
Biophysical and Integrative Characterization of Protein Intrinsic Disorder as a Prime Target for Drug Discovery


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Biophysical Techniques
2.1. Global Conformations via Small-Angle X-ray Scattering (SAXS)
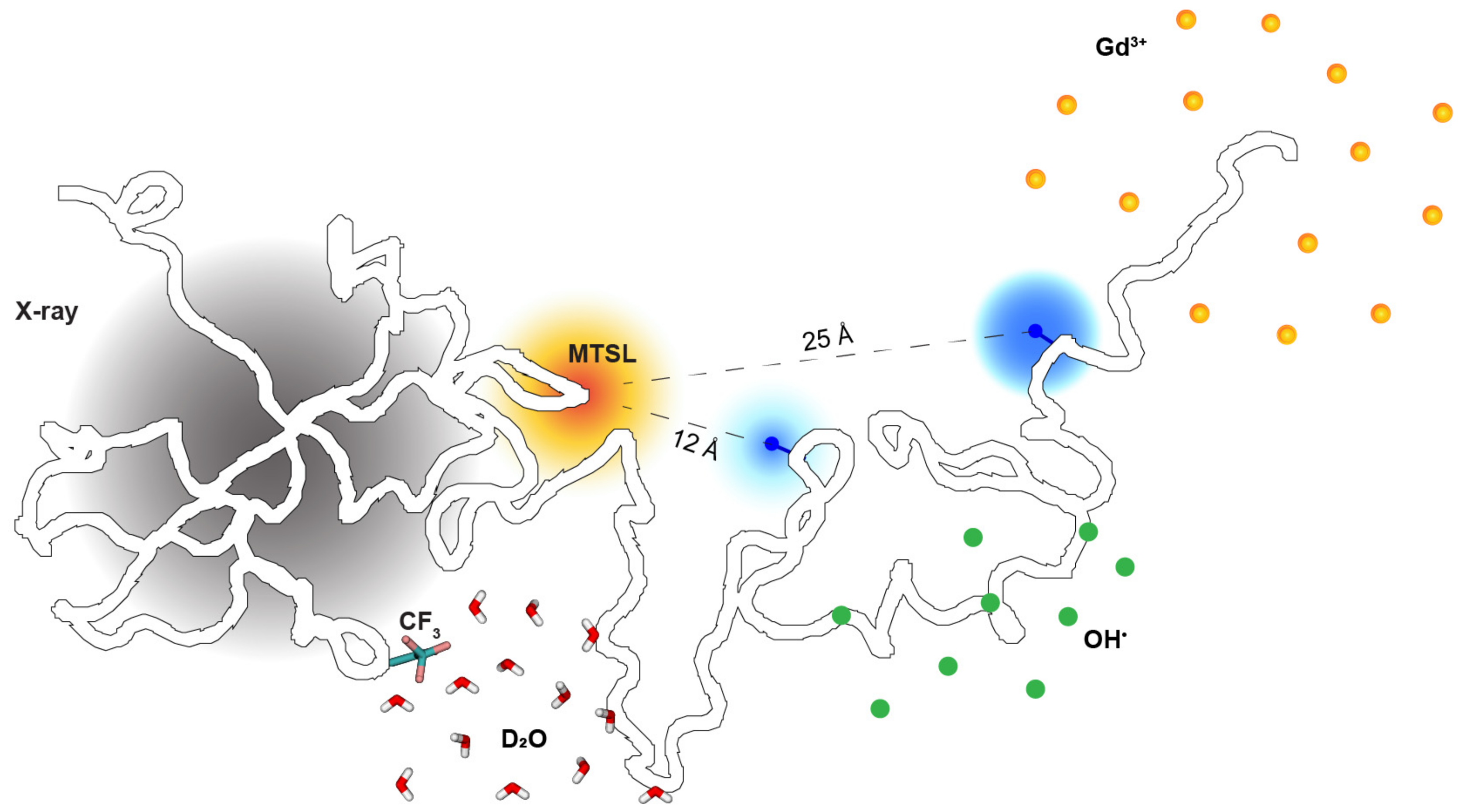
2.2. Site-Specific Solvent Accessibility through the Lens of Three Labeling Techniques
2.3. Probing Single Pairwise Distances between Amino Acids
2.4. Versatile NMR Techniques
3. Theoretical and Computational Biophysical Techniques
3.1. Prediction from the IDP’s Primary Amino Acid Sequence
3.2. Polymer Models for Interpreting Experimental Measurements
3.3. Molecular Simulations and Modeling Methods
3.4. Computational Strategies for Combining Multiple Experimental Measurements
4. Targeting Protein Intrinsic Disorder as a New Frontier of Drug Discovery
5. Perspectives: Chaotic Life of Protein Intrinsic Disorder at a Crossroads
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Bernado, P.; Svergun, D.I. Structural analysis of intrinsically disordered proteins by small-angle X-ray scattering. Mol. Biosyst. 2012, 8, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Kohn, J.E.; Millett, I.S.; Jacob, J.; Zagrovic, B.; Dillon, T.M.; Cingel, N.; Dothager, R.S.; Seifert, S.; Thiyagarajan, P.; Sosnick, T.R.; et al. Random-coil behavior and the dimensions of chemically unfolded proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 12491–12496. [Google Scholar] [CrossRef]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA; London, UK, 1953. [Google Scholar]
- Peng, Y.; Cao, S.; Kiselar, J.; Xiao, X.; Du, Z.; Hsieh, A.; Ko, S.; Chen, Y.; Agrawal, P.; Zheng, W.; et al. A metastable contact and structural disorder in the estrogen receptor transactivation domain. Structure 2019, 27, 229–240.e4. [Google Scholar] [CrossRef]
- Belorusova, A.; Osz, J.; Petoukhov, M.V.; Peluso-Iltis, C.; Kieffer, B.; Svergun, D.I.; Rochel, N. Solution behavior of the intrinsically disordered n-terminal domain of retinoid x receptor alpha in the context of the full-length protein. Biochemistry 2016, 55, 1741–1748. [Google Scholar] [CrossRef]
- Johansen, D.; Trewhella, J.; Goldenberg, D.P. Fractal dimension of an intrinsically disordered protein: Small-angle X-ray scattering and computational study of the bacteriophage lambda n protein. Protein Sci. 2011, 20, 1955–1970. [Google Scholar] [CrossRef]
- Riback, J.A.; Bowman, M.A.; Zmyslowski, A.M.; Knoverek, C.R.; Jumper, J.M.; Hinshaw, J.R.; Kaye, E.B.; Freed, K.F.; Clark, P.L.; Sosnick, T.R. Innovative scattering analysis shows that hydrophobic disordered proteins are expanded in water. Science 2017, 358, 238–241. [Google Scholar] [CrossRef]
- Hofmann, H.; Soranno, A.; Borgia, A.; Gast, K.; Nettels, D.; Schuler, B. Polymer scaling laws of unfolded and intrinsically disordered proteins quantified with single-molecule spectroscopy. Proc. Natl. Acad. Sci. USA 2012, 109, 16155–16160. [Google Scholar] [CrossRef]
- Koch, M.H.J.; Vachette, P.; Svergun, D.I. Small-angle scattering: A view on the properties, structures and structural changes of biological macromolecules in solution. Q. Rev. Biophys. 2003, 36, 147–227. [Google Scholar] [CrossRef]
- Putnam, C.D.; Hammel, M.; Hura, G.L.; Tainer, J.A. X-ray solution scattering. (saxs) combined with crystallography and computation: Defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 2007, 40, 191–285. [Google Scholar] [CrossRef]
- Chandler, D. Introduction to Modern Statistical Mechanics; Oxford University Press: New York, NY, USA, 1987. [Google Scholar]
- Svergun, D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992, 25, 495–503. [Google Scholar] [CrossRef]
- Yang, S. Methods for saxs-based structure determination of biomolecular complexes. Adv. Mater. 2014, 26, 7902–7910. [Google Scholar] [CrossRef]
- Perez, J.; Nishino, Y. Advances in X-ray scattering: From solution saxs to achievements with coherent beams. Curr. Opin. Struct. Biol. 2012, 22, 670–678. [Google Scholar] [CrossRef]
- Englander, S.W.; Kallenbach, N.R. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q. Rev. Biophys. 1983, 16, 521–655. [Google Scholar] [CrossRef]
- Bai, Y.; Milne, J.S.; Mayne, L.; Englander, S.W. Primary structure effects on peptide group hydrogen exchange. Proteins 1993, 17, 75–86. [Google Scholar] [CrossRef]
- Goswami, D.; Devarakonda, S.; Chalmers, M.J.; Pascal, B.D.; Spiegelman, B.M.; Griffin, P.R. Time window expansion for hdx analysis of an intrinsically disordered protein. J. Am. Soc. Mass Spectrom. 2013, 24, 1584–1592. [Google Scholar] [CrossRef]
- Xu, G.; Chance, M.R. Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chem. Rev. 2007, 107, 3514–3543. [Google Scholar] [CrossRef]
- Ralston, C.Y.; Sharp, J.S. Structural investigation of therapeutic antibodies using hydroxyl radical protein footprinting methods. Antibodies 2022, 11, 71. [Google Scholar] [CrossRef]
- Hambly, D.M.; Gross, M.L. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J. Am. Soc. Mass Spectrom. 2005, 16, 2057–2063. [Google Scholar] [CrossRef]
- Johnson, D.T.; Jones, L.M. Hydroxyl radical protein footprinting for analysis of higher order structure. Trends Biochem. Sci. 2022, 47, 989–991. [Google Scholar] [CrossRef] [PubMed]
- McKenzie-Coe, A.; Montes, N.S.; Jones, L.M. Hydroxyl radical protein footprinting: A mass spectrometry-based structural method for studying the higher order structure of proteins. Chem. Rev. 2022, 122, 7532–7561. [Google Scholar] [CrossRef] [PubMed]
- Sharp, J.S.; Chea, E.E.; Misra, S.K.; Orlando, R.; Popov, M.; Egan, R.W.; Holman, D.; Weinberger, S.R. Flash oxidation. (fox) system: A novel laser-free fast photochemical oxidation protein footprinting platform. J. Am. Soc. Mass Spectrom. 2021, 32, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Kitevski-LeBlanc, J.L.; Prosser, R.S. Current applications of 19f nmr to studies of protein structure and dynamics. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 62, 1–33. [Google Scholar] [CrossRef]
- Chrisman, I.M.; Nemetchek, M.D.; de Vera, I.M.S.; Shang, J.; Heidari, Z.; Long, Y.; Reyes-Caballero, H.; Galindo-Murillo, R.; Cheatham, T.E., 3rd; Blayo, A.L.; et al. Defining a conformational ensemble that directs activation of ppargamma. Nat. Commun. 2018, 9, 1794. [Google Scholar] [CrossRef]
- Chance, M.R.; Farquhar, E.R.; Yang, S.; Lodowski, D.T.; Kiselar, J. Protein footprinting: Auxiliary engine to power the structural biology revolution. J. Mol. Biol. 2020, 432, 2973–2984. [Google Scholar] [CrossRef]
- Liu, X.R.; Zhang, M.M.; Gross, M.L. Mass spectrometry-based protein footprinting for higher-order structure analysis: Fundamentals and applications. Chem. Rev. 2020, 120, 4355–4454. [Google Scholar] [CrossRef]
- Liu, J.J.; Horst, R.; Katritch, V.; Stevens, R.C.; Wuthrich, K. Biased signaling pathways in beta(2)-adrenergic receptor characterized by f-19-nmr. Science 2012, 335, 1106–1110. [Google Scholar] [CrossRef]
- Didenko, T.; Liu, J.J.; Horst, R.; Stevens, R.C.; Wuthrich, K. Fluorine-19 nmr of integral membrane proteins illustrated with studies of gpcrs. Curr. Opin. Struc. Biol. 2013, 23, 740–747. [Google Scholar] [CrossRef]
- Matei, E.; Gronenborn, A.M. (19)f paramagnetic relaxation enhancement: A valuable tool for distance measurements in proteins. Angew. Chem. Int. Ed. Engl. 2016, 55, 150–154. [Google Scholar] [CrossRef]
- Evanics, F.; Kitevski, J.L.; Bezsonova, I.; Forman-Kay, J.; Prosser, R.S. F-19 nmr studies of solvent exposure and peptide binding to an sh3 domain. BBA Gen. Subjects 2007, 1770, 221–230. [Google Scholar] [CrossRef]
- Gerig, J.T. Fluorine nmr of proteins. Prog. Nucl. Mag. Res. Sp. 1994, 26, 293–370. [Google Scholar] [CrossRef]
- Kaur, P.; Kiselar, J.; Yang, S.; Chance, M.R. Quantitative protein topography analysis and high-resolution structure prediction using hydroxyl radical labeling and tan.ndem-ion mass spectrometry. (ms). Mol. Cell Proteom. 2015, 14, 1159–1168. [Google Scholar] [CrossRef]
- Kiselar, J.; Chance, M.R. High-resolution hydroxyl radical protein footprinting: Biophysics tool for drug discovery. Annu. Rev. Biophys. 2018, 47, 315–333. [Google Scholar] [CrossRef]
- Gupta, S.; Chen, Y.; Petzold, C.J.; DePonte, D.P.; Ralston, C.Y. Development of container free sample exposure for synchrotron X-ray footprinting. Anal. Chem. 2020, 92, 1565–1573. [Google Scholar] [CrossRef]
