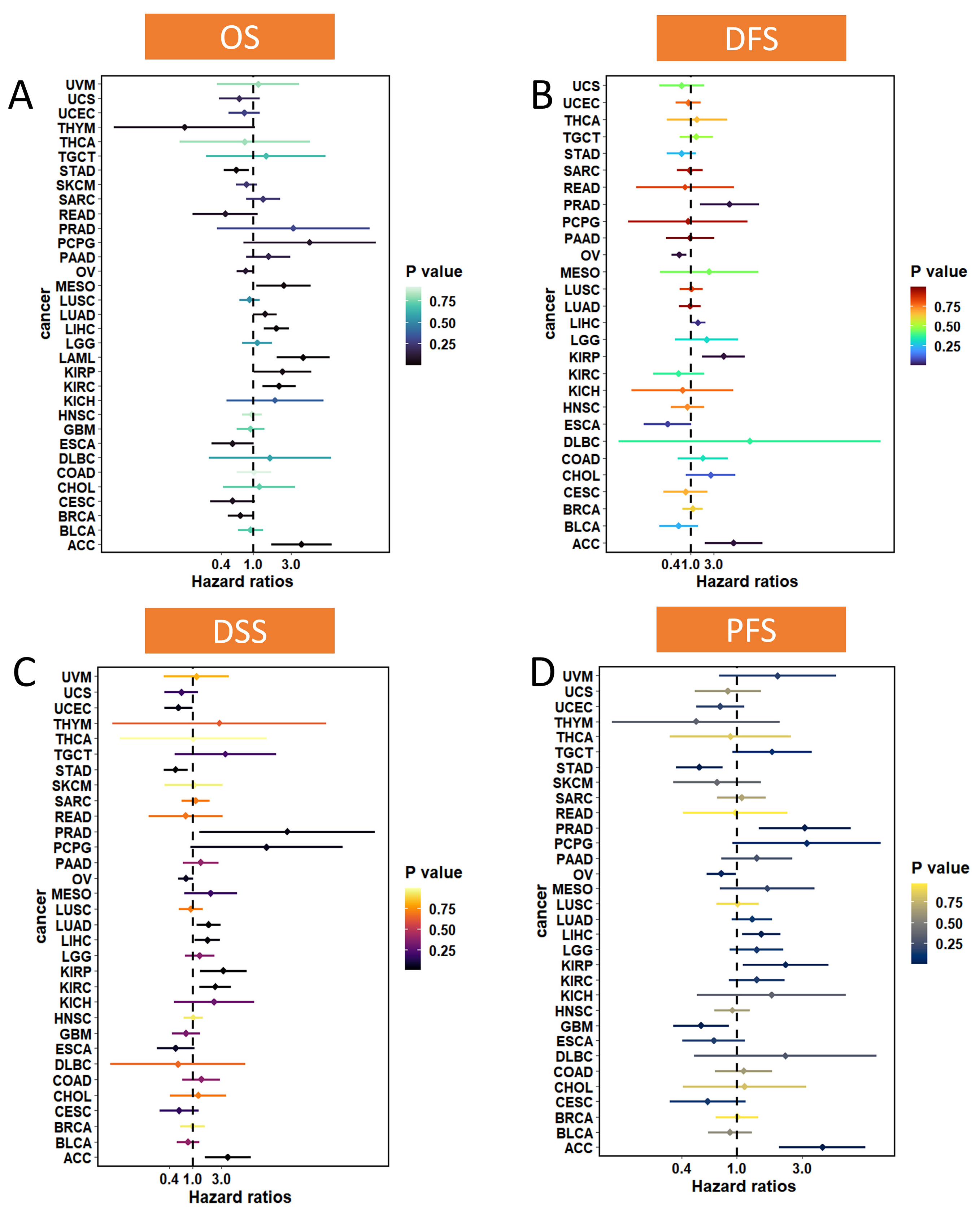
3.2. Prognostic Value of MTA2 in Pan-Cancer
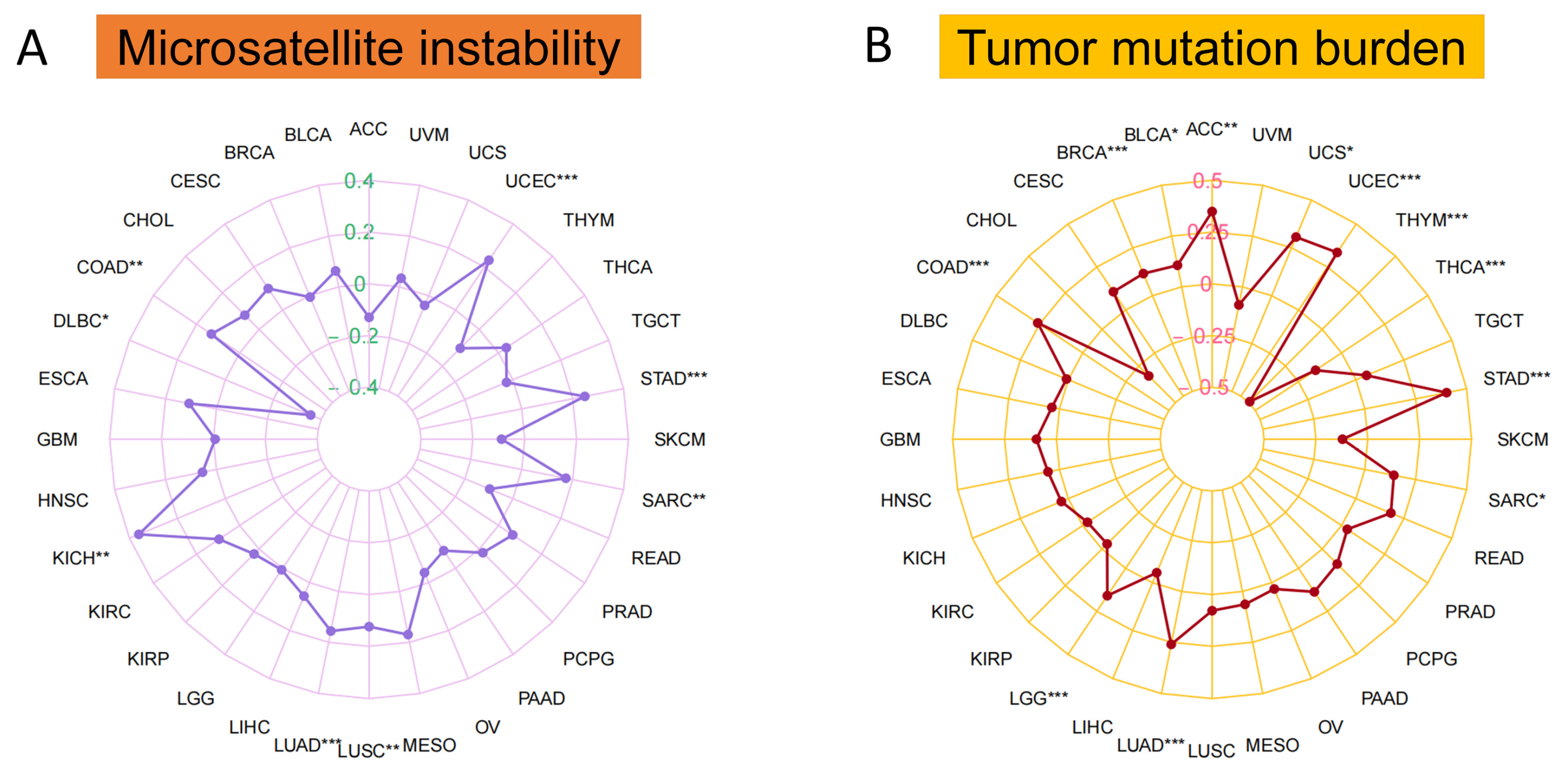
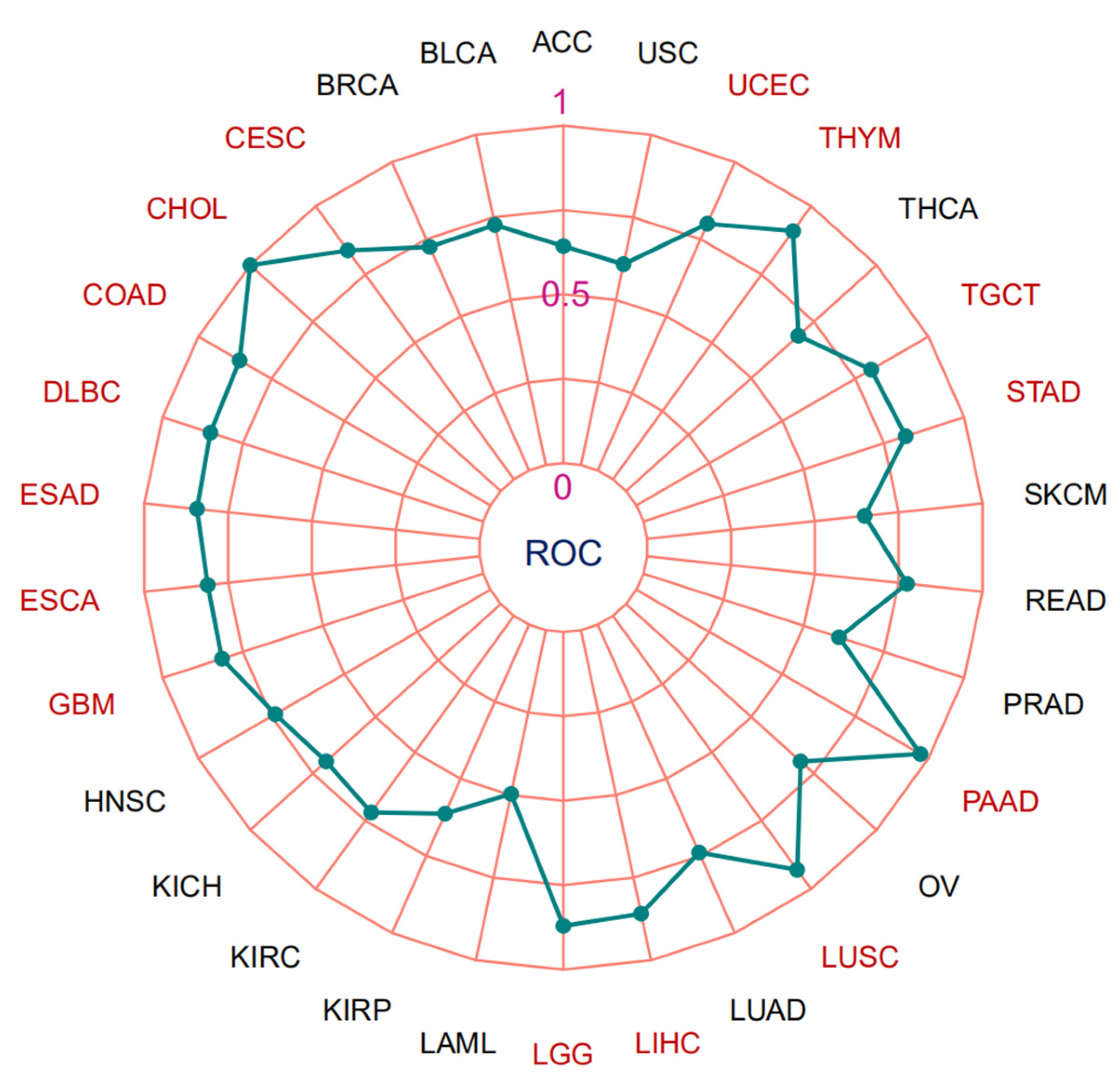
3.3. Microsatellite Instability, Tumor Mutation Burden and Area under the Curve (AUC) Values of MTA2 in Pan-Cancer
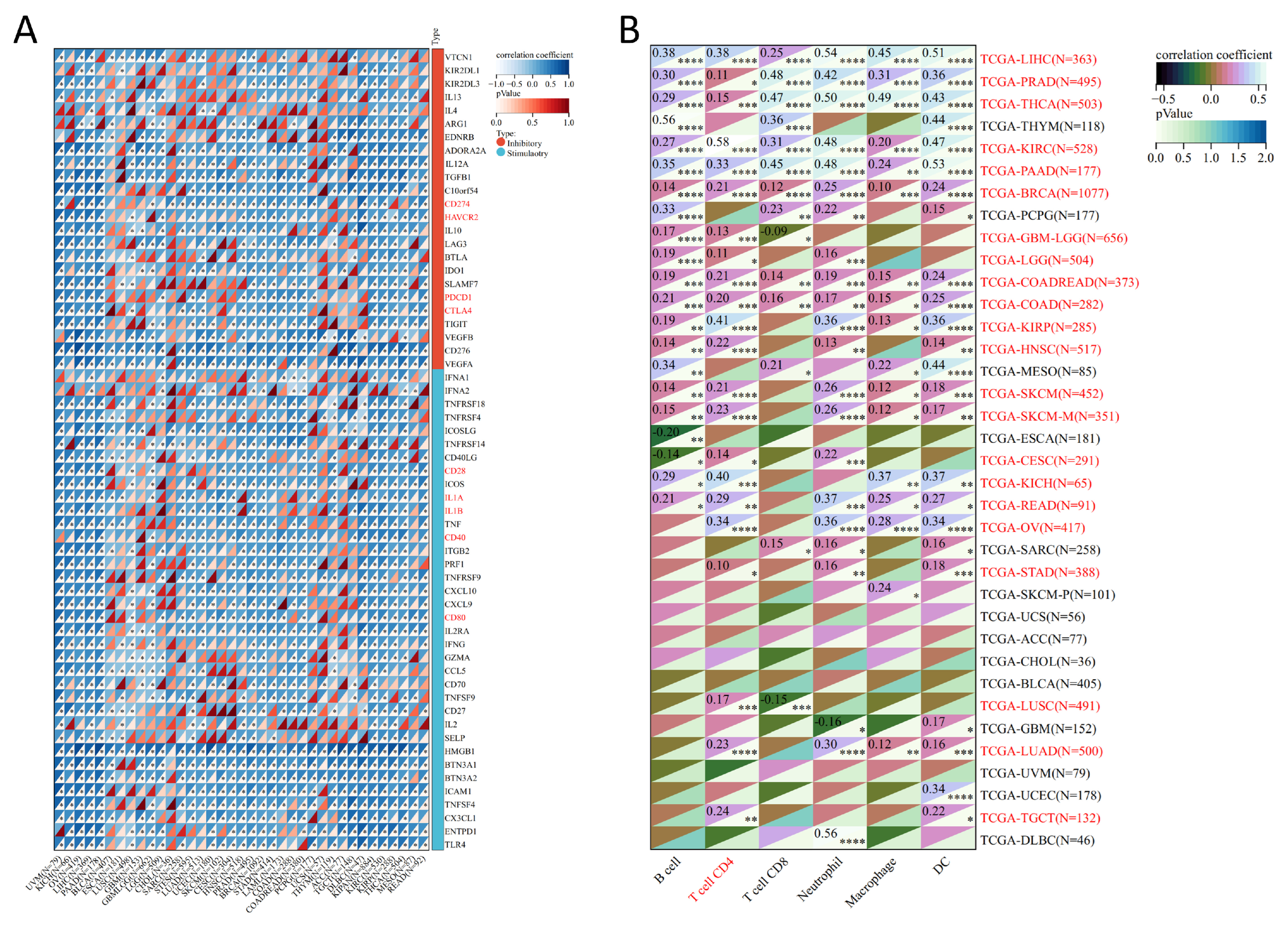
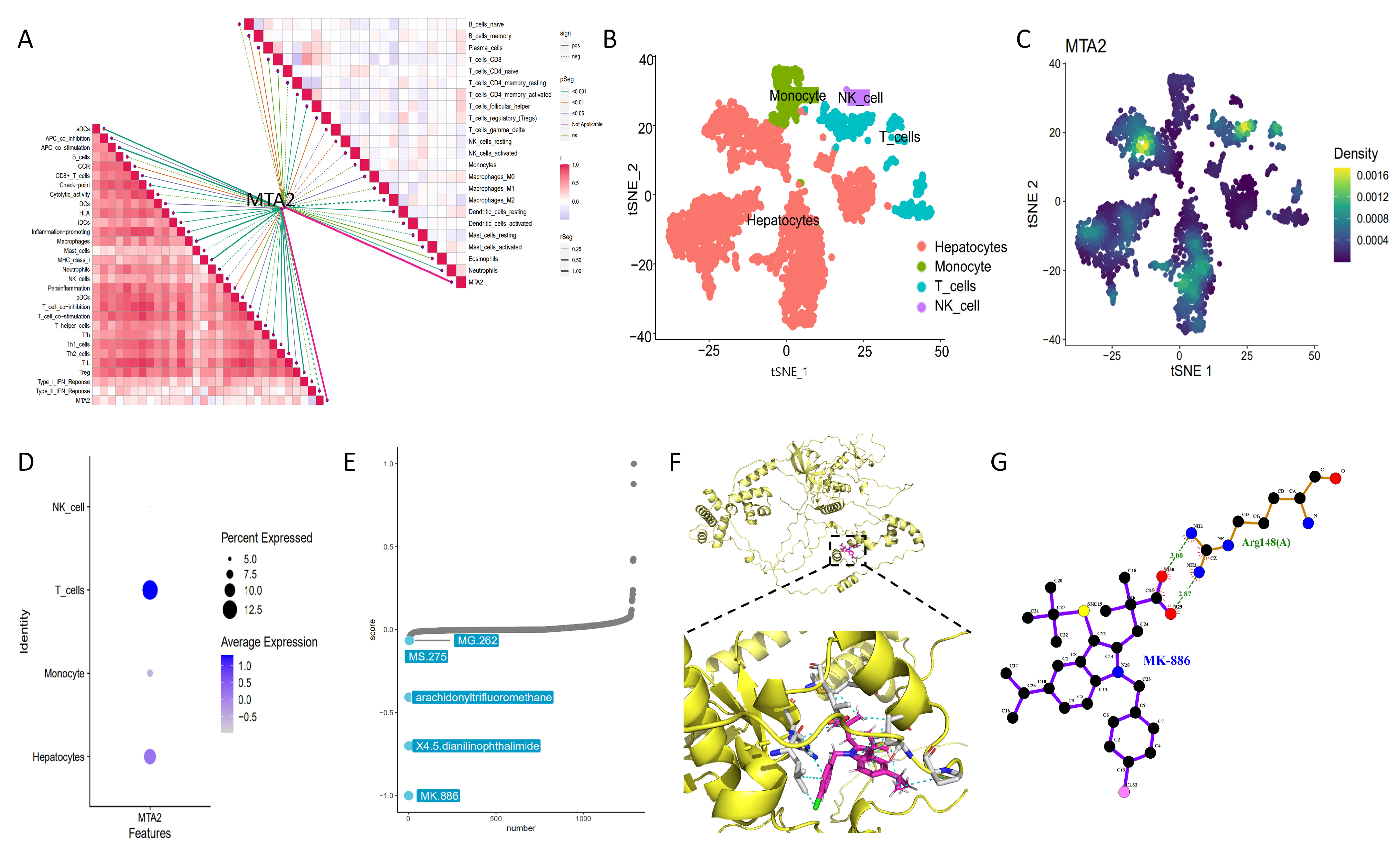
3.4. Analysis of Genes Co-Expressed with MTA2 in LIHC, Pan-Cancer Extent of Immune Infiltration and Enrichment Analysis
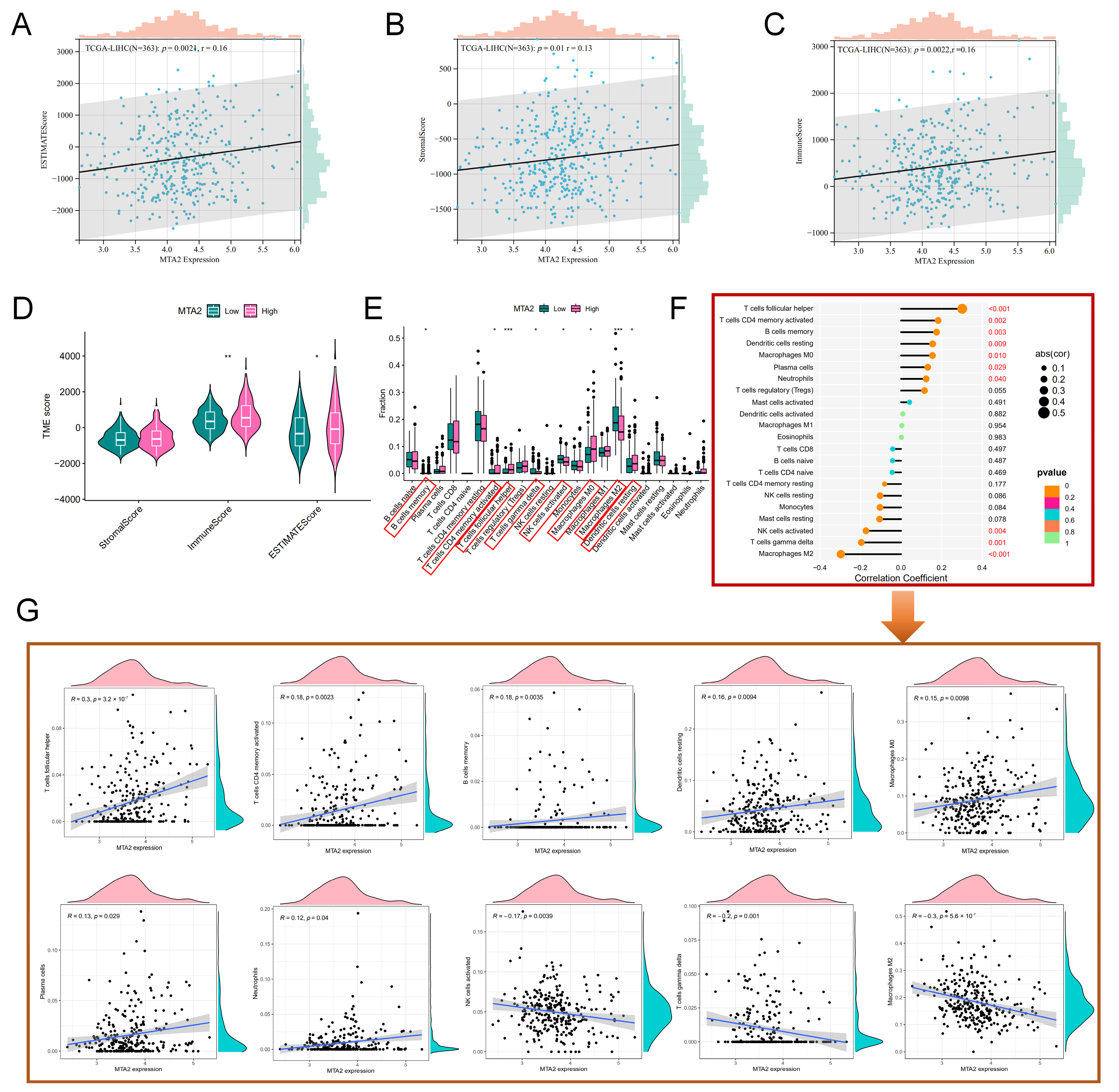
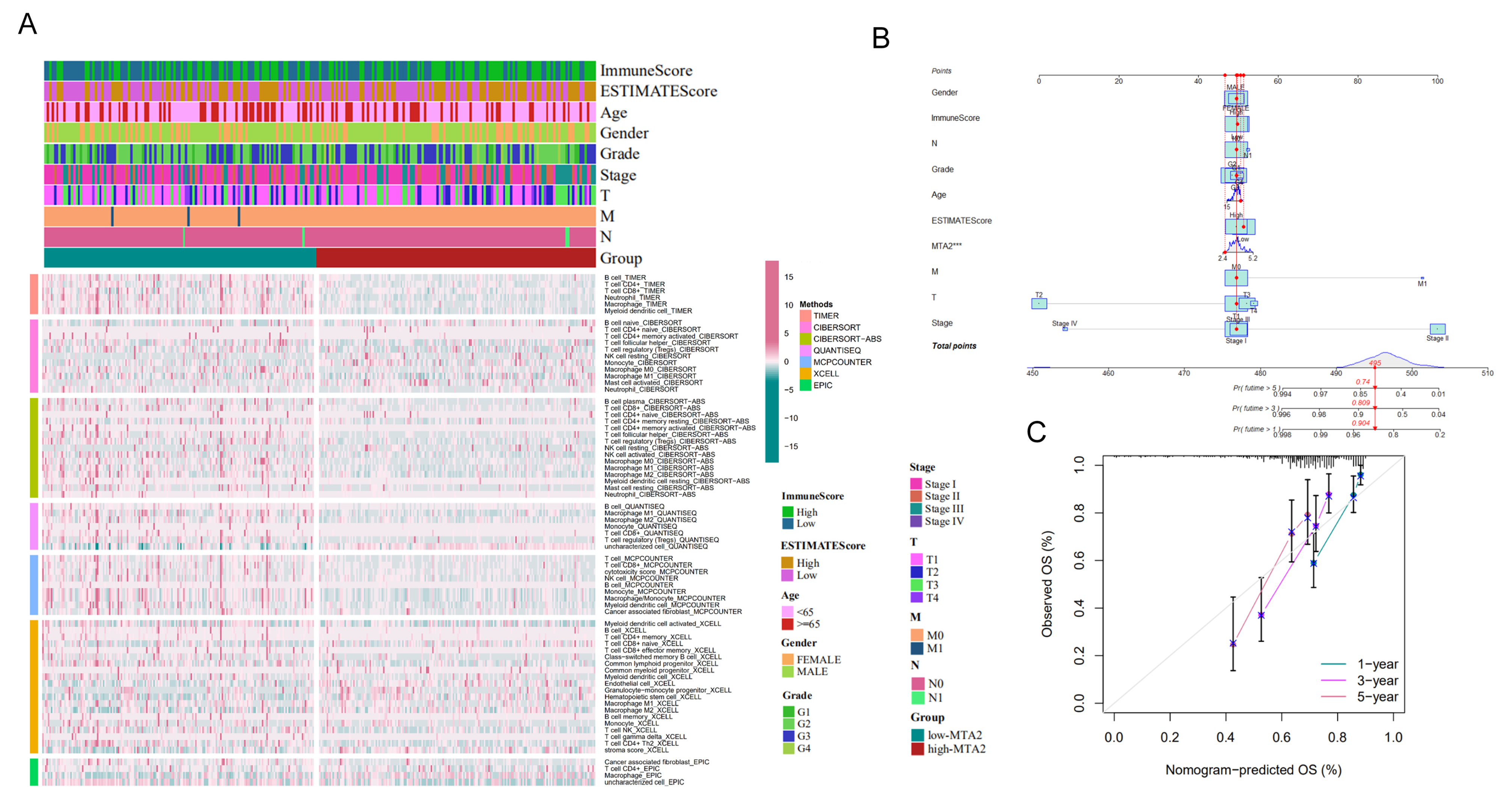
3.5. Analysis of High-MTA2 and Low-MTA2 Groups in LIHC with Immune Checkpoints, TME Score, IPS and Correlation with Immune Cells
3.6. Single Cell Sequencing and Drug Therapy in HCC
3.7. Molecular Docking
3.8. Construction of the Nomogram and Validation on the Basis of Clinical Information in LIHC
3.9. Analysis of Mutations in High and Low MTA2 Expression Groups in HCC
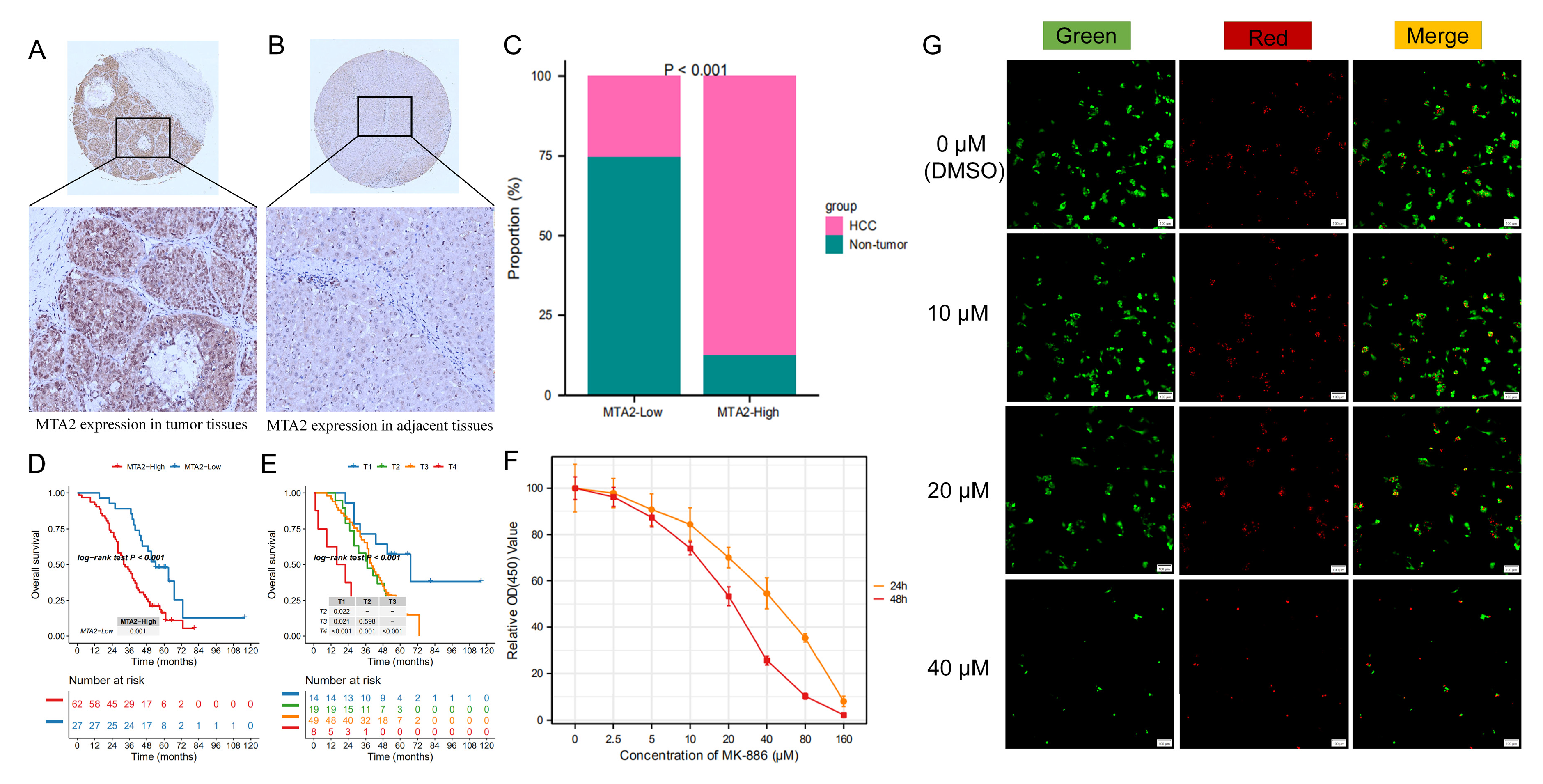
3.10. MTA2 Protein Is Overexpressed and Associated with Poor Prognosis in Human HCC
3.11. Growth Inhibition Effects of MK-886 on Proliferation of HCC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviations | Full Name |
| ACC | Adrenocortical Cancer |
| ATP8A2 | ATPase phospholipid transporting 8A2 |
| BLCA | Bladder Cancer |
| BRCA | Breast Cancer |
| CHD4 | Chromodomain helicase DNA binding protein 4 |
| CLTC | Clathrin heavy chain |
| COL7A1 | Collagen type VII alpha 1 chain |
| CTNNA2 | Catenin alpha 2 |
| CESC | Cervical Cancer |
| CHOL | Bile Duct Cancer |
| COAD | Colon Cancer |
| DCs | Dendritic cells |
| DNAH10 | Dynein axonemal heavy chain 10 |
| DNAH14 | Dynein axonemal heavy chain 14 |
| DLBC | Large B-cell Lymphoma |
| ESCA | Esophageal Cancer |
| FAM47A | Family with sequence similarity 47 member A |
| GBM | Glioblastoma |
| GEO | Gene Expression Omnibus |
| GTEx | Genotype-Tissue Expression Project |
| HAVCR2 | Hepatitis A virus cellular receptor 2 |
| HDAC2 | Histone deacetylase 2 |
| HNSC | Head and Neck Cancer |
| IL6ST | Interleukin 6 cytokine family signal transducer |
| KICH | Kidney Chromophobe |
| KIRC | Kidney Clear Cell Carcinoma |
| KIRP | Kidney Papillary Cell Carcinoma |
| KPRP | Keratinocyte proline rich protein |
| LRP1B | LDL receptor related protein 1B |
| LAML | Acute Myeloid Leukemia |
| LGG | Lower Grade Glioma |
| LIHC | Liver hepatocellular carcinoma |
| LUAD | Lung Adenocarcinoma |
| LUSC | Lung Squamous Cell Carcinoma |
| MBD3 | Methyl-CpG binding domain protein 3 |
| MESO | Mesothelioma |
| MSigDB | Molecular Signatures Database |
| NBEA | Neurobeachin |
| NK cells | Natural killer cells |
| NLRP12 | NLR family pyrin domain containing 12 |
| OBSCN | Bscurin, cytoskeletal calmodulin and titin-interacting RhoGEF |
| OV | Ovarian Cancer |
| PAAD | Pancreatic Cancer |
| PCPG | Pheochromocytoma & Paraganglioma |
| PRAD | Prostate Cancer |
