
LRRK2 Structure-Based Activation Mechanism and Pathogenesis



Abstract
1. Introduction
2. Molecular Structure of LRRK2
2.1. From Bacterial Roco to Human Full-Length LRRK2 Structures
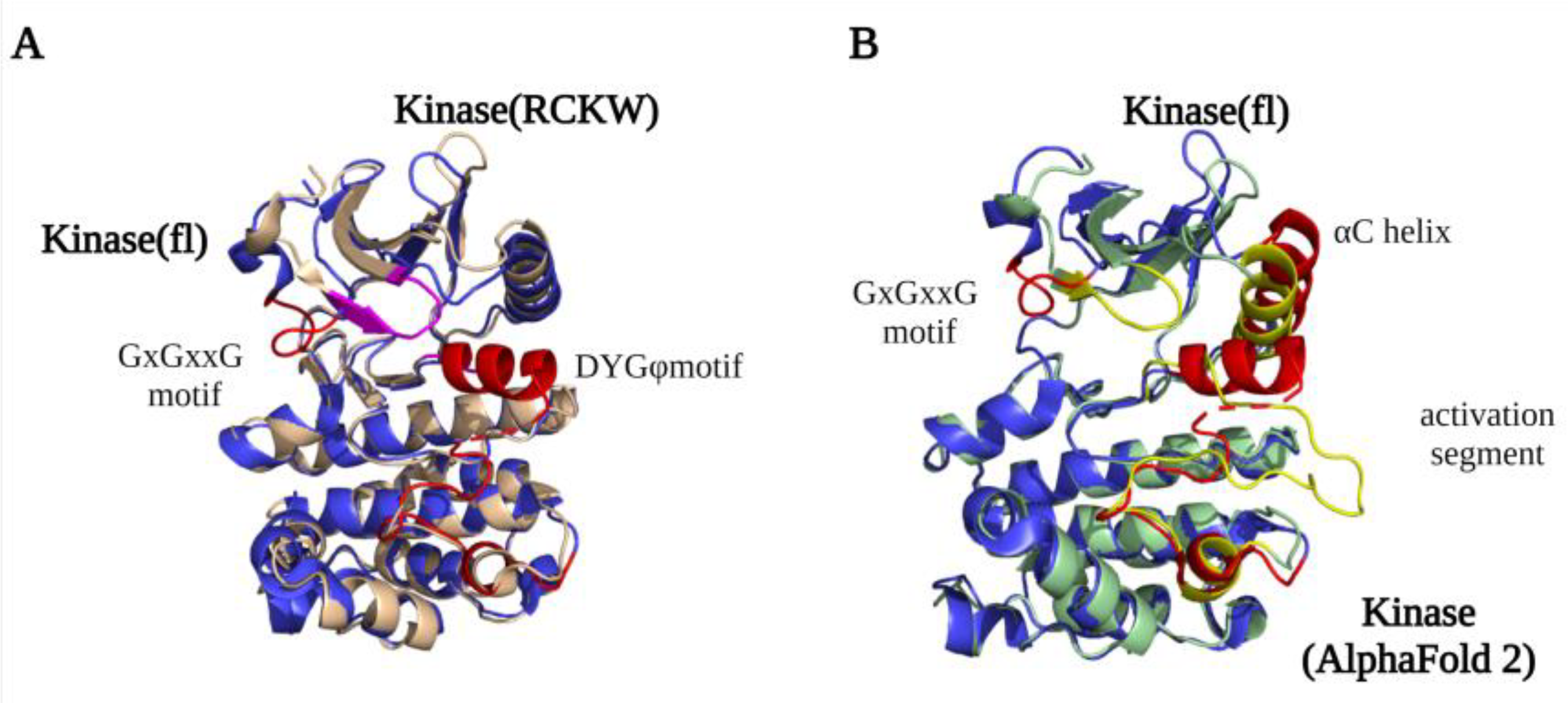
2.2. Kinase Domain Structure
2.3. RocCOR Structure
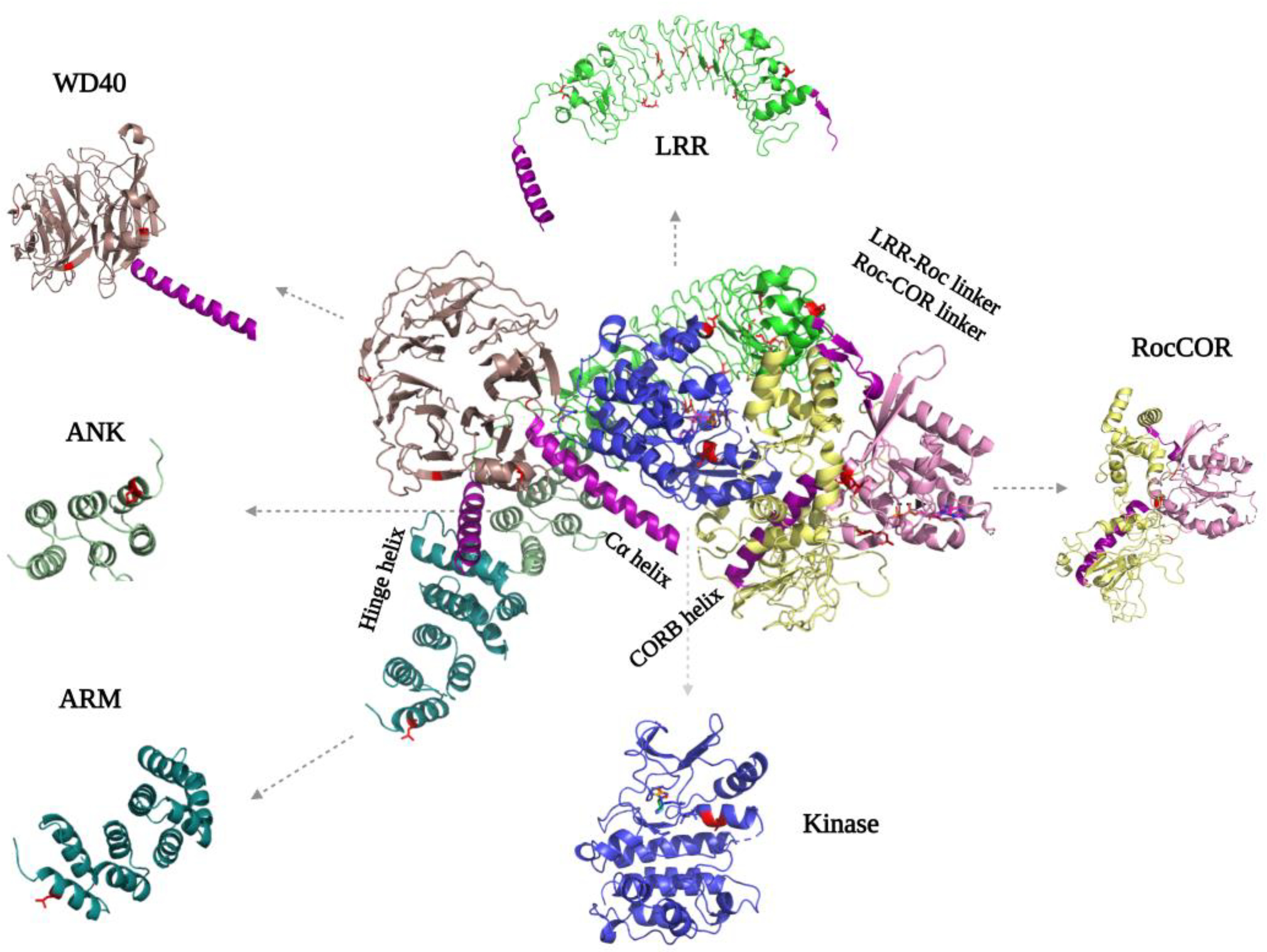
2.4. N-Terminal and C-Terminal Scaffold Structure
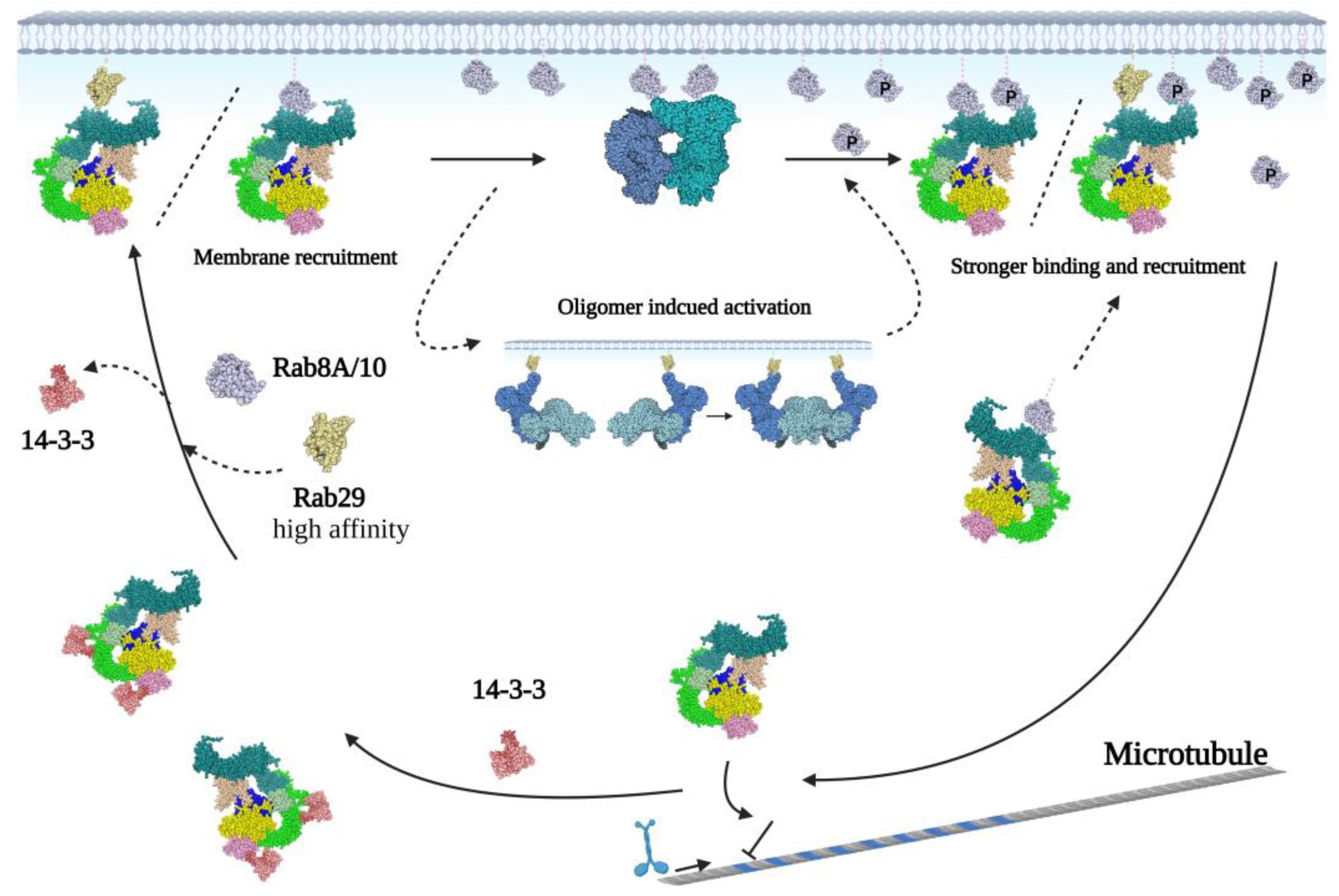
3. Structural Studies to Understand the LRRK2 Activation Mechanism
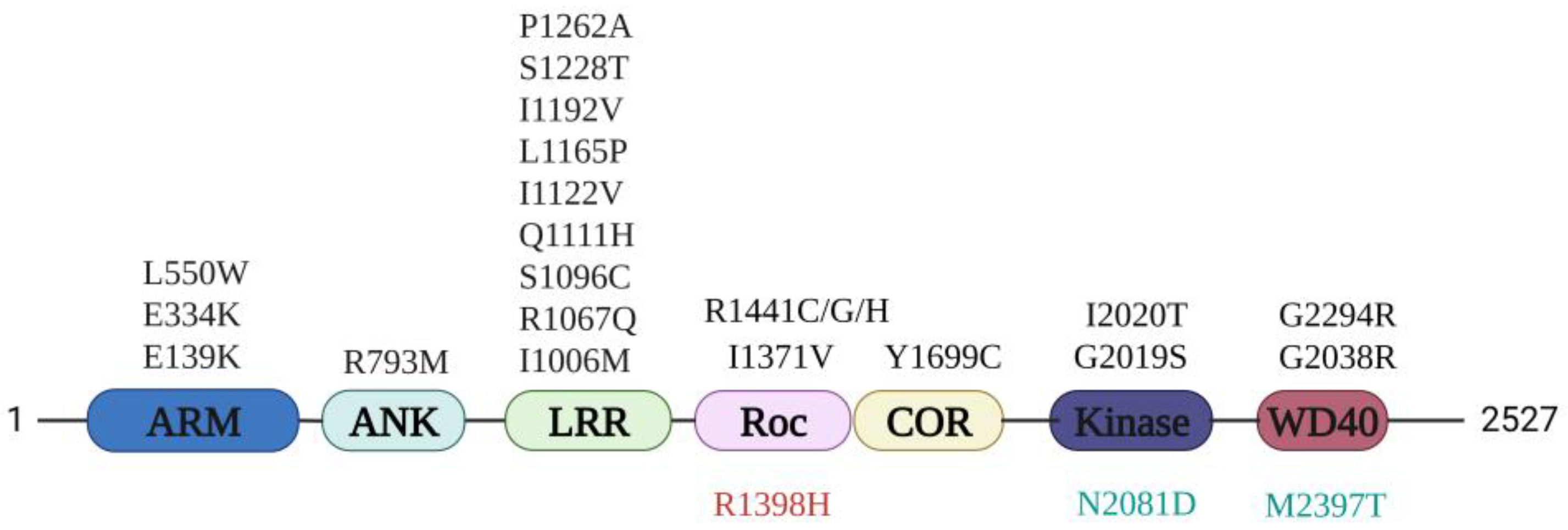
4. PD Mutation Localization and Potential Pathogenesis
5. Perspective
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dorsey, E.; Sherer, T.; Okun, M.; Bloem, B. The Rise of Parkinson’s Disease. Am. Sci. 2020, 108, 176. [Google Scholar] [CrossRef]
- Lee, F.J.S.; Liu, F. Genetic Factors Involved in the Pathogenesis of Parkinson’s Disease. Brain Res. Rev. 2008, 58, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Day, J.O.; Mullin, S. The Genetics of Parkinson’s Disease and Implications for Clinical Practice. Genes 2021, 12, 1006. [Google Scholar] [CrossRef]
- Chandler, R.J.; Cogo, S.; Lewis, P.A.; Kevei, E. Modelling the Functional Genomics of Parkinson’s Disease in Caenorhabditis Elegans: LRRK2 and Beyond. Biosci. Rep. 2021, 41, BSR20203672. [Google Scholar] [CrossRef]
- Di Maio, R.; Hoffman, E.K.; Rocha, E.M.; Keeney, M.T.; Sanders, L.H.; De Miranda, B.R.; Zharikov, A.; Van Laar, A.; Stepan, A.F.; Lanz, T.A.; et al. LRRK2 Activation in Idiopathic Parkinson’s Disease. Sci. Transl. Med. 2018, 10, 451. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Q.; Tan, L.; Yu, J.T. The Role of the LRRK2 Gene in Parkinsonism. Mol. Neurodegener. 2014, 9, 47. [Google Scholar] [CrossRef]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics Reveals That Parkinson’s Disease Kinase LRRK2 Regulates a Subset of Rab GTPases. Elife 2016, 5. [Google Scholar] [CrossRef]
- Taylor, M.; Alessi, D.R. Advances in Elucidating the Function of Leucine-Rich Repeat Protein Kinase-2 in Normal Cells and Parkinson’s Disease. Curr. Opin. Cell Biol. 2020, 63, 102–113. [Google Scholar] [CrossRef]
- Madureira, M.; Connor-Robson, N.; Wade-Martins, R. LRRK2: Autophagy and Lysosomal Activity. Front. Neurosci. 2020, 14, 498. [Google Scholar] [CrossRef]
- Lewis, P.A.; Greggio, E.; Beilina, A.; Jain, S.; Baker, A.; Cookson, M.R. The R1441C Mutation of LRRK2 Disrupts GTP Hydrolysis. Biochem. Biophys. Res. Commun. 2007, 357, 668–671. [Google Scholar] [CrossRef]
- Daniẽls, V.; Vancraenenbroeck, R.; Law, B.M.H.; Greggio, E.; Lobbestael, E.; Gao, F.; De Maeyer, M.; Cookson, M.R.; Harvey, K.; Baekelandt, V.; et al. Insight into the Mode of Action of the LRRK2 Y1699C Pathogenic Mutant. J. Neurochem. 2011, 116, 304–315. [Google Scholar] [CrossRef] [PubMed]
