Abstract
Mitochondria are widely considered the “power hub” of the cell because of their pivotal roles in energy metabolism and oxidative phosphorylation. However, beyond the production of ATP, which is the major source of chemical energy supply in eukaryotes, mitochondria are also central to calcium homeostasis, reactive oxygen species (ROS) balance, and cell apoptosis. The mitochondria also perform crucial multifaceted roles in biosynthetic pathways, serving as an important source of building blocks for the biosynthesis of fatty acid, cholesterol, amino acid, glucose, and heme. Since mitochondria play multiple vital roles in the cell, it is not surprising that disruption of mitochondrial function has been linked to a myriad of diseases, including neurodegenerative diseases, cancer, and metabolic disorders. In this review, we discuss the key physiological and pathological functions of mitochondria and present bioactive compounds with protective effects on the mitochondria and their mechanisms of action. We highlight promising compounds and existing difficulties limiting the therapeutic use of these compounds and potential solutions. We also provide insights and perspectives into future research windows on mitochondrial modulators.
1. Introduction
Mitochondria, widely understood to be the “power hub” of the cell, are organelles in eukaryotes responsible for most of the chemical energy supply required to fuel the cells’ complex web of biochemical reactions [1,2]. The mitochondria also perform crucial multifaceted roles in biosynthetic pathways, serving as an important source of building blocks for fatty acid, cholesterol, amino acid, glucose, and heme synthesis [3]. The mitochondria use fuels consumed by cells in the form of sugars, fatty acids, and amino acids to generate chemical energy [3,4]. For example, the mitochondria are an important hub for synthesizing amino acids such as glutamine, glutamate, alanine, proline, and aspartate. In addition, the initial step of gluconeogenesis, during which pyruvate carboxylase oxaloacetate is converted to malate, occurs in the mitochondria [3].
As shown in Figure 1, the mitochondria generate small molecule storage of chemical energy known as adenosine triphosphate (ATP) via electron transport-linked phosphorylation, otherwise known as oxidative phosphorylation (OXPHOS). The OXPHOS pathway utilizes five enzyme complexes in the inner membrane of the mitochondria to produce ATP as it progresses through the respiratory chain. These complexes include Complex I (NADH: ubiquinone oxidoreductase), Complex II (succinate dehydrogenase), Complex III (ubiquinol-cytochrome c oxidoreductase), Complex IV (cytochrome c oxidase), and Complex V (ATP synthase) [2,5,6,7]. In addition to its role at the core of energy metabolism, the mitochondrion is also an important site for calcium ion (Ca2+) storage and homeostasis while also playing a crucial role in cell apoptosis [2,8]. Furthermore, mitochondria are known to play a major role in the generation of reactive oxygen species (ROS), most of which are produced by Complex I and Complex III (on a smaller scale). Almost 90% of ROS generated in the mitochondria are essentially a by-product of the OXPHOS pathway [5,6,9]. The role of mitochondria in ROS generation is certainly noteworthy, especially with mounting research evidence establishing a link between disease progression in several neurodegenerative diseases and increased ROS production [10].
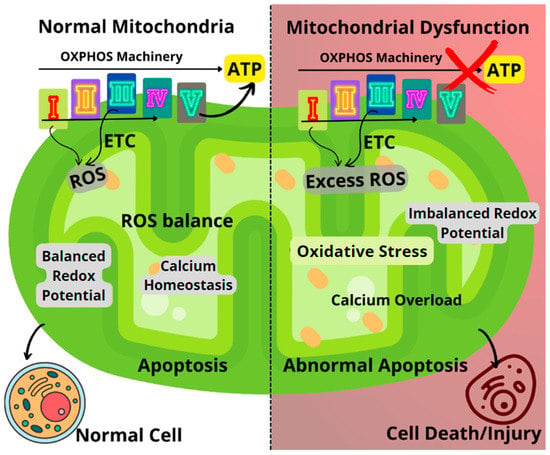
Figure 1.