| RB1 | RB transcriptional corepressor 1 |
| READ | Rectal Cancer |
| SYCP2 | Synaptonemal complex protein 2 |
| SI | Sucrase-isomaltase |
| SPEG | Striated muscle enriched protein kinase |
| SARC | Sarcoma |
| SKCM | Skin Cutaneous Melanoma |
| STAD | Stomach Cancer |
| TSC2 | TSC complex subunit 2 |
| TP53 | Tumor protein p53 |
| TCGA | The Cancer Genome Atlas |
| TCGT | Testicular Cancer |
| TCIA | The Cancer Imaging Archive |
| THCA | Thyroid Cancer |
| TENM2 | Teneurin transmembrane protein 2 |
| THYM | Thymoma |
| UCEC | Uterine Corpus Endometrial Carcinoma |
| UCS | Uterine Carcinosarcoma |
| UCSC | University of California Santa Cruz |
| UVM | Uveal Melanoma |
References
- Cao, W.; Chen, H.-D.; Yu, Y.-W.; Li, N.; Chen, W.-Q. Changing Profiles of Cancer Burden Worldwide and in China: A Secondary Analysis of the Global Cancer Statistics 2020. Chin. Med. J. 2021, 134, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wei, C. Advances in the Early Diagnosis of Hepatocellular Carcinoma. Genes Dis. 2020, 7, 308–319. [Google Scholar] [CrossRef]
- Wu, W.-T.; Li, Y.-J.; Feng, A.-Z.; Li, L.; Huang, T.; Xu, A.-D.; Lyu, J. Data Mining in Clinical Big Data: The Frequently Used Databases, Steps, and Methodological Models. Mil. Med. Res. 2021, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer Immunotherapy Using Checkpoint Blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Wu, Y.; Yu, S.; Qiao, H. Understanding the Functional Inflammatory Factors Involved in Therapeutic Response to Immune Checkpoint Inhibitors for Pan-Cancer. Front. Pharmacol. 2022, 13, 990445. [Google Scholar] [CrossRef] [PubMed]
- Jardim, D.L.; Goodman, A.; de Melo Gagliato, D.; Kurzrock, R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell 2021, 39, 154–173. [Google Scholar] [CrossRef] [PubMed]
- Mu, Q.; Najafi, M. Modulation of the Tumor Microenvironment (Tme) by Melatonin. Eur. J. Pharmacol. 2021, 907, 174365. [Google Scholar] [CrossRef]
- Bejarano, L.; Jordāo, M.J.C.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell 2020, 78, 1019–1033. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Cancer Metabolism: Looking Forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef]
- Henriksen, A.; Dyhl-Polk, A.; Chen, I.; Nielsen, D. Checkpoint Inhibitors in Pancreatic Cancer. Cancer Treat. Rev. 2019, 78, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Borch, T.H.; Donia, M.; Andersen, M.H.; Svane, I.M. Reorienting the Immune System in the Treatment of Cancer by Using Anti-Pd-1 and Anti-Pd-L1 Antibodies. Drug Discov. Today 2015, 20, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Hutchinson, R.; Wong, H.-L.; Tie, J.; Putoczki, T.; Tran, B.; Gibbs, P.; Christie, M. Emerging Biomarkers for Immunomodulatory Cancer Treatment of Upper Gastrointestinal, Pancreatic and Hepatic Cancers. Semin. Cancer Biol. 2018, 52 Pt 2, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Schildberg, F.A.; Klein, S.R.; Freeman, G.J.; Sharpe, A.H. Coinhibitory Pathways in the B7-Cd28 Ligand-Receptor Family. Immunity 2016, 44, 955–972. [Google Scholar] [CrossRef]
