

Maternal Immune Activation Induces Adolescent Cognitive Deficits Preceded by Developmental Perturbations in Cortical Reelin Signalling

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Design
2.2. Tissue Processing
2.2.1. Nucleic Acid Isolation
2.2.2. Protein Isolation
2.3. Gene Expression Analysis by RT-qPCR
2.4. DNA Methylation Analysis by Pyrosequencing
2.4.1. Assay Design
2.4.2. Bisulphite Treatment and Bisulphite PCR
2.4.3. Pyrosequencing
2.5. Western Blotting
2.6. Behavioural Testing
2.6.1. Object Location
2.6.2. Object in Place (OPT)
2.7. Statistical Analysis and Sample Size
3. Results
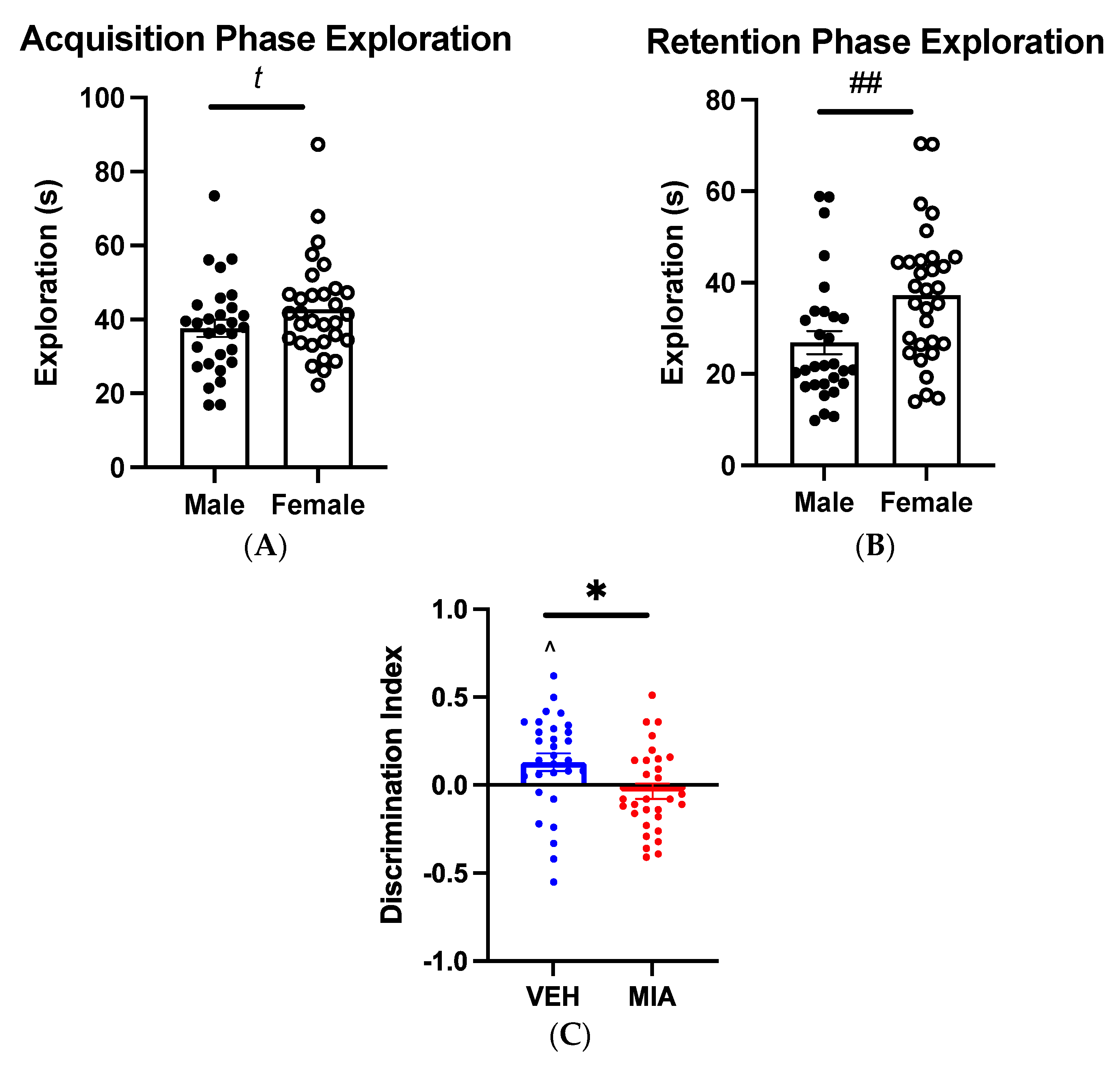
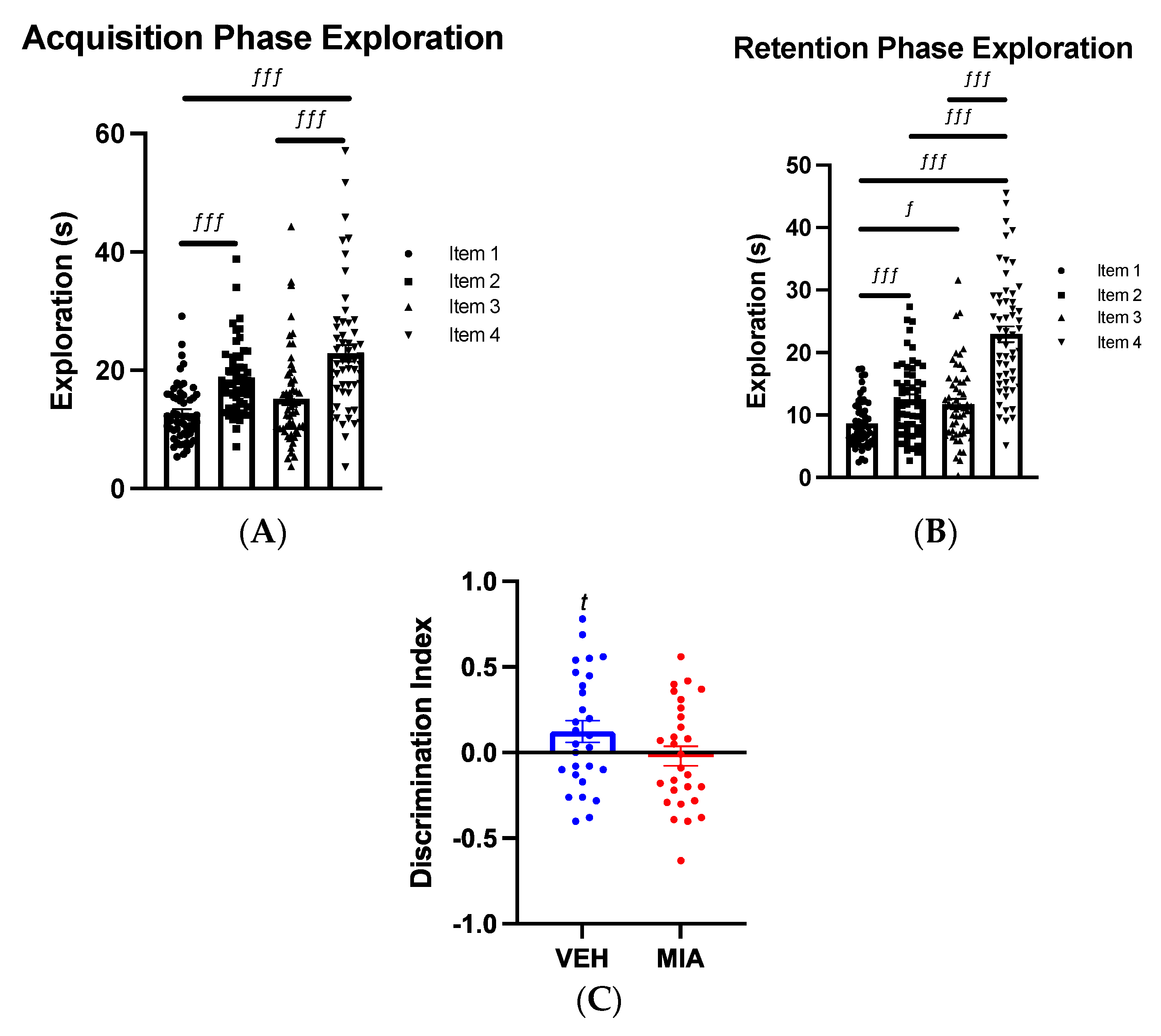
3.1. MIA Selectively Affects Spatial Object Memory
3.1.1. Object-Location Task
3.1.2. Object-in-Place Task
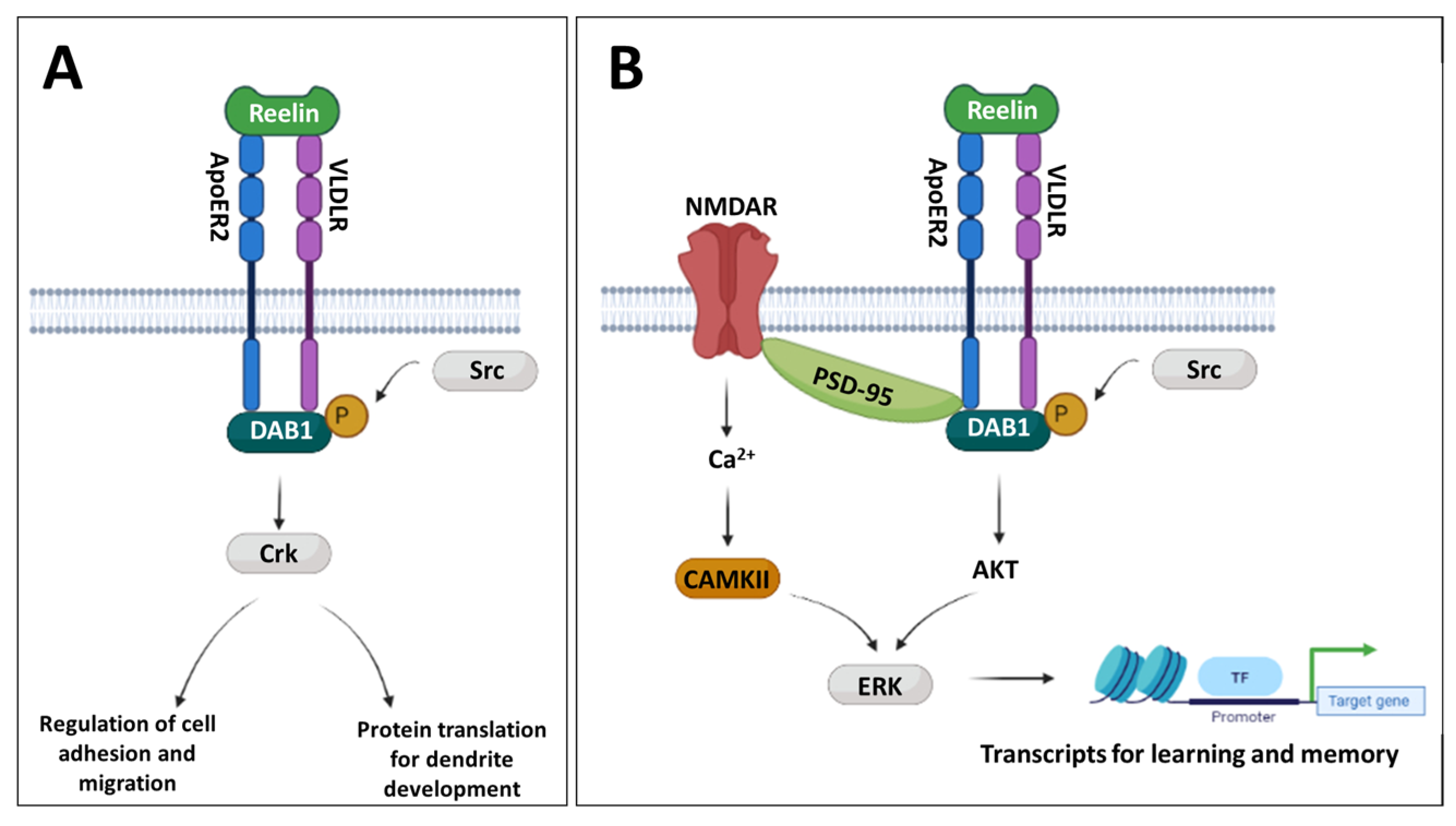
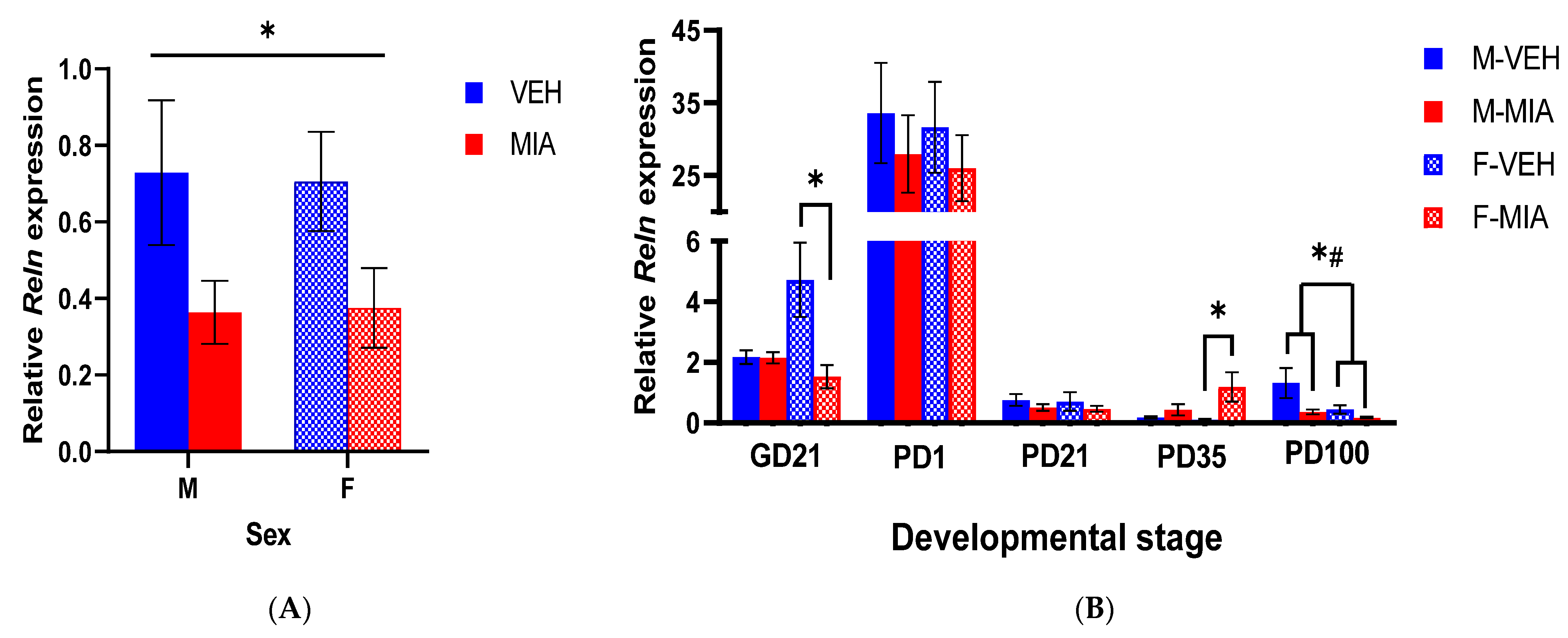
3.2. MIA Induces Reductions in Reln Gene Expression Prenatally and in the Adult Prefrontal Cortex
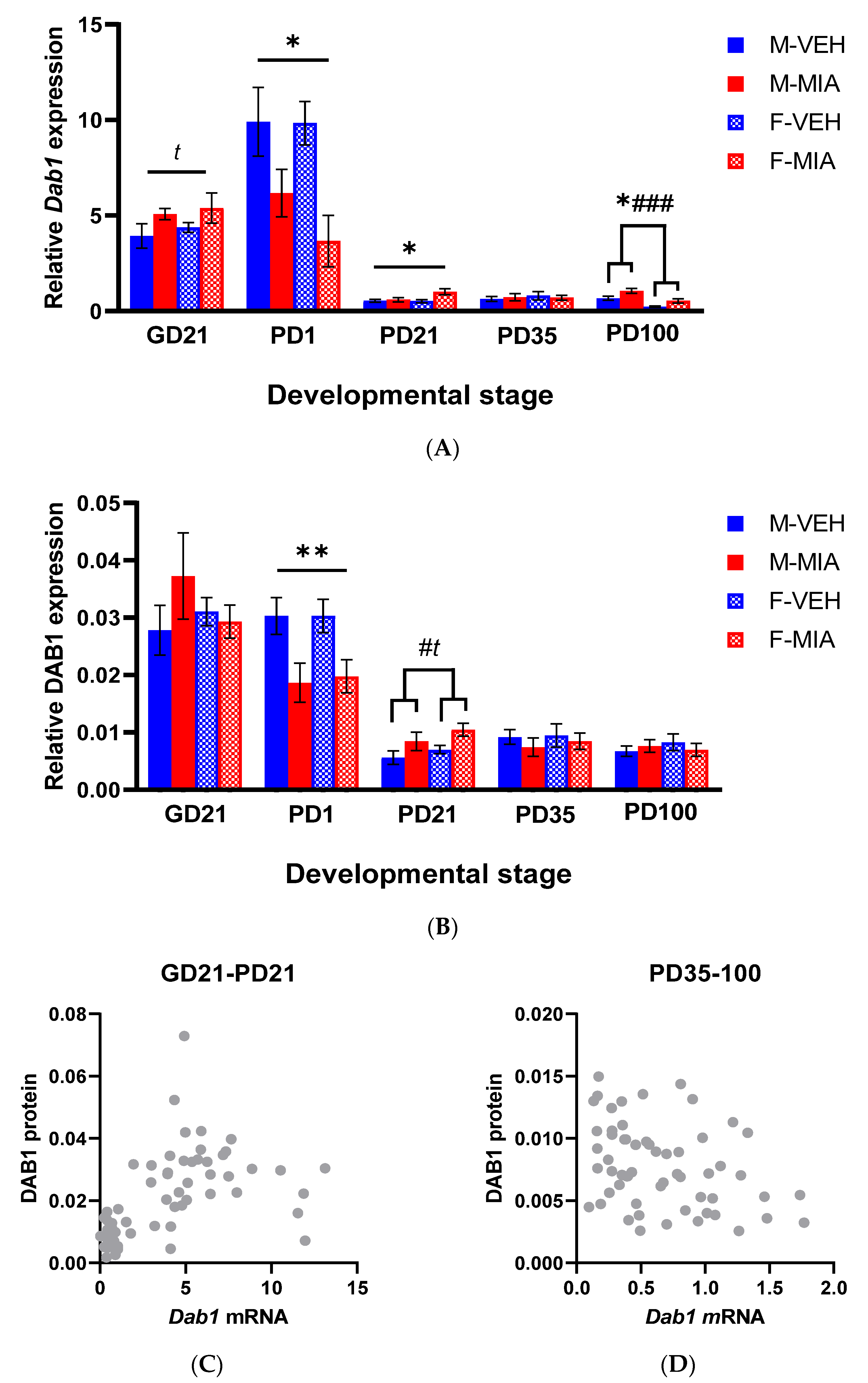
3.3. Developmental Disturbances in Dab1 mRNA and DAB1 Protein Expression Occur Subsequent to MIA-Induced Reelin Dysregulation
3.3.1. Dab1 mRNA
3.3.2. DAB1 Protein
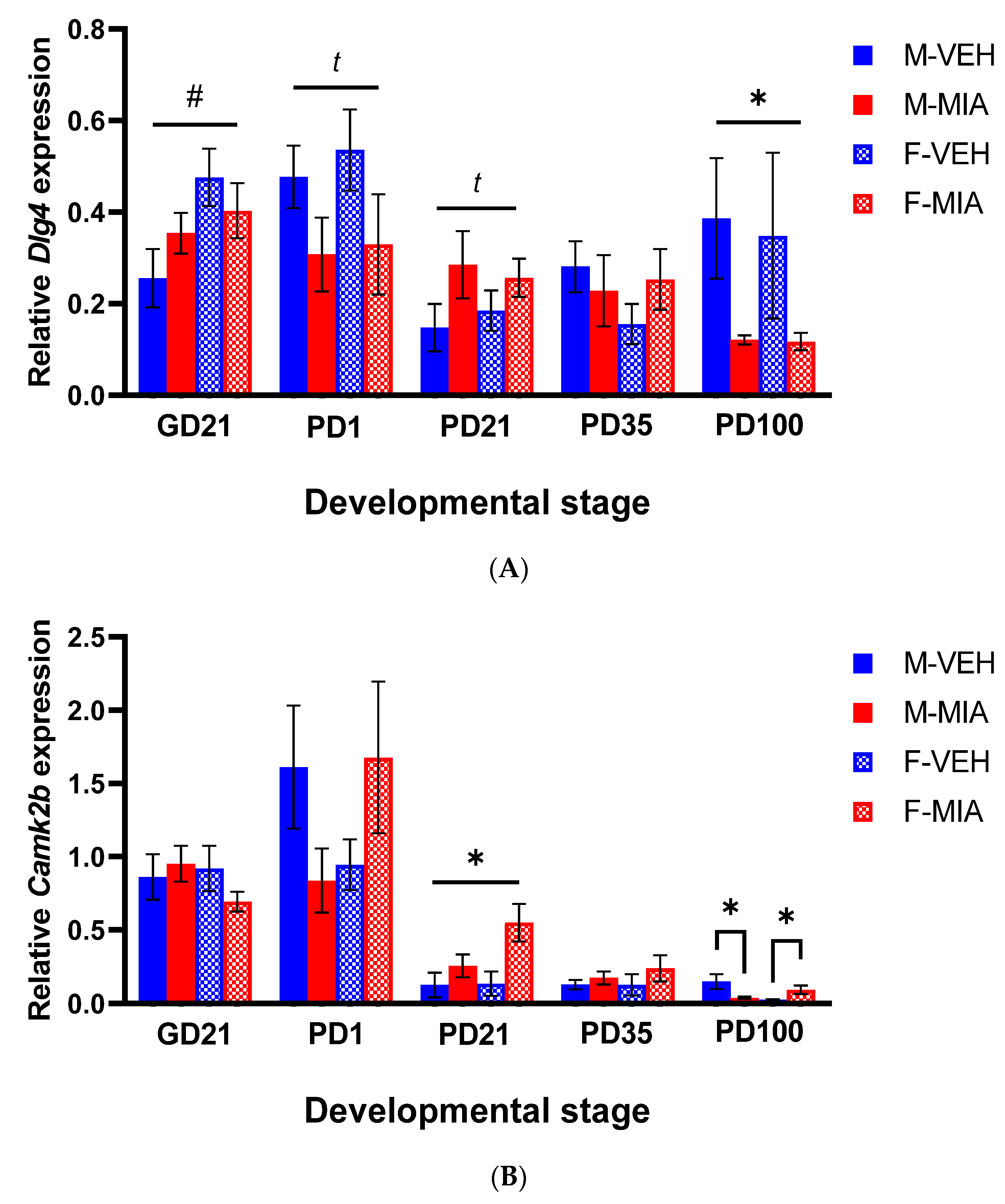
3.4. Changes to PSD-95 and CAMKII Expression Are Observed in the Later Postnatal Period Following MIA
3.4.1. Dlg4 (PSD-95)
3.4.2. Camk2b (CAMKII)
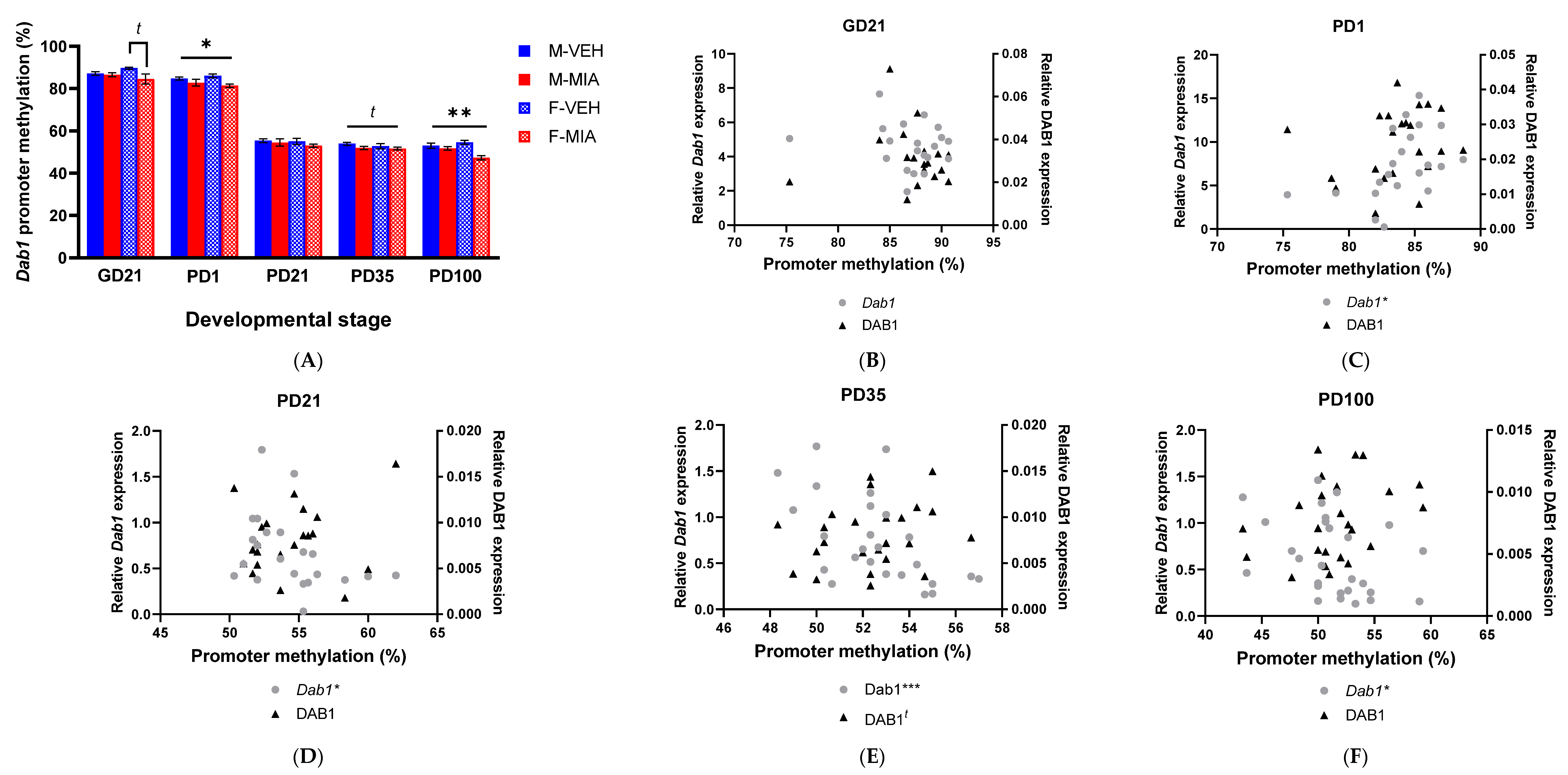
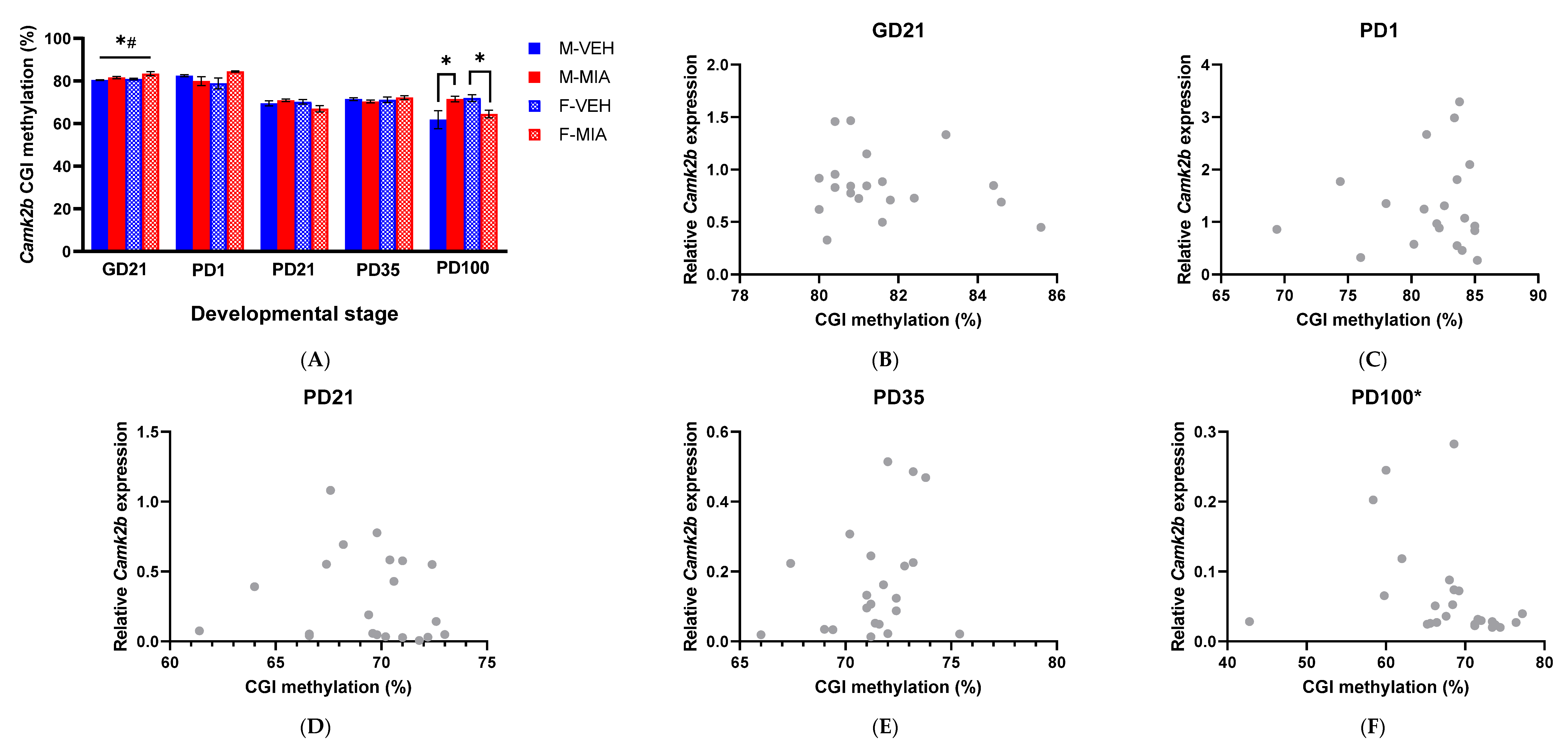
3.5. MIA-Induced Changes in Gene Expression Precede Developmental Changes to Dab1 and Camk2b DNA Methylation
3.5.1. Dab1 Promoter Methylation
3.5.2. Camk2b CGI Methylation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Charlson, F.J.; Ferrari, A.J.; Santomauro, D.F.; Diminic, S.; Stockings, E.; Scott, J.G.; McGrath, J.J.; Whiteford, H.A. Global epidemiology and burden of schizophrenia: Findings from the global burden of disease study 2016. Schizophr. Bull. 2018, 44, 1195–1203. [Google Scholar] [CrossRef]
- Tamminga, C.A. Domains of dysfunction in schizophrenia: Implications for diagnosis. World Psychiatry 2008, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, E.; Mérette, C.; Jomphe, V.; Émond, C.; Rouleau, N.; Bouchard, R.H.; Roy, M.A.; Paccalet, T.; Maziade, M. Cluster analysis of cognitive deficits may mark heterogeneity in schizophrenia in terms of outcome and response to treatment. Eur. Arch. Psychiatry Clin. Neurosci. 2014, 264, 333–343. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, R.A.; Krystal, J.H.; Howes, O.D. Dopamine and glutamate in schizophrenia: Biology, symptoms and treatment. World Psychiatry 2020, 19, 15–33. [Google Scholar] [CrossRef]
- Keefe, R.S. The longitudinal course of cognitive impairment in schizophrenia: An examination of data from premorbid through posttreatment phases of illness. J. Clin. Psychiatry 2014, 75, 10562. [Google Scholar] [CrossRef]
- O’Grada, C.; Dinan, T. Executive function in schizophrenia: What impact do antipsychotics have? Hum. Psychopharmacol. Clin. Exp. 2007, 22, 397–406. [Google Scholar] [CrossRef]