- Shcherbakova, I.; Mitra, S.; Beer, R.H.; Brenowitz, M. Fast fenton footprinting: A laboratory-based method for the time-resolved analysis of DNA, rna and proteins. Nucleic Acids Res. 2006, 34, e48. [Google Scholar] [CrossRef]
- Gupta, S.; Celestre, R.; Petzold, C.J.; Chance, M.R.; Ralston, C. Development of a microsecond X-ray protein footprinting facility at the advanced light source. J. Synchrotron Radiat. 2014, 21, 690–699. [Google Scholar] [CrossRef]
- Chen, J.; Rempel, D.L.; Gross, M.L. Temperature jump and fast photochemical oxidation probe submillisecond protein folding. J. Am. Chem. Soc. 2010, 132, 15502–15504. [Google Scholar] [CrossRef]
- Huang, W.; Ravikumar, K.M.; Chance, M.R.; Yang, S. Quantitative mapping of protein structure by hydroxyl radical footprinting-mediated structural mass spectrometry: A protection factor analysis. Biophys. J. 2015, 108, 107–115. [Google Scholar] [CrossRef]
- Zheng, W.; Du, Z.; Ko, S.B.; Wickramasinghe, N.P.; Yang, S. Incorporation of d(2)o-induced fluorine chemical shift perturbations into ensemble-structure characterization of the eralpha disordered region. J. Phys. Chem. B 2022, 126, 9176–9186. [Google Scholar] [CrossRef]
- Schuler, B.; Soranno, A.; Hofmann, H.; Nettels, D. Single-molecule fret spectroscopy and the polymer physics of unfolded and intrinsically disordered proteins. Annu. Rev. Biophys. 2016, 45, 207–231. [Google Scholar] [CrossRef]
- Drescher, M. Epr in protein science: Intrinsically disordered proteins. Top. Curr. Chem. 2012, 321, 91–119. [Google Scholar]
- Schiemann, O.; Heubach, C.A.; Abdullin, D.; Ackermann, K.; Azarkh, M.; Bagryanskaya, E.G.; Drescher, M.; Endeward, B.; Freed, J.H.; Galazzo, L.; et al. Benchmark test and guidelines for deer/peldor experiments on nitroxide-labeled biomolecules. J. Am. Chem. Soc. 2021, 143, 17875–17890. [Google Scholar] [CrossRef]
- Lapidus, L.J.; Eaton, W.A.; Hofrichter, J. Measuring the rate of intramolecular contact formation in polypeptides. Proc. Natl. Acad. Sci. USA 2000, 97, 7220–7225. [Google Scholar] [CrossRef] [PubMed]
- Förster, T. Zwischenmolekulare energiewanderung und fluoreszenz. Ann. Phys. 1948, 6, 55–75. [Google Scholar] [CrossRef]
- Trexler, A.J.; Rhoades, E. Single molecule characterization of alpha-synuclein in aggregation-prone states. Biophys. J. 2010, 99, 3048–3055. [Google Scholar] [CrossRef] [PubMed]
- Wiggers, F.; Wohl, S.; Dubovetskyi, A.; Rosenblum, G.; Zheng, W.; Hofmann, H. Diffusion of a disordered protein on its folded ligand. Proc. Natl. Acad. Sci. USA 2021, 118, e2106690118. [Google Scholar] [CrossRef]
- Chiang, Y.W.; Borbat, P.P.; Freed, J.H. The determination of pair distance distributions by pulsed esr using tikhonov regularization. J. Magn. Reson. 2005, 172, 279–295. [Google Scholar] [CrossRef]
- Buscaglia, M.; Kubelka, J.; Eaton, W.A.; Hofrichter, J. Determination of ultrafast protein folding rates from loop formation dynamics. J. Mol. Biol. 2005, 347, 657–664. [Google Scholar] [CrossRef]
- Sizemore, S.M.; Cope, S.M.; Roy, A.; Ghirlanda, G.; Vaiana, S.M. Slow internal dynamics and charge expansion in the disordered protein cgrp: A comparison with amyl.lin. Biophys. J. 2015, 109, 1038–1048. [Google Scholar] [CrossRef]
- Zerze, G.H.; Mittal, J.; Best, R.B. Diffusive dynamics of contact formation in disordered polypeptides. Phys. Rev. Lett. 2016, 116, 068102. [Google Scholar] [CrossRef]
- Lum, J.K.; Neuweiler, H.; Fersht, A.R. Long-range modulation of chain motions within the intrinsically disordered transactivation domain of tumor suppressor p53. J. Am. Chem. Soc. 2012, 134, 1617–1622. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Nmr illuminates intrinsic disorder. Curr. Opin. Struct. Biol. 2021, 70, 44–52. [Google Scholar] [CrossRef]
- Prestel, A.; Bugge, K.; Staby, L.; Hendus-Altenburger, R.; Kragelund, B.B. Characterization of dynamic idp complexes by nmr spectroscopy. Methods Enzym. 2018, 611, 193–226. [Google Scholar]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Konrat, R. Nmr contributions to structural dynamics studies of intrinsically disordered proteins. J. Magn. Reson. 2014, 241, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.F.; Feng, H.; Zhou, Z.; Bai, Y.; Kay, L.E. Selective characterization of microsecond motions in proteins by nmr relaxation. J. Am. Chem. Soc. 2009, 131, 16257–16265. [Google Scholar] [CrossRef] [PubMed]
- Kay, L.E.; Torchia, D.A.; Bax, A. Backbone dynamics of proteins as studied by 15n inverse detected heteronuclear nmr spectroscopy: Application to staphylococcal nuclease. Biochemistry 1989, 28, 8972–8979. [Google Scholar] [CrossRef]
- Yuwen, T.; Skrynnikov, N.R. Proton-decoupled cpmg: A better experiment for measuring. (15)n r2 relaxation in disordered proteins. J. Magn. Reson. 2014, 241, 155–169. [Google Scholar] [CrossRef]
- Klein-Seetharaman, J.; Oikawa, M.; Grimshaw, S.B.; Wirmer, J.; Duchardt, E.; Ueda, T.; Imoto, T.; Smith, L.J.; Dobson, C.M.; Schwalbe, H. Long-range interactions within a nonnative protein. Science 2002, 295, 1719–1722. [Google Scholar] [CrossRef]
- Martin, E.W.; Holehouse, A.S.; Peran, I.; Farag, M.; Incicco, J.J.; Bremer, A.; Grace, C.R.; Soranno, A.; Pappu, R.V.; Mittag, T. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science 2020, 367, 694–699. [Google Scholar] [CrossRef]
- Yu, L.; Bruschweiler, R. Quantitative prediction of ensemble dynamics, shapes and contact propensities of intrinsically disordered proteins. PLoS Comput. Biol. 2022, 18, e1010036. [Google Scholar] [CrossRef] [PubMed]
- Clore, G.M. Practical aspects of paramagnetic relaxation enhancement in biological macromolecules. Methods Enzym. 2015, 564, 485–497. [Google Scholar]
- Battiste, J.L.; Wagner, G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry 2000, 39, 5355–5365. [Google Scholar] [CrossRef]
- Sjodt, M.; Clubb, R.T. Nitroxide labeling of proteins and the determination of paramagnetic relaxation derived distance restraints for nmr studies. Bio. Protoc. 2017, 7, e2207. [Google Scholar] [CrossRef]
- Clore, G.M.; Gronenborn, A.M. Determination of three-dimensional structures of proteins and nucleic acids in solution by nuclear magnetic resonance spectroscopy. Crit. Rev. Biochem. Mol. Biol. 1989, 24, 479–564. [Google Scholar] [CrossRef]
- Iwahara, J.; Tang, C.; Marius Clore, G. Practical aspects of. (1)h transverse paramagnetic relaxation enhancement measurements on macromolecules. J. Magn. Reson. 2007, 184, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Iwahara, J.; Clore, G.M. Visualization of transient encounter complexes in protein-protein association. Nature 2006, 444, 383–386. [Google Scholar] [CrossRef]
- Lietzow, M.A.; Jamin, M.; Dyson, H.J.; Wright, P.E. Mapping long-range contacts in a highly unfolded protein. J. Mol. Biol. 2002, 322, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Salmon, L.; Nodet, G.; Ozenne, V.; Yin, G.; Jensen, M.R.; Zweckstetter, M.; Blackledge, M. Nmr characterization of long-range order in intrinsically disordered proteins. J. Am. Chem. Soc. 2010, 132, 8407–8418. [Google Scholar] [CrossRef]
- Senicourt, L.; le Maire, A.; Allemand, F.; Carvalho, J.E.; Guee, L.; Germain, P.; Schubert, M.; Bernado, P.; Bourguet, W.; Sibille, N. Structural insights into the interaction of the intrinsically disordered co-activator tif2 with retinoic acid receptor heterodimer. (rxr/rar). J. Mol. Biol. 2021, 433, 166899. [Google Scholar] [CrossRef] [PubMed]
- Bertoncini, C.W.; Jung, Y.S.; Fernandez, C.O.; Hoyer, W.; Griesinger, C.; Jovin, T.M.; Zweckstetter, M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 1430–1435. [Google Scholar] [CrossRef]
- Mittag, T.; Marsh, J.; Grishaev, A.; Orlicky, S.; Lin, H.; Sicheri, F.; Tyers, M.; Forman-Kay, J.D. Structure/function implications in a dynamic complex of the intrinsically disordered sic1 with the cdc4 subunit of an scf ubiquitin ligase. Structure 2010, 18, 494–506. [Google Scholar] [CrossRef]
- Mosure, S.A.; Munoz-Tello, P.; Kuo, K.-T.; MacTavish, B.; Yu, X.; Scholl, D.; Williams, C.C.; Strutzenberg, T.S.; Bass, J.; Brust, R.; et al. Structural basis of interdomain communication in pparγ. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kurzbach, D.; Vanas, A.; Flamm, A.G.; Tarnoczi, N.; Kontaxis, G.; Maltar-Strmecki, N.; Widder, K.; Hinderberger, D.; Konrat, R. Detection of correlated conformational fluctuations in intrinsically disordered proteins through paramagnetic relaxation interference. Phys. Chem. Chem. Phys. 2016, 18, 5753–5758. [Google Scholar] [CrossRef] [PubMed]