- Fu, J.; Wang, H. Precision Diagnosis and Treatment of Liver Cancer in China. Cancer Lett. 2018, 412, 283–288. [Google Scholar] [CrossRef]
- Suhail, Y.; Cain, M.P.; Vanaja, K.; Kurywchak, P.A.; Levchenko, A.; Kalluri, R. Systems Biology of Cancer Metastasis. Cell Syst. 2019, 9, 109–127. [Google Scholar] [CrossRef]
- Covington, K.R.; Fuqua, S.A.W. Role of Mta2 in Human Cancer. Cancer Metastasis Rev. 2014, 33, 921–928. [Google Scholar] [CrossRef]
- Kumar, R.; Wang, R.-A. Structure, Expression and Functions of Mta Genes. Gene 2016, 582, 112–121. [Google Scholar] [CrossRef]
- Lee, H.; Ryu, S.H.; Hong, S.S.; Seo, D.D.; Min, H.J.; Jang, M.K.; Kwon, H.J.; Yu, E.; Chung, Y.-H.; Kim, K.-W. Overexpression of Metastasis-Associated Protein 2 Is Associated with Hepatocellular Carcinoma Size and Differentiation. J. Gastroenterol. Hepatol. 2009, 24, 1445–1450. [Google Scholar] [CrossRef]
- Guan, C.; Chang, Z.; Gu, X.; Liu, R. Mta2 Promotes Hcc Progression through Repressing Frmd6, a Key Upstream Component of Hippo Signaling Pathway. Biochem. Biophys. Res. Commun. 2019, 515, 112–118. [Google Scholar] [CrossRef]
- Si, W.; Liu, X.; Wei, R.; Zhang, Y.; Zhao, Y.; Cui, L.; Hong, T. Mta2-Mediated Inhibition of Pten Leads to Pancreatic Ductal Adenocarcinoma Carcinogenicity. Cell Death Dis. 2019, 10, 206. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-S.; Hung, T.-W.; Su, S.-C.; Lin, C.-L.; Yang, S.-F.; Lee, C.-C.; Yeh, C.-F.; Hsieh, Y.-H.; Tsai, J.-P. Mta2 as a Potential Biomarker and Its Involvement in Metastatic Progression of Human Renal Cancer by Mir-133b Targeting Mmp-9. Cancers 2019, 11, 1851. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Wei, S.; Zhang, C.; Li, X.; Liu, Y.; Ma, M.; Lv, H.; Zhang, Z.; Zhao, L.; Shan, B. Mta2 Promotes the Metastasis of Esophageal Squamous Cell Carcinoma Via Eif4e-Twist Feedback Loop. Cancer Sci. 2021, 112, 1060–1074. [Google Scholar] [CrossRef]
- Zhou, C.; Ji, J.; Cai, Q.; Shi, M.; Chen, X.; Yu, Y.; Liu, B.; Zhu, Z.; Zhang, J. Mta2 Promotes Gastric Cancer Cells Invasion and Is Transcriptionally Regulated by Sp1. Mol. Cancer 2013, 12, 102. [Google Scholar] [CrossRef]
- Hsu, W.-H.; Chiou, H.-L.; Lin, C.-L.; Kao, S.-H.; Lee, H.-L.; Liu, C.-J.; Hsieh, Y.-H. Metastasis-Associated Protein 2 Regulates Human Hepatocellular Carcinoma Metastasis Progression through Modulating P38mapk/Mmp2 Pathways. J. Cancer 2019, 10, 6716–6725. [Google Scholar] [CrossRef] [PubMed]