- Dahoun, T.; Trossbach, S.V.; Brandon, N.J.; Korth, C.; Howes, O.D. The impact of Disrupted-in-Schizophrenia 1 (DISC1) on the dopaminergic system: A systematic review. Transl. Psychiatry 2017, 7, e1015. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Derkits, E.J. Prenatal infection and schizophrenia: A review of epidemiologic and translational studies. Am. J. Psychiatry 2010, 167, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Woods, R.M.; Lorusso, J.M.; Potter, H.G.; Neill, J.C.; Glazier, J.D.; Hager, R. Maternal immune activation in rodent models: A systematic review of neurodevelopmental changes in gene expression and epigenetic modulation in the offspring brain. Neurosci. Biobehav. Rev. 2021, 129, 389–421. [Google Scholar] [CrossRef] [PubMed]
- Young, J.W.; Powell, S.B.; Risbrough, V.; Marston, H.M.; Geyer, M.A. Using the MATRICS to guide development of a preclinical cognitive test battery for research in schizophrenia. Pharmacol. Ther. 2009, 122, 150–202. [Google Scholar] [CrossRef]
- Czepielewski, L.S.; Alliende, L.M.; Castañeda, C.P.; Castro, M.; Guinjoan, S.M.; Massuda, R.; Berberian, A.A.; Fonseca, A.O.; Gadelha, A.; Bressan, R.; et al. Effects of socioeconomic status in cognition of people with schizophrenia: Results from a Latin American collaboration network with 1175 subjects. Psychol. Med. 2022, 52, 2177–2188. [Google Scholar] [CrossRef] [PubMed]
- Estes, M.L.; McAllister, A.K. Maternal immune activation: Implications for neuropsychiatric disorders. Science 2016, 353, 772–777. [Google Scholar] [CrossRef]
- Bao, M.; Hofsink, N.; Plösch, T. LPS versus Poly I: C model: Comparison of long-term effects of bacterial and viral maternal immune activation on the offspring. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2022, 322, R99–R111. [Google Scholar] [CrossRef] [PubMed]
- Kowash, H.M.; Potter, H.G.; Edye, M.E.; Prinssen, E.P.; Bandinelli, S.; Neill, J.C.; Hager, R.; Glazier, J.D. Poly(I:C) source, molecular weight and endotoxin contamination affect dam and prenatal outcomes, implications for models of maternal immune activation. Brain Behav. Immun. 2019, 82, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.M.; Edye, M.E.; Manca, M.; Vernon, A.C.; Oladipo, J.M.; Fasolino, V.; Harte, M.K.; Mason, V.; Grayson, B.; McHugh, P.C.; et al. Evolution of a maternal immune activation (mIA) model in rats: Early developmental effects. Brain Behav. Immun. 2019, 75, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Kowash, H.M.; Potter, H.G.; Woods, R.M.; Ashton, N.; Hager, R.; Neill, J.C.; Glazier, J.D. Maternal immune activation in rats induces dysfunction of placental leucine transport and alters fetal brain growth. Clin. Sci. 2022, 136, 1117–1137. [Google Scholar] [CrossRef] [PubMed]
- Potter, H.G.; Kowash, H.M.; Woods, R.M.; Revill, G.; Grime, A.; Deeney, B.; Burgess, M.A.; Aarons, T.; Glazier, J.D.; Neill, J.C.; et al. Maternal behaviours and adult offspring behavioural deficits are predicted by maternal TNFα concentration in a rat model of neurodevelopmental disorders. Brain Behav. Immun. 2023, 108, 162–175. [Google Scholar] [CrossRef]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef]
