

Strain-Dependent Morphology of Reactive Astrocytes in Human- and Animal-Vole-Adapted Prions
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. Prion Strains
2.3. Samples
2.4. Neuropathology and Lesion Profile
2.5. Paraffin-Embedded Tissue Blot (PET-Blot)
2.6. Immunohistochemistry
2.7. Immunofluorescence
2.8. Cellular Area Measurement with Fiji Software
2.9. Statistical Analysis
3. Results
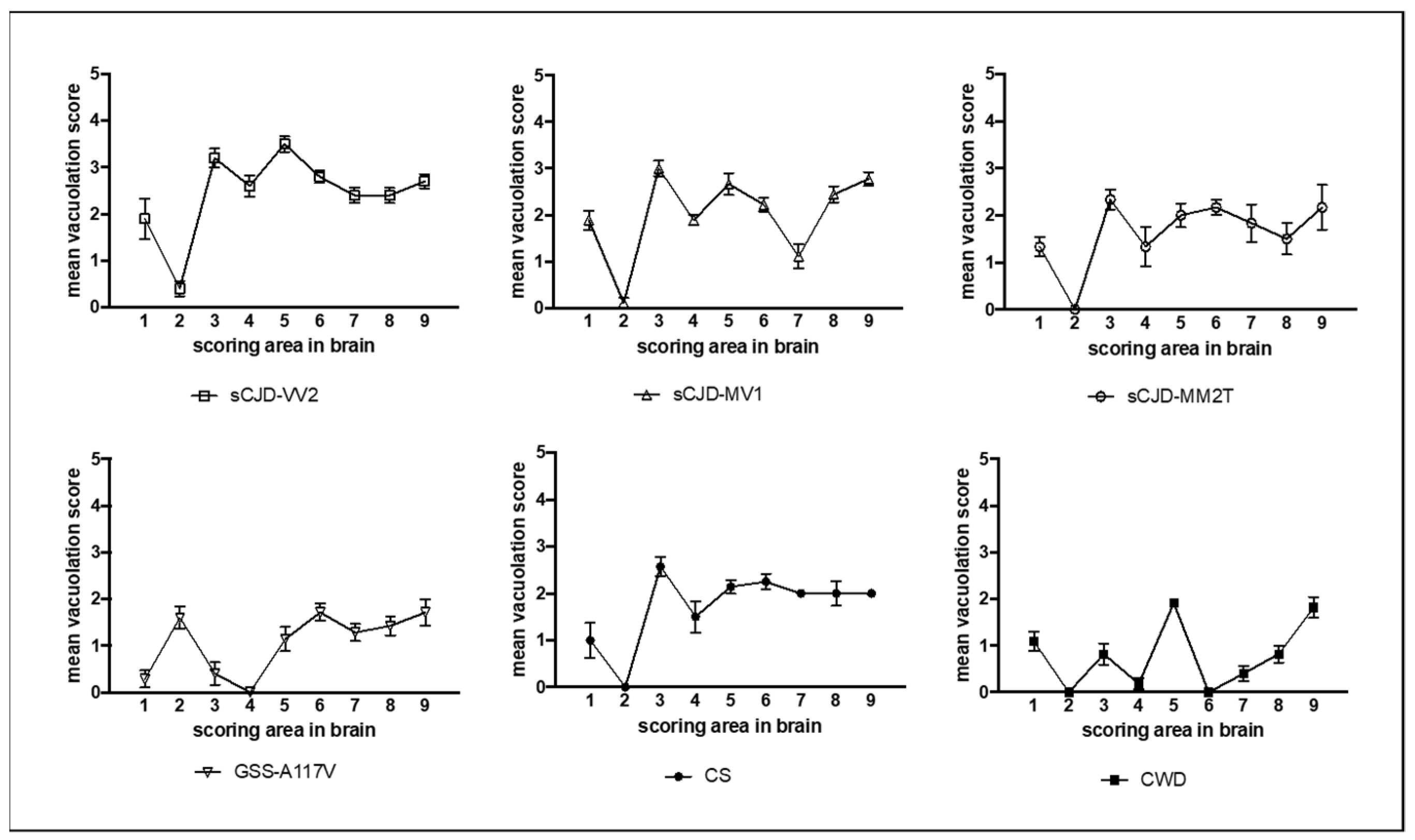
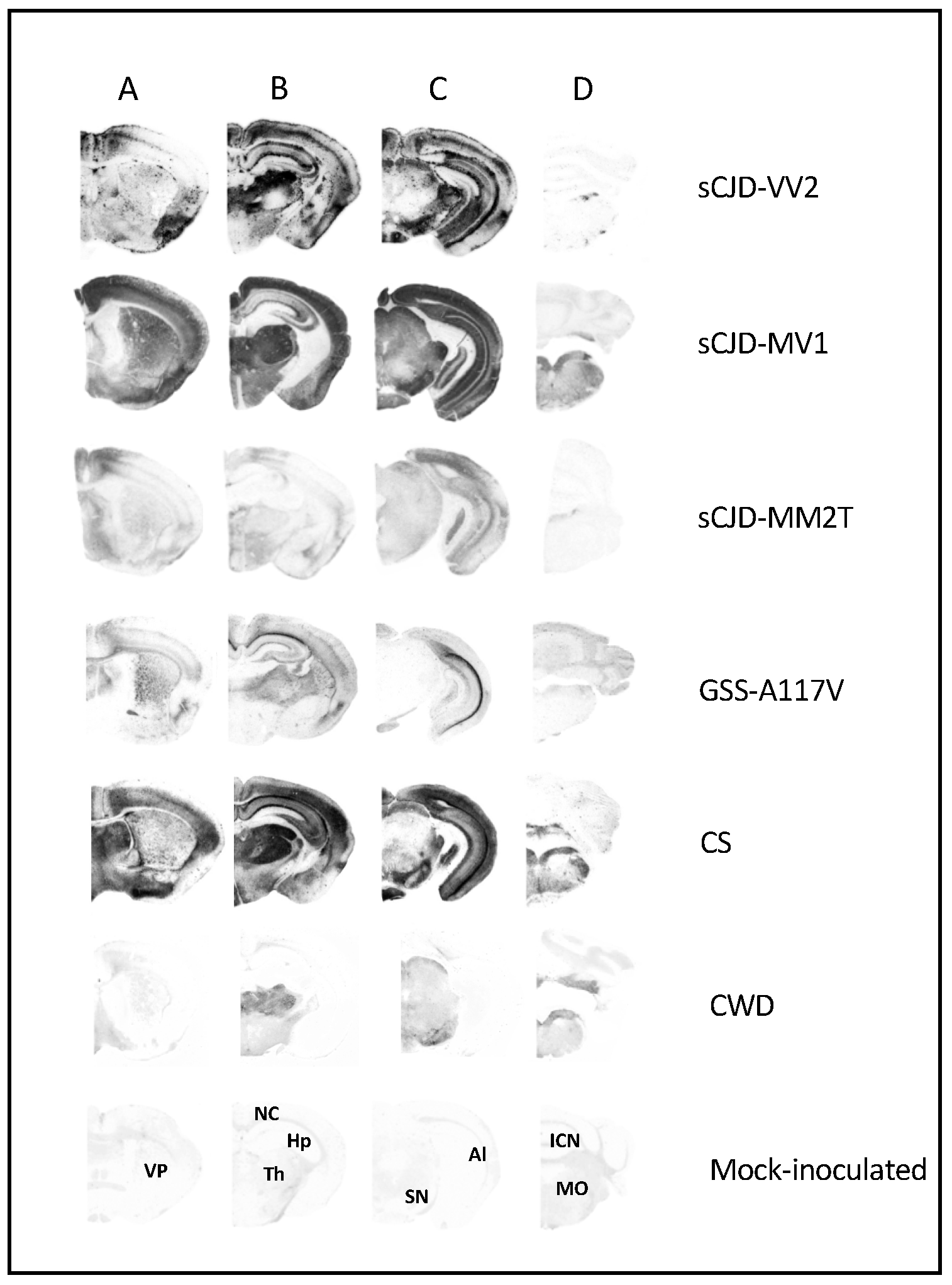
3.1. Characterization of Vole-Adapted Strains
3.2. Comparison of Astrocyte Response in Vole-Adapted Strains
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baiardi, S.; Rossi, M.; Capellari, S.; Parchi, P. Recent Advances in the Histo-Molecular Pathology of Human Prion Disease. Brain Pathol. 2019, 29, 278–300. [Google Scholar] [CrossRef] [PubMed]
- Babelhadj, B.; Di Bari, M.A.; Pirisinu, L.; Chiappini, B.; Gaouar, S.B.S.; Riccardi, G.; Marcon, S.; Agrimi, U.; Nonno, R.; Vaccari, G. Prion Disease in Dromedary Camels, Algeria. Emerg. Infect. Dis. 2018, 24, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef]
- Bruce, M.E.; McConnell, I.; Fraser, H.; Dickinson, A.G. The Disease Characteristics of Different Strains of Scrapie in Sinc Congenic Mouse Lines: Implications for the Nature of the Agent and Host Control of Pathogenesis. J. Gen. Virol. 1991, 72, 595–603. [Google Scholar] [CrossRef]
- Telling, G.C.; Parchi, P.; DeArmond, S.J.; Cortelli, P.; Montagna, P.; Gabizon, R.; Mastrianni, J.; Lugaresi, E.; Gambetti, P.; Prusiner, S.B. Evidence for the Conformation of the Pathologic Isoform of the Prion Protein Enciphering and Propagating Prion Diversity. Science 1996, 274, 2079–2082. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.; Wille, H.; Itri, V.; Groth, D.; Serban, H.; Torchia, M.; Cohen, F.E.; Prusiner, S.B. Eight Prion Strains Have PrPSc Molecules with Different Conformations. Nat. Med. 1998, 4, 1157–1165. [Google Scholar] [CrossRef]
- Rossi, M.; Baiardi, S.; Parchi, P. Understanding Prion Strains: Evidence from Studies of the Disease Forms Affecting Humans. Viruses 2019, 11, 309. [Google Scholar] [CrossRef]
- Hernández, R.S.; Sarasa, R.; Toledano, A.; Badiola, J.J.; Monzón, M. Morphological Approach to Assess the Involvement of Astrocytes in Prion Propagation. Cell Tissue Res. 2014, 358, 57–63. [Google Scholar] [CrossRef]
- Carroll, J.A.; Striebel, J.F.; Rangel, A.; Woods, T.; Phillips, K.; Peterson, K.E.; Race, B.; Chesebro, B. Prion Strain Differences in Accumulation of PrPSc on Neurons and Glia Are Associated with Similar Expression Profiles of Neuroinflammatory Genes: Comparison of Three Prion Strains. PLoS Pathog. 2016, 12, e1005551. [Google Scholar] [CrossRef]