- Kurzbach, D.; Beier, A.; Vanas, A.; Flamm, A.G.; Platzer, G.; Schwarz, T.C.; Konrat, R. Nmr probing and visualization of correlated structural fluctuations in intrinsically disordered proteins. Phys. Chem. Chem. Phys. 2017, 19, 10651–10656. [Google Scholar] [CrossRef]
- Kawasaki, R.; Tate, S.I. Impact of the hereditary p301l mutation on the correlated conformational dynamics of human tau protein revealed by the paramagnetic relaxation enhancement nmr experiments. Int. J. Mol. Sci. 2020, 21, 3920. [Google Scholar] [CrossRef]
- Hocking, H.G.; Zangger, K.; Madl, T. Studying the structure and dynamics of biomolecules by using soluble paramagnetic probes. Chemphyschem A Eur. J. Chem. Phys. Phys. Chem. 2013, 14, 3082–3094. [Google Scholar] [CrossRef]
- Gong, Z.; Gu, X.H.; Guo, D.C.; Wang, J.; Tang, C. Protein structural ensembles visualized by solvent paramagnetic relaxation enhancement. Angew. Chem. Int. Ed. Engl. 2017, 56, 1002–1006. [Google Scholar] [CrossRef]
- Kooshapur, H.; Schwieters, C.D.; Tjandra, N. Conformational ensemble of disordered proteins probed by solvent paramagnetic relaxation enhancement. (spre). Angew. Chem. Int. Ed. Engl. 2018, 57, 13519–13522. [Google Scholar] [CrossRef]
- Spreitzer, E.; Usluer, S.; Madl, T. Probing surfaces in dynamic protein interactions. J. Mol. Biol. 2020, 432, 2949–2972. [Google Scholar] [CrossRef]
- Hartlmuller, C.; Spreitzer, E.; Gobl, C.; Falsone, F.; Madl, T. Nmr characterization of solvent accessibility and transient structure in intrinsically disordered proteins. J. Biomol. Nmr. 2019, 73, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Pletka, C.C.; Pettitt, B.M.; Iwahara, J. De novo determination of near-surface electrostatic potentials by NMR. Proc. Natl. Acad. Sci. USA 2021, 118, e2104020118. [Google Scholar] [CrossRef]
- Toyama, Y.; Rangadurai, A.K.; Forman-Kay, J.D.; Kay, L.E. Mapping the per-residue surface electrostatic potential of caprin1 along its phase-separation trajectory. Proc. Natl. Acad. Sci. USA 2022, 119, e2210492119. [Google Scholar] [CrossRef] [PubMed]
- Rangadurai, A.K.; Toyama, Y.; Kay, L.E. Practical considerations for the measurement of near-surface electrostatics based on solvent paramagnetic relaxation enhancements. J. Magn. Reson. 2023, 349, 107400. [Google Scholar] [CrossRef]
- Sigler, P.B. Transcriptional activation. Acid blobs and negative noodles. Nature 1988, 333, 210–212. [Google Scholar] [CrossRef] [PubMed]
- Struhl, K. Promoters, activator proteins, and the mechanism of transcriptional initiation in yeast. Cell 1987, 49, 295–297. [Google Scholar] [CrossRef]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are "natively unfolded" proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Borgia, A.; Borgia, M.B.; Bugge, K.; Kissling, V.M.; Heidarsson, P.O.; Fernandes, C.B.; Sottini, A.; Soranno, A.; Buholzer, K.J.; Nettels, D.; et al. Extreme disorder in an ultrahigh-affinity protein complex. Nature 2018, 555, 61–66. [Google Scholar] [CrossRef]
- Mao, A.H.; Crick, S.L.; Vitalis, A.; Chicoine, C.L.; Pappu, R.V. Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 8183–8188. [Google Scholar] [CrossRef]
- Huang, F.; Oldfield, C.J.; Xue, B.; Hsu, W.L.; Meng, J.; Liu, X.; Shen, L.; Romero, P.; Uversky, V.N.; Dunker, A. Improving protein order-disorder classification using charge-hydropathy plots. BMC Bioinform. 2014, 15, S4. [Google Scholar] [CrossRef]
- Kapcha, L.H.; Rossky, P.J. A simple atomic-level hydrophobicity scale reveals protein interfacial structure. J. Mol. Biol. 2014, 426, 484–498. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Sormanni, P.; Camilloni, C.; Fariselli, P.; Vendruscolo, M. The s2d method: Simultaneous sequence-based prediction of the statistical populations of ordered and disordered regions in proteins. J. Mol. Biol. 2015, 427, 982–996. [Google Scholar] [CrossRef]
- Muñoz, V.; Serrano, L. Elucidating the folding problem of helical peptides using empirical paramters. Nat. Struct. Biol. 1994, 1, 399–409. [Google Scholar] [CrossRef]
- Petersen, B.; Petersen, T.N.; Andersen, P.; Nielsen, M.; Lundegaard, C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 2009, 9, 51. [Google Scholar] [CrossRef]
- Lin, Y.; Currie, S.L.; Rosen, M.K. Intrinsically disordered sequences enable modulation of protein phase separation through distributed tyrosine motifs. J. Biol. Chem. 2017, 292, 19110–19120. [Google Scholar] [CrossRef]
- Mateos, B.; Conrad-Billroth, C.; Schiavina, M.; Beier, A.; Kontaxis, G.; Konrat, R.; Felli, I.C.; Pierattelli, R. The ambivalent role of proline residues in an intrinsically disordered protein: From disorder promoters to compaction facilitators. J. Mol. Biol. 2020, 432, 3093–3111. [Google Scholar] [CrossRef]
- Cohan, M.C.; Shinn, M.K.; Lalmansingh, J.M.; Pappu, R.V. Uncovering non-random binary patterns within sequences of intrinsically disordered proteins. J. Mol. Biol. 2022, 434, 167373. [Google Scholar] [CrossRef]
- He, B.; Wang, K.; Liu, Y.; Xue, B.; Uversky, V.N.; Dunker, A.K. Predicting intrinsic disorder in proteins: An overview. Cell Res. 2009, 19, 929–949. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Quaglia, F.; Meszaros, B.; Salladini, E.; Hatos, A.; Pancsa, R.; Chemes, L.B.; Pajkos, M.; Lazar, T.; Pena-Diaz, S.; Santos, J.; et al. Disprot in 2022: Improved quality and accessibility of protein intrinsic disorder annotation. Nucleic Acids Res. 2022, 50, D480–D487. [Google Scholar] [CrossRef] [PubMed]
- Greber, B.J.; Remis, J.; Ali, S.; Nogales, E. 2.5 a-resolution structure of human cdk-activating kinase bound to the clinical inhibitor icec0942. Biophys. J. 2021, 120, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.J.; Richardson, D.C.; Richardson, J.S. The importance of residue-level filtering and the top2018 best-parts dataset of high-quality protein residues. Protein Sci. 2022, 31, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zerze, G.H.; Borgia, A.; Mittal, J.; Schuler, B.; Best, R.B. Inferring properties of disordered chains from fret transfer efficiencies. J. Chem. Phys. 2018, 148, 123329. [Google Scholar] [CrossRef]
- Piovesan, D.; Tabaro, F.; Micetic, I.; Necci, M.; Quaglia, F.; Oldfield, C.J.; Aspromonte, M.C.; Davey, N.E.; Davidovic, R.; Dosztanyi, Z.; et al. Disprot 7.0: A major update of the database of disordered proteins. Nucleic Acids Res. 2017, 45, D219–D227. [Google Scholar] [CrossRef]
- Fukuchi, S.; Amemiya, T.; Sakamoto, S.; Nobe, Y.; Hosoda, K.; Kado, Y.; Murakami, S.D.; Koike, R.; Hiroaki, H.; Ota, M. Ideal in 2014 illustrates interaction networks composed of intrinsically disordered proteins and their binding partners. Nucleic Acids Res. 2014, 42, D320–D325. [Google Scholar] [CrossRef]
- Piovesan, D.; Del Conte, A.; Clementel, D.; Monzon, A.M.; Bevilacqua, M.; Aspromonte, M.C.; Iserte, J.A.; Orti, F.E.; Marino-Buslje, C.; Tosatto, S.C.E. Mobidb: 10 years of intrinsically disordered proteins. Nucleic Acids Res. 2023, 51, D438–D444. [Google Scholar] [CrossRef]
- Necci, M.; Piovesan, D.; Predictors, C.; DisProt, C.; Tosatto, S.C.E. Critical assessment of protein intrinsic disorder prediction. Nat. Methods 2021, 18, 472–481. [Google Scholar] [CrossRef]
- Hanson, J.; Paliwal, K.K.; Litfin, T.; Zhou, Y. Spot-disorder2: Improved protein intrinsic disorder prediction by ensembled deep learning. Genom. Proteom. Bioinform. 2019, 17, 645–656. [Google Scholar] [CrossRef]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. Pondr-fit: A meta-predictor of intrinsically disordered amino acids. BBA Proteins Proteom. 2010, 1804, 996–1010. [Google Scholar] [CrossRef]
- Hu, G.; Katuwawala, A.; Wang, K.; Wu, Z.; Ghadermarzi, S.; Gao, J.; Kurgan, L. Fldpnn: Accurate intrinsic disorder prediction with putative propensities of disorder functions. Nat. Commun. 2021, 12, 4438. [Google Scholar] [CrossRef]
- Erdos, G.; Pajkos, M.; Dosztanyi, Z. Iupred3: Prediction of protein disorder enhanced with unambiguous experimental annotation and visualization of evolutionary conservation. Nucleic Acids Res. 2021, 49, W297–W303. [Google Scholar] [CrossRef]
- Basu, S.; Kihara, D.; Kurgan, L. Computational prediction of disordered binding regions. Comput. Struct. Biotechnol. J. 2023, 21, 1487–1497. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Romero, P.; Uversky, V.N.; Dunker, A.K. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry 2005, 44, 12454–12470. [Google Scholar] [CrossRef]