- Brenet, F.; Moh, M.; Funk, P.; Feierstein, E.; Viale, A.J.; Socci, N.D.; Scandura, J.M. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS ONE 2011, 6, e14524. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Villicaña, S.; Bell, J.T. Genetic impacts on DNA methylation: Research findings and future perspectives. BMC Genome Biol. 2021, 22, 127. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestart, M.L.; Heine-Suner, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.Y.; Caspi, A.; Williams, B.; Craig, I.W.; Houts, R.; Ambler, A.; Moffitt, T.E.; Mill, J. A longitudinal study of epigenetic variation in twins. Epigenetics 2010, 5, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Bianco-Miotto, T.; Craig, J.M.; Gasser, Y.P.; van Dijk, S.J.; Ozanne, S.E. Epigenetics and DOHaD: From basics to birth and beyond. J. Dev. Orig. Health Dis. 2017, 8, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Rutten, B.P.F. Neuroepigenetics of Mental Illness: The Inside Outs of the Outside Within. Prog. Mol. Biol. Transl. Sci. 2018, 158, 1–13. [Google Scholar]
- Braun, P.R.; Han, S.; Hing, B.; Nagahama, Y.; Gaul, L.N.; Heinzman, J.T.; Grossbach, A.J.; Close, L.; Dlouhy, B.J.; Howard, I.I.I.; et al. Genome-wide DNA methylation comparison between live human brain and peripheral tissues within individuals. Transl. Psychiatry 2019, 9, 47. [Google Scholar] [CrossRef]
- Pries, L.K.; Gülöksüz, S.; Kenis, G. DNA Methylation in Schizophrenia. Adv. Exp. Med. Biol. 2017, 978, 211–236. [Google Scholar]
- Lee, G.H.; D’Arcangelo, G. New Insights into Reelin-Mediated Signaling Pathways. Front. Cell. Neuorsci. 2016, 10, 122. [Google Scholar] [CrossRef]
- Guidotti, A.; Grayson, D.R.; Caruncho, H.J. Epigenetic RELN Dysfunction in Schizophrenia and Related Neuropsychiatric Disorders. Front. Cell. Neurosci. 2016, 10, 89. [Google Scholar] [CrossRef]
- Fikri, R.M.N.; Norlelawati, A.T.; Nour El-Huda, A.R.; Hanisah, M.N.; Kartini, A.; Norsidah, K.; Nor Zamzila, A. Reelin (RELN) DNA methylation in the peripheral blood of schizophrenia. J. Psychiatr. Res. 2017, 88, 28–37. [Google Scholar] [CrossRef]
- Yasui, N.; Nogi, T.; Takagi, J. Structural basis for specific recognition of reelin by its receptors. Structure 2010, 18, 320–331. [Google Scholar] [CrossRef]
- Trotter, J.; Lee, G.H.; Kazdoba, T.M.; Crowell, B.; Domogauer, J.; Mahoney, H.M.; Franco, S.J.; Müller, U.; Weeber, E.J.; D’Arcangelo, G. Dab1 is required for synaptic plasticity and associative learning. J. Neurosci. 2013, 33, 15652–15668. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Ochalski, P.G.; Tran, T.S.; Sahir, N.; Schubert, M.; Pramatarova, A.; Howell, B.W. Interaction between Dab1 and CrkII is promoted by Reelin signaling. J. Cell Sci. 2004, 117, 4527–4536. [Google Scholar] [CrossRef] [PubMed]
- Howell, B.W.; Herrick, T.M.; Cooper, J.A. Reelin-induced tyrosine phosphorylation of disabled 1 during neuronal positioning. Genes Dev. 1999, 13, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Park, T.J.; Curran, T. Crk and Crk-like play essential overlapping roles downstream of disabled-1 in the Reelin pathway. J. Neurosci. 2008, 28, 13551–13562. [Google Scholar] [CrossRef]
