

KinFams: De-Novo Classification of Protein Kinases Using CATH Functional Units

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
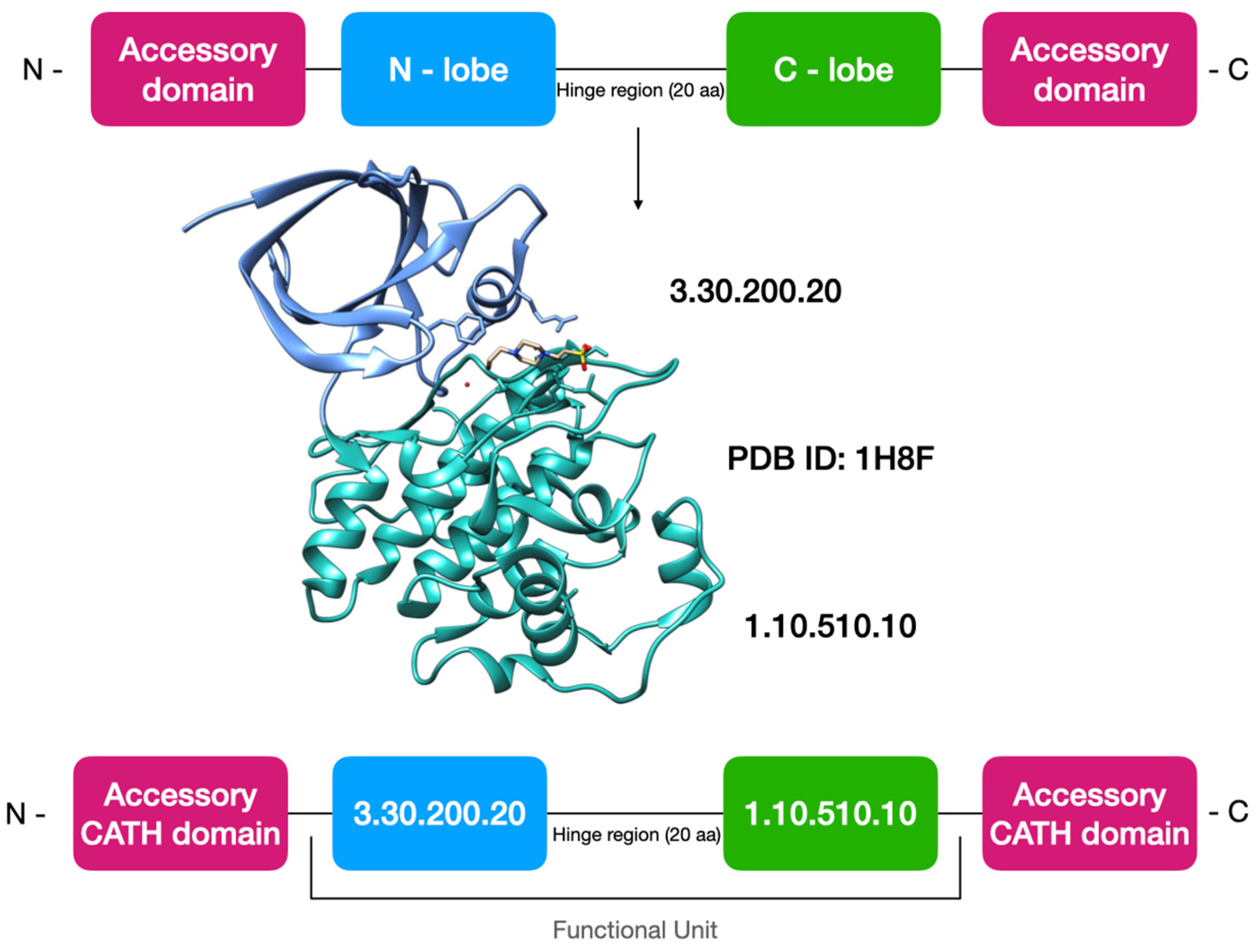
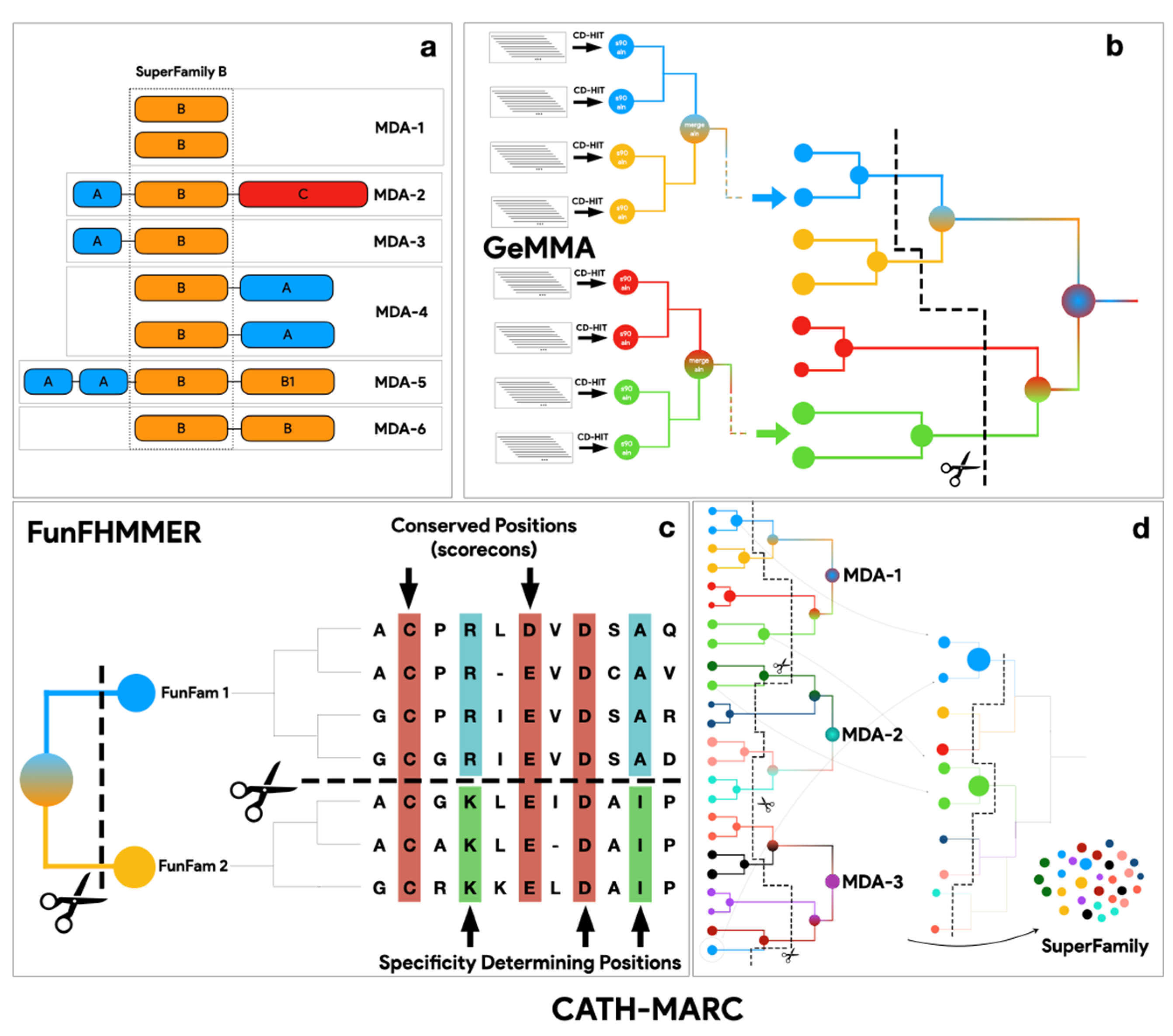
2.1. Generating CATH-KinFams
2.2. Updating Kinase Domain Sequences in the CATH Family Classification and Generating the Kinase Functional Unit
2.3. Running the FunFam-MARC Algorithm
2.4. Assessing the Functional Coherence of KinFams and KinBase Classifications Using the Enzyme Classification
2.5. Mapping of the CATH KinFams and KinBase Classifications
2.6. Mapping Drug Information from ChEMBL to Human KinFams
2.7. Obtaining 3D Structures (PDB and AlphaFold2) for Human-KinFams
3. Results and Discussion
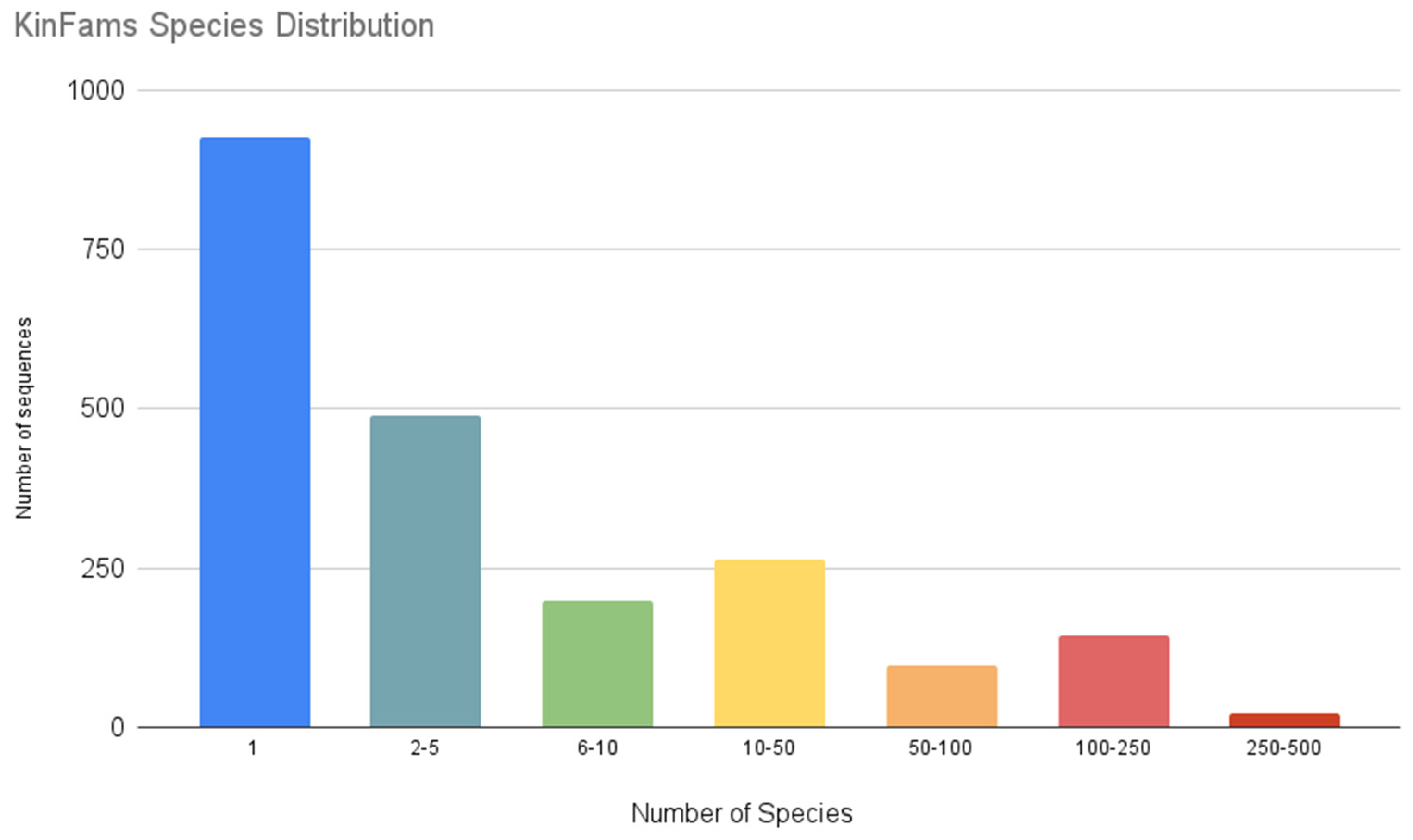
3.1. Updating the CATH Kinase Superfamily
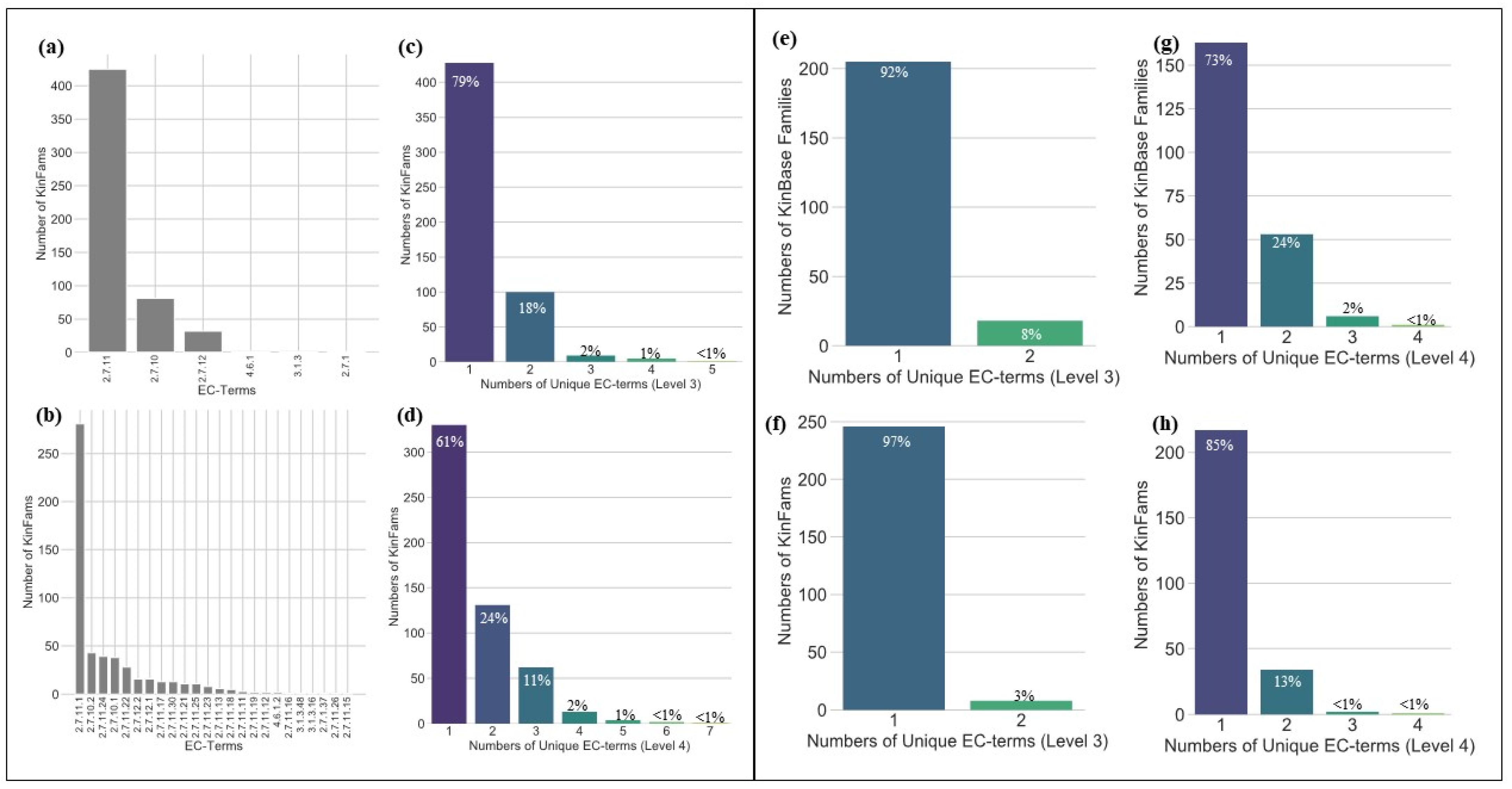
3.2. Assessing the Functional Coherence of the KinFams
3.3. Example Illustrating KinFam Sub-Classification of the KinBase JAK Family
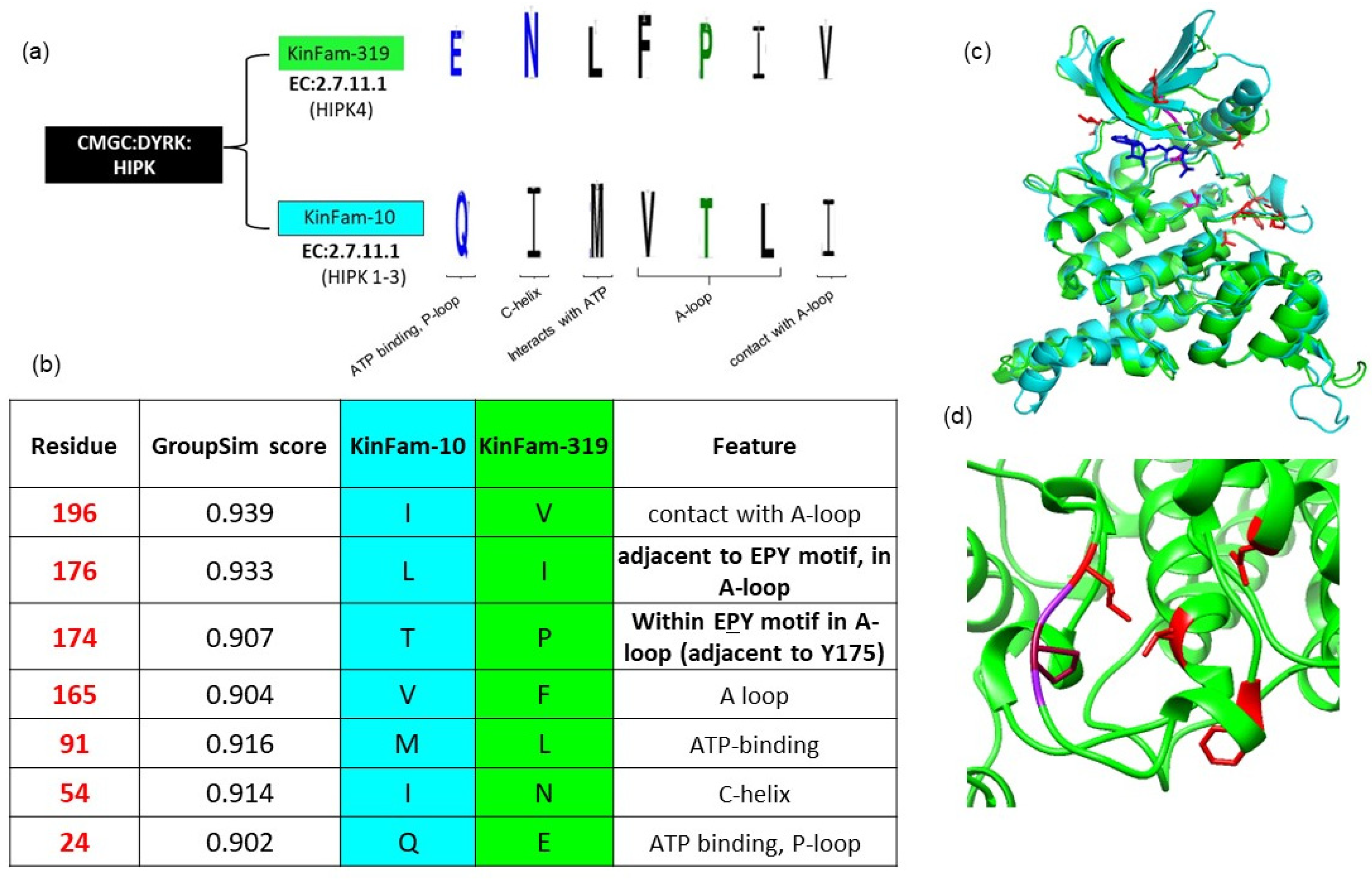
3.4. KinFam Subclassification of the KinBase HIPK Subfamily
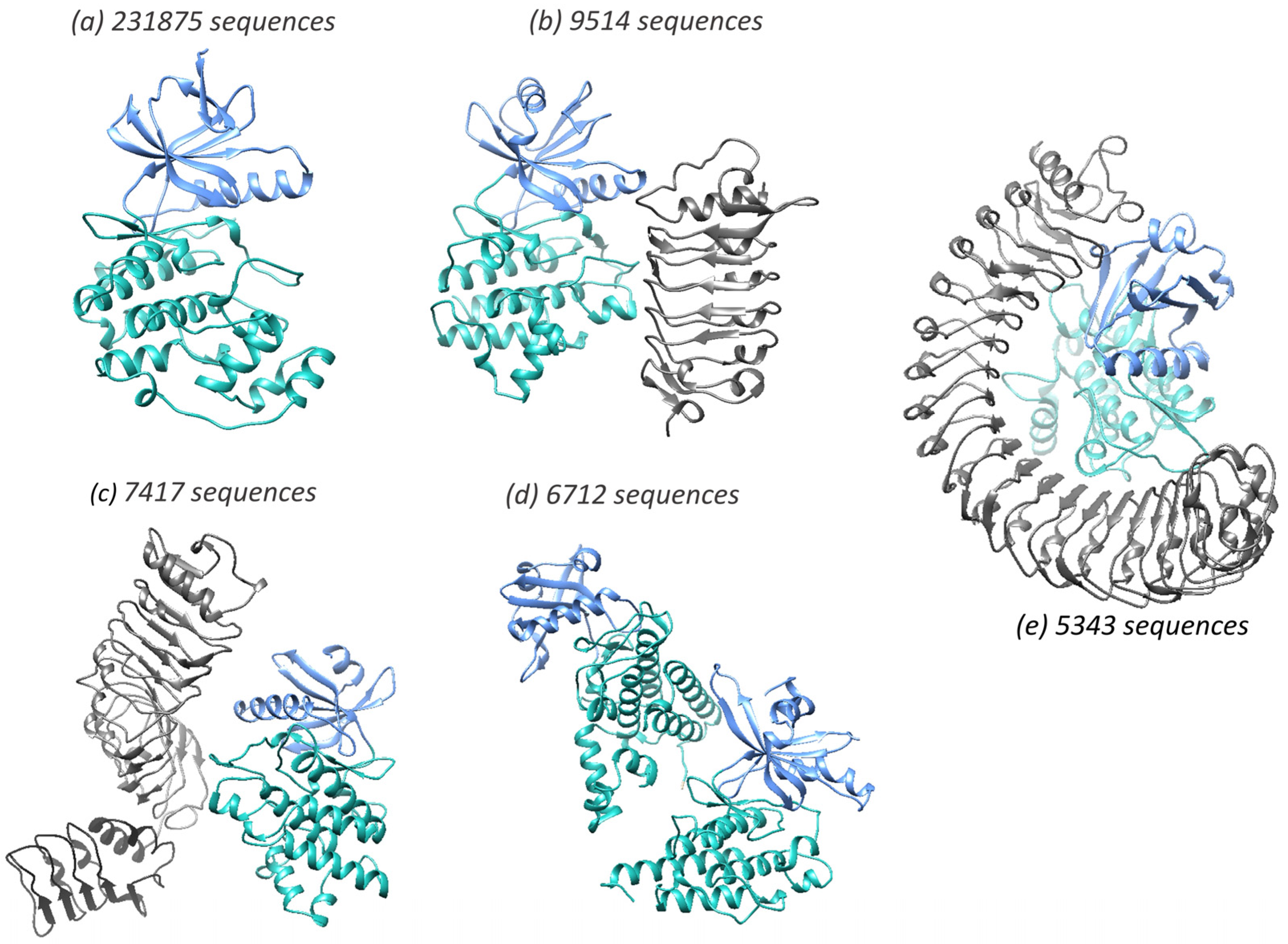
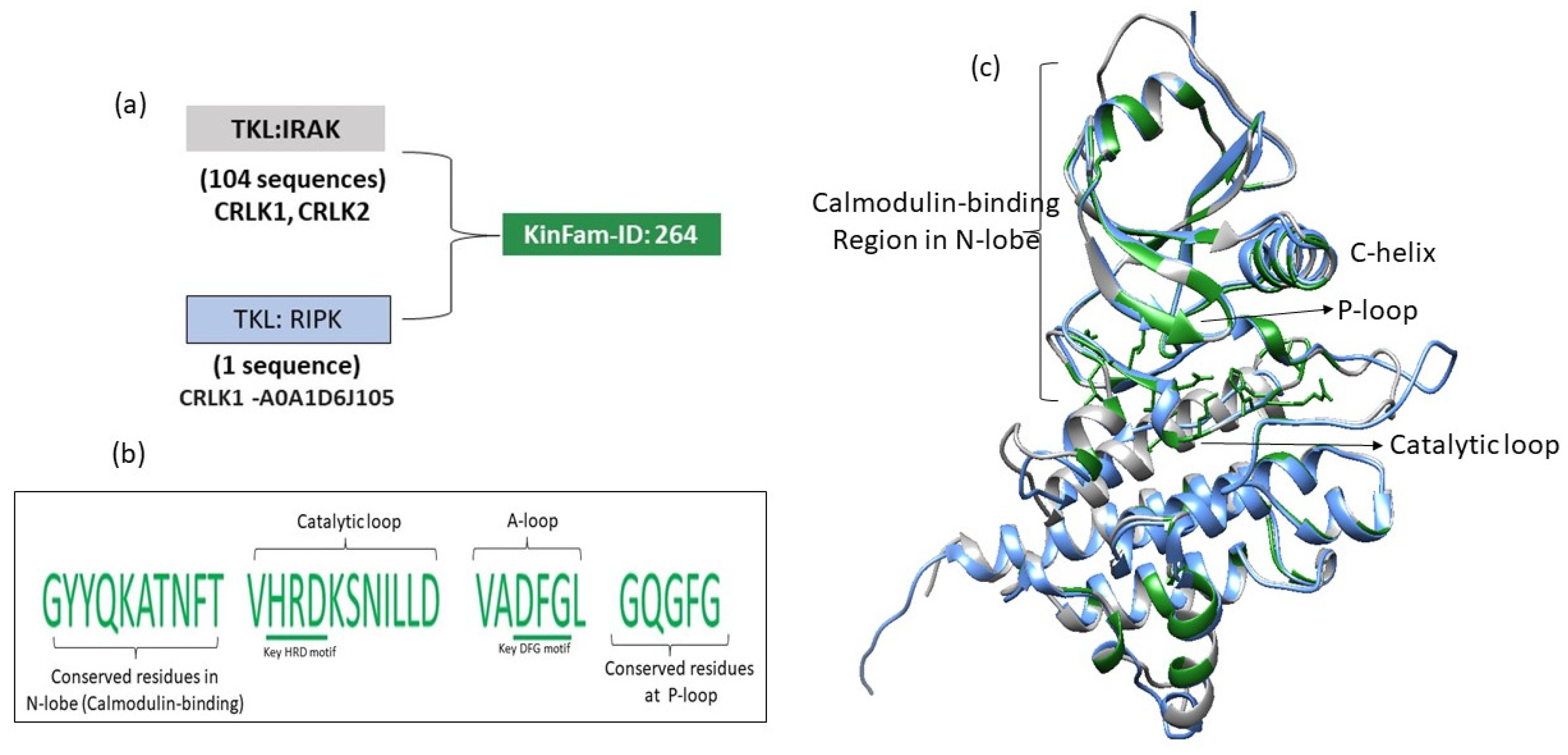
3.5. Merging of KinBase Groupings by KinFams
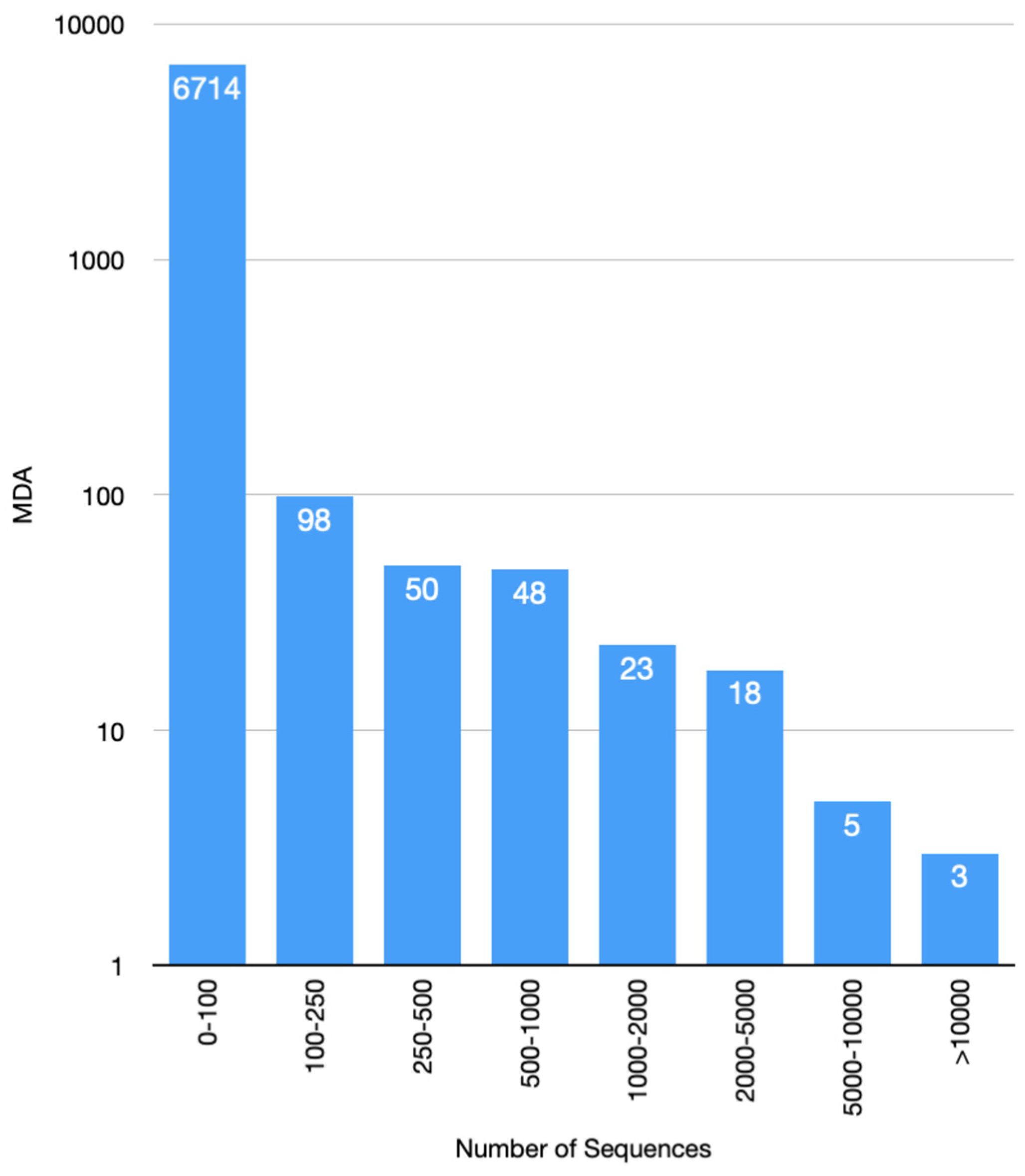
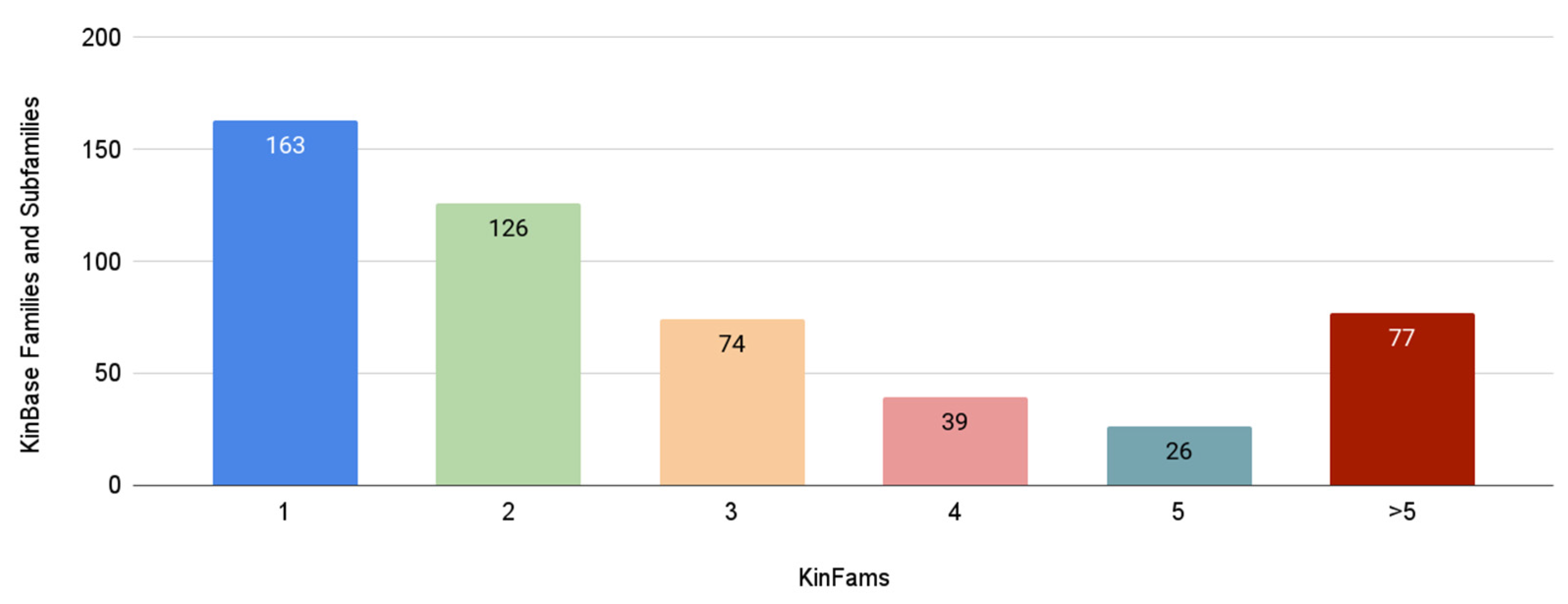
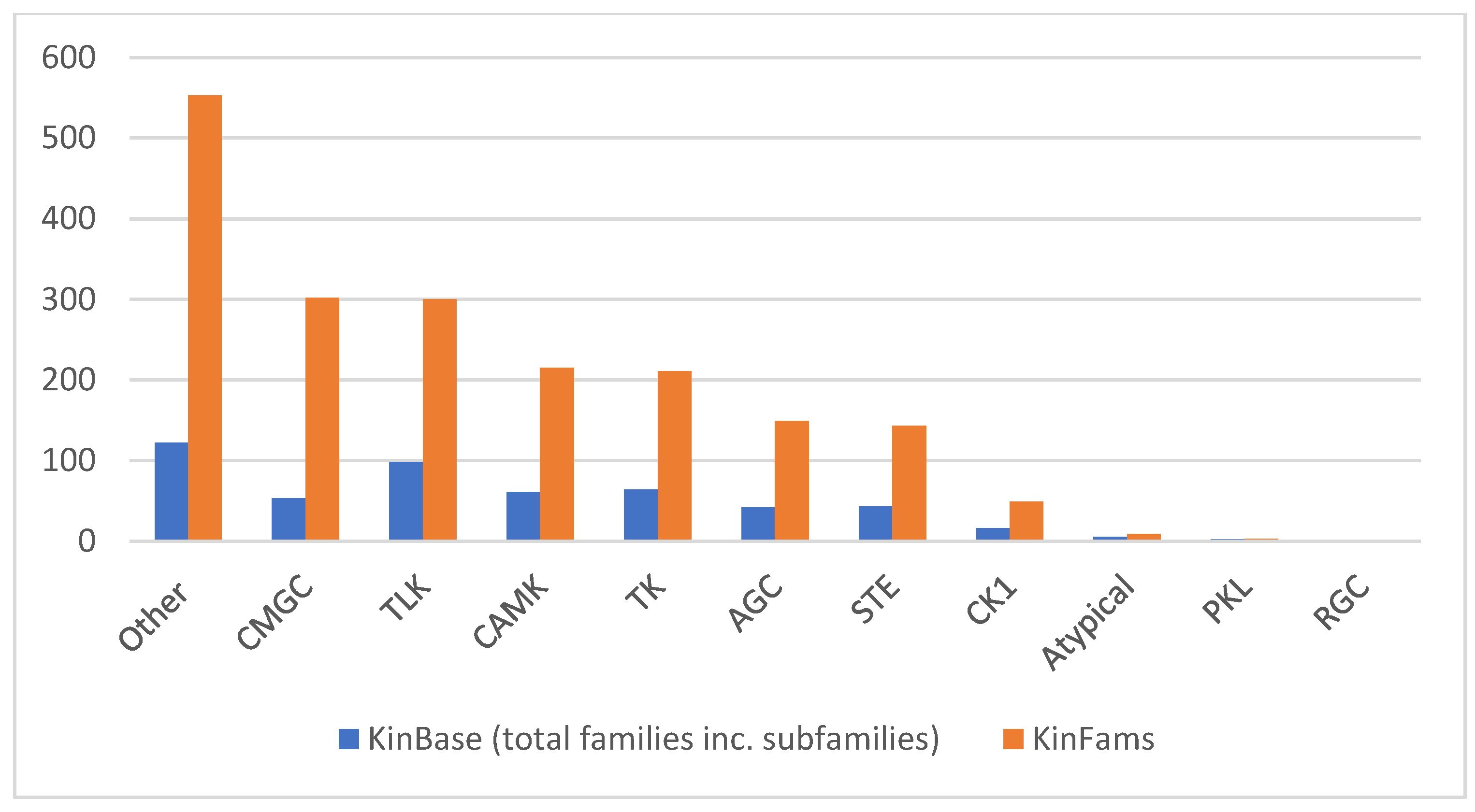
3.6. Increase in Kinase Family Space in KinFams Relative to KinBase
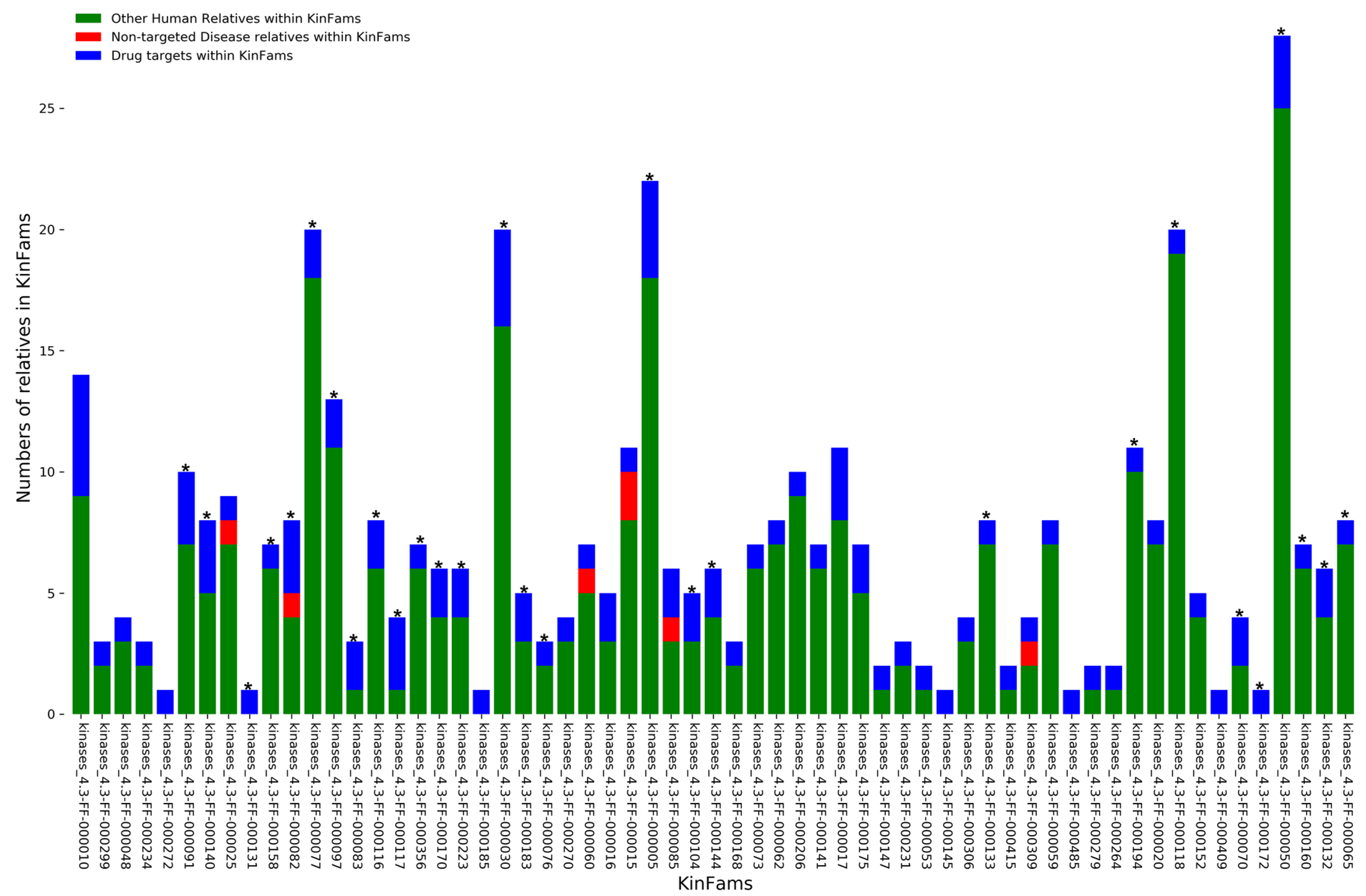
3.7. Identifying Druggable KinFams
4. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fabbro, D.; Cowan-Jacob, S.W.; Moebitz, H. Ten things you should know about protein kinases: IUPHAR Review 14. Br. J. Pharmacol. 2015, 172, 2675–2700. [Google Scholar] [CrossRef] [PubMed]