- Blum, A.; Wang, P.; Zenklusen, J.C. Snapshot: Tcga-Analyzed Tumors. Cell 2018, 173, 530. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The Cbio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. Ncbi Geo: Archive for Functional Genomics Data Sets—Update. Nucleic Acids Res. 2013, 41, D991–D9955. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (Msigdb) Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. Pubchem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Linde, M.E.; Colquhoun, D.R.; Mohien, C.U.; Kole, T.; Aquino, V.; Cotter, R.; Edwards, N.; Hildreth, J.E.; Graham, D.R. The Conserved Set of Host Proteins Incorporated into Hiv-1 Virions Suggests a Common Egress Pathway in Multiple Cell Types. J. Proteome Res. 2013, 12, 2045–2054. [Google Scholar] [CrossRef]
- Subbannayya, Y.; Pinto, S.M.; Bösl, K.; Prasad, T.S.K.; Kandasamy, R.K. Dynamics of Dual Specificity Phosphatases and Their Interplay with Protein Kinases in Immune Signaling. Int. J. Mol. Sci. 2019, 20, 86. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Imai, K. Microsatellite Instability: An Update. Arch. Toxicol. 2015, 89, 899–921. [Google Scholar] [CrossRef] [PubMed]
- Palmeri, M.; Mehnert, J.; Silk, A.; Jabbour, S.; Ganesan, S.; Popli, P.; Riedlinger, G.; Stephenson, R.; de Meritens, A.; Leiser, A.; et al. Real-World Application of Tumor Mutational Burden-High (Tmb-High) and Microsatellite Instability (Msi) Confirms Their Utility as Immunotherapy Biomarkers. ESMO Open 2022, 7, 100336. [Google Scholar] [CrossRef] [PubMed]
- Schober, P.; Boer, C.; Schwarte, L.A. Correlation Coefficients: Appropriate Use and Interpretation. Anesth. Analg. 2018, 126, 1763–1768. [Google Scholar] [CrossRef]
- Xu, Q.; Xu, H.; Deng, R.; Wang, Z.; Li, N.; Qi, Z.; Zhao, J.; Huang, W. Multi-Omics Analysis Reveals Prognostic Value of Tumor Mutation Burden in Hepatocellular Carcinoma. Cancer Cell Int. 2021, 21, 342. [Google Scholar] [CrossRef]
- Yi, M.; Li, A.; Zhou, L.; Chu, Q.; Luo, S.; Wu, K. Immune Signature-Based Risk Stratification and Prediction of Immune Checkpoint Inhibitor’s Efficacy for Lung Adenocarcinoma. Cancer Immunol. Immunother. 2021, 70, 1705–1719. [Google Scholar] [CrossRef]
- Sedgwick, P. Spearman’s Rank Correlation Coefficient. BMJ 2018, 362, k4131. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, H.; Chen, M.; Wang, S.; Qian, R.; Zhang, L.; Huang, X.; Wang, J.; Liu, Z.; Qin, W.; et al. A Survey of Optimal Strategy for Signature-Based Drug Repositioning and an Application to Liver Cancer. eLife 2022, 11, e71880. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Sun, Y.; Xu, F.; Liang, Y.; Liu, H.; Yi, Y. Annexin A2 Silencing Inhibits Proliferation and Epithelial-to-Mesenchymal Transition through P53-Dependent Pathway in Nsclcs. J. Cancer 2019, 10, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Fitzgerald, R.C.; Antoniou, A.C.; Fruk, L.; Rosenfeld, N. The Future of Early Cancer Detection. Nat. Med. 2022, 28, 666–677. [Google Scholar] [CrossRef]