- Beffert, U.; Weeber, E.J.; Durudas, A.; Qiu, S.; Masiulis, I.; Sweatt, J.D.; Li, W.P.; Adelmann, G.; Frotscher, M.; Hammer, R.E.; et al. Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor ApoER2. Neuron 2005, 47, 567–579. [Google Scholar] [CrossRef]
- Chen, Y.; Beffert, U.; Ertunc, M.; Tang, T.S.; Kavalali, E.T.; Bezprozvanny, I.; Herz, J. Reelin modulates NMDA receptor activity in cortical neurons. J. Neurosci. 2005, 25, 8209–8216. [Google Scholar] [CrossRef]
- Lee, G.H.; Chhangawala, Z.; von Daake, S.; Savas, J.N.; Yates, J.R., III; Comoletti, D.; D’Arcangelo, G. Reelin induces Erk1/2 signaling in cortical neurons through a non-canonical pathway. J. Biol. Chem. 2014, 289, 20307–20317. [Google Scholar] [CrossRef] [PubMed]
- Ventruti, A.; Kazdoba, T.M.; Niu, S.; D’Arcangelo, G. Reelin deficiency causes specific defects in the molecular composition of the synapses in the adult brain. Neuroscience 2011, 189, 32–42. [Google Scholar] [CrossRef]
- Brigman, J.L.; Padukiewicz, K.E.; Sutherland, M.L.; Rothblat, L.A. Executive functions in the heterozygous reeler mouse model of schizophrenia. Behav. Neurosci. 2006, 120, 984–988. [Google Scholar] [CrossRef]
- Krueger, D.D.; Howell, J.L.; Hebert, B.F.; Olausson, P.; Taylor, J.R.; Nairn, A.C. Assessment of cognitive function in the heterozygous reeler mouse. Psychopharmacology 2006, 189, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Larson, J.; Hoffman, J.S.; Guidotti, A.; Costa, E. Olfactory discrimination learning deficit in heterozygous reeler mice. Brain Res. 2003, 971, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Trommsdorff, M.; Gotthardt, M.; Hiesberger, T.; Shelton, J.; Stockinger, W.; Nimpf, J.; Hammer, R.E.; Richardson, J.A.; Herz, J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 1999, 97, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Weeber, E.J.; Beffert, U.; Jones, C.; Christian, J.M.; Forster, E.; Sweatt, J.D.; Herz, J. Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J. Biol. Chem. 2002, 277, 39944–39952. [Google Scholar] [CrossRef] [PubMed]
- Howell, B.W.; Herrick, T.M.; Hildebrand, J.D.; Zhang, Y.; Cooper, J.A. Dab1 tyrosine phosphorylation sites relay positional signals during mouse brain development. Curr. Biol. 2000, 10, 877–885. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Emamian, E.S.; Kist, D.; Sidwell, R.W.; Nakajima, K.; Akhter, P.; Shier, A.; Sheikh, S.; Bailey, K. Defective corticogenesis and reduction in Reelin immunoreactivity in cortex and hippocampus of prenatally infected neonatal mice. Mol. Psychiatry 1999, 4, 145–154. [Google Scholar] [CrossRef]
- Nakamura, J.P.; Schoeder, A.; Gibbons, A.; Sundram, A.; Hill, R.A. Timing of maternal immune activation and sex influence schizophrenia-relevant cognitive constructs and neuregulin and GABAergic pathways. Brain Behav. Immun. 2022, 100, 70–82. [Google Scholar] [CrossRef]