- Monzón, M.; Hernández, R.S.; Garcés, M.; Sarasa, R.; Badiola, J.J. Glial Alterations in Human Prion Diseases. Medicine 2018, 97, e0320. [Google Scholar] [CrossRef]
- Smith, H.L.; Freeman, O.J.; Butcher, A.J.; Holmqvist, S.; Humoud, I.; Schätzl, T.; Hughes, D.T.; Verity, N.C.; Swinden, D.P.; Hayes, J.; et al. Astrocyte Unfolded Protein Response Induces a Specific Reactivity State That Causes Non-Cell-Autonomous Neuronal Degeneration. Neuron 2020, 105, 855–866.e5. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, R.; Sinha, A.; Makarava, N.; Molesworth, K.; Baskakov, I.V. Non-Cell Autonomous Astrocyte-Mediated Neuronal Toxicity in Prion Diseases. Acta Neuropathol. Commun. 2021, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Mychko, O.; Chen, J.; Chang, Y.; Molesworth, K.; Baskakov, I. V The Degree of Astrocyte Activation Is Predictive of the Incubation Time to Prion Disease. Acta Neuropathol. Commun. 2021, 9, 87. [Google Scholar] [CrossRef] [PubMed]
- González, L.; Martin, S.; Begara-McGorum, I.; Hunter, N.; Houston, F.; Simmons, M.; Jeffrey, M. Effects of Agent Strain and Host Genotype on PrP Accumulation in the Brain of Sheep Naturally and Experimentally Affected with Scrapie. J. Comp. Pathol. 2002, 126, 17–29. [Google Scholar] [CrossRef]
- Cronier, S.; Laude, H.; Peyrin, J.-M. Prions Can Infect Primary Cultured Neurons and Astrocytes and Promote Neuronal Cell Death. Proc. Natl. Acad. Sci. USA 2004, 101, 12271–12276. [Google Scholar] [CrossRef] [PubMed]
- Kovács, G.G.; Preusser, M.; Strohschneider, M.; Budka, H. Subcellular Localization of Disease-Associated Prion Protein in the Human Brain. Am. J. Pathol. 2005, 166, 287–294. [Google Scholar] [CrossRef]
- Hollister, J.R.; Lee, K.S.; Dorward, D.W.; Baron, G.S. Efficient Uptake and Dissemination of Scrapie Prion Protein by Astrocytes and Fibroblasts from Adult Hamster Brain. PLoS ONE 2015, 10, e0115351. [Google Scholar] [CrossRef]
- Krejciova, Z.; Alibhai, J.; Zhao, C.; Krencik, R.; Rzechorzek, N.M.; Ullian, E.M.; Manson, J.; Ironside, J.W.; Head, M.W.; Chandran, S. Human Stem Cell-Derived Astrocytes Replicate Human Prions in a PRNP Genotype-Dependent Manner. J. Exp. Med. 2017, 214, 3481–3495. [Google Scholar] [CrossRef]
- Tahir, W.; Abdulrahman, B.; Abdelaziz, D.H.; Thapa, S.; Walia, R.; Schätzl, H.M. An Astrocyte Cell Line That Differentially Propagates Murine Prions. J. Biol. Chem. 2020, 295, 11572–11583. [Google Scholar] [CrossRef]