- West, A.B.; Moore, D.J.; Choi, C.; Andrabi, S.A.; Li, X.; Dikeman, D.; Biskup, S.; Zhang, Z.; Lim, K.L.; Dawson, V.L.; et al. Parkinson’s Disease-Associated Mutations in LRRK2 Link Enhanced GTP-Binding and Kinase Activities to Neuronal Toxicity. Hum. Mol. Genet. 2007, 16, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Van Limbergen, J.; Wilson, D.C.; Satsangi, J. The Genetics of Crohn’s Disease. Annu. Rev. Genomics Hum. Genet. 2009, 10, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.-R.; Huang, W.; Chen, S.-M.; Sun, L.-D.; Liu, H.; Li, Y.; Cui, Y.; Yan, X.-X.; Yang, H.-T.; Yang, R.-D.; et al. Genomewide Association Study of Leprosy. N. Engl. J. Med. 2009, 361, 2609–2618. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.Y.; Fernandez-Hernandez, H.; Hu, J.; Schaffner, A.; Pankratz, N.; Hsu, N.Y.; Chuang, L.S.; Carmi, S.; Villaverde, N.; Li, X.; et al. Functional Variants in the LRRK2 Gene Confer Shared Effects on Risk for Crohn’s Disease and Parkinson’s Disease. Sci. Transl. Med. 2018, 10, 423. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Lewis, P.A.; Greggio, E.; Sluch, E.; Beilina, A.; Cookson, M.R. Structure of the ROC Domain from the Parkinson’s Disease-Associated Leucine-Rich Repeat Kinase 2 Reveals a Dimeric GTPase. Proc. Natl. Acad. Sci. USA 2008, 105, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Fan, Y.; Ru, H.; Wang, L.; Magupalli, V.G.; Taylor, S.S.; Alessi, D.R.; Wu, H. Crystal Structure of the WD40 Domain Dimer of LRRK2. Proc. Natl. Acad. Sci. USA 2019, 116, 1579–1584. [Google Scholar] [CrossRef]
- Merk, A.; Bartesaghi, A.; Banerjee, S.; Falconieri, V.; Rao, P.; Davis, M.I.; Pragani, R.; Boxer, M.B.; Earl, L.A.; Milne, J.L.S.; et al. Breaking Cryo-EM Resolution Barriers to Facilitate Drug Discovery. Cell 2016, 165, 1698–1707. [Google Scholar] [CrossRef]
- Lyumkis, D. Challenges and Opportunities in Cryo-EM Single-Particle Analysis. J. Biol. Chem. 2019, 294, 5181–5197. [Google Scholar] [CrossRef]
- Benjin, X.; Ling, L. Developments, Applications, and Prospects of Cryo-Electron Microscopy. Protein Sci. 2020, 29, 872–882. [Google Scholar] [CrossRef]
- Guaitoli, G.; Raimondi, F.; Gilsbach, B.K.; Gómez-Llorente, Y.; Deyaert, E.; Renzi, F.; Li, X.; Schaffner, A.; Jagtap, P.K.A.; Boldt, K.; et al. Structural Model of the Dimeric Parkinson’s Protein LRRK2 Reveals a Compact Architecture Involving Distant Interdomain Contacts. Proc. Natl. Acad. Sci. USA 2016, 113, E4357–E4366. [Google Scholar] [CrossRef] [PubMed]
- Sejwal, K.; Chami, M.; Rémigy, H.; Vancraenenbroeck, R.; Sibran, W.; Sütterlin, R.; Baumgartner, P.; McLeod, R.; Chartier-Harlin, M.C.; Baekelandt, V.; et al. Cryo-EM Analysis of Homodimeric Full-Length LRRK2 and LRRK1 Protein Complexes. Sci. Rep. 2017, 7, 8667. [Google Scholar] [CrossRef] [PubMed]
- Deniston, C.K.; Salogiannis, J.; Mathea, S.; Snead, D.M.; Lahiri, I.; Matyszewski, M.; Donosa, O.; Watanabe, R.; Böhning, J.; Shiau, A.K.; et al. Structure of LRRK2 in Parkinson’s Disease and Model for Microtubule Interaction. Nature 2020, 588, 344–349. [Google Scholar] [CrossRef]