Comparison between healthy and dysfunctional mitochondria, highlighting the key mechanisms of mitochondrial dysfunction. ETC: Electron transport chain; ROS: Reactive oxygen species.
Oxidative stress resulting from the dysregulated generation of ROS can lead to impairments in the OXPHOS machinery, causing an imbalance in the mitochondrial redox potential and significant loss of mitochondrial functions (Figure 1). Impairments to mitochondrial function could result in decreased ATP generation, calcium overload, and unbalanced apoptosis [2,6,9]. Furthermore, disease conditions such as Huntington’s, Alzheimer’s, Parkinson’s, diabetes, several cancers, seizures, kidney failure, cardiomyopathy, and brain disorders, as well as normal aging processes, have been linked to failing mitochondrial functions [1,9,11,12,13,14].
As illustrated in Figure 2, the OXPHOS pathway starts with the entry of electrons into the respiratory chain via Complexes I and II, which are subsequently transferred to Complexes III and IV, respectively, and then used by Complex V to generate energy. [1,5,11,15].
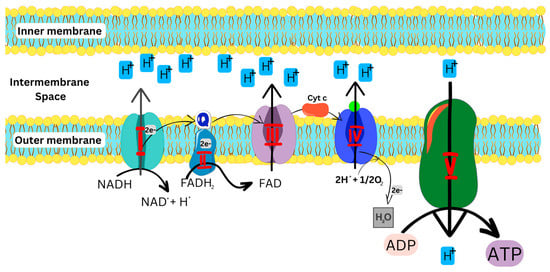
Figure 2.
OXPHOS pathway, showing the transfer of electrons in the ETC to produce ATP. NADH: Reduced nicotinamide adenine dinucleotide; FADH: Reduced flavin adenine dinucleotide; ADP: Adenosine diphosphate; ATP: Adenosine triphosphate.
Complex I, an L-shaped multimeric enzyme, catalyzes the first step of OXPHOS in the electron transport chain (Figure 2). Complex I binds and oxidizes NADH to generate two electrons which are used to reduce ubiquinone (coenzyme Q) to ubiquinol, an electron-rich form of coenzyme Q, which further transfers the electrons to Complex III [6,7]. Alongside complex I, complex II also binds and oxidizes FADH2 to generate two electrons, which are then transferred to ubiquinone [8,16]. Complex III transfers the electrons to cytochrome c (cyt c)and finally to Complex IV, which reduces O2 to H2O. The energy generated and released during this cascade of processes is then used to move protons from the mitochondrial matrix into the intermembrane space to generate an electrochemical potential utilized by Complex V to produce ATP from ADP and phosphate for cellular energy [6,7,11].
1.1. Mitochondria in Human Diseases
As previously enumerated, mitochondria play prominent roles in several processes at the cellular level. Consequently, the slightest alterations to any of these processes can ultimately lead to diseases and maladies in the human body. As a result, many vigorous attempts have been undertaken to explore the role of mitochondria in the pathogenesis of different diseases [2,9]. Despite the immense progress made to date, there are crucial things yet to be understood when it comes to the structure and function of the mitochondria as related to human diseases. Highlighted in the subsequent paragraphs is available evidence of mitochondrial dysfunction in selected human diseases.
1.1.1. Mitochondria in Neurodegenerative Diseases and Ageing
Neurodegenerative diseases are a class of incurable diseases characterized by progressive degeneration and/or death of neurons, leading to the destruction of the nervous system. Neurons are well known to be high-energy demanding cells; hence it is not surprising that their health and functionality are closely linked to the mitochondria, which are the powerhouses of the cell [2,9,12]. Each neuron contains hundreds to thousands of mitochondria, and the central nervous system relies on normal mitochondrial functions for its high metabolic needs [17,18,19,20]. Although the etiology and pathogenesis of neurodegenerative diseases largely remain a mystery, research evidence has identified mitochondrial dysfunction as one of the key features of neurodegenerative disorders, such as Parkinson’s and Alzheimer’s diseases [17,21]. Therefore, a better understanding of mitochondrial function is critical to understanding and detangling the mysteries of neurological disorders.
