
LRRK2 Kinase Inhibition Attenuates Astrocytic Activation in Response to Amyloid β1-42 Fibrils

 , , , ,
, , , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Primary Astrocytes
2.2. Aβ1-42 Fibril Generation and Validation
2.3. Reactive Oxygen Species (ROS) Detection
2.4. Compound and Cell Treatment
2.5. Cell Immunofluorescence and Imaging
2.6. Aβ1-42 Intracellular Quantification
2.7. Cell Lysis and Western Blotting
2.8. Statistical Analysis
3. Results
3.1. Astrocytic Activation in Response to Aβ1-42 Fibrils Priming
3.2. LRRK2 Kinase Inhibition Attenuates Aβ1-42 Fibril-Mediated Astrocytic Activation
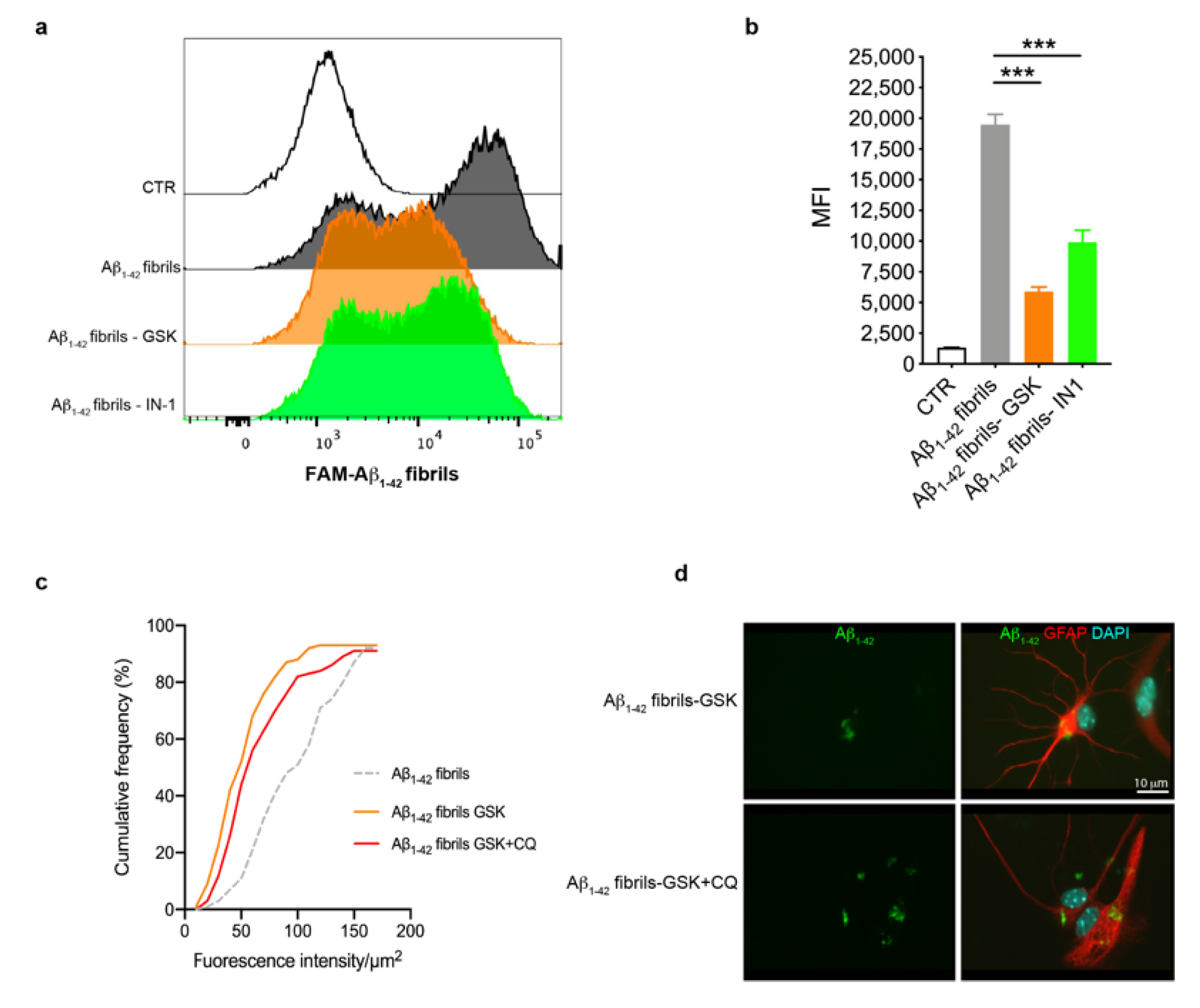
3.3. Astrocytes with LRRK2 Kinase Inhibition Exhibited Increased Clearance of Aβ1-42 Fibrils
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| LRRK2 | leucine-rich repeat kinase 2 |
| PD | Parkinson’s disease |
| GSK | GSK2578215A |
| IN-1 | inhibitor-1 |
| α-syn | α-synuclein |
| NF-κB | nuclear factor kappa-B |
| LPS | lipopolysaccharide |
| COX-2 | cyclooxygenase -2 |
| IL-1β | interleukin-1β |
| TNF- α | tumor necrosis factor-α |
| IL-6 | interleukin-6 |
| DMEM | Dulbecco’s Modified Eagle Medium |
| FBS | fetal bovine serum |
| GFAP | glial fibrillar acidic protein |
| HFIP | hexafluoroisopropanol |
| DMSO | dimethylsulfoxide |
| ThioT | thioflavin T |
| TEM | transmission electron microscopy |
| ROS | reactive oxygen species |
| FACS | fluorescence-activated cell sorting |
| PBS | phosphate-buffered saline |
| RT | room temperature |
| PFA | paraformaldehyde |
| CQ | chloroquine |
| SDS | sodium dodecyl sulphate |
| PVDF | polyvinylidene |
| TBST | TBS-tween |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| HRP | horseradish peroxidase |
| EDTA | ethylenediaminetetraacetic acid |
| MFI | median fluorescence intensity |
| LPS | lipopolysaccharide |
| PKA | protein kinase A |
References
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011, 7, 137–152. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G. A century of Alzheimer’s disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef]
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of microglia and astrocytes: A roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease—A double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar]
- Uddin, M.S.; Lim, L.W. Glial cells in Alzheimer’s disease: From neuropathological changes to therapeutic implications. Ageing Res. Rev. 2022, 78, 101622. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, B.; Cruz-Martins, N.; Kumar, D. Microglia in Alzheimer’s Disease: An Unprecedented Opportunity as Prospective Drug Target. Mol. Neurobiol. 2022, 59, 2678–2693. [Google Scholar] [CrossRef]
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia development and function. Annu. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef]
- Mirzaei, N.; Davis, N.; Chau, T.W.; Sastre, M. Astrocyte Reactivity in Alzheimer’s Disease: Therapeutic Opportunities to Promote Repair. Curr. Alzheimer Res. 2022, 19, 1–15. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Rodrigues, J.J.; Pivoriunas, A.; Zorec, R.; Semyanov, A. Astroglial atrophy in Alzheimer’s disease. Pflugers Arch. 2019, 471, 1247–1261. [Google Scholar] [CrossRef] [PubMed]