- Xue, B.; Dunker, A.K.; Uversky, V.N. Retro-morfs: Identifying protein binding sites by normal and reverse alignment and intrinsic disorder prediction. Int. J. Mol. Sci. 2010, 11, 3725–3747. [Google Scholar] [CrossRef]
- Sharma, R.; Raicar, G.; Tsunoda, T.; Patil, A.; Sharma, A. Opal: Prediction of morf regions in intrinsically disordered protein sequences. Bioinformatics 2018, 34, 1850–1858. [Google Scholar] [CrossRef]
- Jones, D.T.; Cozzetto, D. Disopred3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef]
- Hanson, J.; Litfin, T.; Paliwal, K.; Zhou, Y. Identifying molecular recognition features in intrinsically disordered regions of proteins by transfer learning. Bioinformatics 2020, 36, 1107–1113. [Google Scholar] [CrossRef]
- Krystkowiak, I.; Davey, N.E. Slimsearch: A framework for proteome-wide discovery and annotation of functional modules in intrinsically disordered regions. Nucleic Acids Res. 2017, 45, W464–W469. [Google Scholar] [CrossRef]
- O’Brien, K.T.; Haslam, N.J.; Shields, D.C. Slimscape: A protein short linear motif analysis plugin for cytoscape. BMC Bioinform. 2013, 14, 224. [Google Scholar] [CrossRef] [PubMed]
- Palopoli, N.; Lythgow, K.T.; Edwards, R.J. Qslimfinder: Improved short linear motif prediction using specific query protein data. Bioinformatics 2015, 31, 2284–2293. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Michael, S.; Alvarado-Valverde, J.; Meszaros, B.; Samano-Sanchez, H.; Zeke, A.; Dobson, L.; Lazar, T.; Ord, M.; Nagpal, A.; et al. The eukaryotic linear motif resource: 2022 release. Nucleic Acids Res. 2022, 50, D497–D508. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, B.; Simon, I.; Dosztanyi, Z. Prediction of protein binding regions in disordered proteins. PLoS Comput. Biol. 2009, 5, e1000376. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.T.C.; Gsponer, J. Predicting protein-protein interfaces that bind intrinsically disordered protein regions. J. Mol. Biol. 2019, 431, 3157–3178. [Google Scholar] [CrossRef]
- Peng, Z.; Kurgan, L. High-throughput prediction of rna, DNA and protein binding regions mediated by intrinsic disorder. Nucleic Acids Res. 2015, 43, e121. [Google Scholar] [CrossRef]
- Katuwawala, A.; Zhao, B.; Kurgan, L. Disolippred: Accurate prediction of disordered lipid-binding residues in protein sequences with deep recurrent networks and transfer learning. Bioinformatics 2021, 38, 115–124. [Google Scholar] [CrossRef]
- Schad, E.; Ficho, E.; Pancsa, R.; Simon, I.; Dosztanyi, Z.; Meszaros, B. Dibs: A repository of disordered binding sites mediating interactions with ordered proteins. Bioinformatics 2018, 34, 535–537. [Google Scholar] [CrossRef]
- Miskei, M.; Antal, C.; Fuxreiter, M. Fuzdb: Database of fuzzy complexes, a tool to develop stochastic structure-function relationships for protein complexes and higher-order assemblies. Nucleic Acids Res. 2017, 45, D228–D235. [Google Scholar] [CrossRef]
- Gonzalez-Foutel, N.S.; Glavina, J.; Borcherds, W.M.; Safranchik, M.; Barrera-Vilarmau, S.; Sagar, A.; Estana, A.; Barozet, A.; Garrone, N.A.; Fernandez-Ballester, G.; et al. Conformational buffering underlies functional selection in intrinsically disordered protein regions. Nat. Struct. Mol. Biol. 2022, 29, 781–790. [Google Scholar] [CrossRef]
- Bugge, K.; Brakti, I.; Fernandes, C.B.; Dreier, J.E.; Lundsgaard, J.E.; Olsen, J.G.; Skriver, K.; Kragelund, B.B. Interactions by disorder-a matter of context. Front. Mol. Biosci. 2020, 7, 110. [Google Scholar] [CrossRef]
- Das, R.K.; Pappu, R.V. Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. USA 2013, 110, 13392–13397. [Google Scholar] [CrossRef]
- Sawle, L.; Ghosh, K. A theoretical method to compute sequence dependent configurational properties in charged polymers and proteins. J. Chem. Phys. 2015, 143, 085101. [Google Scholar] [CrossRef]
- Samanta, H.S.; Chakraborty, D.; Thirumalai, D. Charge fluctuation effects on the shape of flexible polyampholytes with applications to intrinsically disordered proteins. J. Chem. Phys. 2018, 149, 163323. [Google Scholar] [CrossRef]
- Zheng, W.; Dignon, G.; Brown, M.; Kim, Y.C.; Mittal, J. Hydropathy patterning complements charge patterning to describe conformational preferences of disordered proteins. J. Phys. Chem. Lett. 2020, 11, 3408–3415. [Google Scholar] [CrossRef]
- Amin, A.N.; Lin, Y.H.; Das, S.; Chan, H.S. Analytical theory for sequence-specific binary fuzzy complexes of charged intrinsically disordered proteins. J. Phys. Chem. B 2020, 124, 6709–6720. [Google Scholar] [CrossRef]
- Yamazaki, H.; Takagi, M.; Kosako, H.; Hirano, T.; Yoshimura, S.H. Cell cycle-specific phase separation regulated by protein charge blockiness. Nat. Cell Biol. 2022, 24, 625–632. [Google Scholar] [CrossRef]
- Lyons, H.; Veettil, R.T.; Pradhan, P.; Fornero, C.; De La Cruz, N.; Ito, K.; Eppert, M.; Roeder, R.G.; Sabari, B.R. Functional partitioning of transcriptional regulators by patterned charge blocks. Cell 2023, 186, 327–345.e28. [Google Scholar] [CrossRef]
- Ruff, K.M.; Pappu, R.V. Alphafold and implications for intrinsically disordered proteins. J. Mol. Biol. 2021, 433, 167208. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with alphafold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Lancaster, A.K.; Nutter-Upham, A.; Lindquist, S.; King, O.D. Plaac: A web and command-line application to identify proteins with prion-like amino acid composition. Bioinformatics 2014, 30, 2501–2502. [Google Scholar] [CrossRef] [PubMed]
- Orlando, G.; Raimondi, D.; Tabaro, F.; Codice, F.; Moreau, Y.; Vranken, W.F. Computational identification of prion-like rna-binding proteins that form liquid phase-separated condensates. Bioinformatics 2019, 35, 4617–4623. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.Y.; Khaodeuanepheng, N.P.; Amarasekara, D.L.; Correia, J.J.; Lewis, K.A.; Fitzkee, N.C.; Hough, L.E.; Whitten, S.T. Intrinsically disordered regions that drive phase separation form a robustly distinct protein class. J. Biol. Chem. 2023, 299, 102801. [Google Scholar] [CrossRef] [PubMed]
- Vernon, R.M.; Chong, P.A.; Tsang, B.; Kim, T.H.; Bah, A.; Farber, P.; Lin, H.; Forman-Kay, J.D. Pi-pi contacts are an overlooked protein feature relevant to phase separation. Elife 2018, 7, e31486. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Sun, T.; Li, Q.; Xu, Y.; Zhang, Z.; Lai, L.; Pei, J. Prediction of liquid-liquid phase separating proteins using machine learning. BMC Bioinform. 2022, 23, 72. [Google Scholar] [CrossRef]
- Vendruscolo, M.; Fuxreiter, M. Sequence determinants of the aggregation of proteins within condensates generated by liquid-liquid phase separation. J. Mol. Biol. 2022, 434, 167201. [Google Scholar] [CrossRef]
- Hatos, A.; Tosatto, S.C.E.; Vendruscolo, M.; Fuxreiter, M. Fuzdrop on alphafold: Visualizing the sequence-dependent propensity of liquid-liquid phase separation and aggregation of proteins. Nucleic Acids Res. 2022, 50, W337–W344. [Google Scholar] [CrossRef]
- Mentes, A.; Magyar, C.; Ficho, E.; Simon, I. Analysis of heterodimeric “mutual synergistic folding”-complexes. Int. J. Mol. Sci. 2019, 20, 5136. [Google Scholar] [CrossRef]
- Marsh, J.A.; Forman-Kay, J.D. Sequence determinants of compaction in intrinsically disordered proteins. Biophys. J. 2010, 98, 2383–2390. [Google Scholar] [CrossRef]
- Borgia, A.; Zheng, W.; Buholzer, K.; Borgia, M.B.; Schuler, A.; Hofmann, H.; Soranno, A.; Nettels, D.; Gast, K.; Grishaev, A.; et al. Consistent view of polypeptide chain expansion in chemical denaturants from multiple experimental methods. J. Am. Chem. Soc. 2016, 138, 11714–11726. [Google Scholar] [CrossRef]
- des Cloizeaux, J. Langrangian theory for a self-avoiding random chain. Phys. Rev. A 1974, 10, 1665–1669. [Google Scholar] [CrossRef]
- Le Guillou, J.C.; Zinn-Justin, J. Critical exponents for n-vector model in 3 dimensions from field-theory. Phys. Rev. Lett. 1977, 39, 95–98. [Google Scholar] [CrossRef]
- Fisher, M.E. Shape of a self-avoiding walk or polymer chain. J. Chem. Phys. 1966, 44, 616–622. [Google Scholar] [CrossRef]
- Witten, T.A.; Schäfer, L. Two critical ratios in polymer solutions. J. Phys. A 1978, 11, 1843–1854. [Google Scholar] [CrossRef]
- Zheng, W.; Best, R.B. An extended guinier analysis for intrinsically disordered proteins. J. Mol. Biol. 2018, 430, 2540–2553. [Google Scholar] [CrossRef]