- Myasnikov, A.; Zhu, H.; Hixson, P.; Xie, B.; Yu, K.; Pitre, A.; Peng, J.; Sun, J. Structural Analysis of the Full-Length Human LRRK2. Cell 2021, 184, 3519–3527.e10. [Google Scholar] [CrossRef]
- Snead, D.M.; Matyszewski, M.; Dickey, A.M.; Lin, Y.X.; Leschziner, A.E.; Reck-Peterson, S.L. Structural Basis for Parkinson’s Disease-Linked LRRK2’s Binding to Microtubules. Nat. Struct. Mol. Biol. 2022, 29, 1196–1207. [Google Scholar] [CrossRef]
- Watanabe, R.; Buschauer, R.; Böhning, J.; Audagnotto, M.; Lasker, K.; Lu, T.W.; Boassa, D.; Taylor, S.; Villa, E. The In Situ Structure of Parkinson’s Disease-Linked LRRK2. Cell 2020, 182, 1508–1518.e16. [Google Scholar] [CrossRef] [PubMed]
- Skolnick, J.; Gao, M.; Zhou, H.; Singh, S. AlphaFold 2: Why It Works and Its Implications for Understanding the Relationships of Protein Sequence, Structure, and Function. J. Chem. Inf. Model. 2021, 61, 4827–4831. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Gotthardt, K.; Weyand, M.; Kortholt, A.; Van Haastert, P.J.M.; Wittinghofer, A. Structure of the Roc-COR Domain Tandem of C. Tepidum, a Prokaryotic Homologue of the Human LRRK2 Parkinson Kinase. EMBO J. 2008, 27, 2239–2249. [Google Scholar] [CrossRef]
- Gilsbach, B.K.; Ho, F.Y.; Vetter, I.R.; Van Haastert, P.J.M.; Wittinghofer, A.; Kortholt, A. Roco Kinase Structures Give Insights into the Mechanism of Parkinson Disease-Related Leucine-Rich-Repeat Kinase 2 Mutations. Proc. Natl. Acad. Sci. USA 2012, 109, 10322–10327. [Google Scholar] [CrossRef]
- Terheyden, S.; Ho, F.Y.; Gilsba, B.K.; Wittinghofer, A.; Kortholt, A. Revisiting the Roco G-Protein Cycle. Biochem. J. 2015, 465, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Deyaert, E.; Leemans, M.; Singh, R.K.; Gallardo, R.; Steyaert, J.; Kortholt, A.; Lauer, J.; Versées, W. Structure and Nucleotide-Induced Conformational Dynamics of the Chlorobium Tepidum Roco Protein. Biochem. J. 2019, 476, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Steger, M.; Diez, F.; Dhekne, H.S.; Lis, P.; Nirujogi, R.S.; Karayel, O.; Tonelli, F.; Martinez, T.N.; Lorentzen, E.; Pfeffer, S.R.; et al. Systematic Proteomic Analysis of LRRK2-Mediated Rab GTPase Phosphorylation Establishes a Connection to Ciliogenesis. Elife 2017, 6, e31012. [Google Scholar] [CrossRef]
- Liu, Z.; Mobley, J.A.; Delucas, L.J.; Kahn, R.A.; West, A.B. LRRK2 Autophosphorylation Enhances Its GTPase Activity. FASEB J. 2016, 30, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Webber, P.J.; Smith, A.D.; Sen, S.; Renfrow, M.B.; Mobley, J.A.; West, A.B. Autophosphorylation in the Leucine-Rich Repeat Kinase 2 (LRRK2) GTPase Domain Modifies Kinase and GTP-Binding Activities. J. Mol. Biol. 2011, 412, 94–110. [Google Scholar] [CrossRef]
- Krebs, E.G. An Accidental Biochemist. Annu. Rev. Biochem. 1998, 67, xiii–xxxii. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Taylor, S.S.; Kornev, A.P. Protein Kinases: Evolution of Dynamic Regulatory Proteins. Trends Biochem. Sci. 2011, 36, 65–776. [Google Scholar] [CrossRef]
