
Reversibility and Low Commitment to Forward Catalysis in the Conjugation of Lipid Alkenals by Glutathione Transferase A4-4

Abstract
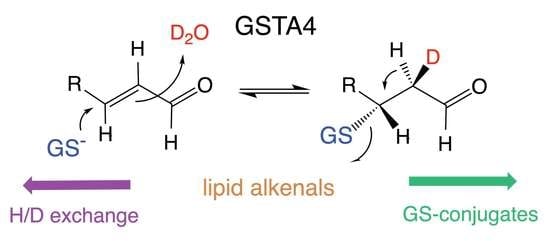
1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. UV-Vis Assays for Forward and Reverse Reactions
2.3. GS-NE Synthesis
2.4. LC-Mass Spectrometry
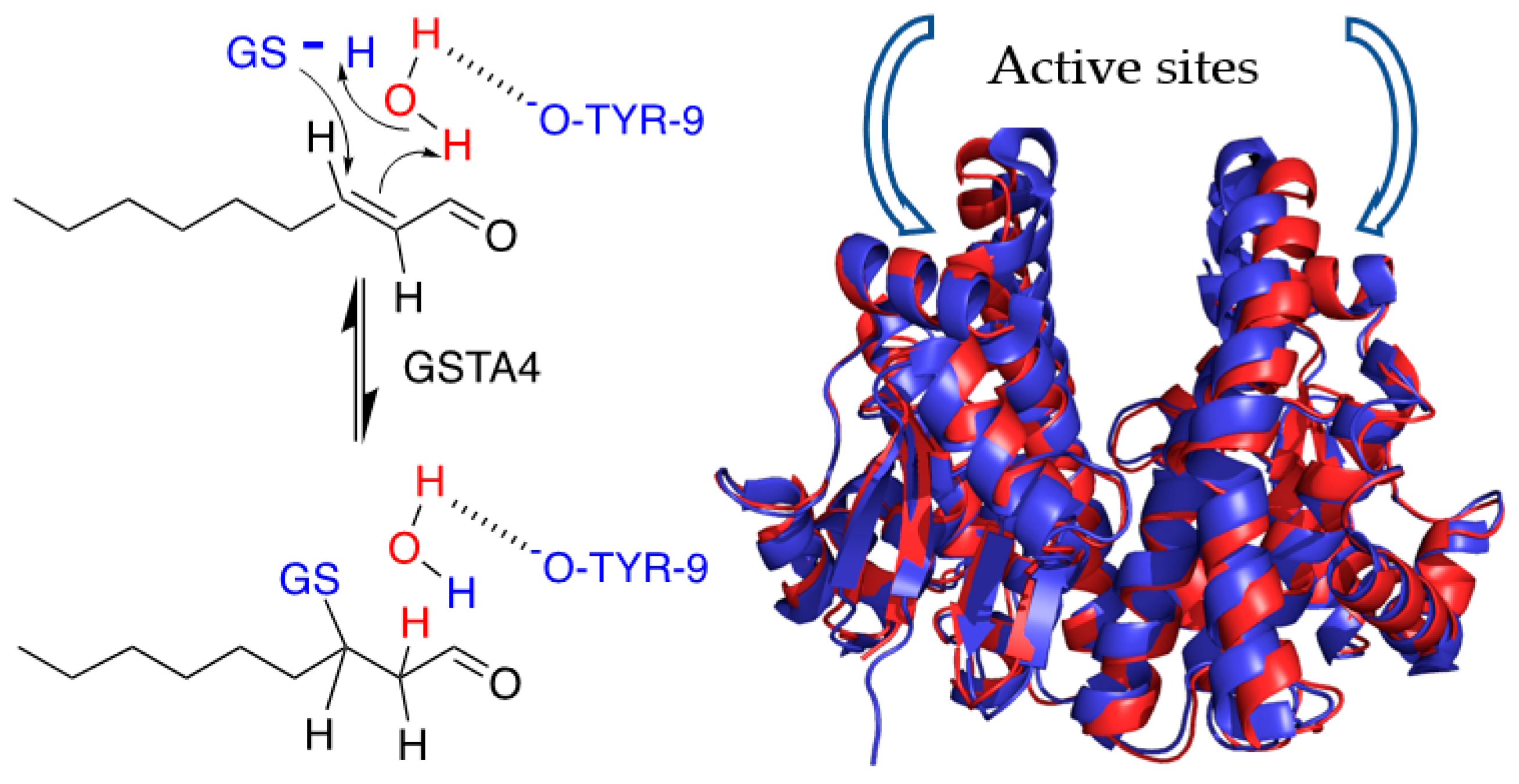
2.5. NMR Assignment of NE
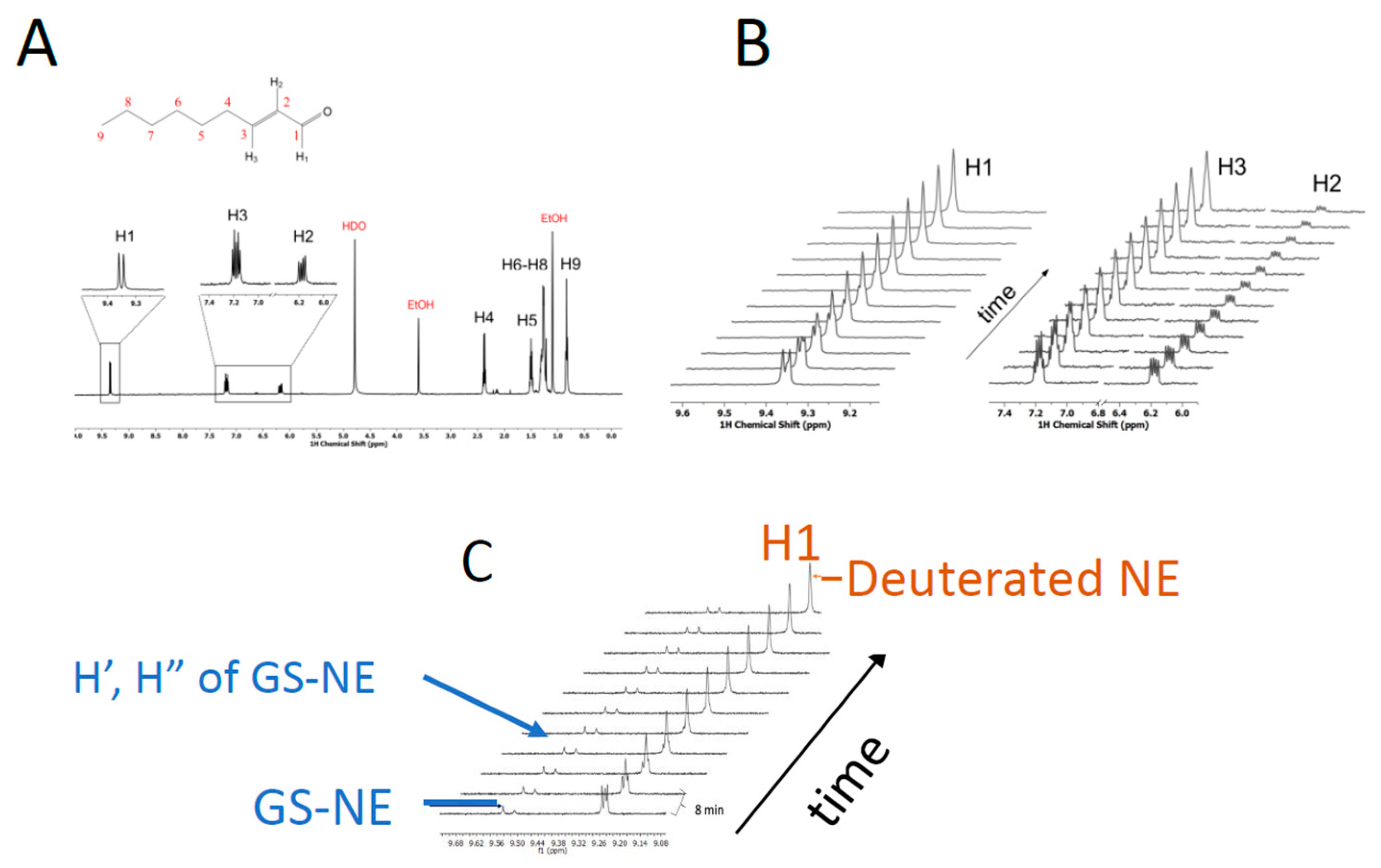
2.6. The GSH-Dependence of Deuterium Exchange
2.7. Equilibrium of GSH, NE and GS-NE by NMR
2.8. Deuterium NMR of NE Recovered from Enzyme Incubation
3. Results
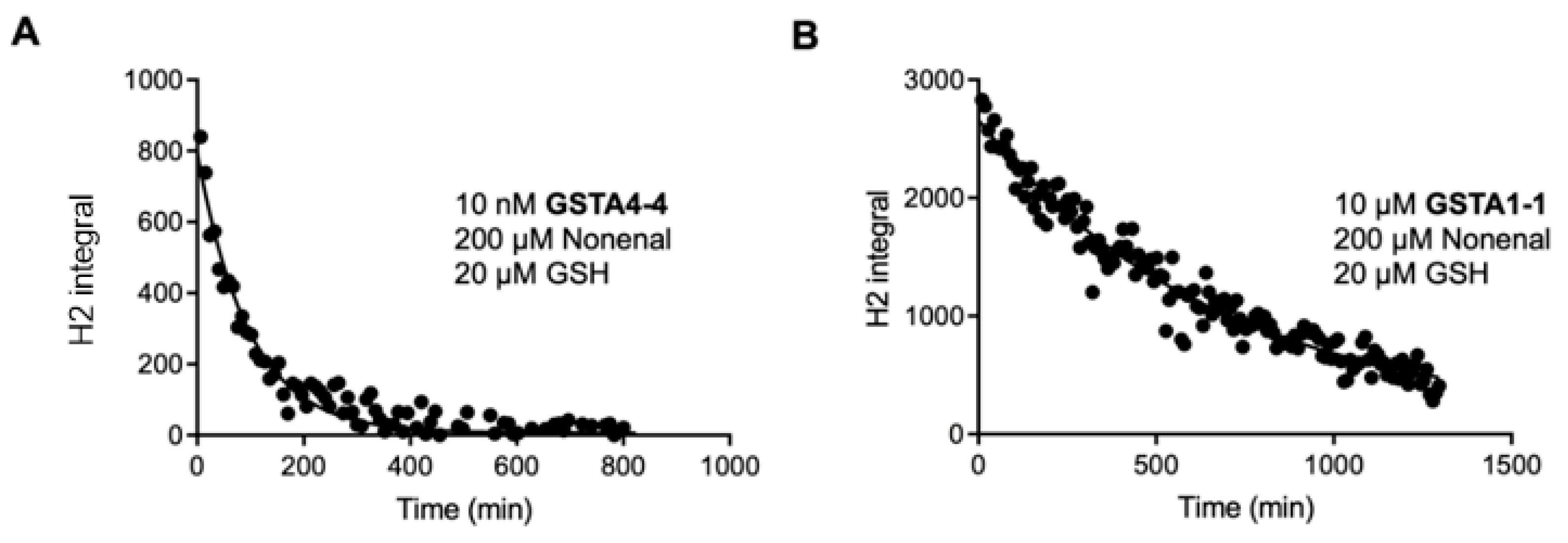
3.1. Steady State Forward and Reverse Reaction
3.2. Solvent Exchange with NE by NMR
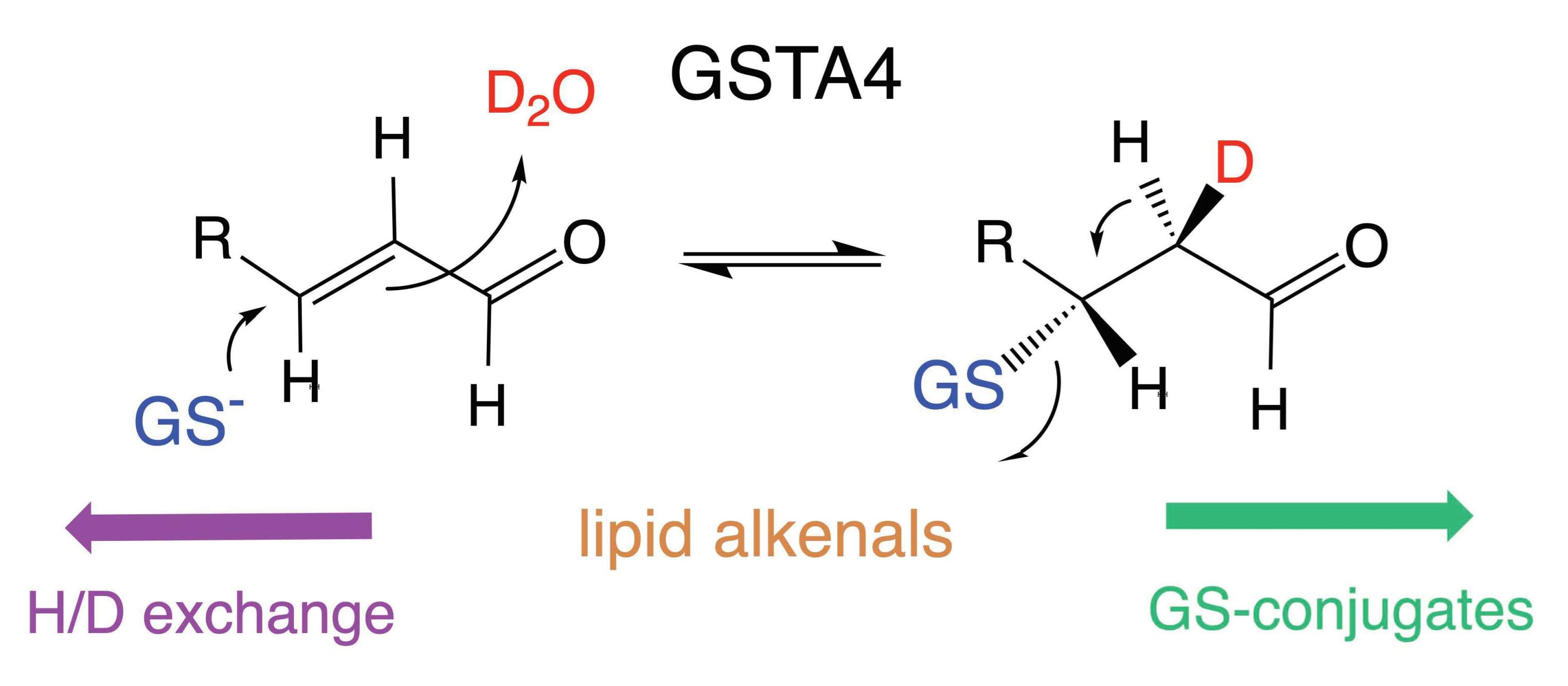
3.3. The Site of Deuterium Incorporation from Solvent Exchange
3.4. Solvent Exchange Requires GSH
3.5. Equilibrium Constant for the Reaction
3.6. Estimating Relative Rates of Deuterium Exchange into NE and Formation of GS-NE
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hubatsch, I.; Ridderström, M.; Mannervik, B. Human glutathione transferase A4-4: An alpha class enzyme with high catalytic efficiency in the conjugation of 4-hydroxyNE and other genotoxic products of lipid peroxidation. Biochem. J. 1998, 330 Pt 1, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Board, P.G. Identification of cDNAs encoding two human alpha class glutathione transferases (GSTA3 and GSTA4-4) and the heterologous expression of GSTA4-4. Biochem. J. 1998, 330 Pt 2, 827–831. [Google Scholar] [CrossRef] [PubMed]
- Sousa, B.C.; Pitt, A.R.; Spickett, C.M. Chemistry and analysis of HNE and other prominent carbonyl-containing lipid oxidation compounds. Free. Radic. Biol. Med. 2017, 111, 294–308. [Google Scholar] [CrossRef] [PubMed]
- LoPachin, R.M.; Gavin, T.; Petersen, D.R.; Barber, D.S. Molecular Mechanisms of 4-Hydroxy-2-nonenal and Acrolein Toxicity: Nucleophilic Targets and Adduct Formation. Chem. Res. Toxicol. 2009, 22, 1499–1508. [Google Scholar] [CrossRef]