- Dignon, G.L.; Zheng, W.; Best, R.B.; Kim, Y.C.; Mittal, J. Relation between single-molecule properties and phase behavior of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2018, 115, 9929–9934. [Google Scholar] [CrossRef]
- Gruet, A.; Dosnon, M.; Blocquel, D.; Brunel, J.; Gerlier, D.; Das, R.K.; Bonetti, D.; Gianni, S.; Fuxreiter, M.; Longhi, S.; et al. Fuzzy regions in an intrinsically disordered protein impair protein-protein interactions. FEBS J. 2016, 283, 576–594. [Google Scholar] [CrossRef]
- Staby, L.; Due, A.D.; Kunze, M.B.A.; Jorgensen, M.L.M.; Skriver, K.; Kragelund, B.B. Flanking disorder of the folded alphaalpha-hub domain from radical induced cell death1 affects transcription factor binding by ensemble redistribution. J. Mol. Biol. 2021, 433, 167320. [Google Scholar] [CrossRef]
- Wang, R.Y.; Han, Y.; Krassovsky, K.; Sheffler, W.; Tyka, M.; Baker, D. Modeling disordered regions in proteins using rosetta. PLoS ONE 2011, 6, e22060. [Google Scholar] [CrossRef]
- Zheng, W.; Dignon, G.L.; Jovic, N.; Xu, X.; Regy, R.M.; Fawzi, N.L.; Kim, Y.C.; Best, R.B.; Mittal, J. Molecular details of protein condensates probed by microsecond long atomistic simulations. J. Phys. Chem. B 2020, 124, 11671–11679. [Google Scholar] [CrossRef]
- Zheng, W.; Hofmann, H.; Schuler, B.; Best, R.B. Origin of internal friction in disordered proteins depends on solvent quality. J. Phys. Chem. B 2018, 122, 11478–11487. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Borgia, A.; Buholzer, K.; Grishaev, A.; Schuler, B.; Best, R.B. Probing the action of chemical denaturant on an intrinsically disordered protein by simulation and experiment. J. Am. Chem. Soc. 2016, 138, 11702–11713. [Google Scholar] [CrossRef]
- Best, R.B.; Zheng, W.; Mittal, J. Balanced protein-water interactions improve properties of disordered proteins and non-specific protein association. J. Chem. Theory Comput. 2014, 10, 5113–5124. [Google Scholar] [CrossRef] [PubMed]
- Piana, S.; Donchev, A.G.; Robustelli, P.; Shaw, D.E. Water dispersion interactions strongly influence simulated structural properties of disordered protein states. J. Phys. Chem. B 2015, 119, 5113–5123. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D. Charmm36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef]
- Song, D.; Luo, R.; Chen, H.F. The idp-specific force field ff14idpsff improves the conformer sampling of intrinsically disordered proteins. J. Chem. Inf. Model. 2017, 57, 1166–1178. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.; Mittal, J.; Feig, M.; MacKerell, A.D., Jr. Optimization of the additive charmm all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Bottaro, S.; Lindorff-Larsen, K.; Best, R.B. Variational optimization of an all-atom implicit solvent force field to match explicit solvent simulation data. J. Chem. Theory Comput. 2013, 9, 5641–5652. [Google Scholar] [CrossRef]
- Vitalis, A.; Pappu, R.V. Absinth: A new continuum solvation model for simulations of polypeptides in aqueous solutions. J. Comput. Chem. 2008, 30, 673–699. [Google Scholar] [CrossRef]
- Choi, J.M.; Pappu, R.V. Improvements to the absinth force field for proteins based on experimentally derived amino acid specific backbone conformational statistics. J. Chem. Theory Comput. 2019, 15, 1367–1382. [Google Scholar] [CrossRef]
- Robustelli, P.; Piana, S.; Shaw, D.E. Mechanism of coupled folding-upon-binding of an intrinsically disordered protein. J. Am. Chem. Soc. 2020, 142, 11092–11101. [Google Scholar] [CrossRef]
- Strodel, B. Amyloid aggregation simulations: Challenges, advances and perspectives. Curr. Opin. Struct. Biol. 2021, 67, 145–152. [Google Scholar] [CrossRef]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics methods for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Liu, P.; Kim, B.; Friesner, R.A.; Berne, B.J. Replica exchange with solute tempering: A method for sampling biological systems in explicit water. Proc. Natl. Acad. Sci. USA 2005, 102, 13749–13754. [Google Scholar] [CrossRef]
- Liu, X.; Chen, J. Residual structures and transient long-range interactions of p53 transactivation domain: Assessment of explicit solvent protein force fields. J. Chem. Theory Comput. 2019, 15, 4708–4720. [Google Scholar] [CrossRef]
- Miao, Y.; Feher, V.A.; McCammon, J.A. Gaussian accelerated molecular dynamics: Unconstrained enhanced sampling and free energy calculation. J. Chem. Theory Comput. 2015, 11, 3584–3595. [Google Scholar] [CrossRef]
- Hamelberg, D.; Mongan, J.; McCammon, J.A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120, 11919–11929. [Google Scholar] [CrossRef]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. Plumed 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Non-physical sampling distributions in monte-carlo free-energy estimation. J. Comp. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Shaw, D.E.; Deneroff, M.M.; Dror, R.O.; Kuskin, J.S.; Larson, R.H.; Salmon, J.K.; Young, C.; Batson, B.; Bowers, K.J.; Chao, J.C.; et al. Anton, a special-purpose machine for molecular dynamics simulation. In Isca’07: 34th Annual International Symposium on Computer Architecture, Conference Proceedings 1–12; Assoc Computing Machinery: New York, NY, USA, 2007. [Google Scholar]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E., III; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. Amber, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comp. Phys. Comm. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van der Spoel, D.; Lindahl, E. Gromacs4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.G.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with namd. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Chen, X.; Jin, S.; Chen, M.; Bueno, C.; Wolynes, P.G. The marionette mechanism of domain-domain communication in the antagonist, agonist, and coactivator responses of the estrogen receptor. Proc. Natl. Acad. Sci. USA 2023, 120, e2216906120. [Google Scholar] [CrossRef]
- Wu, H.; Wolynes, P.G.; Papoian, G.A. Awsem-idp: A coarse-grained force field for intrinsically disordered proteins. J. Phys. Chem. B 2018, 122, 11115–11125. [Google Scholar] [CrossRef]
- Latham, A.P.; Zhang, B. Improving coarse-grained protein force fields with small-angle X-ray scattering data. J. Phys. Chem. B 2019, 123, 1026–1034. [Google Scholar] [CrossRef]
- Ozenne, V.; Bauer, F.; Salmon, L.; Huang, J.R.; Jensen, M.R.; Segard, S.; Bernado, P.; Charavay, C.; Blackledge, M. Flexible-meccano: A tool for the generation of explicit ensemble descriptions of intrinsically disordered proteins and their associated experimental observables. Bioinformatics 2012, 28, 1463–1470. [Google Scholar] [CrossRef]
- Dignon, G.L.; Zheng, W.W.; Kim, Y.C.; Best, R.B.; Mittal, J. Sequence determinants of protein phase behavior from a coarse-grained model. PLoS Comput. Biol. 2018, 14, e1005941. [Google Scholar] [CrossRef]
- Debye, P.; Hückel, E. De la theorie des electrolytes. I. Abaissement du point de congelation et phenomenes associes. Phys. Z. 1923, 24, 185–206. [Google Scholar]
- Ashbaugh, H.S.; Hatch, H.W. Natively unfolded protein stability as a coil-to-globule transition in charge/hydropathy space. J. Am. Chem. Soc. 2008, 130, 9536–9542. [Google Scholar] [CrossRef]
- Joseph, J.A.; Reinhardt, A.; Aguirre, A.; Chew, P.Y.; Russell, K.O.; Espinosa, J.R.; Garaizar, A.; Collepardo-Guevara, R. Physics-driven coarse-grained model for biomolecular phase separation with near-quantitative accuracy. Nat. Comput. Sci. 2021, 1, 732–743. [Google Scholar] [CrossRef]
- Wang, X.; Ramirez-Hinestrosa, S.; Dobnikar, J.; Frenkel, D. The lennard-jones potential: When. (not) to use it. Phys. Chem. Chem. Phys. 2020, 22, 10624–10633. [Google Scholar] [CrossRef]
- Kim, Y.C.; Hummer, G. Coarse-grained models for simulations of multiprotein complexes: Application to ubiquitin binding. J. Mol. Biol. 2008, 375, 1416–1433. [Google Scholar] [CrossRef]
- Ravikumar, K.M.; Huang, W.; Yang, S. Coarse-grained simulations of protein-protein association: An energy landscape perspective. Biophys. J. 2012, 103, 837–845. [Google Scholar] [CrossRef]
- Dannenhoffer-Lafage, T.; Best, R.B. A data-driven hydrophobicity scale for predicting liquid-liquid phase separation of proteins. J. Phys. Chem. B 2021, 125, 4046–4056. [Google Scholar] [CrossRef]
- Regy, R.M.; Thompson, J.; Kim, Y.C.; Mittal, J. Improved coarse-grained model for studying sequence dependent phase separation of disordered proteins. Protein Sci. 2021, 30, 1371–1379. [Google Scholar] [CrossRef]