- Shudler, M.; Niv, M.Y. Blockmaster: Partitioning Protein Kinase Structures Using Normal-Mode Analysis. J. Phys. Chem. A 2009, 113, 7528–7534. [Google Scholar] [CrossRef]
- Modi, V.; Dunbrack, R.L. Defining a New Nomenclature for the Structures of Active and Inactive Kinases. Proc. Natl. Acad. Sci. USA 2019, 116, 6818–6827. [Google Scholar] [CrossRef]
- Schmidt, S.H.; Weng, J.H.; Aoto, P.C.; Boassa, D.; Mathea, S.; Silletti, S.; Hu, J.; Wallbott, M.; Komives, E.A.; Knapp, S.; et al. Conformation and Dynamics of the Kinase Domain Drive Subcellular Location and Activation of LRRK2. Proc. Natl. Acad. Sci. USA 2021, 118, e2100844118. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Kaila-Sharma, P.; Weng, J.H.; Aoto, P.; Schmidt, S.H.; Knapp, S.; Mathea, S.; Herberg, F.W. Kinase Domain Is a Dynamic Hub for Driving LRRK2 Allostery. Front. Mol. Neurosci. 2020, 13, 538219. [Google Scholar] [CrossRef]
- Schmidt, S.H.; Knape, M.J.; Boassa, D.; Mumdey, N.; Kornev, A.P.; Ellisman, M.H.; Taylor, S.S.; Herberg, F.W. The Dynamic Switch Mechanism That Leads to Activation of LRRK2 Is Embedded in the DFGψ Motif in the Kinase Domain. Proc. Natl. Acad. Sci. USA 2019, 116, 14979–14988. [Google Scholar] [CrossRef] [PubMed]
- Mills, R.D.; Liang, L.Y.; Lio, D.S.S.; Mok, Y.F.; Mulhern, T.D.; Cao, G.; Griffin, M.; Kenche, V.B.; Culvenor, J.G.; Cheng, H.C. The Roc-COR Tandem Domain of Leucine-Rich Repeat Kinase 2 Forms Dimers and Exhibits Conventional Ras-like GTPase Properties. J. Neurochem. 2018, 147, 409–428. [Google Scholar] [CrossRef]
- Paduch, M.; Jeleń, F.; Otlewski, J. Structure of Small G Proteins and Their Regulators. Acta Biochim. Pol. 2001, 48, 829–850. [Google Scholar] [CrossRef] [PubMed]
- Wittinghofer, A.; Vetter, I.R. Structure-Function Relationships of the G Domain, a Canonical Switch Motif. Annu. Rev. Biochem. 2011, 80, 943–971. [Google Scholar] [CrossRef] [PubMed]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase Superfamily: Conserved Structure and Molecular Mechanism. Nature 1991, 349, 125–132. [Google Scholar] [CrossRef]
- Mccormick, F.; Clark, B.F.C.; La Cour, T.F.M.; Kjeldgaard, M.; Norskov-Lauritsen, L.; Nyborg, J. A Model for the Tertiary Structure of P21, the Product of the Ras Oncogene. Science 1985, 230, 78–82. [Google Scholar] [CrossRef]
- Liao, J.; Wu, C.X.; Burlak, C.; Zhang, S.; Sahm, H.; Wang, M.; Zhang, Z.Y.; Vogel, K.W.; Federici, M.; Riddle, S.M.; et al. Parkinson Disease-Associated Mutation R1441H in LRRK2 Prolongs the “Active State” of Its GTPase Domain. Proc. Natl. Acad. Sci. USA 2014, 111, 4055–4060. [Google Scholar] [CrossRef]
- Deyaert, E.; Wauters, L.; Guaitoli, G.; Konijnenberg, A.; Leemans, M.; Terheyden, S.; Petrovic, A.; Gallardo, R.; Nederveen-Schippers, L.M.; Athanasopoulos, P.S.; et al. A Homologue of the Parkinson’s Disease-Associated Protein LRRK2 Undergoes a Monomer-Dimer Transition during GTP Turnover. Nat. Commun. 2017, 8, 1008. [Google Scholar] [CrossRef]