- Olabarria, M.; Noristani, H.N.; Verkhratsky, A.; Rodríguez, J.J. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 2010, 58, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 1–12. [Google Scholar] [CrossRef]
- Garwood, C.J.; Pooler, A.M.; Atherton, J.; Hanger, D.P.; Noble, W. Astrocytes are important mediators of Aβ-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011, 2, e167. [Google Scholar] [CrossRef]
- Griffin, W.S.T.; Liu, L.; Li, Y.; Mrak, R.E.; Barger, S.W. Interleukin-1 mediates Alzheimer and Lewy body pathologies. J. Neuroinflammation 2006, 3, 5. [Google Scholar] [CrossRef]
- Sastre, M.; Dewachter, I.; Landreth, G.E.; Wilson, T.M.; Klockgether, T.; Van Leuven, F.; Heneka, M.T. Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-γ agonists modulate immunostimulated processing of amyloid precursor protein through regulation of β-secretase. J. Neurosci. 2003, 23, 9796–9804. [Google Scholar] [CrossRef]
- Liaoi, Y.F.; Wang, B.J.; Cheng, H.T.; Kuo, L.H.; Wolfe, M.S. Tumor necrosis factor-α, interleukin-1β, and interferon-γ stimulate γ-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J. Biol. Chem. 2004, 279, 49523–49532. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Paisán-Ruiz, C.; Lewis, P.A.; Singleton, A.B. LRRK2: Cause, risk, and mechanism. J. Park. Dis. 2013, 3, 85–103. [Google Scholar] [CrossRef]
- Filippini, A.; Gennarelli, M.; Russo, I. Leucine-rich repeat kinase 2-related functions in GLIA: An update of the last years. Biochem. Soc. Trans. 2021, 49, 1375–1384. [Google Scholar] [CrossRef]
- Russo, I.; Kaganovich, A.; Ding, J.; Landeck, N.; Mamais, A.; Varanita, T.; Biosa, A.; Tessari, I.; Bubacco, L.; Greggio, E.; et al. Transcriptome analysis of LRRK2 knock-out microglia cells reveals alterations of inflammatory- and oxidative stress-related pathways upon treatment with α-synuclein fibrils. Neurobiol. Dis. 2019, 129, 67–78. [Google Scholar] [CrossRef]
- Russo, I.; Berti, G.; Plotegher, N.; Bernardo, G.; Filograna, R.; Bubacco, L.; Greggio, E. Leucine-rich repeat kinase 2 positively regulates inflammation and down-regulates NF-κB p50 signaling in cultured microglia cells. J. Neuroinflammation 2015, 12, 230. [Google Scholar] [CrossRef]
- Kim, B.; Yang, M.S.; Choi, D.; Kim, J.H.; Kim, H.S.; Seol, W.; Choi, S.; Jou, I.; Kim, E.Y.; Joe, E.H. Impaired inflammatory responses in murine lrrk2-knockdown brain microglia. PLoS ONE 2012, 7, e34693. [Google Scholar] [CrossRef] [PubMed]
- Moehle, M.S.; Webber, P.J.; Tse, T.; Sukar, N.; Standaert, D.G.; Desilva, T.M.; Cowell, R.M.; West, A.B. LRRK2 inhibition attenuates microglial inflammatory responses. J. Neurosci. 2012, 32, 1602–1611. [Google Scholar] [CrossRef]