- Schwenke, K.A.; Wälzlein, J.-H.; Bauer, A.; Thomzig, A.; Beekes, M. Primary Glia Cells from Bank Vole Propagate Multiple Rodent-Adapted Scrapie Prions. Sci. Rep. 2022, 12, 2190. [Google Scholar] [CrossRef]
- Baskakov, I.V. On the Reactive States of Astrocytes in Prion Diseases. Prion 2021, 15, 87–93. [Google Scholar] [CrossRef]
- Tahir, W.; Thapa, S.; Schatzl, H. Astrocyte in Prion Disease: A Double-Edged Sword. Neural Regen. Res. 2022, 17, 1659. [Google Scholar] [CrossRef]
- Bushong, E.A.; Martone, M.E.; Jones, Y.Z.; Ellisman, M.H. Protoplasmic Astrocytes in CA1 Stratum Radiatum Occupy Separate Anatomical Domains. J. Neurosci. 2002, 22, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Höft, S.; Griemsmann, S.; Seifert, G.; Steinhäuser, C. Heterogeneity in Expression of Functional Ionotropic Glutamate and GABA Receptors in Astrocytes across Brain Regions: Insights from the Thalamus. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130602. [Google Scholar] [CrossRef] [PubMed]
- Haim, L.B.; Rowitch, D.H. Functional Diversity of Astrocytes in Neural Circuit Regulation. Nat. Rev. Neurosci. 2017, 18, 31–41. [Google Scholar] [CrossRef]
- Lanjakornsiripan, D.; Pior, B.J.; Kawaguchi, D.; Furutachi, S.; Tahara, T.; Katsuyama, Y.; Suzuki, Y.; Fukazawa, Y.; Gotoh, Y. Layer-Specific Morphological and Molecular Differences in Neocortical Astrocytes and Their Dependence on Neuronal Layers. Nat. Commun. 2018, 9, 1623. [Google Scholar] [CrossRef]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic Analysis of Reactive Astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Pekna, M.; Messing, A.; Steinhäuser, C.; Lee, J.-M.; Parpura, V.; Hol, E.M.; Sofroniew, M.V.; Verkhratsky, A. Astrocytes: A Central Element in Neurological Diseases. Acta Neuropathol. 2016, 131, 323–345. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Chang, J.C.Y.; Kushwaha, R.; Baskakov, I.V. Region-Specific Response of Astrocytes to Prion Infection. Front. Neurosci. 2019, 13, 1048. [Google Scholar] [CrossRef]
- Areškevičiūtė, A.; Litman, T.; Broholm, H.; Melchior, L.C.; Nielsen, P.R.; Green, A.; Eriksen, J.O.; Smith, C.; Lund, E.L. Regional Differences in Neuroinflammation-Associated Gene Expression in the Brain of Sporadic Creutzfeldt–Jakob Disease Patients. Int. J. Mol. Sci. 2021, 22, 140. [Google Scholar] [CrossRef] [PubMed]
- Bradford, B.M.; Wijaya, C.A.W.; Mabbott, N.A. Discrimination of Prion Strain Targeting in the Central Nervous System via Reactive Astrocyte Heterogeneity in CD44 Expression. Front. Cell. Neurosci. 2019, 13, 411. [Google Scholar] [CrossRef] [PubMed]