- Tesei, G.; Schulze, T.K.; Crehuet, R.; Lindorff-Larsen, K. Accurate model of liquid-liquid phase behavior of intrinsically disordered proteins from optimization of single-chain properties. Proc. Natl. Acad. Sci. USA 2021, 118, e2111696118. [Google Scholar] [CrossRef]
- Dignon, G.L.; Zheng, W.; Kim, Y.C.; Mittal, J. Temperature-controlled liquid-liquid phase separation of disordered proteins. ACS Cent. Sci. 2019, 5, 821–830. [Google Scholar] [CrossRef]
- Wohl, S.; Jakubowski, M.; Zheng, W. Salt-dependent conformational changes of intrinsically disordered proteins. J. Phys. Chem. Lett. 2021, 12, 6684–6691. [Google Scholar] [CrossRef]
- Rizuan, A.; Jovic, N.; Phan, T.M.; Kim, Y.C.; Mittal, J. Developing bonded potentials for a coarse-grained model of intrinsically disordered proteins. J. Chem. Inf. Model. 2022, 62, 4474–4485. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wu, J.; Sternke-Hoffmann, R.; Zheng, W.; Morman, C.; Luo, J. Multivariate effects of ph, salt, and zn(2+) ions on abeta(40) fibrillation. Commun. Chem. 2022, 5, 171. [Google Scholar] [CrossRef] [PubMed]
- Regy, R.M.; Dignon, G.L.; Zheng, W.; Kim, Y.C.; Mittal, J. Sequence dependent phase separation of protein-polynucleotide mixtures elucidated using molecular simulations. Nucleic Acids Res. 2020, 48, 12593–12603. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Vendruscolo, M. Determination of ensembles of protein structures consistent with nmr order parameters. J. Am. Chem. Soc. 2004, 126, 8090–8091. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Best, R.B.; Depristo, M.A.; Dobson, C.M.; Vendruscolo, M. Simultaneous determination of protein structure and dynamics. Nature 2005, 433, 128–132. [Google Scholar] [CrossRef]
- Jensen, M.R.; Salmon, L.; Nodet, G.; Blackledge, M. Defining conformational ensembles of intrinsically disordered and partially folded proteins directly from chemical shifts. J. Am. Chem. Soc. 2010, 132, 1270–1272. [Google Scholar] [CrossRef]
- Kofinger, J.; Stelzl, L.S.; Reuter, K.; Allande, C.; Reichel, K.; Hummer, G. Efficient ensemble refinement by reweighting. J. Chem. Theory Comput. 2019, 15, 3390–3401. [Google Scholar] [CrossRef]
- Brookes, D.H.; Head-Gordon, T. Experimental inferential structure determination of ensembles for intrinsically disordered proteins. J. Am. Chem. Soc. 2016, 138, 4530–4538. [Google Scholar] [CrossRef]
- Gomes, G.W.; Krzeminski, M.; Namini, A.; Martin, E.W.; Mittag, T.; Head-Gordon, T.; Forman-Kay, J.D.; Gradinaru, C.C. Conformational ensembles of an intrinsically disordered protein consistent with nmr, saxs, and single-molecule fret. J. Am. Chem. Soc. 2020, 142, 15697–15710. [Google Scholar] [CrossRef]
- Hsieh, A.; Lu, L.; Chance, M.R.; Yang, S. A practical guide to ispot modeling: An integrative structural biology platform. Biol. Small Angle Scatt. Tech. Strateg. Tips 2017, 1009, 229–238. [Google Scholar]
- Huang, W.; Ravikumar, K.M.; Parisien, M.; Yang, S. Theoretical modeling of multiprotein complexes by ispot: Integration of small-angle X-ray scattering, hydroxyl radical footprinting, and computational docking. J. Struct. Biol. 2016, 196, 340–349. [Google Scholar] [CrossRef]
- Yang, S.; Bernado, P. Integrative biophysics: Protein interaction and disorder. J. Mol. Biol. 2020, 432, 2843–2845. [Google Scholar] [CrossRef]
- Tong, D.; Yang, S.; Lu, L. Accurate optimization of amino acid form factors for computing small-angle X-ray scattering intensity of atomistic protein structures. J. Appl. Crystallogr. 2016, 49, 1148–1161. [Google Scholar] [CrossRef]
- Ravikumar, K.M.; Huang, W.; Yang, S. Fast-saxs-pro: A unified approach to computing saxs profiles of DNA, rna, protein, and their complexes. J. Chem. Phys. 2013, 138, 024112. [Google Scholar] [CrossRef]
- Niebling, S.; Bjorling, A.; Westenhoff, S. Martini bead form factors for the analysis of time-resolved X-ray scattering of proteins. J. Appl. Crystallogr. 2014, 47, 1190–1198. [Google Scholar] [CrossRef]
- Svergun, D.; Barberato, C.; Koch, M.H.J. Crysol-a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 1995, 28, 768–773. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Hammel, M.; Tainer, J.A.; Sali, A. Accurate saxs profile computation and its assessment by contrast variation experiments. Biophys. J. 2013, 105, 962–974. [Google Scholar] [CrossRef]
- Gong, Z.; Schwieters, C.D.; Tang, C. Theory and practice of using solvent paramagnetic relaxation enhancement to characterize protein conformational dynamics. Methods 2018, 148, 48–56. [Google Scholar] [CrossRef]
- Schwieters, C.D.; Kuszewski, J.J.; Tjandra, N.; Clore, G.M. The xplor-nih nmr molecular structure determination package. J. Magn. Reson. 2003, 160, 65–73. [Google Scholar] [CrossRef]
- Qi, Y.; Lee, J.; Cheng, X.; Shen, R.; Islam, S.M.; Roux, B.; Im, W. Charmm-gui deer facilitator for spin-pair distance distribution calculations and preparation of restrained-ensemble molecular dynamics simulations. J. Comput. Chem. 2020, 41, 415–420. [Google Scholar] [CrossRef]
- Worswick, S.G.; Spencer, J.A.; Jeschke, G.; Kuprov, I. Deep neural network processing of deer data. Sci. Adv. 2018, 4, eaat5218. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.M.; Stein, R.A.; McHaourab, H.S.; Roux, B. Structural refinement from restrained-ensemble simulations based on epr/deer data: Application to t4 lysozyme. J. Phys. Chem. B 2013, 117, 4740–4754. [Google Scholar] [CrossRef] [PubMed]
- Roux, B.; Weare, J. On the statistical equivalence of restrained-ensemble simulations with the maximum entropy method. J. Chem. Phys. 2013, 138, 084107. [Google Scholar] [CrossRef] [PubMed]
- Dannenhoffer-Lafage, T.; White, A.D.; Voth, G.A. A direct method for incorporating experimental data into multiscale coarse-grained models. J. Chem. Theory Comput. 2016, 12, 2144–2153. [Google Scholar] [CrossRef]
- White, A.D.; Voth, G.A. Efficient and minimal method to bias molecular simulations with experimental data. J. Chem. Theory Comput. 2014, 10, 3023–3030. [Google Scholar] [CrossRef]
- Pitera, J.W.; Chodera, J.D. On the use of experimental observations to bias simulated ensembles. J. Chem. Theory Comput. 2012, 8, 3445–3451. [Google Scholar] [CrossRef]
- Hermann, M.R.; Hub, J.S. Saxs-restrained ensemble simulations of intrinsically disordered proteins with commitment to the principle of maximum entropy. J. Chem. Theory Comput. 2019, 15, 5103–5115. [Google Scholar] [CrossRef]
- Hub, J.S. Interpreting solution X-ray scattering data using molecular simulations. Curr. Opin. Struct. Biol. 2018, 49, 18–26. [Google Scholar] [CrossRef]
- Shen, R.; Han, W.; Fiorin, G.; Islam, S.M.; Schulten, K.; Roux, B. Structural refinement of proteins by restrained molecular dynamics simulations with non-interacting molecular fragments. PLoS Comput. Biol. 2015, 11, e1004368. [Google Scholar] [CrossRef]
- Biehn, S.E.; Lindert, S. Accurate protein structure prediction with hydroxyl radical protein footprinting data. Nat. Commun. 2021, 12, 341. [Google Scholar] [CrossRef]
- Nath, A.; Sammalkorpi, M.; DeWitt, D.C.; Trexler, A.J.; Elbaum-Garfinkle, S.; O’Hern, C.S.; Rhoades, E. The conformational ensembles of alpha-synuclein and tau: Combining single-molecule fret and simulations. Biophys. J. 2012, 103, 1940–1949. [Google Scholar] [CrossRef]
- Tang, C.; Gong, Z. Integrating non-nmr distance restraints to augment nmr depiction of protein structure and dynamics. J. Mol. Biol. 2020, 432, 2913–2929. [Google Scholar] [CrossRef]
- Delhommel, F.; Gabel, F.; Sattler, M. Current approaches for integrating solution nmr spectroscopy and small-angle scattering to study the structure and dynamics of biomolecular complexes. J. Mol. Biol. 2020, 432, 2890–2912. [Google Scholar] [CrossRef]
- Schindler, C.E.M.; de Vries, S.J.; Sasse, A.; Zacharias, M. Saxs data alone can generate high-quality models of protein-protein complexes. Structure 2016, 24, 1387–1397. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The cluspro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Xia, B.; Mamonov, A.; Leysen, S.; Allen, K.N.; Strelkov, S.V.; Paschalidis, I.; Vajda, S.; Kozakov, D. Accounting for observed small angle X-ray scattering profile in the protein-protein docking server cluspro. J. Comput. Chem. 2015, 36, 1568–1572. [Google Scholar] [CrossRef]