- Russo, I.; Bubacco, L.; Greggio, E. LRRK2 as a target for modulating immune system responses. Neurobiol. Dis. 2022, 169, 105724. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Di Benedetto, G.; Kaganovich, A.; Ding, J.; Mercatelli, D.; Morari, M.; Cookson, M.R.; Bubacco, L.; Greggio, E. Leucine-rich repeat kinase 2 controls protein kinase A activation state through phosphodiesterase 4. J. Neuroinflammation 2018, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Munoz, L.; Kavanagh, M.E.; Phoa, A.F.; Heng, B.; Dzamko, N.; Chen, E.-J.; Doddareddy, M.R.; Guillemin, G.J.; Kassiou, M. Optimisation of LRRK2 inhibitors and assessment of functional efficacy in cell-based models of neuroinflammation. Eur. J. Med. Chem. 2015, 95, 29–34. [Google Scholar] [CrossRef]
- Ho, D.H.; Seol, W.; Eun, J.H.; Son, I.H. Phosphorylation of p53 by LRRK2 induces microglial tumor necrosis factor α-mediated neurotoxicity. Biochem. Biophys. Res. Commun. 2017, 482, 1088–1094. [Google Scholar] [CrossRef] [PubMed]
- Sonninen, T.-M.; Hämäläinen, R.H.; Koskuvi, M.; Oksanen, M.; Shakirzyanova, A.; Wojciechowski, S.; Puttonen, K.; Naumenko, N.; Goldsteins, G.; Laham-Karam, N.; et al. Metabolic alterations in Parkinson’s disease astrocytes. Sci. Rep. 2020, 10, 14474. [Google Scholar] [CrossRef] [PubMed]
- Marín, I. The Parkinson disease gene LRRK2: Evolutionary and structural insights. Mol. Biol. Evol. 2006, 23, 2423–2433. [Google Scholar] [CrossRef] [PubMed]
- Marchand, A.; Drouyer, M.; Sarchione, A.; Chartier-Harlin, M.C.; Taymans, J.M. LRRK2 Phosphorylation, More Than an Epiphenomenon. Front. Neurosci. 2020, 14, 527. [Google Scholar] [CrossRef] [PubMed]
- Chia, R.; Haddock, S.; Beilina, A.; Rudenko, I.N.; Mamais, A.; Kaganovich, A.; Li, Y.; Kumaran, R.; Nalls, M.A.; Cookson, M.R. Phosphorylation of LRRK2 by casein kinase 1α regulates trans-Golgi clustering via differential interaction with ARHGEF7. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dzamko, N.; Inesta-Vaquera, F.; Zhang, J.; Xie, C.; Cai, H.; Arthur, S.; Tan, L.; Choi, H.; Gray, N.; Cohen, P.; et al. The IkappaB kinase family phosphorylates the Parkinson’s disease kinase LRRK2 at Ser935 and Ser910 during Toll-like receptor signaling. PLoS ONE 2012, 7, e39132. [Google Scholar] [CrossRef]
- Dzamko, N.; Deak, M.; Hentati, F.; Reith, A.D.; Prescott, A.R.; Alessi, D.R.; Nichols, R.J. Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14-3-3 binding and altered cytoplasmic localization. Biochem. J. 2010, 430, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Vancraenenbroeck, R.; De Raeymaecker, J.; Lobbestael, E.; Gao, F.; De Maeyer, M.; Voet, A.; Baekelandt, V.; Taymans, J.M. In silico, in vitro and cellular analysis with a kinome-wide inhibitor panel correlates cellular LRRK2 dephosphorylation to inhibitor activity on LRRK2. Front. Mol. Neurosci. 2014, 7, 51. [Google Scholar] [CrossRef]
- Familian, A.; Eikelenboom, P.; Veerhuis, R. Minocycline does not affect amyloid β phagocytosis by human microglial cells. Neurosci. Lett. 2007, 416, 87–91. [Google Scholar] [CrossRef]
- Montoliu-Gaya, L.; Mulder, S.D.; Veerhuis, R.; Villegas, S. Effects of an Aβ-antibody fragment on Aβ aggregation and astrocytic uptake are modulated by apolipoprotein E and J mimetic peptides. PLoS ONE 2017, 12, e0188191. [Google Scholar] [CrossRef]