- Ugalde, C.L.; Lewis, V.; Stehmann, C.; McLean, C.A.; Lawson, V.A.; Collins, S.J.; Hill, A.F. Markers of A1 Astrocytes Stratify to Molecular Sub-Types in Sporadic Creutzfeldt–Jakob Disease Brain. Brain Commun. 2020, 2, fcaa029. [Google Scholar] [CrossRef]
- Makarava, N.; Chang, J.C.-Y.Y.; Molesworth, K.; Baskakov, I.V. Region-Specific Glial Homeostatic Signature in Prion Diseases Is Replaced by a Uniform Neuroinflammation Signature, Common for Brain Regions and Prion Strains with Different Cell Tropism. Neurobiol. Dis. 2020, 137, 104783. [Google Scholar] [CrossRef]
- Nonno, R.; Di Bari, M.A.; Cardone, F.; Vaccari, G.; Fazzi, P.; Dell’Omo, G.; Cartoni, C.; Ingrosso, L.; Boyle, A.; Galeno, R.; et al. Efficient Transmission and Characterization of Creutzfeldt-Jakob Disease Strains in Bank Voles. PLoS Pathog. 2006, 2, e12. [Google Scholar] [CrossRef] [PubMed]
- Nonno, R.; Notari, S.; Di Bari, M.A.; Cali, I.; Pirisinu, L.; D’agostino, C.; Cracco, L.; Kofskey, D.; Vanni, I.; Lavrich, J.; et al. Variable Protease-Sensitive Prionopathy Transmission to Bank Voles. Emerg. Infect. Dis. 2019, 25, 73–81. [Google Scholar] [CrossRef]
- Nonno, R.; Di Bari, M.A.; Pirisinu, L.; D’Agostino, C.; Vanni, I.; Chiappini, B.; Marcon, S.; Riccardi, G.; Tran, L.; Vikøren, T.; et al. Studies in Bank Voles Reveal Strain Differences between Chronic Wasting Disease Prions from Norway and North America. Proc. Natl. Acad. Sci. USA 2020, 117, 31417–31426. [Google Scholar] [CrossRef] [PubMed]
- Agrimi, U.; Nonno, R.; Dell’Omo, G.; Di Bari, M.A.; Conte, M.; Chiappini, B.; Esposito, E.; Di Guardo, G.; Windl, O.; Vaccari, G.; et al. Prion Protein Amino Acid Determinants of Differential Susceptibility and Molecular Feature of Prion Strains in Mice and Voles. PLoS Pathog. 2008, 4, e1000113. [Google Scholar] [CrossRef] [PubMed]
- Di Bari, M.A.; Chianini, F.; Vaccari, G.; Esposito, E.; Conte, M.; Eaton, S.L.; Hamilton, S.; Finlayson, J.; Steele, P.J.; Dagleish, M.P.; et al. The Bank Vole (Myodes Glareolus) as a Sensitive Bioassay for Sheep Scrapie. J. Gen. Virol. 2008, 89, 2975–2985. [Google Scholar] [CrossRef]
- Di Bari, M.A.; Nonno, R.; Castilla, J.; D’Agostino, C.; Pirisinu, L.; Riccardi, G.; Conte, M.; Richt, J.; Kunkle, R.; Langeveld, J.; et al. Chronic Wasting Disease in Bank Voles: Characterisation of the Shortest Incubation Time Model for Prion Diseases. PLoS Pathog. 2013, 9, e1003219. [Google Scholar] [CrossRef]