- Mills, R.D.; Mulhern, T.D.; Cheng, H.C.; Culvenor, J.G. Analysis of LRRK2 Accessory Repeat Domains: Prediction of Repeat Length, Number and Sites of Parkinson’s Disease Mutations. Biochem. Soc. Trans. 2012, 40, 1086–1089. [Google Scholar] [CrossRef]
- Rosenbusch, K.E.; Kortholt, A. Activation Mechanism of LRRK2 and Its Cellular Functions in Parkinson’s Disease. Parkinsons. Dis. 2016, 2016, 7351985. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, G.; Onofri, F.; Cirnaru, M.D.; Kaiser, C.J.O.; Jagtap, P.; Kastenmüller, A.; Pischedda, F.; Marte, A.; von Zweydorf, F.; Vogt, A.; et al. Leucine-Rich Repeat Kinase 2 Binds to Neuronal Vesicles through Protein Interactions Mediated by Its C-Terminal WD40 Domain. Mol. Cell. Biol. 2014, 34, 2147–2161. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Weis, W.I. Structure of the Armadillo Repeat Domain of Plakophilin 1. J. Mol. Biol. 2005, 346, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Cardona, F.; Tormos-Pérez, M.; Pérez-Tur, J. Structural and Functional in Silico Analysis of LRRK2 Missense Substitutions. Mol. Biol. Rep. 2014, 41, 2529–2542. [Google Scholar] [CrossRef] [PubMed]
- Michaely, P.; Tomchick, D.R.; Machius, M.; Anderson, R.G.W. Crystal Structure of a 12 ANK Repeat Stack from Human AnkyrinR. EMBO J. 2002, 21, 6387–6396. [Google Scholar] [CrossRef] [PubMed]
- Mosavi, L.K.; Cammett, T.J.; Desrosiers, D.C.; Peng, Z. The Ankyrin Repeat as Molecular Architecture for Protein Recognition. Protein Sci. 2004, 13, 1435–1448. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Mahajan, A.; Tsai, M.D. Ankyrin Repeat: A Unique Motif Mediating Protein-Protein Interactions. Biochemistry 2006, 45, 15168–15178. [Google Scholar] [CrossRef]
- Berger, Z.; Smith, K.A.; Lavoie, M.J. Membrane Localization of LRRK2 Is Associated with Increased Formation of the Highly Active Lrrk2 Dimer and Changes in Its Phosphorylation. Biochemistry 2010, 49, 5511–5523. [Google Scholar] [CrossRef]
- Manschwetus, J.T.; Wallbott, M.; Fachinger, A.; Obergruber, C.; Pautz, S.; Bertinetti, D.; Schmidt, S.H.; Herberg, F.W. Binding of the Human 14-3-3 Isoforms to Distinct Sites in the Leucine-Rich Repeat Kinase 2. Front. Neurosci. 2020, 14, 302. [Google Scholar] [CrossRef]
- McGrath, E.; Waschbüsch, D.; Baker, B.M.; Khan, A.R. LRRK2 Binds to the Rab32 Subfamily in a GTP-Dependent Manner via Its Armadillo Domain. Small GTPases 2021, 12, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, N.; Vlachakis, D.; Memou, A.; Leandrou, E.; Valkimadi, P.E.; Melachroinou, K.; Re, D.B.; Przedborski, S.; Dauer, W.T.; Stefanis, L.; et al. A Motif within the Armadillo Repeat of Parkinson’s-Linked LRRK2 Interacts with FADD to Hijack the Extrinsic Death Pathway. Sci. Rep. 2018, 8, 3455. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Goldberg, M.S. Regulation of LRRK2 Stability by the E3 Ubiquitin Ligase CHIP. PLoS ONE 2009, 4, e5949. [Google Scholar] [CrossRef]