- Nielsen, H.M.; Mulder, S.D.; Beliën, J.A.M.; Musters, R.J.P.; Eikelenboom, P.; Veerhuis, R. Astrocytic A β 1-42 uptake is determined by A β-aggregation state and the presence of amyloid-associated proteins. Glia 2010, 58, 1235–1246. [Google Scholar] [CrossRef]
- Smith, A.M.; Davey, K.; Tsartsalis, S.; Khozoie, C.; Fancy, N.; Tang, S.S.; Liaptsi, E.; Weinert, M.; McGarry, A.; Muirhead, R.C.J.; et al. Diverse human astrocyte and microglial transcriptional responses to Alzheimer’s pathology. Acta Neuropathol. 2022, 143, 75–91. [Google Scholar] [CrossRef]
- Benzing, W.C.; Wujek, J.R.; Ward, E.K.; Shaffer, D.; Ashe, K.H.; Younkin, S.G.; Brunden, K.R. Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol. Aging 1999, 20, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.P.; Yang, F.; Chu, T.; Chen, P.; Beech, W.; Teter, B.; Tran, T.; Ubeda, O.; Ashe, K.H.; Frautschy, S.A.; et al. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J. Neurosci. 2000, 20, 5709–5714. [Google Scholar] [CrossRef] [PubMed]
- Venegas, C.; Heneka, M.T. Inflammasome-mediated innate immunity in Alzheimer’s disease. FASEB J. 2019, 33, 13075–13084. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.S.T.; Sheng, J.G.; Royston, M.C.; Gentleman, S.M.; McKenzie, J.E.; Graham, D.I.; Roberts, G.W.; Mrak, R.E. Glial-neuronal interactions in Alzheimer’s disease: The potential role of a “cytokine cycle” in disease progression. Brain Pathol. 1998, 8, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Nuutinen, T.; Suuronen, T.; Kauppinen, A.; Salminen, A. Clusterin: A forgotten player in Alzheimer’s disease. Brain Res. Rev. 2009, 61, 89–104. [Google Scholar] [CrossRef]
- Marker, D.F.; Puccini, J.M.; Mockus, T.E.; Barbieri, J.; Lu, S.M.; Gelbard, H.A. LRRK2 kinase inhibition prevents pathological microglial phagocytosis in response to HIV-1 Tat protein. J. Neuroinflammation 2012, 9, 261. [Google Scholar] [CrossRef]
- Kim, J.; Pajarillo, E.; Rizor, A.; Son, D.S.; Lee, J.; Aschner, M.; Lee, E. LRRK2 kinase plays a critical role in manganese-induced inflammation and apoptosis in microglia. PLoS ONE 2019, 14, e0210248. [Google Scholar] [CrossRef]
- Schapansky, J.; Nardozzi, J.D.; Felizia, F.; LaVoie, M.J. Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum. Mol. Genet. 2014, 23, 4201–4214. [Google Scholar] [CrossRef]
- Puccini, J.M.; Marker, D.F.; Fitzgerald, T.; Barbieri, J.; Kim, C.S.; Miller-Rhodes, P.; Lu, S.M.; Dewhurst, S.; Gelbard, H.A. Leucine-rich repeat kinase 2 modulates neuroinflammation and neurotoxicity in models of human immunodeficiency virus 1-associated neurocognitive disorders. J. Neurosci. 2015, 35, 5271–5283. [Google Scholar] [CrossRef]
- Reith, A.D.; Bamborough, P.; Jandu, K.; Andreotti, D.; Mensah, L.; Dossang, P.; Choi, H.G.; Deng, X.; Zhang, J.; Alessi, D.R.; et al. GSK2578215A; a potent and highly selective 2-arylmethyloxy-5-substitutent-N-arylbenzamide LRRK2 kinase inhibitor. Bioorganic Med. Chem. Lett. 2012, 22, 5625–5629. [Google Scholar] [CrossRef]