- Milanesi, L.; Petrillo, M.; Sepe, L.; Boccia, A.; D’Agostino, N.; Passamano, M.; di Nardo, S.; Tasco, G.; Casadio, R.; Paolella, G. Systematic analysis of human kinase genes: A large number of genes and alternative splicing events result in functional and structural diversity. BMC Bioinform. 2005, 6 (Suppl. S4), S20. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef] [PubMed]
- Hanks, S.K.; Hunter, T. The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. FASEB J. 1995, 9, 576–596. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Kornev, A.P. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem. Sci. 2011, 36, 65–77. [Google Scholar] [CrossRef]
- Kobe, B.; Kemp, B.E. Chapter 74—Principles of Kinase Regulation. In Handbook of Cell Signaling, 2nd ed.; Academic Press: Cambridge, MA, USA, 2010; pp. 559–563. [Google Scholar] [CrossRef]
- Hanks, S.; Quinn, A.; Hunter, T. The protein kinase family: Conserved features and deduced phylogeny of the catalytic domains. Science 1988, 241, 42–52. [Google Scholar] [CrossRef]
- Dreher, J.; Baumann, K. Comparison of ATP binding sites using structure-based similarity methods and molecular interaction fields. J. Cheminform. 2011, 3 (Suppl. S1), P34. [Google Scholar] [CrossRef]
- Smith, C.M.; Shindyalov, I.N.; Veretnik, S.; Gribskov, M.; Taylor, S.S.; Ten Eyck, L.F.; Bourne, P.E. The protein kinase resource. Trends Biochem. Sci. 1997, 22, 444–446. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Miranda-Saavedra, D.; Barton, G.J. Classification and functional annotation of eukaryotic protein kinases. Proteins 2007, 68, 893–914. [Google Scholar] [CrossRef]
- Bairoch, A.; Boeckmann, B.; Ferro, S.; Gasteiger, E. Swiss-Prot: Juggling between evolution and stability. Brief. Bioinform. 2004, 5, 39–55. [Google Scholar] [CrossRef]
- Martin, D.M.; Miranda-Saavedra, D.; Barton, G.J. Kinomer v. 1.0: A database of systematically classified eukaryotic protein kinases. Nucleic Acids Res. 2009, 37, D244–D250. [Google Scholar] [CrossRef]