- Cheng, C.-Y.; Chou, Y.-E.; Ko, C.-P.; Yang, S.-F.; Hsieh, S.-C.; Lin, C.-L.; Hsieh, Y.-H.; Chen, K.-C. Metastasis Tumor-Associated Protein-2 Knockdown Suppresses the Proliferation and Invasion of Human Glioma Cells in Vitro and in Vivo. J. Neurooncol. 2014, 120, 273–281. [Google Scholar] [CrossRef]
- El-Nikhely, N.; Karger, A.; Sarode, P.; Singh, I.; Weigert, A.; Wietelmann, A.; Stiewe, T.; Dammann, R.; Fink, L.; Grimminger, F.; et al. Metastasis-Associated Protein 2 Represses Nf-Κb to Reduce Lung Tumor Growth and Inflammation. Cancer Res. 2020, 80, 4199–4211. [Google Scholar] [CrossRef]
- Isaksson, H.S.; Sorbe, B.; Nilsson, T.K. Whole Blood Rna Expression Profiles in Ovarian Cancer Patients with or without Residual Tumors after Primary Cytoreductive Surgery. Oncol. Rep. 2012, 27, 1331–1335. [Google Scholar] [CrossRef]
- Ji, Y.; Zhang, P.; Lu, Y.; Ma, D. Expression of Mta2 Gene in Ovarian Epithelial Cancer and Its Clinical Implication. J. Huazhong Univ. Sci. Technolo. Med. Sci. 2006, 26, 359–362. [Google Scholar]
- Lin, C.-L.; Ying, T.-H.; Yang, S.-F.; Chiou, H.-L.; Chen, Y.-S.; Kao, S.-H.; Hsieh, Y.-H. Mta2 Silencing Attenuates the Metastatic Potential of Cervical Cancer Cells by Inhibiting Ap1-Mediated Mmp12 Expression via the Ask1/Mek3/P38/Yb1 Axis. Cell Death Dis. 2021, 12, 451. [Google Scholar] [CrossRef]
- Lu, J.; Jin, M.-L. Short-Hairpin Rna-Mediated Mta2 Silencing Inhibits Human Breast Cancer Cell Line Mda-Mb231 Proliferation and Metastasis. Asian Pac. J. Cancer Prev. 2014, 15, 5577–5582. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhan, S.; Tan, W.; Cheng, R.; Gong, H.; Zhu, Q. P300 Binds to and Acetylates Mta2 to Promote Colorectal Cancer Cells Growth. Biochem. Biophys. Res. Commun. 2014, 444, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Calvo, A.; Xiao, N.; Kang, J.; Best, C.J.M.; Leiva, I.; Emmert-Buck, M.R.; Jorcyk, C.; Green, J.E. Alterations in Gene Expression Profiles During Prostate Cancer Progression: Functional Correlations to Tumorigenicity and Down-Regulation of Selenoprotein-P in Mouse and Human Tumors. Cancer Res. 2002, 62, 5325–5335. [Google Scholar] [PubMed]
- Luan, S.; Fu, P.; Wang, X.; Gao, Y.; Shi, K.; Guo, Y. Circular Rna Circ-Ncor2 Accelerates Papillary Thyroid Cancer Progression by Sponging Mir-516a-5p to Upregulate Metastasis-Associated Protein 2 Expression. J. Int. Med. Res. 2020, 48, 300060520934659. [Google Scholar] [CrossRef]
- Zhou, C.; Ji, J.; Cai, Q.; Shi, M.; Chen, X.; Yu, Y.; Zhu, Z.; Zhang, J. Mta2 Enhances Colony Formation and Tumor Growth of Gastric Cancer Cells through Il-11. BMC Cancer 2015, 15, 343. [Google Scholar] [CrossRef]
- Fumet, J.-D.; Truntzer, C.; Yarchoan, M.; Ghiringhelli, F. Tumour Mutational Burden as a Biomarker for Immunotherapy: Current Data and Emerging Concepts. Eur. J. Cancer 2020, 131, 40–50. [Google Scholar] [CrossRef]
- Steuer, C.E.; Ramalingam, S.S. Tumor Mutation Burden: Leading Immunotherapy to the Era of Precision Medicine? J. Clin. Oncol. 2018, 36, 631–632. [Google Scholar] [CrossRef]