- Deng, X.; Dzamko, N.; Prescott, A.; Davies, P.; Liu, Q.; Yang, Q.; Lee, J.D.; Patricelli, M.P.; Nomanbhoy, T.K.; Alessi, D.R.; et al. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat. Chem. Biol. 2011, 7, 203–205. [Google Scholar] [CrossRef]
- Streubel-Gallasch, L.; Giusti, V.; Sandre, M.; Tessari, I.; Plotegher, N.; Giusto, E.; Masato, A.; Iovino, L.; Battisti, I.; Arrigoni, G.; et al. Parkinson’s Disease-Associated LRRK2 Interferes with Astrocyte-Mediated Alpha-Synuclein Clearance. Mol. Neurobiol. 2021, 58, 3119–3140. [Google Scholar] [CrossRef]
- Maekawa, T.; Sasaoka, T.; Azuma, S.; Ichikawa, T.; Melrose, H.L.; Farrer, M.J.; Obata, F. Leucine-rich repeat kinase 2 (LRRK2) regulates α-synuclein clearance in microglia. BMC Neurosci. 2016, 17, 77. [Google Scholar] [CrossRef]
- Ravichandran, K.A.; Heneka, M.T. Inflammasome activation in neurodegenerative diseases. Essays Biochem. 2021, 65, 885–904. [Google Scholar] [CrossRef] [PubMed]
- Doty, K.R.; Guillot-Sestier, M.V.; Town, T. The role of the immune system in neurodegenerative disorders: Adaptive or maladaptive? Brain Res. 2015, 1617, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Bubacco, L.; Greggio, E. LRRK2 and neuroinflammation: Partners in crime in Parkinson’s disease? J. Neuroinflammation 2014, 11, 52. [Google Scholar] [CrossRef]
- Craft, J.M.; Watterson, D.M.; Van Eldik, L.J. Human amyloid β-induced neuroinflammation is an early event in neurodegeneration. Glia 2006, 53, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Vignon, A.; Salvador-Prince, L.; Lehmann, S.; Perrier, V.; Torrent, J. Deconstructing Alzheimer’s Disease: How to Bridge the Gap between Experimental Models and the Human Pathology? Int. J. Mol. Sci. 2021, 22, 8769. [Google Scholar] [CrossRef]
- Bronzuoli, M.R.; Iacomino, A.; Steardo, L.; Scuderi, C. Targeting neuroinflammation in Alzheimer’s disease. J. Inflamm. Res. 2016, 9, 199–208. [Google Scholar] [CrossRef]
- Beach, T.G.; McGeer, E.G. Lamina-specific arrangement of astrocytic gliosis and senile plaques in Alzheimer’s disease visual cortex. Brain Res. 1988, 463, 357–361. [Google Scholar] [CrossRef] [PubMed]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef]
- Rodríguez, J.J.; Olabarria, M.; Chvatal, A.; Verkhratsky, A. Astroglia in dementia and Alzheimer’s disease. Cell Death Differ. 2009, 16, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Singh, R.; Ganeshpurkar, A.; Pokle, A.V.; bhushan Singh, R.; Singh, S.K.; Kumar, A. Cellular and molecular influencers of neuroinflammation in Alzheimer’s disease: Recent concepts & roles. Neurochem. Int. 2021, 151, 105212. [Google Scholar] [CrossRef]
- Shaftel, S.S.; Griffin, W.S.T.; Kerry, K.M. The role of interleukin-1 in neuroinflammation and Alzheimer disease: An evolving perspective. J. Neuroinflammation 2008, 5, 7. [Google Scholar] [CrossRef]
- Kim, C.; Beilina, A.; Smith, N.; Li, Y.; Kim, M.; Kumaran, R.; Kaganovich, A.; Mamais, A.; Adame, A.; Iba, M.; et al. LRRK2 mediates microglial neurotoxicity via NFATc2 in rodent models of synucleinopathies. Sci. Transl. Med. 2020, 12, eaay0399. [Google Scholar] [CrossRef] [PubMed]