- Csala, M.; Kardon, T.; Legeza, B.; Lizák, B.; Mandl, J.; Margittai, É.; Puskás, F.; Száraz, P.; Szelényi, P.; Bánhegyi, G. On the role of 4-hydroxynonenal in health and disease. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2015, 1852, 826–838. [Google Scholar] [CrossRef]
- Schaur, R.J.; Siems, W.; Bresgen, N.; Eckl, P.M. 4-Hydroxy-nonenal—A Bioactive Lipid Peroxidation Product. Biomolecules 2015, 5, 2247–2337. [Google Scholar] [CrossRef]
- Forman, H.J.; Fukuto, J.M.; Miller, T.; Zhang, H.; Rinna, A.; Levy, S. The chemistry of cell signaling by reactive oxygen and nitrogen species and 4-hydroxynonenal. Arch. Biochem. Biophys. 2008, 477, 183–195. [Google Scholar] [CrossRef]
- Guéraud, F. 4-Hydroxynonenal metabolites and adducts in pre-carcinogenic conditions and cancer. Free. Radic. Biol. Med. 2017, 111, 196–208. [Google Scholar] [CrossRef]
- Ishino, K.; Wakita, C.; Shibata, T.; Toyokuni, S.; Machida, S.; Matsuda, S.; Matsuda, T.; Uchida, K. Lipid Peroxidation Generates Body Odor Component trans-2-Nonenal Covalently Bound to Protein in Vivo. J. Biol. Chem. 2010, 285, 15302–15313. [Google Scholar] [CrossRef]
- Saito, K.; Tokorodani, Y.; Sakamoto, C.; Kataoka, H. Headspace Solid-Phase Microextraction/Gas Chromatography–Mass Spectrometry for the Determination of 2-Nonenal and Its Application to Body Odor Analysis. Molecules 2021, 26, 5739. [Google Scholar] [CrossRef]
- Wójcik, P.; Gęgotek, A.; Žarković, N.; Skrzydlewska, E. Oxidative Stress and Lipid Mediators Modulate Immune Cell Functions in Autoimmune Diseases. Int. J. Mol. Sci. 2021, 22, 723. [Google Scholar] [CrossRef]
- Gasparovic, A.C.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. Cancer growth regulation by 4-hydroxynonenal. Free. Radic. Biol. Med. 2017, 111, 226–234. [Google Scholar] [CrossRef]
- Noguchi, N. Role of Oxidative Stress in Adaptive Responses in Special Reference to Atherogenesis. J. Clin. Biochem. Nutr. 2008, 43, 131–138. [Google Scholar] [CrossRef]
- Awasthi, Y.C.; Ramana, K.V.; Chaudhary, P.; Srivastava, S.K.; Awasthi, S. Regulatory roles of glutathione-S-transferases and 4-hydroxynonenal in stress-mediated signaling and toxicity. Free. Radic. Biol. Med. 2017, 111, 235–243. [Google Scholar] [CrossRef]