- Krupa, A.; Abhinandan, K.R.; Srinivasan, N. KinG: A database of protein kinases in genomes. Nucleic Acids Res. 2004, 32, D153–D155. [Google Scholar] [CrossRef] [PubMed]
- Gosal, G.; Kochut, K.J.; Kannan, N. ProKinO: An ontology for integrative analysis of protein kinases in cancer. PLoS ONE 2011, 6, e28782. [Google Scholar] [CrossRef]
- Sillitoe, I.; Bordin, N.; Dawson, N.; Waman, V.P.; Ashford, P.; Scholes, H.M.; Pang, C.S.M.; Woodridge, L.; Rauer, C.; Sen, N.; et al. CATH: Increased structural coverage of functional space. Nucleic Acids Res. 2021, 49, D266–D273. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.; Anamika, K.; Srinivasan, N. Classification of Protein Kinases on the Basis of Both Kinase and Non-Kinase Regions. PLoS ONE 2010, 5, e12460. [Google Scholar] [CrossRef] [PubMed]
- McSkimming, D.I.; Dastgheib, S.; Talevich, E.; Narayanan, A.; Katiyar, S.; Taylor, S.S.; Kochut, K.; Kannan, N. ProKinO: A unified resource for mining the cancer kinome. Hum. Mutat. 2015, 36, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.Y.; Lin, C.T.; Huang, J.W.; Hsu, K.C.; Tseng, J.H.; You, S.R.; Yang, J.M. KIDFamMap: A database of kinase-inhibitor-disease family maps for kinase inhibitor selectivity and binding mechanisms. Nucleic Acids Res. 2013, 41, D430–D440. [Google Scholar] [CrossRef]
- Eid, S.; Turk, S.; Volkamer, A.; Rippmann, F.; Fulle, S. KinMap: A web-based tool for interactive navigation through human kinome data. BMC Bioinform. 2017, 18, 16. [Google Scholar] [CrossRef]
- Huang, L.C.; Taujale, R.; Gravel, N.; Venkat, A.; Yeung, W.; Byrne, D.P.; Eyers, P.A.; Kannan, N. KinOrtho: A method for mapping human kinase orthologs across the tree of life and illuminating understudied kinases. BMC Bioinform. 2021, 22, 446. [Google Scholar] [CrossRef]