In Parkinson’s disease (PD), extensive studies using cellular and animal models have implicated mitochondrial dysfunction, increased generation of ROS, and calcium imbalance as pivotal factors in the etiology of PD. In addition, a decline in mitochondrial Complex I activity has been reported in PD patient-sourced olfactory neurosphere-derived (hONS) cells, and involvement of PARK proteins in altered mitochondrial regulation has also been observed [12,15,22,23]. A thorough review discussing the role of mitochondria in PD was recently authored by Zambrano et al. [12].
In Alzheimer’s disease (AD), mitochondrial anomalies have also been identified as a common and consistent feature [21,24]. Accumulation of amyloid-β and phosphorylation of tau protein leading to the formation of neurofibrillary tangles are key hallmarks of AD, both of which have been linked to mitochondrial abnormalities [21,25]. Alterations in the morphology of the mitochondrion, enzyme, and DNA changes have also been observed in the brains of AD patients [21]. Similar to PD, oxidative stress induced by abnormal mitochondrial function is an early feature in AD [21,25]. In addition, partial inhibition of mitochondrial Complex I has been touted as a potential strategy for the treatment of AD [11].
Impairments in mitochondrial activity have also been reported in aging, which is one of the critical risk factors associated with most neurodegenerative diseases. Impairments such as mitochondrial DNA alteration, decreased proteasomal activity, increased ROS generation, and reduced activity of the OXPHOS machinery have all been linked to aging [10,17].
1.1.2. Mitochondria in Metabolic Diseases
Lately, there has been an interesting surge in research findings linking mitochondrial impairments to metabolic diseases such as type II diabetes [25], insulin resistance [26], obesity, metabolic syndrome, stroke, non-alcoholic liver disease, and the list goes on [2,8]. A resounding finding in these reports is that mitochondrial dysfunction contributes significantly to oxidative stress and inflammation, which is a usual commonality in these metabolic diseases [9,27,28,29]. ROS homeostasis is pivotal to aerobic organisms as this ensures the balance between the rate and magnitude of production and subsequent elimination of ROS over time. Any imbalance of ROS homeostasis essentially overwhelms the mitochondrial electron transport chain and, by extension, OXPHOS, leading to a decline in mitochondrial contents and the rate of OXPHOS [9,27]. A decline in mitochondrial contents and the rate of OXPHOS, as well as the modification of mitochondrial dynamics in key organs associated with metabolic diseases, have been implicated in the etiology of several metabolic diseases [9,26].
1.1.3. Mitochondria in Cancer
Although the role of mitochondria in cancer and tumor development is yet to be fully understood, mitochondrial defects have long been implicated in the etiology of cancers and tumors [30]. One important hallmark of cancer is the Warburg effect, which involves the reprogramming of ATP generation via the OXPHOS pathway to aerobic glycolysis [30,31]. In normal cells, the common mechanism for ATP generation is glucose metabolism via the OXPHOS pathway in mitochondria. However, even in the presence of functional mitochondria, most cancer cells bypass the mitochondria and rewire their metabolism to produce needed energy through aerobic glycolysis, which is less efficient and involves a high rate of glucose uptake and glycolysis followed by lactate formation [32,33]. This ‘selfish’ reprogramming enhances the progression and proliferation of cancer and tumor cells through the overexpression of glucose transporters, speedy inefficient production of ATP to meet energy demands, and accumulation of lactate which aids tumor progression and acidosis [33,34].