- Wu, R.P.; Hayashi, T.; Cottam, H.B.; Jin, G.; Yao, S.; Wu, C.C.N.; Rosenbach, M.D.; Corr, M.; Schwab, R.B.; Carson, D.A. Nrf2 responses and the therapeutic selectivity of electrophilic compounds in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 7479–7484. [Google Scholar] [CrossRef]
- Gerhäuser, C.; Klimo, K.; Hümmer, W.; Hölzer, J.; Petermann, A.; Garreta-Rufas, A.; Böhmer, F.-D.; Schreier, P. Identification of 3-hydroxy-β-damascone and related carotenoid-derived aroma compounds as novel potent inducers of Nrf2-mediated phase 2 response with concomitant anti-inflammatory activity. Mol. Nutr. Food Res. 2009, 53, 1237–1244. [Google Scholar] [CrossRef]
- Mol, M.; Regazzoni, L.; Altomare, A.; Degani, G.; Carini, M.; Vistoli, G.; Aldini, G. Enzymatic and non-enzymatic detoxification of 4-hydroxynonenal: Methodological aspects and biological consequences. Free. Radic. Biol. Med. 2017, 111, 328–344. [Google Scholar] [CrossRef]
- Hartley, D.P.; A Ruth, J.; Petersen, D.R. The Hepatocellular Metabolism of 4-Hydroxynonenal by Alcohol Dehydrogenase, Aldehyde Dehydrogenase, and Glutathione S-Transferase. Arch. Biochem. Biophys. 1995, 316, 197–205. [Google Scholar] [CrossRef]
- Nath, A.; Atkins, W.M. A quantitative index of substrate promiscuity. Biochemistry. 2008, 47, 157–166. [Google Scholar] [CrossRef]
- Bruns, C.M.; Hubatsch, I.; Ridderström, M.; Mannervik, B.; Tainer, J. Human glutathione transferase A4-4 crystal structures and mutagenesis reveal the basis of high catalytic efficiency with toxic lipid peroxidation products. J. Mol. Biol. 1999, 288, 427–439. [Google Scholar] [CrossRef]
- Nilsson, L.O.; Gustafsson, A.; Mannervik, B. Redesign of substrate-selectivity determining modules of glutathione transferase A1–1 installs high catalytic efficiency with toxic alkenal products of lipid peroxidation. Proc. Natl. Acad. Sci. USA 2000, 97, 9408–9412. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Honaker, M.T.; Shireman, L.M.; Balogh, L.M.; Roberts, A.G.; Ng, K.