- Chan, T.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.; Stenzinger, A.; Peters, S. Development of Tumor Mutation Burden as an Immunotherapy Biomarker: Utility for the Oncology Clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Lee, D.-W.; Han, S.-W.; Bae, J.M.; Jang, H.; Han, H.; Kim, H.; Bang, D.; Jeong, S.-Y.; Park, K.J.; Kang, G.H.; et al. Tumor Mutation Burden and Prognosis in Patients with Colorectal Cancer Treated with Adjuvant Fluoropyrimidine and Oxaliplatin. Clin. Cancer Res. 2019, 25, 6141–6147. [Google Scholar] [CrossRef]
- Xiao, Y.; Yu, D. Tumor Microenvironment as a Therapeutic Target in Cancer. Pharmacol. Ther. 2021, 221, 107753. [Google Scholar] [CrossRef]
- Zhang, H.; Chang, Z.; Qin, L.-N.; Liang, B.; Han, J.-X.; Qiao, K.-L.; Yang, C.; Liu, Y.-R.; Zhou, H.-G.; Sun, T. Mta2 Triggered R-Loop Trans-Regulates Bdh1-Mediated Β-Hydroxybutyrylation and Potentiates Propagation of Hepatocellular Carcinoma Stem Cells. Signal Transduct. Target. Ther. 2021, 6, 135. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Chu, C.-S.; Fang, T.; Rayon-Estrada, V.; Fang, F.; Patke, A.; Qian, Y.; Clarke, S.H.; Melnick, A.M.; Zhang, Y.; et al. Mta2/Nurd Regulates B Cell Development and Cooperates with Oca-B in Controlling the Pre-B to Immature B Cell Transition. Cell Rep. 2019, 28, 472–485.e5. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Kovalev, G.I.; Chang, H.; Kallin, E.; Knudsen, G.; Xia, L.; Mishra, N.; Ruiz, P.; Li, E.; Su, L.; et al. Inactivation of Nurd Component Mta2 Causes Abnormal T Cell Activation and Lupus-Like Autoimmune Disease in Mice. J. Biol. Chem. 2008, 283, 13825–13833. [Google Scholar] [CrossRef]
- Olgen, S. Overview on Anticancer Drug Design and Development. Curr. Med. Chem. 2018, 25, 1704–1719. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Zhang, Y.; Shang, Y.; Mai, J.; Shi, S.; Lu, M.; Bu, C.; Zhang, Z.; Zhang, Z.; Li, Y.; et al. Cancerscem: A Database of Single-Cell Expression Map across Various Human Cancers. Nucleic Acids Res. 2022, 50, D1147–D1155. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.M.; Alrefai, W.A.; Bonomi, P.A.; Anderson, C.A.; Dudeja, P.; Harris, J.E. A Genomic Response of H-358 Bronchiolar Carcinoma Cells to Mk 886, an Inhibitor of 5-Lipoxygenase, Assessed with a Cdna Array. Anticancer Res. 2000, 20, 2433–2439. [Google Scholar] [PubMed]
- Anderson, K.M.; Rubenstein, M.; Tsui, P.; Harris, J.E. Disparate Forms of Mk 886-Induced Programmed Death in Bcl-2 (+) Blood and Bcl-2 (-) Solid Cancer Cells and a Putative “Nuclear” Ca2+ Channel: “Soil” Trumps “Seed”? Med. Hypotheses 2005, 64, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Avis, I.; Martínez, A.; Tauler, J.; Zudaire, E.; Mayburd, A.; Abu-Ghazaleh, R.; Ondrey, F.; Mulshine, J.L. Inhibitors of the Arachidonic Acid Pathway and Peroxisome Proliferator-Activated Receptor Ligands Have Superadditive Effects on Lung Cancer Growth Inhibition. Cancer Res. 2005, 65, 4181–4190. [Google Scholar] [CrossRef]