Since the 1920s, when the Warburg effect was first documented, several studies have reported defective mitochondrial respiration and mutated or low copies of mitochondrial DNAs in various cancers, including adenocarcinoma, breast, colon, prostate, head, and neck cancers [35,36,37]. Another widely reported hallmark of cancer is the excessive generation of ROS, which are commonly a by-product of the mitochondria-mediated metabolic process [30,37,38,39]. The mitochondria have also been established as a proven target for cancer treatment with a handful of FDA-approved mitochondrial-targeted compounds and several others at different stages of preclinical and clinical trials. Notable examples include metformin, mitoxantrone, cisplatin, and ME344 [30].
1.1.4. Mitochondria and infectious diseases
Beyond their conventional role as the cell’s energy hub, mitochondria also play a crucial role as a signaling platform for innate immunity against infectious microbes, and the role of mitochondria in infectious disease has been extensively documented [40,41,42,43]. Major host responses against infections depend on mitochondrial functions, and receptors of the innate immune system can detect compromises in mitochondrial functions, subsequently triggering an immune response [41,42]. In addition, pathogens exploit mitochondrial functions to influence their survival and evade immunity by affecting OXPHOS and mitochondrial dynamics and disrupting communication between the mitochondria and nucleus [40,41,43].
During infection, pathogens are detected by pattern-recognition receptors (PRRs), which can recognize pathogen-associated molecular patterns (PAMPs) such as flagellins, liposaccharides, proteins, mannose, and nucleic acids, as well as danger-associated molecular motifs (DAMPs) such as cardiolipin, ROS, mitochondrial DNA and n-formyl peptide [40,42]. Mitochondrial DAMPs are released into the cytosol as a result of infections, injuries, or loss of mitochondrial homeostasis [40,42].
DAMPs and PAMPs can be detected by PRRs to trigger innate immune responses against viruses, bacteria, and other infectious pathogens [42,44]. PRRs are classed into four families, which include toll-like receptors TLRs, (NOD)-like receptors (NLRs), C-type lectin receptors (CLRs), and retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) [42]. In viral infections such as influenza, PAMPs are recognized by RLRs, which interact with mitochondria antiviral signaling protein (MAVs) in the mitochondrial membrane to trigger the production of pro-inflammatory cytokines and type 1 interferon as an immune response [40,42,43]. During bacterial infection, TLRs are stimulated by bacterial PAMPs to induce the release of mitochondrial ROS to initiate antibacterial defense, resulting in the killing of pathogenic bacteria [42,45].
1.2. Materials and methods
The aim of this review is to provide a comprehensive insight into compounds with therapeutic potential on mitochondrial functions and their mechanisms of action, with a focus on compounds that can modulate the mitochondria such that mitochondrial dysfunction is mitigated or prevented altogether. To achieve this, an extensive literature search was conducted on PubMed, Science-Direct, and Google-Scholar databases using the following search terms:
“Mitochondria,” “Mitochondrial Complex,” “Mitochondria Health Disease,” “Mitochondrial Dysfunction,” “Mitochondrial Dysfunction Compounds.”
As a result of our search, we found 61 compounds (Table 1) with protective effects on the mitochondria. Further searches were conducted exclusively on PubMed using the name of each of the 61 compounds and mitochondria as keywords. This was performed to discover multiple mitochondrial modulating activities of any compound that might not have been covered in the first round of searches and to explore detailed mechanistic studies of each compound.

Table 1.
Mitochondrial Modulators.
All 61 compounds were gathered from articles published in the last 20 years, and all consulted articles were thoroughly read to extract relevant information, such as the disease model used for the bioactivity study, the dose administered, mitochondria-related activities, and mechanisms of action. In Table 1, we present all 61 compounds, the disease model, effective doses, mitochondria-related targets, mode of action, and references for each compound.