-C.; Nath, A.; Atkins, W.M. Functional Promiscuity Correlates with Conformational Heterogeneity in A-class Glutathione S-Transferases. J. Biol. Chem. 2007, 282, 23264–23274. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, C.A.; Chowdhury, P.; Petrich, J.W.; Atkins, W.M. The Anomalous pK of Tyr-9 in Glutathione S-Transferase A1-1 Catalyzes Product Release. J. Biol. Chem. 2003, 278, 19257–19265. [Google Scholar] [CrossRef] [PubMed]
- Grahn, E.; Novotny, M.; Jakobsson, E.; Gustafsson, A.; Grehn, L.; Olin, B.; Madsen, D.; Wahlberg, M.; Mannervik, B.; Kleywegt, G.J. New crystal structures of human glutathione transferase A1-1 shed light on glutathione binding and the conformation of the C-terminal helix. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62 Pt 2, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Balogh, L.M.; Le Trong, I.; Kripps, K.A.; Tars, K.; Stenkamp, R.E.; Mannervik, B.; Atkins, W.M. Structural Analysis of a Glutathione Transferase A1-1 Mutant Tailored for High Catalytic Efficiency with Toxic Alkenals. Biochemistry 2009, 48, 7698–7704. [Google Scholar] [CrossRef]
- Zhan, Y.; Rule, G.S. Glutathione induces helical formation in the carboxy terminus of human glutathione transferase A1-1. Biochemistry. 2004, 43, 7244–7254. [Google Scholar] [CrossRef]
- Sinning, I.; Kleywegt, G.J.; Cowan, S.W.; Reinemer, P.; Dirr, H.; Huber, R.; Gilliland, G.L.; Armstrong, R.N.; Ji, X.; Board, P.G.; et al. Structure Determination and Refinement of Human Alpha Class Glutathione Transferase A1-1, and a Comparison with the Mu and Pi Class Enzymes. J. Mol. Biol. 1993, 232, 192–212. [Google Scholar] [CrossRef]
- Adman, E.T.; Le Trong, I.; Stenkamp, R.E.; Nieslanik, B.S.; Dietze, E.C.; Tai, G.; Ibarra, C.; Atkins, W.M. Localization of the C-terminus of rat glutathione S-transferase A1-1: Crystal structure of mutants W21F and W21F/F220Y. Proteins Struct. Funct. Bioinform. 2000, 42, 192–200. [Google Scholar] [CrossRef]