- Soleymani, S.; Gravel, N.; Huang, L.-C.; Yeung, W.; Bozorgi, E.; Bendzunas, N.G.; Kochut, K.J.; Kannan, N. Dark kinase annotation, mining and visualization using the Protein Kinase Ontology. bioRxiv 2022. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Thomas, P.D.; Ebert, D.; Muruganujan, A.; Mushayahama, T.; Albou, L.; Mi, H. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 2022, 31, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Lee, D.; Sillitoe, I.; Dawson, N.L.; Lees, J.G.; Orengo, C.A. Functional classification of CATH superfamilies: A domain-based approach for protein function annotation. Bioinformatics 2015, 31, 3460–3467. [Google Scholar] [CrossRef]
- Das, S.; Sillitoe, I.; Lee, D.; Lees, J.G.; Dawson, N.L.; Ward, J.; Orengo, C.A. CATH FunFHMMer web server: Protein functional annotations using functional family assignments. Nucleic Acids Res. 2015, 43, W148–W153. [Google Scholar] [CrossRef]
- Zhou, N.; Jiang, Y.; Bergquist, T.R.; Lee, A.J.; Kacsoh, B.Z.; Crocker, A.W.; Lewis, K.A.; Georghiou, G.; Nguyen, H.N.; Hamid, N.; et al. The CAFA challenge reports improved protein function prediction and new functional annotations for hundreds of genes through experimental screens. Genome Biol. 2019, 20, 244. [Google Scholar] [CrossRef]
- Moya-García, A.; Adeyelu, T.; Kruger, F.A.; Dawson, N.L.; Lees, J.G.; Overington, J.P.; Orengo, C.; Ranea, J.A.G. Structural and Functional View of Polypharmacology. Sci. Rep. 2017, 7, 10102. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Lewis, T.E.; Sillitoe, I.; Lees, J.G. Cath-resolve-hits: A new tool that resolves domain matches suspiciously quickly. Bioinformatics 2019, 35, 1766–1767. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.A.; Rentzsch, R.; Orengo, C. GeMMA: Functional subfamily classification within superfamilies of predicted protein structural domains. Nucleic Acids Res. 2010, 38, 720–737. [Google Scholar] [CrossRef] [PubMed]