2. Mitochondrial Modulators, Mechanisms, and Targets
In Table 1, we present 61 mitochondrial modulators which are able to protect the mitochondria from toxic insults and/or improve mitochondrial function. This collection of compounds includes 52 natural products (Figure 3, Figure 4, Figure 5, Figure 6 and Figure 7) and nine synthetic compounds (Figure 8). It is noteworthy that 31 of the 52 natural products discussed are phenolic compounds, which represent 50.8% of the total (Figure 3 and Figure 4). This is unsurprising given that phenolic compounds are renowned for their excellent antioxidant activity and the fact that oxidative stress is one of the major indicators of mitochondrial dysfunction [166,167]. Others include four alkaloids (Figure 5), eight terpenes (Figure 6), one organic acid, one amine, one cyclic polyketide, one lactone, one benzochromone, and one coumarin derivative (Figure 7).
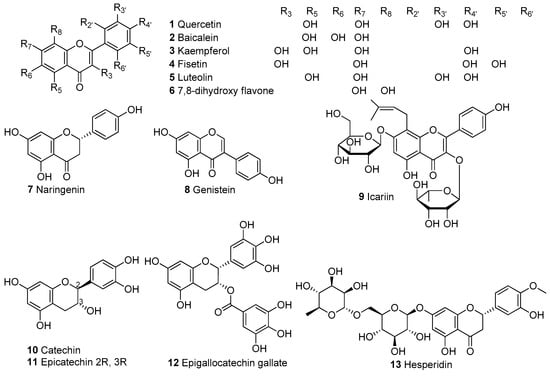
Figure 3.
Chemical structures of flavonoids.
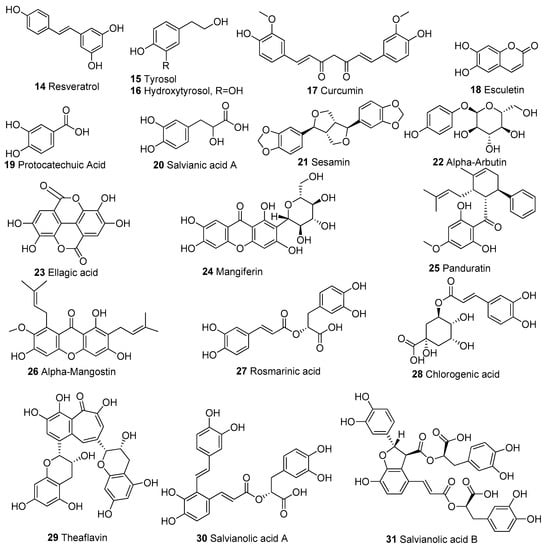
Figure 4.
Chemical structures of other phenolic compounds apart from flavonoids.
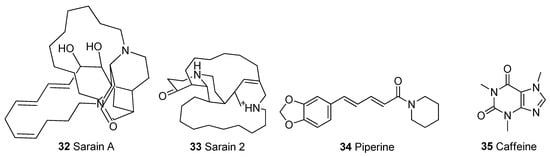
Figure 5.
Chemical structures of alkaloids.
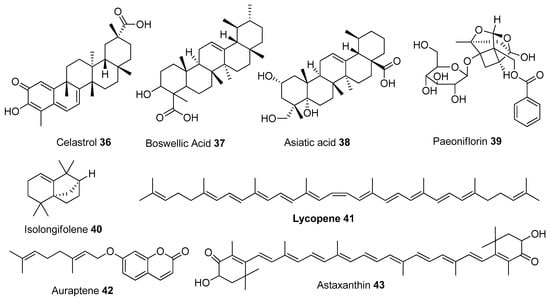
Figure 6.
Chemical structures of terpenes.
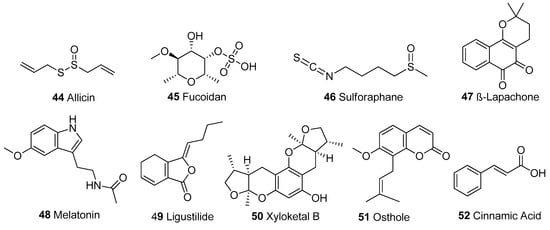
Figure 7.