- Honaker, M.T.; Acchione, M.; Zhang, W.; Mannervik, B.; Atkins, W.M. Enzymatic Detoxication, Conformational Selection, and the Role of Molten Globule Active Sites. J. Biol. Chem. 2013, 288, 18599–18611. [Google Scholar] [CrossRef]
- Honaker, M.; Acchione, M.; Sumida, J.P.; Atkins, W.M. Ensemble perspective for catalytic promiscuity: Calorimetric analysis of the active site conformational landscape of a detoxification enzyme. J. Biol. Chem. 2011, 286, 42770–42776. [Google Scholar] [CrossRef]
- Hubatsch, I.; Mannervik, B. A Highly Acidic Tyrosine 9 and a Normally Titrating Tyrosine 212 Contribute to the Catalytic Mechanism of Human Glutathione Transferase A4-4. Biochem. Biophys. Res. Commun. 2001, 280, 878–882. [Google Scholar] [CrossRef]
- Atkins, W.M.; Wang, R.W.; Bird, A.W.; Newton, D.J.; Lu, A.Y. The catalytic mechanism of glutathione S-transferase (GST). Spectroscopic determination of the pKa of Tyr-9 in rat alpha 1-1 GST. J. Biol. Chem. 1993, 268, 19188–19191. [Google Scholar] [CrossRef]
- Gustafsson, A.; Etahadieh, M.; Jemth, P.; Mannervik, B. The C-terminal region of human glutathione transferase A1-1 affects the rate of glutathione binding and the ionization of the active-site Tyr9. Biochemistry 1999, 38, 16268–16275. [Google Scholar] [CrossRef]
- Dietze, E.C.; Ibarra, C.; Dabrowski, M.J.; Bird, A.; Atkins, W.M. Rational modulation of the catalytic activity of A1-1 glutathione S-transferase: Evidence for incorporation of an on-face (pi...HO-Ar) hydrogen bond at tyrosine-9. Biochemistry 1996, 35, 11938–11944. [Google Scholar] [CrossRef]
- Johnson, K.A. Role of induced fit in enzyme specificity: A molecular forward/reverse switch. J. Biol. Chem. 2008, 283, 26297–26301. [Google Scholar] [CrossRef]
- Alary, J.; Fernandez, Y.; Debrauwer, L.; Perdu, E.; Guéraud, F. Identification of Intermediate Pathways of 4-HydroxyNE Metabolism in the Rat. Chem. Res. Toxicol. 2003, 16, 320–327. [Google Scholar] [CrossRef]
- Habig, W.H.; Pabst, M.J.; Jakoby, W.B. Glutathione S-Transferases: THE FIRST ENZYMATIC STEP IN MERCAPTURIC ACID FORMATION. J. Biol. Chem. 1974, 249, 7130–7139. [Google Scholar] [CrossRef]