- Wallings, R.L.; Tansey, M.G. LRRK2 regulation of immune-pathways and inflammatory disease. Biochem. Soc. Trans. 2019, 47, 1581–1595. [Google Scholar] [CrossRef]
- Filippini, A.; Gennarelli, M.; Russo, I. α-Synuclein and Glia in Parkinson’s Disease: A Beneficial or a Detrimental Duet for the Endo-Lysosomal System? Cell. Mol. Neurobiol. 2019, 39, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Y.; McLaurin, J. Clearance of amyloid-β peptides by microglia and macrophages: The issue of what, when and where. Future Neurol. 2012, 7, 165–176. [Google Scholar] [CrossRef]
- Ren, X.; Yao, L.; Wang, Y.G.; Mei, L.; Xiong, W.C. Microglial VPS35 deficiency impairs Aβ phagocytosis and Aβ-induced disease-associated microglia, and enhances Aβ associated pathology. J. Neuroinflammation 2022, 19, 1–17. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, Z.; Rudyk, C.; Thompson, A.; Farmer, K.; Fenner, B.; Fortin, T.; Derksen, A.; Sun, H.; Hayley, S. Leucine-rich repeat kinase-2 (LRRK2) modulates microglial phenotype and dopaminergic neurodegeneration. Neurobiol. Aging 2020, 91, 45–55. [Google Scholar] [CrossRef]
- Henry, A.G.; Aghamohammadzadeh, S.; Samaroo, H.; Chen, Y.; Mou, K.; Needle, E.; Hirst, W.D. Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum. Mol. Genet. 2015, 24, 6013–6028. [Google Scholar] [CrossRef] [PubMed]
- di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Muñoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-Specific iPSC-Derived Astrocytes Contribute to Non-Cell-Autonomous Neurodegeneration in Parkinson’s Disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Söllvander, S.; Nikitidou, E.; Brolin, R.; Söderberg, L.; Sehlin, D.; Lannfelt, L.; Erlandsson, A. Accumulation of amyloid-β by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons. Mol. Neurodegener. 2016, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filippini, A.; Salvi, V.; Dattilo, V.; Magri, C.; Castrezzati, S.; Veerhuis, R.; Bosisio, D.; Gennarelli, M.; Russo, I. LRRK2 Kinase Inhibition Attenuates Astrocytic Activation in Response to Amyloid β1-42 Fibrils. Biomolecules 2023, 13, 307. https://doi.org/10.3390/biom13020307
Filippini A, Salvi V, Dattilo V, Magri C, Castrezzati S, Veerhuis R, Bosisio D, Gennarelli M, Russo I. LRRK2 Kinase Inhibition Attenuates Astrocytic Activation in Response to Amyloid β1-42 Fibrils. Biomolecules. 2023; 13(2):307. https://doi.org/10.3390/biom13020307
Chicago/Turabian StyleFilippini, Alice, Valentina Salvi, Vincenzo Dattilo, Chiara Magri, Stefania Castrezzati, Robert Veerhuis, Daniela Bosisio, Massimo Gennarelli, and Isabella Russo. 2023. "LRRK2 Kinase Inhibition Attenuates Astrocytic Activation in Response to Amyloid β1-42 Fibrils" Biomolecules 13, no. 2: 307. https://doi.org/10.3390/biom13020307
APA StyleFilippini, A., Salvi, V., Dattilo, V., Magri, C., Castrezzati, S., Veerhuis, R., Bosisio, D., Gennarelli, M., & Russo, I. (2023). LRRK2 Kinase Inhibition Attenuates Astrocytic Activation in Response to Amyloid β1-42 Fibrils. Biomolecules, 13(2), 307. https://doi.org/10.3390/biom13020307