- Jimenez-Garcia, B.; Bernado, P.; Fernandez-Recio, J. Structural characterization of protein-protein interactions with pydocksaxs. Methods Mol. Biol. 2020, 2112, 131–144. [Google Scholar]
- Vangone, A.; Oliva, R.; Cavallo, L.; Bonvin, A.M.J.J. Prediction of biomolecular complexes. In From Protein Structure to Function with Bioinformatics; Rigden, D.J., Ed.; Springer: Dordrecht, The Netherlands, 2017; pp. 265–292. [Google Scholar]
- Karaca, E.; Bonvin, A.M.J.J. On the usefulness of ion-mobility mass spectrometry and saxs data in scoring docking decoys. Acta Crystallogr. Sect. D 2013, 69, 683–694. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Hammel, M.; Sali, A. Macromolecular docking restrained by a small angle X-ray scattering profile. J. Struct. Biol. 2011, 173, 461–471. [Google Scholar] [CrossRef]
- Huang, W.; Peng, Y.; Kiselar, J.; Zhao, X.; Albaqami, A.; Mendez, D.; Chen, Y.; Chakravarthy, S.; Gupta, S.; Ralston, C.; et al. Multidomain architecture of estrogen receptor reveals interfacial cross-talk between its DNA-binding and ligand-binding domains. Nat. Commun. 2018, 9, 3520. [Google Scholar] [CrossRef]
- Paissoni, C.; Jussupow, A.; Camilloni, C. Martini bead form factors for nucleic acids and their application in the refinement of protein-nucleic acid complexes against saxs data. J. Appl. Crystallogr. 2019, 52, 394–402. [Google Scholar] [CrossRef]
- Pahari, S.; Liu, S.; Lee, C.H.; Akbulut, M.; Kwon, J.S. Saxs-guided unbiased coarse-grained monte carlo simulation for identification of self-assembly nanostructures and dimensions. Soft Matter 2022, 18, 5282–5292. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Kiselar, J.; Zhang, W.; Li, S.S.; Xiong, R.; Liu, Y.; Yang, S.; Lai, L. Integrative structural modeling of a multidomain polo-like kinase. Phys. Chem. Chem. Phys. 2020, 22, 27581–27589. [Google Scholar] [CrossRef] [PubMed]
- Ekimoto, T.; Ikeguchi, M. Hybrid methods for modeling protein structures using molecular dynamics simulations and small-angle X-ray scattering data. In Integrative Structural Biology with Hybrid Methods; Nakamura, H., Kleywegt, G., Burley, S.K., Markley, J.L., Eds.; Springer: Singapore, 2018; pp. 237–258. [Google Scholar]
- Bowerman, S.; Curtis, J.E.; Clayton, J.; Brookes, E.H.; Wereszczynski, J. Bees: Bayesian ensemble estimation from sas. Biophys. J. 2019, 117, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Blachowicz, L.; Makowski, L.; Roux, B. Multidomain assembled states of hck tyrosine kinase in solution. Proc. Natl. Acad. Sci. USA 2010, 107, 15757–15762. [Google Scholar] [CrossRef]
- Bernado, P.; Blackledge, M. Structural biology: Proteins in dynamic equilibrium. Nature 2010, 468, 1046–1048. [Google Scholar] [CrossRef]
- Song, L.; Yang, L.; Meng, J.; Yang, S. Thermodynamics of hydrophobic amino acids in solution: A combined experimental-computational study. J. Phys. Chem. Lett. 2017, 8, 347–351. [Google Scholar] [CrossRef]
- Antonov, L.D.; Olsson, S.; Boomsma, W.; Hamelryck, T. Bayesian inference of protein ensembles from saxs data. Phys. Chem. Chem. Phys. 2016, 18, 5832–5838. [Google Scholar] [CrossRef]
- Jamros, M.A.; Oliveira, L.C.; Whitford, P.C.; Onuchic, J.N.; Adams, J.A.; Blumenthal, D.K.; Jennings, P.A. Proteins at work: A combined small angle X-ray scattering and theoretical determination of the multiple structures involved on the protein kinase functional landscape. J. Biol. Chem. 2010, 285, 36121–36128. [Google Scholar] [CrossRef]
- Zhang, Y.; Wen, B.; Peng, J.; Zuo, X.; Gong, Q.; Zhang, Z. Determining structural ensembles of flexible multi-domain proteins using small-angle X-ray scattering and molecular dynamics simulations. Protein Cell 2015, 6, 619–623. [Google Scholar] [CrossRef]
- Miyashita, O.; Gorba, C.; Tama, F. Structure modeling from small angle X-ray scattering data with elastic network normal mode analysis. J. Struct. Biol. 2011, 173, 451–460. [Google Scholar] [CrossRef]
- Liu, Z.; Gong, Z.; Cao, Y.; Ding, Y.H.; Dong, M.Q.; Lu, Y.B.; Zhang, W.P.; Tang, C. Characterizing protein dynamics with integrative use of bulk and single-molecule techniques. Biochemistry 2018, 57, 305–313. [Google Scholar] [CrossRef]
- Chen, Y.; Pollack, L. Saxs studies of rna: Structures, dynamics, and interactions with partners. Wiley Interdiscip Rev. RNA 2016, 7, 512–526. [Google Scholar] [CrossRef]
- Yang, S.C.; Parisien, M.; Major, F.; Roux, B. Rna structure determination using saxs data. J. Phys. Chem. B 2010, 114, 10039–10048. [Google Scholar] [CrossRef]
- Sun, L.Z.; Zhang, D.; Chen, S.J. Theory and modeling of rna structure and interactions with metal ions and small molecules. Annu. Rev. Biophys. 2017, 46, 227–246. [Google Scholar] [CrossRef]
- Prajapati, J.D.; Onuchic, J.N.; Sanbonmatsu, K.Y. Exploring the energy landscape of riboswitches using collective variables based on tertiary contacts. J. Mol. Biol. 2022, 434, 167788. [Google Scholar] [CrossRef]
- Bernado, P.; Mylonas, E.; Petoukhov, M.V.; Blackledge, M.; Svergun, D.I. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 2007, 129, 5656–5664. [Google Scholar] [CrossRef]
- Sterckx, Y.G.; Volkov, A.N.; Vranken, W.F.; Kragelj, J.; Jensen, M.R.; Buts, L.; Garcia-Pino, A.; Jove, T.; Van Melderen, L.; Blackledge, M.; et al. Small-angle X-ray scattering-and nuclear magnetic resonance-derived conformational ensemble of the highly flexible antitoxin paaa2. Structure 2014, 22, 854–865. [Google Scholar] [CrossRef]
- Bernado, P.; Blanchard, L.; Timmins, P.; Marion, D.; Ruigrok, R.W.; Blackledge, M. A structural model for unfolded proteins from residual dipolar couplings and small-angle X-ray scattering. Proc. Natl. Acad. Sci. USA 2005, 102, 17002–17007. [Google Scholar] [CrossRef]
- Lin, X.; Roy, S.; Jolly, M.K.; Bocci, F.; Schafer, N.P.; Tsai, M.Y.; Chen, Y.; He, Y.; Grishaev, A.; Weninger, K.; et al. Page4 and conformational switching: Insights from molecular dynamics simulations and implications for prostate cancer. J. Mol. Biol. 2018, 430, 2422–2438. [Google Scholar] [CrossRef]
- Receveur-Brechot, V.; Durand, D.H. ow random are intrinsically disordered proteins? A small angle scattering perspective. Curr. Protein Pept. Sci. 2012, 13, 55–75. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.A.; Forman-Kay, J.D. Ensemble modeling of protein disordered states: Experimental restraint contributions and validation. Proteins 2012, 80, 556–572. [Google Scholar] [CrossRef] [PubMed]
- Gomes, G.-N.W.; Namini, A.; Gradinaru, C.C. Integrative conformational ensembles of sic1 using different initial pools and optimization methods. Front. Mol. Biosci. 2022, 9, 910956. [Google Scholar] [CrossRef] [PubMed]
- Aznauryan, M.; Delgado, L.; Soranno, A.; Nettels, D.; Huang, J.R.; Labhardt, A.M.; Grzesiek, S.; Schuler, B. Comprehensive structural and dynamical view of an unfolded protein from the combination of single-molecule fret, nmr, and saxs. Proc. Natl. Acad. Sci. USA 2016, 113, E5389–E5398. [Google Scholar] [CrossRef]
- Rozycki, B.; Kim, Y.C.; Hummer, G. Saxs ensemble refinement of escrt-iii chmp3 conformational transitions. Structure 2011, 19, 109–116. [Google Scholar] [CrossRef]
- Manalastas, K.G.; Svergun, D.I. Molecular dissection of the intrinsically disordered estrogen receptor alpha-ntd. Structure 2019, 27, 207–208. [Google Scholar] [CrossRef]
- Ambadipudi, S.; Zweckstetter, M. Targeting intrinsically disordered proteins in rational drug discovery. Expert Opin. Drug Discov. 2016, 11, 65–77. [Google Scholar] [CrossRef]
- Ruan, H.; Sun, Q.; Zhang, W.; Liu, Y.; Lai, L. Targeting intrinsically disordered proteins at the edge of chaos. Drug Discov. Today 2019, 24, 217–227. [Google Scholar] [CrossRef]
- Metallo, S.J. Intrinsically disordered proteins are potential drug targets. Curr. Opin. Chem. Biol. 2010, 14, 481–488. [Google Scholar] [CrossRef]
- Choi, S.H.; Mahankali, M.; Lee, S.J.; Hull, M.; Petrassi, H.M.; Chatterjee, A.K.; Schultz, P.G.; Jones, K.A.; Shen, W. Targeted disruption of myc-max oncoprotein complex by a small molecule. ACS Chem. Biol. 2017, 12, 2715–2719. [Google Scholar] [CrossRef]
- Zhao, J.; Blayney, A.; Liu, X.; Gandy, L.; Jin, W.; Yan, L.; Ha, J.H.; Canning, A.J.; Connelly, M.; Yang, C.; et al. Egcg binds intrinsically disordered n-terminal domain of p53 and disrupts p53-mdm2 interaction. Nat. Commun. 2021, 12, 986. [Google Scholar] [CrossRef]
- Tatenhorst, L.; Eckermann, K.; Dambeck, V.; Fonseca-Ornelas, L.; Walle, H.; Lopes da Fonseca, T.; Koch, J.C.; Becker, S.; Tonges, L.; Bahr, M.; et al. Fasudil attenuates aggregation of alpha-synuclein in models of parkinson’s disease. Acta Neuropathol. Commun. 2016, 4, 39. [Google Scholar] [CrossRef]