- Zhu, H.; Tonelli, F.; Alessi, D.R.; Sun, J. Structural Basis of Human LRRK2 Membrane Recruitment and Activation. bioRxiv 2022. [Google Scholar] [CrossRef]
- Vides, E.G.; Adhikari, A.; Chiang, C.Y.; Lis, P.; Purlyte, E.; Limouse, C.; Shumate, J.L.; Spinola-Lasso, E.; Dhekne, H.S.; Alessi, D.R.; et al. A Feed-Forward Pathway Drives LRRK2 Kinase Membrane Recruitment and Activation. Elife 2022, 11, e79771. [Google Scholar] [CrossRef] [PubMed]
- Taymans, J.M.; Vancraenenbroeck, R.; Ollikainen, P.; Beilina, A.; Lobbestael, E.; de Maeyer, M.; Baekelandt, V.; Cookson, M.R. LRRK2 Kinase Activity Is Dependent on LRRK2 Gtp Binding Capacity but Independent of LRRK2 GTP Binding. PLoS ONE 2011, 6, e23207. [Google Scholar] [CrossRef] [PubMed]
- Ito, G.; Okai, T.; Fujino, G.; Takeda, K.; Ichijo, H.; Katada, T.; Iwatsubo, T. GTP Binding Is Essential to the Protein Kinase Activity of LRRK2, a Causative Gene Product for Familial Parkinson’s Disease. Biochemistry 2007, 46, 1380–1388. [Google Scholar] [CrossRef]
- Weng, J.H.; Aoto, P.C.; Lorenz, R.; Wu, J.; Schmidt, S.H.; Manschwetus, J.T.; Kaila-Sharma, P.; Silletti, S.; Mathea, S.; Chatterjee, D.; et al. LRRK2 Dynamics Analysis Identifies Allosteric Control of the Crosstalk between Its Catalytic Domains. PLoS Biol. 2022, 20, e3001427. [Google Scholar] [CrossRef] [PubMed]
- Kett, L.R.; Boassa, D.; Ho, C.C.Y.; Rideout, H.J.; Hu, J.; Terada, M.; Ellisman, M.; Dauer, W.T. LRRK2 Parkinson Disease Mutations Enhance Its Microtubule Association. Hum. Mol. Genet. 2012, 21, 890–899. [Google Scholar] [CrossRef]
- Gilsbach, B.K.; Kortholt, A. Structural Biology of the LRRK2 GTPase and Kinase Domains: Implications for Regulation. Front. Mol. Neurosci. 2014, 7, 32. [Google Scholar] [CrossRef]
- Muda, K.; Bertinetti, D.; Gesellchen, F.; Hermann, J.S.; Von Zweydorf, F.; Geerlof, A.; Jacob, A.; Ueffing, M.; Gloeckner, C.J.; Herberg, F.W. Parkinson-Related LRRK2 Mutation R1441C/G/H Impairs PKA Phosphorylation of LRRK2 and Disrupts Its Interaction with 14-3-3. Proc. Natl. Acad. Sci. USA 2014, 111, E34–E43. [Google Scholar] [CrossRef] [PubMed]
- Fraser, K.B.; Moehle, M.S.; Daher, J.P.L.; Webber, P.J.; Williams, J.Y.; Stewart, C.A.; Yacoubian, T.A.; Cowell, R.M.; Dokland, T.; Ye, T.; et al. LRRK2 Secretion in Exosomes Is Regulated by 14-3-3. Hum. Mol. Genet. 2013, 22, 4988–5000. [Google Scholar] [CrossRef]
- Zhao, J.; Molitor, T.P.; Langston, J.W.; Nichols, R.J. LRRK2 Dephosphorylation Increases Its Ubiquitination. Biochem. J. 2015, 469, 107–120. [Google Scholar] [CrossRef]
- Rudi, K.; Ho, F.Y.; Gilsbach, B.K.; Pots, H.; Wittinghofer, A.; Kortholt, A.; Klare, J.P. Conformational Heterogeneity of the Roc Domains in C. Tepidum Roc-COR and Implications for Human LRRK2 Parkinson Mutations. Biosci. Rep. 2015, 35, e00254. [Google Scholar] [CrossRef] [PubMed]