- Ibi, D.; Nakasai, G.; Koide, N.; Sawahata, M.; Kohno, T.; Takaba, R.; Nagai, T.; Hattori, M.; Nabeshima, T.; Yamada, K.; et al. Reelin supplementation into the hippocampus rescues abnormal behavior in a mouse model of neurodevelopmental disorders. Front. Cell. Neurosci. 2020, 14, 285. [Google Scholar] [CrossRef]
- Lorusso, J.M.; Woods, R.M.; McEwan, F.; Glazier, J.D.; Neill, J.C.; Harte, M.; Hager, R. Clustering of cognitive phenotypes identifies susceptible and resilient offspring in a rat model of maternal immune activation and early-life stress. Brain Behav. Immun.-Health 2022, 25, 100514. [Google Scholar] [CrossRef]
- Ballendine, S.A.; Greba, Q.; Dawicki, W.; Zhang, X.; Gordon, J.R.; Howland, J.G. Behavioral alterations in rat offspring following maternal immune activation and ELR-CXC chemokine receptor antagonism during pregnancy: Implications for neurodevelopmental psychiatric disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2015, 57, 155–165. [Google Scholar] [CrossRef]
- Barker, G.R.; Bird, F.; Alexander, V.; Warburton, E.C. Recognition memory for objects, place, and temporal order: A disconnection analysis of the role of the medial prefrontal cortex and perirhinal cortex. J. Neurosci. 2007, 27, 2948–2957. [Google Scholar] [CrossRef] [PubMed]
- Barker, G.R.I.; Warburton, E.C. Object-in-place associative recognition memory depends on glutamate receptor neurotransmission within two defined hippocampal-cortical circuits: A critical role for AMPA and NMDA receptors in the hippocampus, perirhinal, and prefrontal cortices. Cereb. Cortex 2015, 25, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Mueller, F.S.; Polesel, M.; Richetto, J.; Meyer, U.; Weber-Stadlbauer, U. Mouse models of maternal immune activation: Mind your caging system! Brain Behav. Immun. 2018, 73, 643–660. [Google Scholar] [CrossRef]
- Lyst, S.J.; Davis, K.; Gigg, J.; Hager, R. Effects of increased spatial complexity on behavioural development and task performance in Lister Hooded rats. PLoS ONE 2012, 7, e47640. [Google Scholar] [CrossRef]
- Brenes, J.C.; Lackinger, M.; Höglinger, G.U.; Schratt, G.; Schwarting, R.K.; Wöhr, M. Differential effects of social and physical environmental enrichment on brain plasticity, cognition, and ultrasonic communication in rats. J. Comp. Neurol. 2016, 5248, 1586–1607. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Rondón-Ortiz, A.N.; Lima, E.P.; Puracchio, M.; Roderick, R.C.; Kentner, A.C. Therapeutic efficacy of environmental enrichment on behavioral, endocrine, and synaptic alterations in an animal model of maternal immune activation. Brain Behav. Immun. Health 2020, 3, 100043. [Google Scholar] [CrossRef]
- Kentner, A.C.; Khoury, A.; Queiroz, E.L.; MacRae, M. Environmental enrichment rescues the effects of early life inflammation on markers of synaptic transmission and plasticity. Brain Behav. Immun. 2016, 57, 151–160. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 6th ed.; Academic Press: London, UK, 2007. [Google Scholar]
- Kolk, S.M.; Rakic, P. Development of prefrontal cortex. Neuropsychopharmacology 2022, 47, 41–57. [Google Scholar] [CrossRef]
- Vandesompele, J.; De preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, e0034. [Google Scholar] [CrossRef]