- Cianchi, F.; Cortesini, C.; Magnelli, L.; Fanti, E.; Papucci, L.; Schiavone, N.; Messerini, L.; Vannacci, A.; Capaccioli, S.; Perna, F.; et al. Inhibition of 5-Lipoxygenase by Mk886 Augments the Antitumor Activity of Celecoxib in Human Colon Cancer Cells. Mol. Cancer Ther. 2006, 5, 2716–2726. [Google Scholar] [CrossRef]
- Ding, X.; Zhu, C.; Qiang, H.; Zhou, X.; Zhou, G. Enhancing Antitumor Effects in Pancreatic Cancer Cells by Combined Use of Cox-2 and 5-Lox Inhibitors. Biomed. Pharmacother. 2011, 65, 486–490. [Google Scholar] [CrossRef]
- Fan, X.M.; Tu, S.P.; Lam, S.K.; Wang, W.P.; Wu, J.; Wong, W.M.; Yuen, M.F.; Lin, M.C.M.; Kung, H.F.; Wong, B.C.-Y. Five-Lipoxygenase-Activating Protein Inhibitor Mk-886 Induces Apoptosis in Gastric Cancer through Upregulation of P27kip1 and Bax. J. Gastroenterol. Hepatol. 2004, 19, 31–37. [Google Scholar] [CrossRef]
- Przylipiak, A.; Hafner, J.; Przylipiak, J.; Köhn, F.M.; Runnebaum, B.; Rabe, T. Influence of 5-Lipoxygenase on in Vitro Growth of Human Mammary Carcinoma Cell Line Mcf-7. Gynecol. Obstet. Investig. 1998, 46, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Rioux, N.; Castonguay, A. Inhibitors of Lipoxygenase: A New Class of Cancer Chemopreventive Agents. Carcinogenesis 1998, 19, 1393–1400. [Google Scholar] [CrossRef]
- Runarsson, G.; Liu, A.; Mahshid, Y.; Feltenmark, S.; Pettersson, A.; Klein, E.; Björkholm, M.; Claesson, H.-E. Leukotriene B4 Plays a Pivotal Role in Cd40-Dependent Activation of Chronic B Lymphocytic Leukemia Cells. Blood 2005, 105, 1274–1279. [Google Scholar] [CrossRef]
- Stika, J.; Vondrácek, J.; Hofmanová, J.; Simek, V.; Kozubík, A. Mk-886 Enhances Tumour Necrosis Factor-Alpha-Induced Differentiation and Apoptosis. Cancer Lett. 2006, 237, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, W.; Wang, Q.; Zhang, X.; Ye, L. Lipid Metabolism Enzyme 5-Lox and Its Metabolite Ltb4 Are Capable of Activating Transcription Factor Nf-Κb in Hepatoma Cells. Biochem. Biophys. Res. Commun. 2012, 418, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Daglar, G.; Karaca, T.; Yuksek, Y.N.; Gozalan, U.; Akbiyik, F.; Sokmensuer, C.; Gurel, B.; Kama, N.A. Effect of Montelukast and Mk-886 on Hepatic Ischemia-Reperfusion Injury in Rats. J. Surg. Res. 2009, 153, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Matuschak, G.M.; Henry, K.A.; Johanns, C.A.; Lechner, A.J. Liver-Lung Interactions Following Escherichia Coli Bacteremic Sepsis and Secondary Hepatic Ischemia/Reperfusion Injury. Am. J. Respir. Crit. Care Med. 2001, 163, 1002–1009. [Google Scholar] [CrossRef] [PubMed]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer | CI | AUC |
|---|---|---|
| ACC (TCGA) | 0.555~0.731 | 0.643 |
| BLCA (TCGA) | 0.620~0.833 | 0.727 |
| BRCA (TCGA) | 0.687~0.763 | 0.725 |