- Nightingale, A.; Antunes, R.; Alpi, E.; Bursteinas, B.; Gonzales, L.; Liu, W.; Luo, J.; Qi, G.; Turner, E.; Martin, M.-J. The Proteins API: Accessing key integrated protein and genome information. Nucleic Acids Res. 2017, 45, W539–W544. [Google Scholar] [CrossRef]
- Steinegger, M.; Meier, M.; Mirdita, M.; Vöhringer, H.; Haunsberger, S.J.; Söding, J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinform. 2019, 20, 473. [Google Scholar] [CrossRef] [PubMed]
- Capra, J.A.; Singh, M. Characterization and prediction of residues determining protein functional specificity. Bioinformatics 2008, 24, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Littmann, M.; Bordin, N.; Heinzinger, M.; Schütze, K.; Dallago, C.; Orengo, C.; Rost, B. Clustering FunFams using sequence embeddings improves EC purity. Bioinformatics 2021, 37, 3449–3455. [Google Scholar] [CrossRef] [PubMed]
- McDonald, A.G.; Tipton, K.F. Enzyme nomenclature and classification: The state of the art. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Valdar, W.S. Scoring residue conservation. Proteins 2002, 48, 227–241. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Bordin, N.; Sillitoe, I.; Nallapareddy, V.; Rauer, C.; Lam, S.D.; Waman, V.P.; Sen, N.; Heinzinger, M.; Littmann, M.; Kim, S.; et al. AlphaFold2 reveals commonalities and novelties in protein structure space for 21 model organisms. bioRxiv 2022. [Google Scholar] [CrossRef]
- Taylor, W.R.; Orengo, C.A. Protein structure alignment. J. Mol. Biol. 1989, 208, 1–22. [Google Scholar] [CrossRef]