- Espinosa, J.C.; Nonno, R.; Di Bari, M.; Aguilar-Calvo, P.; Pirisinu, L.; Fernández-Borges, N.; Vanni, I.; Vaccari, G.; Marín-Moreno, A.; Frassanito, P.; et al. PrP C Governs Susceptibility to Prion Strains in Bank Vole, While Other Host Factors Modulate Strain Features. J. Virol. 2016, 90, 10660–10669. [Google Scholar] [CrossRef]
- Pirisinu, L.; Di Bari, M.A.; D’Agostino, C.; Marcon, S.; Riccardi, G.; Poleggi, A.; Cohen, M.L.; Appleby, B.S.; Gambetti, P.; Ghetti, B.; et al. Gerstmann-Sträussler-Scheinker Disease Subtypes Efficiently Transmit in Bank Voles as Genuine Prion Diseases. Sci. Rep. 2016, 6, 20443. [Google Scholar] [CrossRef]
- Galeno, R.; Di Bari, M.A.; Nonno, R.; Cardone, F.; Sbriccoli, M.; Graziano, S.; Ingrosso, L.; Fiorini, M.; Valanzano, A.; Pasini, G.; et al. Prion Strain Characterization of a Novel Subtype of Creutzfeldt-Jakob Disease. J. Virol. 2017, 91, e02390-16. [Google Scholar] [CrossRef] [PubMed]
- Pirisinu, L.; Di Bari, M.A.; D’Agostino, C.; Vanni, I.; Riccardi, G.; Marcon, S.; Vaccari, G.; Chiappini, B.; Benestad, S.L.; Agrimi, U.; et al. A Single Amino Acid Residue in Bank Vole Prion Protein Drives Permissiveness to Nor98/Atypical Scrapie and the Emergence of Multiple Strain Variants. PLoS Pathog. 2022, 18, e1010646. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Saverioni, D.; Di Bari, M.; Baiardi, S.; Lemstra, A.W.; Pirisinu, L.; Capellari, S.; Rozemuller, A.; Nonno, R.; Parchi, P. Atypical Creutzfeldt-Jakob Disease with Prpamyloid Plaques in White Matter: Molecular Characterization and Transmission to Bank Voles Show the M1 Strain Signature. Acta Neuropathol. Commun. 2017, 5, 87. [Google Scholar] [CrossRef]
- Zanusso, G.; Polo, A.; Farinazzo, A.; Nonno, R.; Cardone, F.; Di Bari, M.; Ferrari, S.; Principe, S.; Gelati, M.; Fasoli, E.; et al. Novel Prion Protein Conformation and Glycotype in Creutzfeldt-Jakob Disease. Arch. Neurol. 2007, 64, 595–599. [Google Scholar] [CrossRef]
- Vanni, I.; Migliore, S.; Cosseddu, G.M.; Di Bari, M.A.; Pirisinu, L.; D’Agostino, C.; Riccardi, G.; Agrimi, U.; Nonno, R. Isolation of a Defective Prion Mutant from Natural Scrapie. PLoS Pathog. 2016, 12, e1006016. [Google Scholar] [CrossRef]
- Nonno, R.; Marin-Moreno, A.; Carlos Espinosa, J.; Fast, C.; Van Keulen, L.; Spiropoulos, J.; Lantier, I.; Andreoletti, O.; Pirisinu, L.; Di Bari, M.A.; et al. Characterization of Goat Prions Demonstrates Geographical Variation of Scrapie Strains in Europe and Reveals the Composite Nature of Prion Strains. Sci. Rep. 2020, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Vanni, I.; Pirisinu, L.; Acevedo-Morantes, C.; Kamali-Jamil, R.; Rathod, V.; Di Bari, M.A.; D’Agostino, C.; Marcon, S.; Esposito, E.; Riccardi, G.; et al. Isolation of Infectious, Non-Fibrillar and Oligomeric Prions from a Genetic Prion Disease. Brain 2020, 143, 1512–1524. [Google Scholar] [CrossRef]