- Stenberg, G.; Bjo¨rnestedt, R.; Mannervik, B. Heterologous expression of recombinant human glutathione transferase A1-1 from a hepatoma cell line. Protein Expr. Purif. 1992, 3, 80–84. [Google Scholar] [CrossRef]
- Piotto, M.; Saudek, V.; Sklenar, V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 1992, 2, 661–665. [Google Scholar] [CrossRef]
- Nath, A.; Atkins, W.M. A Theoretical Validation of the Substrate Depletion Approach to Determining Kinetic Parameters. Drug Metab. Dispos. 2006, 34, 1433–1435. [Google Scholar] [CrossRef]
- Dietze, E.C.; Grillo, M.P.; Kalhorn, T.; Nieslanik, B.S.; Jochheim, C.M.; Atkins, W.M. Thiol ester hydrolysis catalyzed by glutathione S-transferase A1-1. Biochemistry 1998, 37, 14948–14957. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.J.; Crease, D.J.; Ketterer, B. Forward and reverse catalysis and product sequestration by human glutathione S-transferases in the reaction of GSH with dietary aralkyl isothiocyanates. Biochem. J. 1995, 306 Pt 2, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Armstrong, R.N. Stereoselective catalysis of a retro-Michael reaction by class mu glutathione transferases. Consequences for the internal distribution of products in the active site. Chem. Res. Toxicol. 1995, 8, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, C.; Grillo, M.P.; Bello, M.L.; Nucettelli, M.; Bammler, T.K.; Atkins, W.M. Exploration of in vitro pro-drug activation and futile cycling by glutathione S-transferases: Thiol ester hydrolysis and inhibitor maturation. Arch. Biochem. Biophys. 2003, 414, 303–311. [Google Scholar] [CrossRef]
- Zhang, Y.; Kolm, R.; Mannervik, B.; Talalay, P. Reversible Conjugation of Isothiocyanates with Glutathione Catalyzed by Human Glutathione Transferases. Biochem. Biophys. Res. Commun. 1995, 206, 748–755. [Google Scholar] [CrossRef]
- Slatter, J.G.; Rashed, M.S.; Pearson, P.G.; Han, D.H.; Baillie, T.A. Biotransformation of methyl isocyanate in the rat. Evidence for glutathione conjugation as a major pathway of metabolism and implications for isocyanate-mediated toxicities. Chem. Res. Toxicol. 1991, 4, 157–161. [Google Scholar] [CrossRef]