Other structural classes. Organosulphur compounds: 44-46; benzochromone: 47; Amine: 48; Lactone: 49; Cyclic polyketide: 50; Coumarin derivative: 51, Organic acid: 52.
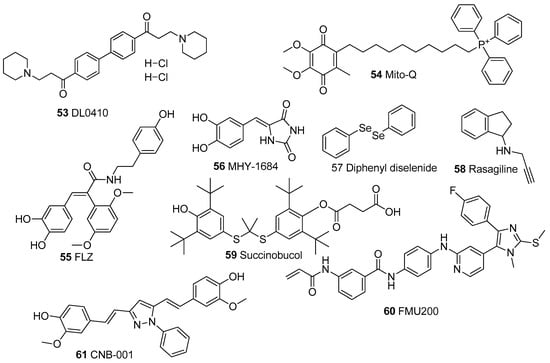
Figure 8.
Chemical Structures of synthetic mitochondrial modulators.
The biological activities of the compounds summarized in Table 1 were evaluated in cellular or animal models or a combination of both. Our search revealed that 83.6% were tested using at least one cell line, with SH-SY5Y cells accounting for 41% of the studies. This is unsurprising because the human neuroblastoma SH-SY5Y cell line is a common in vitro model for PD and other neurodegenerative diseases associated with mitochondrial dysfunctions [168,169,170].
Generally, compounds showed multiple modes of action, exerting their protective effects on the mitochondria by (1) restoring oxidative balance by inhibiting the production of ROS or blocking the harmful effects of ROS and increasing the activity of antioxidant enzymes, (2) modulating apoptotic markers, (3) promoting ATP synthesis, (4) enhancing the activities of mitochondrial complexes, mitochondrial biogenesis and restoration of normal mitochondrial morphology in the presence of mitochondria toxins such as rotenone, 6-OHDA and MPP+ [46,76,82,114,171]. The numerical distribution of the compounds based on structural class and mechanism is shown in Figure 9. Inhibition of ROS is a common feature in all 61 compounds; while 45 of the compounds have anti-apoptotic activity, 33 improved ATP synthesis, 24 increased the activities of complexes, 22 promoted mitochondrial biogenesis, and eight restored normal mitochondrial morphology.
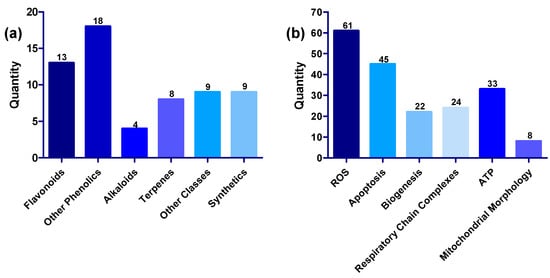
Figure 9.
(a) Distribution of compounds across various structural classes of flavonoids, phenolic compounds, alkaloids, terpenes, other structural classes, and synthetic compounds. (b) Distribution of compounds across various targets, including ROS, apoptosis, biogenesis, respiratory chain complexes, ATP, and mitochondrial morphology.
2.1. Antioxidative Mechanisms of Mitochondrial Modulators
Given that 90% of ROS is generated by mitochondria, oxidative stress is one of the major hallmarks of mitochondrial dysfunction [2,172]. All compounds listed in Table 1 displayed antioxidant activity through multiple mechanisms. In combination with the reduction in ROS generation and lipid peroxidation (LPO), the effect of a compound on the activity of enzymes that regulate free radical scavenging and oxidative balance is a major way to determine its antioxidant activity [129,173]. In normal cellular conditions, super-oxide dismutase (SOD) converts superoxide radical to H2O2, and enzymes such as catalase (CAT) and glutathione peroxidase (GPX) reduce mitochondrial H2O2 by converting it to H2O. Hence the ability of any compound to increase the activity of these enzymes is taken as an indicator that the compound possesses antioxidant potential [114,174].