- Cartoni, C.; Schininà, M.E.; Maras, B.; Nonno, R.; Vaccari, G.; Di Bari, M.; Conte, M.; De Pascalis, A.; Principe, S.; Cardone, F.; et al. Quantitative Profiling of the Pathological Prion Protein Allotypes in Bank Voles by Liquid Chromatography-Mass Spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 849, 302–306. [Google Scholar] [CrossRef]
- Nonno, R.; Angelo Di Bari, M.; Agrimi, U.; Pirisinu, L. Transmissibility of Gerstmann–Sträussler–Scheinker Syndrome in Rodent Models: New Insights into the Molecular Underpinnings of Prion Infectivity. Prion 2016, 10, 421–433. [Google Scholar] [CrossRef]
- Bruno, R.; Pirisinu, L.; Riccardi, G.; D’Agostino, C.; De Cecco, E.; Legname, G.; Cardone, F.; Gambetti, P.; Nonno, R.; Agrimi, U.; et al. Gerstmann–Sträussler–Scheinker Disease with F198S Mutation Induces Independent Tau and Prion Protein Pathologies in Bank Voles. Biomolecules 2022, 12, 1537. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H.; Dickinson, A.G. The Sequential Development of the Brain Lesions of Scrapie in Three Strains of Mice. J. Comp. Pathol. 1968, 78, 301–311. [Google Scholar] [CrossRef]
- Sarasa, R.; Martínez, A.; Monleón, E.; Bolea, R.; Vargas, A.; Badiola, J.J.; Monzón, M. Involvement of Astrocytes in Transmissible Spongiform Encephalopathies: A Confocal Microscopy Study. Cell Tissue Res. 2012, 350, 127–134. [Google Scholar] [CrossRef]
- Guijarro, I.M.; Garcés, M.; Andrés-Benito, P.; Marín, B.; Otero, A.; Barrio, T.; Carmona, M.; Ferrer, I.; Badiola, J.J.; Monzón, M. Assessment of Glial Activation Response in the Progress of Natural Scrapie after Chronic Dexamethasone Treatment. Int. J. Mol. Sci. 2020, 21, 3231. [Google Scholar] [CrossRef] [PubMed]
- Garcés, M.; Toledano, A.; Badiola, J.J.; Monzón, M. Morphological Changes of Glia in Prion and a Prion-Like Disorder. Alzheimer’s Neurodegener. Dis. 2016, 2, 1–4. [Google Scholar] [CrossRef]
- Garcés, M.; Guijarro, I.M.; Ritchie, D.L.; Badiola, J.J.; Monzón, M. Novel Morphological Glial Alterations in the Spectrum of Prion Disease Types: A Focus on Common Findings. Pathogens 2021, 10, 596. [Google Scholar] [CrossRef] [PubMed]
- Bento-Torres, J.; Sobral, L.L.; Reis, R.R.; De Oliveira, R.B.; Anthony, D.C.; Vasconcelos, P.F.C.; Picanço Diniz, C.W. Age and Environment Influences on Mouse Prion Disease Progression: Behavioral Changes and Morphometry and Stereology of Hippocampal Astrocytes. Oxid. Med. Cell. Longev. 2017, 2017, 4504925. [Google Scholar] [CrossRef]