- Baillie, T.A.; Kassahun, K. Reversibility in glutathione-conjugate formation. Adv. Pharmacol. 1994, 27, 163–181. [Google Scholar] [CrossRef]
- Pearson, P.G.; Slatter, J.; Rashed, M.S.; Han, D.-H.; Grillo, M.P.; Baillie, T.A. S-(N-Methylcarbamoyl)glutathione: A reactive S-linked metabolite of methyl isocyanate. Biochem. Biophys. Res. Commun. 1990, 166, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Varma, D.R.; Guest, I. The Bhopal accident and methyl isocyanate toxicity. J. Toxicol. Environ. Health 1993, 40, 513–529. [Google Scholar] [CrossRef]
- Awasthi, Y.C.; Yang, Y.; Tiwari, N.K.; Patrick, B.; Sharma, A.; Li, J.; Awasthi, S. Regulation of 4-hydroxynonenal-mediated signaling by glutathione S-transferases. Free. Radic. Biol. Med. 2004, 37, 607–619. [Google Scholar] [CrossRef]
- Awasthi, Y.C.; Ansari, G.A.; Awasthi, S. Regulation of 4-hydroxynonenal mediated signaling by glutathione S-transferases. Methods Enzymol. 2005, 401, 379–407. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, P.F. Combining solvent isotope effects with substrate isotope effects in mechanistic studies of alcohol and amine oxidation by enzymes. Biochim. Biophys. Acta 2015, 1854, 1746–1755. [Google Scholar] [CrossRef]
- Fernandez, P.L.; Murkin, A.S. Inverse Solvent Isotope Effects in Enzyme-Catalyzed Reactions. Molecules 2020, 25, 1933. [Google Scholar] [CrossRef]
- Schowen, K.B.; Schowen, R.L. Solvent isotope effects of enzyme systems. Methods Enzymol. 1982, 87, 551–606. [Google Scholar]
- Spies, M.A.; Toney, M.D. Intrinsic Primary and Secondary Hydrogen Kinetic Isotope Effects for Alanine Racemase from Global Analysis of Progress Curves. J. Am. Chem. Soc. 2007, 129, 10678–10685. [Google Scholar] [CrossRef]
- Saunders, W.H., Jr.; Edison, D.H. Mechanisms of Elimination Reactions. IV. Deuterium Isotope Effects in E2 Reactions of Some 2-Phenylethyl Derivatives. J. Am. Chem. Soc. 1960, 82, 138–145. [Google Scholar] [CrossRef]