- Heller, G.T.; Aprile, F.A.; Michaels, T.C.T.; Limbocker, R.; Perni, M.; Ruggeri, F.S.; Mannini, B.; Lohr, T.; Bonomi, M.; Camilloni, C.; et al. Small-molecule sequestration of amyloid-beta as a drug discovery strategy for alzheimer’s disease. Sci. Adv. 2020, 6, eabb5924. [Google Scholar] [CrossRef]
- Iconaru, L.I.; Das, S.; Nourse, A.; Shelat, A.A.; Zuo, J.; Kriwacki, R.W. Small molecule sequestration of the intrinsically disordered protein, p27(kip1), within soluble oligomers. J. Mol. Biol. 2021, 433, 167120. [Google Scholar] [CrossRef]
- Myung, J.K.; Banuelos, C.A.; Fernandez, J.G.; Mawji, N.R.; Wang, J.; Tien, A.H.; Yang, Y.C.; Tavakoli, I.; Haile, S.; Watt, K.; et al. An androgen receptor n-terminal domain antagonist for treating prostate cancer. J. Clin. Investig. 2013, 123, 2948–2960. [Google Scholar] [CrossRef]
- Bier, D.; Mittal, S.; Bravo-Rodriguez, K.; Sowislok, A.; Guillory, X.; Briels, J.; Heid, C.; Bartel, M.; Wettig, B.; Brunsveld, L.; et al. The molecular tweezer clr01 stabilizes a disordered protein-protein interface. J. Am. Chem. Soc. 2017, 139, 16256–16263. [Google Scholar] [CrossRef]
- Erkizan, H.V.; Kong, Y.; Merchant, M.; Schlottmann, S.; Barber-Rotenberg, J.S.; Yuan, L.; Abaan, O.D.; Chou, T.H.; Dakshanamurthy, S.; Brown, M.L.; et al. A small molecule blocking oncogenic protein ews-fli1 interaction with rna helicase a inhibits growth of ewing’s sarcoma. Nat. Med. 2009, 15, 750–756. [Google Scholar] [CrossRef]
- Andersen, R.J.; Mawji, N.R.; Wang, J.J.; Wang, G.; Haile, S.; Myung, J.K.; Watt, K.; Tam, T.; Yang, Y.C.; Banuelos, C.A.; et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell 2010, 17, 535–546. [Google Scholar] [CrossRef]
- De Mol, E.; Fenwick, R.B.; Phang, C.T.; Buzon, V.; Szulc, E.; de la Fuente, A.; Escobedo, A.; Garcia, J.; Bertoncini, C.W.; Estebanez-Perpina, E.; et al. Epi-001, a compound active against castration-resistant prostate cancer, targets transactivation unit 5 of the androgen receptor. ACS Chem. Biol. 2016, 11, 2499–2505. [Google Scholar] [CrossRef]
- Peissert, S.; Schlosser, A.; Kendel, R.; Kuper, J.; Kisker, C. Structural basis for cdk7 activation by mat1 and cyclin h. Proc. Natl. Acad. Sci. USA 2020, 117, 26739–26748. [Google Scholar] [CrossRef]
- Chen, D.; Riedl, T.; Washbrook, E.; Pace, P.E.; Coombes, R.C.; Egly, J.M.; Ali, S. Activation of estrogen receptor alpha by s118 phosphorylation involves a ligand-dependent interaction with tfiih and participation of cdk7. Mol. Cell 2000, 6, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.; Tidow, H.; Rutherford, T.J.; Markwick, P.; Jensen, M.R.; Mylonas, E.; Svergun, D.I.; Blackledge, M.; Fersht, A.R. Structure of tumor suppressor p53 and its intrinsically disordered n-terminal transactivation domain. Proc. Natl. Acad. Sci. USA 2008, 105, 5762–5767. [Google Scholar] [CrossRef] [PubMed]
- Vise, P.; Baral, B.; Stancik, A.; Lowry, D.F.; Daughdrill, G.W. Identifying long-range structure in the intrinsically unstructured transactivation domain of p53. Proteins 2007, 67, 526–530. [Google Scholar] [CrossRef]
- Sadar, M.D. Small molecule inhibitors targeting the "achilles’ heel" of androgen receptor activity. Cancer Res. 2011, 71, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Sadar, M.D. Drugging the undruggable: Targeting the n-terminal domain of nuclear hormone receptors. Adv. Exp. Med. Biol. 2022, 1390, 311–326. [Google Scholar]
- Zhu, J.; Salvatella, X.; Robustelli, P. Small molecules targeting the disordered transactivation domain of the androgen receptor induce the formation of collapsed helical states. Nat. Commun. 2022, 13, 6390. [Google Scholar] [CrossRef]
- Lavery, D.N.; McEwan, I.J. Structure and function of steroid receptor af1 transactivation domains: Induction of active conformations. Biochem. J. 2005, 391, 449–464. [Google Scholar] [CrossRef]
- Krois, A.S.; Dyson, H.J.; Wright, P.E. Long-range regulation of p53 DNA binding by its intrinsically disordered n-terminal transactivation domain. Proc. Natl. Acad. Sci. USA 2018, 115, E11302–E11310. [Google Scholar] [CrossRef]
- Warnmark, A.; Wikstrom, A.; Wright, A.P.H.; Gustafsson, J.A.; Hard, T. The n-terminal regions of estrogen receptor alpha and beta are unstructured in vitro and show different tbp binding properties. J. Biol. Chem. 2001, 276, 45939–45944. [Google Scholar] [CrossRef]
- Rajbhandari, P.; Finn, G.; Solodin, N.M.; Singarapu, K.K.; Sahu, S.C.; Markley, J.L.; Kadunc, K.J.; Ellison-Zelski, S.J.; Kariagina, A.; Haslam, S.Z.; et al. Regulation of estrogen receptor alpha n-terminus conformation and function by peptidyl prolyl isomerase pin1. Mol. Cell Biol. 2012, 32, 445–457. [Google Scholar] [CrossRef]
- Patel, H.; Periyasamy, M.; Sava, G.P.; Bondke, A.; Slafer, B.W.; Kroll, S.H.B.; Barbazanges, M.; Starkey, R.; Ottaviani, S.; Harrod, A.; et al. Icec0942, an orally bioavailable selective inhibitor of cdk7 for cancer treatment. Mol. Cancer Ther. 2018, 17, 1156–1166. [Google Scholar] [CrossRef]
- Sava, G.P.; Fan, H.; Coombes, R.C.; Buluwela, L.; Ali, S. Cdk7 inhibitors as anticancer drugs. Cancer Metastasis Rev. 2020, 39, 805–823. [Google Scholar] [CrossRef]
- Limited, C.T. Modular Study to Evaluate ct7001 Alone in Cancer Patients with Advanced Malignancies. 2017. Available online: https://ClinicalTrials.gov/show/NCT03363893 (accessed on 1 February 2023).
- Sammak, S.; Zinzalla, G. Targeting protein-protein interactions. (ppis) of transcription factors: Challenges of intrinsically disordered proteins. (idps) and regions. (idrs). Prog. Biophys. Mol. Biol. 2015, 119, 41–46. [Google Scholar] [CrossRef]
- Choudhary, S.; Lopus, M.; Hosur, R.V. Targeting disorders in unstructured and structured proteins in various diseases. Biophys. Chem. 2022, 281, 106742. [Google Scholar] [CrossRef]
- Qiu, Y.; Li, X.; He, X.; Pu, J.; Zhang, J.; Lu, S. Computational methods-guided design of modulators targeting protein-protein interactions. (ppis). Eur. J. Med. Chem. 2020, 207, 112764. [Google Scholar] [CrossRef]
- Martin, J.; Frezza, E. A dynamical view of protein-protein complexes: Studies by molecular dynamics simulations. Front. Mol. Biosci. 2022, 9, 970109. [Google Scholar] [CrossRef]
- Tompa, P.; Davey, N.E.; Gibson, T.J.; Babu, M.M. A million peptide motifs for the molecular biologist. Mol. Cell 2014, 55, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Davey, N.E.; Van Roey, K.; Weatheritt, R.J.; Toedt, G.; Uyar, B.; Altenberg, B.; Budd, A.; Diella, F.; Dinkel, H.; Gibson, T.J. Attributes of short linear motifs. Mol. Biosyst. 2012, 8, 268–281. [Google Scholar] [CrossRef]
- Kumar, M.; Gouw, M.; Michael, S.; Samano-Sanchez, H.; Pancsa, R.; Glavina, J.; Diakogianni, A.; Valverde, J.A.; Bukirova, D.; Calyseva, J.; et al. Elm-the eukaryotic linear motif resource in 2020. Nucleic Acids Res. 2020, 48, D296–D306. [Google Scholar] [CrossRef]
- Dinkel, H.; Van Roey, K.; Michael, S.; Davey, N.E.; Weatheritt, R.J.; Born, D.; Speck, T.; Kruger, D.; Grebnev, G.; Kuban, M.; et al. The eukaryotic linear motif resource elm: 10 years and counting. Nucleic Acids Res. 2014, 42, D259–D266. [Google Scholar] [CrossRef]
- Kim, M.; Park, J.; Bouhaddou, M.; Kim, K.; Rojc, A.; Modak, M.; Soucheray, M.; McGregor, M.J.; O’Leary, P.; Wolf, D.; et al. A protein interaction landscape of breast cancer. Science 2021, 374, eabf3066. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, S.; Wohl, S.; Zheng, W.; Yang, S. Biophysical and Integrative Characterization of Protein Intrinsic Disorder as a Prime Target for Drug Discovery. Biomolecules 2023, 13, 530. https://doi.org/10.3390/biom13030530
Luo S, Wohl S, Zheng W, Yang S. Biophysical and Integrative Characterization of Protein Intrinsic Disorder as a Prime Target for Drug Discovery. Biomolecules. 2023; 13(3):530. https://doi.org/10.3390/biom13030530
Chicago/Turabian StyleLuo, Shuqi, Samuel Wohl, Wenwei Zheng, and Sichun Yang. 2023. "Biophysical and Integrative Characterization of Protein Intrinsic Disorder as a Prime Target for Drug Discovery" Biomolecules 13, no. 3: 530. https://doi.org/10.3390/biom13030530
APA StyleLuo, S., Wohl, S., Zheng, W., & Yang, S. (2023). Biophysical and Integrative Characterization of Protein Intrinsic Disorder as a Prime Target for Drug Discovery. Biomolecules, 13(3), 530. https://doi.org/10.3390/biom13030530