- Diniz, D.G.; Oliveira, M.A.; Lima, C.M.; Fôro, C.A.R.; Sosthenes, M.C.K.; Bento-Torres, J.; Costa Vasconcelos, P.F.; Anthony, D.C.; Diniz, C.W.P. Age, Environment, Object Recognition and Morphological Diversity of GFAP-Immunolabeled Astrocytes. Behav. Brain Funct. 2016, 12, 28. [Google Scholar] [CrossRef]
- Victoria, G.S.; Arkhipenko, A.; Zhu, S.; Syan, S.; Zurzolo, C. Astrocyte-to-Neuron Intercellular Prion Transfer Is Mediated by Cell-Cell Contact. Sci. Rep. 2016, 6, 20762. [Google Scholar] [CrossRef]
- Choi, Y.P.; Head, M.W.; Ironside, J.W.; Priola, S.A. Uptake and Degradation of Protease-Sensitive and -Resistant Forms of Abnormal Human Prion Protein Aggregates by Human Astrocytes. Am. J. Pathol. 2014, 184, 3299–3307. [Google Scholar] [CrossRef]
- Olabarria, M.; Noristani, H.N.; Verkhratsky, A.; Rodríguez, J.J. Concomitant Astroglial Atrophy and Astrogliosis in a Triple Transgenic Animal Model of Alzheimer’s Disease. Glia 2010, 58, 831–838. [Google Scholar] [CrossRef]
- Rodríguez, J.J.; Olabarria, M.; Chvatal, A.; Verkhratsky, A. Astroglia in Dementia and Alzheimer’s Disease. Cell Death Differ. 2009, 16, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Reichel, J.M.; Han, C.; Zuniga-Hertz, J.P.; Cai, D. Astrocytic Process Plasticity and IKKβ/NF-ΚB in Central Control of Blood Glucose, Blood Pressure, and Body Weight. Cell Metab. 2017, 25, 1091–1102.e4. [Google Scholar] [CrossRef]
- Endo, F.; Kasai, A.; Soto, J.S.; Yu, X.; Qu, Z.; Hashimoto, H.; Gradinaru, V.; Kawaguchi, R.; Khakh, B.S. Molecular Basis of Astrocyte Diversity and Morphology across the CNS in Health and Disease. Science 2022, 378, eadc9020. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive Astrocyte Nomenclature, Definitions, and Future Directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Hartmann, K.; Sepulveda-Falla, D.; Rose, I.V.L.; Madore, C.; Muth, C.; Matschke, J.; Butovsky, O.; Liddelow, S.; Glatzel, M.; Krasemann, S. Complement 3+-Astrocytes Are Highly Abundant in Prion Diseases, but Their Abolishment Led to an Accelerated Disease Course and Early Dysregulation of Microglia. Acta Neuropathol. Commun. 2019, 7, 83. [Google Scholar] [CrossRef]
- Vanni, S.; Moda, F.; Zattoni, M.; Bistaffa, E.; De Cecco, E.; Rossi, M.; Giaccone, G.; Tagliavini, F.; Haïk, S.; Deslys, J.P.; et al. Differential Overexpression of SERPINA3 in Human Prion Diseases. Sci. Rep. 2017, 7, 15637. [Google Scholar] [CrossRef]