- Min, X.; Ungureanu, D.; Maxwell, S.; Hammarén, H.; Thibault, S.; Hillert, E.-K.; Ayres, M.; Greenfield, B.; Eksterowicz, J.; Gabel, C.; et al. Structural and Functional Characterization of the JH2 Pseudokinase Domain of JAK Family Tyrosine Kinase 2 (TYK2). J. Biol. Chem. 2015, 290, 27261–27270. [Google Scholar] [CrossRef] [PubMed]
- Lupardus, P.J.; Ultsch, M.; Wallweber, H.; Kohli, P.B.; Johnson, A.R.; Eigenbrot, C. Structure of the pseudokinase-kinase domains from protein kinase TYK2 reveals a mechanism for Janus kinase (JAK) autoinhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 8025–8030. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Choi, C.Y.; Lee, S.-J.; Conti, M.A.; Kim, Y. Homeodomain-interacting protein kinases, a novel family of co-repressors for homeodomain transcription factors. J. Biol. Chem. 1998, 273, 25875–25879. [Google Scholar] [CrossRef] [PubMed]
- Van der Laden, J.; Soppa, U.; Becker, W. Effect of tyrosine autophosphorylation on catalytic activity and subcellular localisation of homeodomain-interacting protein kinases (HIPK). Cell Commun. Signal. 2015, 13, 3. [Google Scholar] [CrossRef]
- Arai, S.; Matsushita, A.; Du, K.; Yagi, K.; Okazaki, Y.; Kurokawa, R. Novel homeodomain-interacting protein kinase family member, HIPK4, phosphorylates human p53 at serine 9. FEBS Lett. 2007, 581, 5649–5657. [Google Scholar] [CrossRef]
- He, Q.; Shi, J.; Sun, H.; An, J.; Huang, Y.; Sheikh, M.S. Characterization of Human Homeodomain-interacting Protein Kinase 4 (HIPK4) as a Unique Member of the HIPK Family. Mol. Cell. Pharmacol. 2010, 2, 61–68. [Google Scholar]
- Rinaldo, C.; Siepi, F.; Prodosmo, A.; Soddu, S. HIPKs: Jack of all trades in basic nuclear activities. Biochim. Biophys. Acta 2008, 1783, 2124–2129. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zang, C.; Wu, Y.; Meng, R.; Chen, Y.; Jiang, T.; Wang, C.; Yang, X.; Guo, Y.; Situ, C.; et al. Homeodomain-interacting protein kinase HIPK4 regulates phosphorylation of manchette protein RIMBP3 during spermiogenesis. J. Biol. Chem. 2022, 298, 102327. [Google Scholar] [CrossRef] [PubMed]