- Nixon-Abell, J.; Berwick, D.C.; Grannó, S.; Spain, V.A.; Blackstone, C.; Harvey, K. Protective LRRK2 R1398H Variant Enhances GTPase and Wnt Signaling Activity. Front. Mol. Neurosci. 2016, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.H.; Jang, J.; Joe, E.H.; Son, I.; Seo, H.; Seol, W. G2385R and I2020T Mutations Increase LRRK2 GTPase Activity. Biomed. Res. Int. 2016, 2016, 7917128. [Google Scholar] [CrossRef] [PubMed]
- Di Fonzo, A.; Wu-Chou, Y.H.; Lu, C.S.; Van Doeselaar, M.; Simons, E.J.; Rohé, C.F.; Chang, H.C.; Chen, R.S.; Weng, Y.H.; Vanacore, N.; et al. A Common Missense Variant in the LRRK2 Gene, Gly2385Arg, Associated with Parkinson’s Disease Risk in Taiwan. Neurogenetics 2006, 7, 133–138. [Google Scholar] [CrossRef]
- Rudenko, I.N.; Kaganovich, A.; Hauser, D.N.; Beylina, A.; Chia, R.; Ding, J.; Maric, D.; Jaffe, H.; Cookson, M.R. The G2385R Variant of Leucine-Rich Repeat Kinase 2 Associated with Parkinson’s Disease Is a Partial Loss-of-Function Mutation. Biochem. J. 2012, 446, 99–111. [Google Scholar] [CrossRef]
- Carrion, M.D.P.; Marsicano, S.; Daniele, F.; Marte, A.; Pischedda, F.; Di Cairano, E.; Piovesana, E.; Von Zweydorf, F.; Kremmer, E.; Gloeckner, C.J.; et al. The LRRK2 G2385R Variant Is a Partial Loss-of-Function Mutation That Affects Synaptic Vesicle Trafficking through Altered Protein Interactions. Sci. Rep. 2017, 7, 5377. [Google Scholar] [CrossRef]
- Ogata, J.; Hirao, K.; Nishioka, K.; Hayashida, A.; Li, Y.; Yoshino, H.; Shimizu, S.; Hattori, N.; Imai, Y. A Novel Lrrk2 Variant p.G2294r in the Wd40 Domain Identified in Familial Parkinson’s Disease Affects Lrrk2 Protein Levels. Int. J. Mol. Sci. 2021, 22, 3708. [Google Scholar] [CrossRef]
- Liu, Z.; Lee, J.; Krummey, S.; Lu, W.; Cai, H.; Lenardo, M.J. The Kinase LRRK2 Is a Regulator of the Transcription Factor NFAT That Modulates the Severity of Inflammatory Bowel Disease. Nat. Immunol. 2011, 12, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Perez Carrion, M.; Pischedda, F.; Biosa, A.; Russo, I.; Straniero, L.; Civiero, L.; Guida, M.; Gloeckner, C.J.; Ticozzi, N.; Tiloca, C.; et al. The LRRK2 Variant E193K Prevents Mitochondrial Fission upon MPP+ Treatment by Altering LRRK2 Binding to DRP1. Front. Mol. Neurosci. 2018, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Marku, A.; Carrion, M.D.P.; Pischedda, F.; Marte, A.; Casiraghi, Z.; Marciani, P.; von Zweydorf, F.; Gloeckner, C.J.; Onofri, F.; Perego, C.; et al. The LRRK2 N-Terminal Domain Influences Vesicle Trafficking: Impact of the E193K Variant. Sci. Rep. 2020, 10, 3799. [Google Scholar] [CrossRef]
- Berg, D.; Schweitzer, K.; Leitner, P.; Zimprich, A.; Lichtner, P.; Belcredi, P.; Brussel, T.; Schulte, C.; Maass, S.; Nagele, T. Type and Frequency of Mutations in the LRRK2 Gene in Familial and Sporadic Parkinson’s Disease*. Brain 2005, 128, 3000–3011. [Google Scholar] [CrossRef] [PubMed]