- Carroll, J.A.; Race, B.; Williams, K.; Striebel, J.; Chesebro, B. RNA-Seq and Network Analysis Reveal Unique Glial Gene Expression Signatures during Prion Infection. Mol. Brain 2020, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Sorce, S.; Nuvolone, M.; Russo, G.; Chincisan, A.; Heinzer, D.; Avar, M.; Pfammatter, M.; Schwarz, P.; Delic, M.; Müller, M.; et al. Genome-Wide Transcriptomics Identifies an Early Preclinical Signature of Prion Infection. PLoS Pathog. 2020, 16, e1008653. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.C.; Condello, C.; Stöhr, J.; Oehler, A.; Lee, J.; DeArmond, S.J.; Lannfelt, L.; Ingelsson, M.; Giles, K.; Prusiner, S.B. Serial Propagation of Distinct Strains of Aβ Prions from Alzheimer’s Disease Patients. Proc. Natl. Acad. Sci. USA 2014, 111, 10323–10328. [Google Scholar] [CrossRef] [PubMed]
- Peelaerts, W.; Bousset, L.; Van Der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van Den Haute, C.; Melki, R.; Baekelandt, V. α-Synuclein Strains Cause Distinct Synucleinopathies after Local and Systemic Administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating α-Synuclein Strains in Parkinson’s Disease and Multiple System Atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruno, R.; Riccardi, G.; Iacobone, F.; Chiarotti, F.; Pirisinu, L.; Vanni, I.; Marcon, S.; D’Agostino, C.; Giovannelli, M.; Parchi, P.; et al. Strain-Dependent Morphology of Reactive Astrocytes in Human- and Animal-Vole-Adapted Prions. Biomolecules 2023, 13, 757. https://doi.org/10.3390/biom13050757
Bruno R, Riccardi G, Iacobone F, Chiarotti F, Pirisinu L, Vanni I, Marcon S, D’Agostino C, Giovannelli M, Parchi P, et al. Strain-Dependent Morphology of Reactive Astrocytes in Human- and Animal-Vole-Adapted Prions. Biomolecules. 2023; 13(5):757. https://doi.org/10.3390/biom13050757
Chicago/Turabian StyleBruno, Rosalia, Geraldina Riccardi, Floriana Iacobone, Flavia Chiarotti, Laura Pirisinu, Ilaria Vanni, Stefano Marcon, Claudia D’Agostino, Matteo Giovannelli, Piero Parchi, and et al. 2023. "Strain-Dependent Morphology of Reactive Astrocytes in Human- and Animal-Vole-Adapted Prions" Biomolecules 13, no. 5: 757. https://doi.org/10.3390/biom13050757
APA StyleBruno, R., Riccardi, G., Iacobone, F., Chiarotti, F., Pirisinu, L., Vanni, I., Marcon, S., D’Agostino, C., Giovannelli, M., Parchi, P., Agrimi, U., Nonno, R., & Di Bari, M. A. (2023). Strain-Dependent Morphology of Reactive Astrocytes in Human- and Animal-Vole-Adapted Prions. Biomolecules, 13(5), 757. https://doi.org/10.3390/biom13050757