- Crapster, J.A.; Rack, P.G.; Hellmann, Z.J.; Le, A.D.; Adams, C.M.; Leib, R.D.; E Elias, J.; Perrino, J.; Behr, B.; Li, Y.; et al. HIPK4 is essential for murine spermiogenesis. Elife 2020, 9, e50209. [Google Scholar] [CrossRef] [PubMed]
- Kaltheuner, I.H.; Anand, K.; Moecking, J.; Düster, R.; Wang, J.; Gray, N.S.; Geyer, M. Abemaciclib is a potent inhibitor of DYRK1A and HIP kinases involved in transcriptional regulation. Nat. Commun. 2021, 12, 6607. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Gan, L.; Wang, E.; Wang, J. Pocket-based drug design: Exploring pocket space. AAPS J. 2013, 15, 228–241. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Das, S.; Khan, I.; Kihara, D.; Orengo, C. Exploring Structure–Function Relationships in Moonlighting Proteins. In Moonlighting Proteins: Novel Virulence Factors in Bacterial Infections, 1st ed.; Henderson, B., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; pp. 21–43. [Google Scholar]
- Eddy, S.R. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009, 23, 205–211. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Name of Family Classification/ Database | Number of Groups/Families/ Subfamilies | Organisms/ Version of Uniprot or Swissprot Used | Website | Reference |
|---|---|---|---|---|---|
| 1995 | Hanks and Hunter | 5 groups, 55 subfamilies | Model organisms | Not available | [7,8] |
| 1997 | PKR—Protein kinase resource | 9 groups, 81 families, 238 subfamilies | SwissProt (2004) | http://pkr.sdsc.edu/html/index.shtml (not unavailable), accessed on 24 January 2023 | [9] |
| 2002 | KinBase | KinBase 2014 version 14 Groups, 240 families, 339 Subfamilies | 15 organisms | http://www.kinase.com/kinbase, accessed on 24 January 2023 | [10] |