- Friard, O.; Gamba, M. BORIS: A free, versatile open-source event-logging software for video/audio coding and live observations. Methods Ecol. Evol. 2016, 7, 1325–1330. [Google Scholar] [CrossRef]
- Reincke, S.A.; Hanganu-Opatz, I.L. Early-life stress impairs recognition memory and perturbs the functional maturation of prefrontal-hippocampal-perirhinal networks. Sci. Rep. 2017, 7, 42042. [Google Scholar] [CrossRef] [PubMed]
- Schulz, K.M.; Pearson, J.N.; Neeley, E.W.; Berger, R.; Leonard, S.; Adams, C.E.; Stevens, K.E. Maternal stress during pregnancy causes sex-specific alterations in offspring memory performance, social interactions, indices of anxiety, and body mass. Physiol. Behav. 2011, 104, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Luck, D.; Foucher, J.R.; Offerlin-Meyer, I.; Lepage, M.; Danion, J.M. Assessment of single and bound features in a working memory task in schizophrenia. Schizophr. Res. 2008, 100, 153–160. [Google Scholar] [CrossRef]
- Grayson, B.; Leger, M.; Piercy, C.; Adamson, L.; Harte, M.; Neill, J.C. Assessment of disease-related cognitive impairments using the novel object recognition (NOR) task in rodents. Behav. Brain Res. 2015, 285, 176–193. [Google Scholar] [CrossRef]
- Burglen, F.; Marczewski, P.; Mitchell, K.J.; Van der Linden, M.; Johnson, M.K.; Danion, J.M.; Salame, P. Impaired performance in a working memory binding task in patients with schizophrenia. Psychiatry Res. 2004, 125, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Hannula, D.E.; Ranganath, C.; Ramsay, I.S.; Solomon, M.; Yoon, J.; Niendam, T.A.; Carter, C.S.; Ragland, J.D. Use of eye movement monitoring to examine item and relational memory in schizophrenia. Biol. Psychiatry 2010, 68, 610–616. [Google Scholar] [CrossRef]
- Alcántara, S.; Ruiz, M.; D’Arcangelo, G.; Ezan, F.; de Lecea, L.; Curran, T.; Sotelo, C.; Soriano, E. Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. J. Neurosci. 1998, 18, 7779–7799. [Google Scholar] [CrossRef] [PubMed]
- Ragland, J.D.; Layher, E.; Hannula, D.E.; Niendam, T.A.; Lesh, T.A.; Solomon, M.; Carter, C.S.; Ranganath, C. Impact of schizophrenia on anterior and posterior hippocampus during memory for complex scenes. NeuroImage Clin. 2017, 13, 82–88. [Google Scholar] [CrossRef]
- Howland, J.G.; Cazakoff, B.N.; Zhang, Y. Altered object-in-place recognition memory, prepulse inhibition, and locomotor activity in the offspring of rats exposed to a viral mimetic during pregnancy. Neuroscience 2012, 201, 184–198. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Rusiana, I.; Trotter, J.; Zhao, L.; Donaldson, E.; Pak, D.T.; Babus, L.W.; Peters, M.; Banko, J.L.; Chavis, P.; et al. Reelin supplementation enhances cognitive ability, synaptic plasticity, and dendritic spine density. Learn. Mem. 2011, 18, 558–564. [Google Scholar] [CrossRef]