| CESC (TCGA) | 0.766~0.909 | 0.838 |
| CHOL (TCGA) | 1.000~1.000 | 1.000 |
| COAD (TCGA) | 0.821~0.896 | 0.859 |
| DLBC (TCGA+GTEx) | 0.810~0.892 | 0.851 |
| ESAD (TCGA+GTEx) | 0.682~1.000 | 0.843 |
| ESCA (TCGA) | 0.639~0.984 | 0.811 |
| GBM (TCGA+GTEx) | 0.785~0.845 | 0.815 |
| HNSC (TCGA) | 0.666~0.808 | 0.737 |
| KICH (TCGA) | 0.586~0.809 | 0.697 |
| KIRC (TCGA) | 0.662~0.779 | 0.720 |
| KIRP (TCGA) | 0.525~0.701 | 0.613 |
| LAML (TCGA+GTEx) | 0.426~0.567 | 0.496 |
| LGG (TCGA+GTEx) | 0.854~0.887 | 0.871 |
| LIHC (TCGA) | 0.812~0.905 | 0.859 |
| LUAD (TCGA) | 0.690~0.787 | 0.739 |
| LUSC (TCGA) | 0.907~0.954 | 0.930 |
| OV (TCGA+GTEx) | 0.646~0.747 | 0.697 |
| PAAD (TCGA+GTEx) | 0.954~0.991 | 0.973 |
| PRAD (TCGA) | 0.528~0.692 | 0.610 |
| READ (TCGA) | 0.642~0.908 | 0.775 |
| SKCM (TCGA+GTEx) | 0.617~0.681 | 0.649 |
| STAD (TCGA) | 0.745~0.890 | 0.818 |
| TGCT (TCGA+GTEx) | 0.752~0.857 | 0.804 |
| THCA (TCGA) | 0.627~0.750 | 0.688 |
| THYM (TCGA+GTEx) | 0.885~0.932 | 0.909 |
| UCEC (TCGA) | 0.749~0.850 | 0.800 |
| USC (TCGA+GTEx) | 0.504~0.711 | 0.608 |
| Characteristics | Number of Patients (n = 89) | MTA2 Expression | p-Value | |
|---|---|---|---|---|
| Low (n = 27) | High (n = 62) | |||
| Age | ||||
| <50 | 32 (36.0%) | 12 (13.5%) | 20 (22.5%) | |
| ≥50 | 57 (64.0%) | 15 (16.9%) | 42 (47.2%) | 0.389 |
| Gender | ||||
| Female | 11 (12.4%) | 1 (1.1%) | 10 (11.2%) | |
| Male | 78 (87.6%) | 26 (29.2%) | 52 (58.4%) | 0.198 |
| Clinical stage | ||||
| I + II | 33 (37.1%) | 14 (15.7%) | 19 (21.4%) | |
| III + IV | 56 (62.9%) | 13 (14.6%) | 43 (48.3%) | 0.096 |
| Tumor | ||||
| T1 + T2 | 33 (37.1%) | 14 (15.7%) | 19 (21.4%) | |
| T3 + T4 | 56 (62.9%) | 13 (14.6%) | 43 (48.3%) | 0.096 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, X.; Tan, J.; Chen, M.; Zheng, W.; Zou, S.; Ye, X.; Li, Y.; Wu, M. Prognostic, Immunological, and Mutational Analysis of MTA2 in Pan-Cancer and Drug Screening for Hepatocellular Carcinoma. Biomolecules 2023, 13, 883. https://doi.org/10.3390/biom13060883
Huang X, Tan J, Chen M, Zheng W, Zou S, Ye X, Li Y, Wu M. Prognostic, Immunological, and Mutational Analysis of MTA2 in Pan-Cancer and Drug Screening for Hepatocellular Carcinoma. Biomolecules. 2023; 13(6):883. https://doi.org/10.3390/biom13060883
Chicago/Turabian StyleHuang, Xueshan, Jingyi Tan, Mei Chen, Weirang Zheng, Shanyang Zou, Xiaoxia Ye, Yutong Li, and Minhua Wu. 2023. "Prognostic, Immunological, and Mutational Analysis of MTA2 in Pan-Cancer and Drug Screening for Hepatocellular Carcinoma" Biomolecules 13, no. 6: 883. https://doi.org/10.3390/biom13060883
APA StyleHuang, X., Tan, J., Chen, M., Zheng, W., Zou, S., Ye, X., Li, Y., & Wu, M. (2023). Prognostic, Immunological, and Mutational Analysis of MTA2 in Pan-Cancer and Drug Screening for Hepatocellular Carcinoma. Biomolecules, 13(6), 883. https://doi.org/10.3390/biom13060883