| 2004, 2010 | KinG database | PKR (as above) | >2000 organisms UniProt (2019) | http://king.mbu.iisc.ernet.in/, accessed on 24 January 2023 | [14] |
| 2007, 2009 | Kinomer v.1 | 8 groups from KinBase (2008) | 43 eukaryotic organisms | http://www.compbio.dundee.ac.uk/kinomer/index.html, accessed on 24 January 2023 | [13] |
| 2011, 2015 | PrOKiNO | KinBase (2012 version) 14 groups, 273 Families and 359 Subfamilies | 1321 organisms UniProt (2021) | http://vulcan.cs.uga.edu/prokino/about/prokino, accessed on 24 January 2023 | [15] |
| 2022 | KinFams (CATH v4.3) | 2210 KinFams | a 13,981 organisms (from UniProt 2018) b 34,475 organisms (from UniProt 2022) | https://www.cathdb.info/, accessed on 24 January 2023 | [16] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adeyelu, T.; Bordin, N.; Waman, V.P.; Sadlej, M.; Sillitoe, I.; Moya-Garcia, A.A.; Orengo, C.A. KinFams: De-Novo Classification of Protein Kinases Using CATH Functional Units. Biomolecules 2023, 13, 277. https://doi.org/10.3390/biom13020277
Adeyelu T, Bordin N, Waman VP, Sadlej M, Sillitoe I, Moya-Garcia AA, Orengo CA. KinFams: De-Novo Classification of Protein Kinases Using CATH Functional Units. Biomolecules. 2023; 13(2):277. https://doi.org/10.3390/biom13020277
Chicago/Turabian StyleAdeyelu, Tolulope, Nicola Bordin, Vaishali P. Waman, Marta Sadlej, Ian Sillitoe, Aurelio A. Moya-Garcia, and Christine A. Orengo. 2023. "KinFams: De-Novo Classification of Protein Kinases Using CATH Functional Units" Biomolecules 13, no. 2: 277. https://doi.org/10.3390/biom13020277
APA StyleAdeyelu, T., Bordin, N., Waman, V. P., Sadlej, M., Sillitoe, I., Moya-Garcia, A. A., & Orengo, C. A. (2023). KinFams: De-Novo Classification of Protein Kinases Using CATH Functional Units. Biomolecules, 13(2), 277. https://doi.org/10.3390/biom13020277