- Niu, S.; Renfro, A.; Quattrocchi, C.C.; Sheldon, M.; D’Arcangelo, G. Reelin promotes hippocampal dendrite development through the VLDLR/ApoER2-Dab1 pathway. Neuron 2004, 41, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Selemon, L. A role for synaptic plasticity in the adolescent development of executive function. Transl. Psychiatry 2013, 3, e238. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.S.; Sheldon, M.; D’Arcangelo, G.; Nakajima, K.; Goldowitz, D.; Curran, T. Disabled-1 acts downstream of Reelin in a signaling pathway that controls laminar organization in the mammalian brain. Development 1998, 125, 3719–3729. [Google Scholar] [CrossRef]
- Coiro, P.; Pollak, D.D. Sex and gender bias in the experimental neurosciences: The case of the maternal immune activation model. Transl. Psychiatry 2019, 9, 90. [Google Scholar] [CrossRef]
- Berchtold, N.C.; Cribbs, D.H.; Coleman, P.D.; Rogers, J.; Head, E.; Kim, R.; Beach, T.; Miller, C.; Troncoso, J.; Trojanowski, J.Q.; et al. Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc. Natl. Acad. Sci. USA 2008, 105, 15605–15610. [Google Scholar] [CrossRef] [PubMed]
- Shifman, S.; Johannesson, M.; Bronstein, M.; Chen, S.X.; Collier, D.A.; Craddock, N.J.; Kendler, K.S.; Li, T.; O’Donovan, M.; O’Neill, F.A.; et al. Genome-wide association identifies a common variant in the reelin gene that increases the risk of schizophrenia only in women. PLoS Genet. 2008, 4, e28. [Google Scholar] [CrossRef]
- Sozuguzel, M.D.; Sazci, A.; Yildiz, M. Female gender specific association of the Reelin (RELN) gene rs7341475 variant with schizophrenia. Mol. Biol. Rep. 2019, 46, 3411–3416. [Google Scholar] [CrossRef] [PubMed]
- Zalcman, G.; Federman, N.; Romano, A. CaMKII Isoforms in Learning and Memory: Localization and Function. Front. Mol. Neurosci. 2018, 11, 445. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Huang, G.Z.; Woolley, C.S. Latent Sex Differences in Molecular Signaling That Underlies Excitatory Synaptic Potentiation in the Hippocampus. J. Neurosci. 2019, 39, 1552–1565. [Google Scholar] [CrossRef]
- Rafa-Zabłocka, K.; Kreiner, G.; Bagińska, M.; Nalepa, I. The influence of CaMKII and ERK phosphorylation on BDNF changes observed in mice selectively devoid of CREB in serotonergic or noradrenergic neurons. Pharmacol. Rep. 2019, 71, 753–761. [Google Scholar] [CrossRef]
- Yin, Y.; Qian, S.; Chen, Y.; Sun, Y.; Li, Y.; Yu, Y.; Li, J.; Wu, Z.; Yu, X.; Ge, R.; et al. Latent Sex Differences in CaMKII-nNOS Signaling That Underlie Antidepressant-Like Effects of Yueju-Ganmaidazao Decoction in the Hippocampus. Front. Behav. Neurosci. 2021, 15, 640258. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Liu, C.; Sodhi, M.; Lu, H. Meta-analysis of sex differences in gene expression in schizophrenia. BMC Syst. Biol. 2016, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, C.M.; Masachs, N.; Muhaisen, A.; Bosch, C.; Pérez-Martínez, J.; Howell, B.; Soriano, E. Transient downregulation of Dab1 protein levels during development leads to behavioral and structural deficits: Relevance for psychiatric disorders. Neuropsychopharmacology 2014, 39, 556–568. [Google Scholar] [CrossRef] [PubMed]
- Coley, A.A.; Gao, W.J. PSD-95 deficiency disrupts PFC-associated function and behavior during neurodevelopment. Sci. Rep. 2019, 9, 9486. [Google Scholar] [CrossRef]