- Skipper, L.; Shen, H.; Chua, E.; Bonnard, C.; Kolatkar, P.; Tan, L.C.S.; Jamora, R.D.; Puvan, K.; Puong, K.Y.; Zhao, Y.; et al. Analysis of LRRK2 Functional Domains in Nondominant Parkinson Disease. Neurology 2005, 65, 1319–1321. [Google Scholar] [CrossRef]
- Vancraenenbroeck, R.; Lobbestael, E.; Weeks, S.D.; Strelkov, S.V.; Baekelandt, V.; Taymans, J.M.; De Maeyer, M. Expression, Purification and Preliminary Biochemical and Structural Characterization of the Leucine Rich Repeat Namesake Domain of Leucine Rich Repeat Kinase 2. Biochim. Biophys. Acta Proteins Proteom. 2012, 1824, 450–460. [Google Scholar] [CrossRef]
- De Wit, J.; Hong, W.; Luo, L.; Ghosh, A. Role of Leucine-Rich Repeat Proteins in the Development and Function of Neural Circuits. Annu. Rev. Cell Dev. Biol. 2011, 27, 697–729. [Google Scholar] [CrossRef]
- Helton, L.G.; Soliman, A.; Von Zweydorf, F.; Kentros, M.; Manschwetus, J.T.; Hall, S.; Gilsbach, B.; Ho, F.Y.; Athanasopoulos, P.S.; Singh, R.K.; et al. Allosteric Inhibition of Parkinson’s-Linked LRRK2 by Constrained Peptides. ACS Chem. Biol. 2021, 16, 2326–2338. [Google Scholar] [CrossRef]
- Soliman, A.; Cankara, F.N.; Kortholt, A. Allosteric Inhibition of LRRK2, Where Are We Now. Biochem. Soc. Trans. 2020, 48, 2185–2194. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Protein | Technology | Resolution | Expression host |
|---|---|---|---|---|
| 2008 [16] | Hs LRRK2 Roc | X-ray crystallography | 2.0 Å | E. coli |
| 2008 [29] | Ct RocCOR | X-ray crystallography | 2.9 Å | E. coli |
| 2012 [30] | Dd Roco4 Kinase | X-ray crystallography | 1.8 Å | E. coli |
| 2015 [31] | Mb RocCORA | X-ray crystallography | 2.9 Å | E. coli |
| 2016 [21] | Hs LRRK2 fl | Negative stain EM | 33 Å | HEK293F |
| 2017 [22] | Hs LRRK2 fl | Cryo-EM | 24.2 Å | HEK293FT |
| 2018 [32] | Ct LRR-RocCOR | X-ray crystallography | 3.29 Å | E. coli |
| 2019 [17] | Hs LRRK2 WD40 | X-ray crystallography | 2.6 Å | Sf9 insect cell |
| 2020 [23] | Hs LRRK2 RCKW | Cryo-EM | 3.5 Å | Sf9 insect cell |
| 2020 [26] | Hs LRRK2 fl | Cryo-ET | 14 Å | HEK293T |
| 2021 [24] | Hs LRRK2 fl | Cryo-EM | 3.7 Å | HEK293F |
| 2022 [25] | Hs LRRK2 RCKW | Cryo-EM | 5.2 Å | Sf9 insect cell |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Kortholt, A. LRRK2 Structure-Based Activation Mechanism and Pathogenesis. Biomolecules 2023, 13, 612. https://doi.org/10.3390/biom13040612
Zhang X, Kortholt A. LRRK2 Structure-Based Activation Mechanism and Pathogenesis. Biomolecules. 2023; 13(4):612. https://doi.org/10.3390/biom13040612
Chicago/Turabian StyleZhang, Xiaojuan, and Arjan Kortholt. 2023. "LRRK2 Structure-Based Activation Mechanism and Pathogenesis" Biomolecules 13, no. 4: 612. https://doi.org/10.3390/biom13040612
APA StyleZhang, X., & Kortholt, A. (2023). LRRK2 Structure-Based Activation Mechanism and Pathogenesis. Biomolecules, 13(4), 612. https://doi.org/10.3390/biom13040612