- Balogh, L.M.; Le Trong, I.; Kripps, K.A.; Shireman, L.M.; Stenkamp, R.E.; Zhang, W.; Mannervik, B.; Atkins, W.M. Substrate specificity combined with stereopromiscuity in glutathione transferase A4-4-dependent metabolism of 4-hydroxyNE. Biochemistry 2010, 9, 1541–1548. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forward Reaction | Reverse Reaction | ||||||
|---|---|---|---|---|---|---|---|
| GSTA Isoform (Assay) | KM,GSH (μM) | KM,NE (μM) | kcat,f (s−1) | kcat,f/KM (μM−1s−1) | KM,GS-NE (μM) | kcat,r (s−1) | kcat,r/KM,GS-NE (μM−1s−1) |
| GSTA4-4 (UV) | 530 ± 100 | NA | 89 ± 6 | 0.17 | 19 ± 5 | 1.2 ± 0.1 | 0.06 |
| GSTA4-4 (LC-MS) | NA | 195 ± 49 | 184 ± 14 | 0.95 | NA | NA | NA |
| GSTA1-1 (LC-MS) | NA | 71 ± 19 | 0.35 ± 0.1 | 0.0042 | ND | ND | ND |
| GSTA Isoform | H/D Exchange, kex (s−1) | GS-NE Formation, kcat,f (s−1) | Ratio kcat,f/kex |
|---|---|---|---|
| GSTA4-4 | 63 ± 17 | 184 ± 14 | 2.9 |
| GSTA1-1 | 0.017 ± 0.18 | 0.35 ± 0.1 | 20.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scian, M.; Paço, L.; Murphree, T.A.; Shireman, L.M.; Atkins, W.M. Reversibility and Low Commitment to Forward Catalysis in the Conjugation of Lipid Alkenals by Glutathione Transferase A4-4. Biomolecules 2023, 13, 329. https://doi.org/10.3390/biom13020329
Scian M, Paço L, Murphree TA, Shireman LM, Atkins WM. Reversibility and Low Commitment to Forward Catalysis in the Conjugation of Lipid Alkenals by Glutathione Transferase A4-4. Biomolecules. 2023; 13(2):329. https://doi.org/10.3390/biom13020329
Chicago/Turabian StyleScian, Michele, Lorela Paço, Taylor A. Murphree, Laura M. Shireman, and William M. Atkins. 2023. "Reversibility and Low Commitment to Forward Catalysis in the Conjugation of Lipid Alkenals by Glutathione Transferase A4-4" Biomolecules 13, no. 2: 329. https://doi.org/10.3390/biom13020329
APA StyleScian, M., Paço, L., Murphree, T. A., Shireman, L. M., & Atkins, W. M. (2023). Reversibility and Low Commitment to Forward Catalysis in the Conjugation of Lipid Alkenals by Glutathione Transferase A4-4. Biomolecules, 13(2), 329. https://doi.org/10.3390/biom13020329