- Labouesse, M.A.; Lassalle, O.; Richetto, J.; Iafrati, J.; Weber-Stadlbauer, U.; Notter, T.; Gschwind, T.; Pujadas, L.; Soriano, E.; Reichelt, A.C.; et al. Hypervulnerability of the adolescent prefrontal cortex to nutritional stress via reelin deficiency. Mol. Psychiatry 2017, 22, 961–971. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Target | Primer | Sequence (5′-3′) | Sequence to Analyse | Score |
|---|---|---|---|---|
| Dab1 | FP | TGAAATGTTTTTGTTGGTGTATGT | TATGTGYGGTTYGGGGTGTTTTTT TTGAAGGGAGGAGTTTTTTTTTTG GAGAGGATTTTYGATGAGTT TGGTTAAGGTT | 71% |
| RP * | AACCCCCAACCTTAACCAAACTC | |||
| SP | TGTAGTATTTAGATAGAGTGAATGA | |||
| Camk2b | FP * | TTTTTGGGGGGTAAATTTAAGTG | CRAAACCRTCAACAACR ACRACTTTAAAAC CTATACRTAAATCTCCC | 81% |
| RP | ACCTAATAATCATCCCTATTTTCTCC | |||
| SP | ACCACAAAACAACTCA |
| Acquisition (Mean Difference, Y-X) | Retention (Mean Difference, Y-X) | |||||||
|---|---|---|---|---|---|---|---|---|
| Item 1 (Can/Bottle) | Item 2 (White Bottle) | Item 3 (Ceramic Pot) | Item 4 (Ramekin) | Item 1 (Can/Bottle) | Item 2 (White Bottle) | Item 3 (Ceramic Pot) | Item 4 (Ramekin) | |
| Item 1 (Can/Bottle) | −5.587, p < 0.001 *** | −2.350, p = 0.319 | −10.045, p < 0.001 *** | −3.873, p < 0.001 *** | −2.965, p = 0.039 * | −14.544, p < 0.001 *** | ||
| Item 2 (White Bottle) | 5.587, p < 0.001 *** | 3.237, p = 0.086 | −4.457, p = 0.085 | 3.873, p < 0.001 *** | 0.909, p = 1.000 | −10.670, p < 0.001 *** | ||
| Item 3 (Ceramic Pot) | 2.350, p = 0.319 | −3.237, p = 0.086 | −7.696, p < 0.001 *** | 2.965, p = 0.039 * | −0.909, p = 1.000 | −11.579, p < 0.001 *** | ||
| Item 4 (Ramekin) | 10.045, p < 0.001 *** | 4.458, p = 0.085 | 7.696, p < 0.001 *** | 14.544, p < 0.001 *** | 10.670, p < 0.001 *** | 11.579, p < 0.001 *** | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woods, R.M.; Lorusso, J.M.; Harris, I.; Kowash, H.M.; Murgatroyd, C.; Neill, J.C.; Glazier, J.D.; Harte, M.; Hager, R. Maternal Immune Activation Induces Adolescent Cognitive Deficits Preceded by Developmental Perturbations in Cortical Reelin Signalling. Biomolecules 2023, 13, 489. https://doi.org/10.3390/biom13030489
Woods RM, Lorusso JM, Harris I, Kowash HM, Murgatroyd C, Neill JC, Glazier JD, Harte M, Hager R. Maternal Immune Activation Induces Adolescent Cognitive Deficits Preceded by Developmental Perturbations in Cortical Reelin Signalling. Biomolecules. 2023; 13(3):489. https://doi.org/10.3390/biom13030489
Chicago/Turabian StyleWoods, Rebecca M., Jarred M. Lorusso, Isabella Harris, Hager M. Kowash, Christopher Murgatroyd, Joanna C. Neill, Jocelyn D. Glazier, Michael Harte, and Reinmar Hager. 2023. "Maternal Immune Activation Induces Adolescent Cognitive Deficits Preceded by Developmental Perturbations in Cortical Reelin Signalling" Biomolecules 13, no. 3: 489. https://doi.org/10.3390/biom13030489
APA StyleWoods, R. M., Lorusso, J. M., Harris, I., Kowash, H. M., Murgatroyd, C., Neill, J. C., Glazier, J. D., Harte, M., & Hager, R. (2023). Maternal Immune Activation Induces Adolescent Cognitive Deficits Preceded by Developmental Perturbations in Cortical Reelin Signalling. Biomolecules, 13(3), 489. https://doi.org/10.3390/biom13030489