Nuclear E2-related factor 2 (Nrf2) is one of the most pivotal cell defense mechanisms against stressors. It is a transcription factor that signals the expression of oxidative enzymes and stimulates the increased expression of antioxidant genes in response to oxidative stress. Consequently, any disruption to the activation of the Nrf2-mediated antioxidant response exposes the cells and renders the mitochondria more sensitive to deleterious pro-oxidants and electrophiles [175]. As a result of its crucial cytoprotective role against the effects of oxidative stress, Nrf2 is now a well-known drug target in many neurodegenerative diseases, most of which are associated with mitochondrial dysfunction [23,175,176]. Hence, the ability of a compound of interest to activate Nrf2-mediated antioxidative response is relevant for assessing its antioxidant capacity as a mitochondrial modulator [114,171]. Compounds 1, 2, 7–9, 14, 16, 18, 21, 23, 24, 27, 29, 32, 37, 42, 46, 53, and 54 activate the Nrf2-mediated antioxidative response.
Excessive ROS generation is known to cause disruptions to the OXPHOS pathway and electron transport chain, leading to defects in mitochondrial respiration, ATP production, depletion of mitochondrial complexes, and collapse of mitochondrial membrane potential (ΔΨm) [23,175,177]. Nrf2, when activated, amplifies ATP production and ΔΨm by boosting substrate availability for OXPHOS, leading to enhanced activity of the mitochondrial complexes [175]. The ability of a metabolite to induce blockage of the mitochondrial permeability transition pore (mPTP) is also a very instructive parameter when measuring the extent of mitochondrial oxidative stress. This can be achieved by inhibiting cyclophilin D (CYPD), a key enzyme that regulates the opening and closing of mPTP [114,178]. The opening of mPTP leads to mitochondrial swelling and has been implicated as one of the causes of the loss of mitochondrial function in neurodegeneration. The opening of mPTP occurs as a result of the collapse of the ΔΨm, leading to the continuous burst of ROS into mitochondria, causing oxidative stress [88,178,179,180] and mitochondrial permeability transition (MPT) driven necrosis. MPT-driven necrosis is a type of regulated cell death characterized by uncontrolled loss of post-mitotic cells and can be delayed by inhibition of CYPD [181,182]. All the compounds listed in Table 1, except boswellic acid (37) and MHY-1684 (56), were reported to restore ΔΨm while sarain A (32), ellagic acid (23), salvianolic acid A (30), kaempferol (3), lycopene (41), FLZ (55) induced the blockage of the mPTP. Although not listed in Table 1 because it is out of the 20-year coverage of this review, it is pivotal to mention that cyclosporin A; an FDA-approved immunosuppressant medication is a proven and well-known MPT inhibitor and a promising potential mitochondrial-targeted neuroprotective agent [183,184].
2.2. Inhibition of Apoptosis
Inhibition of apoptotic pathways through the modulation of apoptotic markers is another mechanism commonly reported in compounds discussed in this review. This is unsurprising because oxidative stress is a known precursor to apoptosis, so mitochondria play a key role in maintaining cellular apoptotic balance [2]. Typically in cells, apoptosis is triggered by activating pro-apoptotic proteins belonging to the Bcl-2 family, e.g., Bax and Bak, which translocate to mitochondria to induce the release of cytochrome c into the cytosol [185]. The release of cytochrome c promotes the activation of caspase-3, -6, -7, and -9, which subsequently initiates cell death [115,185]. However, the Bcl-2 family includes several anti-apoptotic members such as Bcl-2, Bcl-xL, Mcl-1, and Bcl-w. For the maintenance of proper cellular function, it is important to establish a steady balance between pro-apoptotic and anti-apoptotic markers [115,185]. Compounds 2–5, 8–9, 12–15, 17–21, 23–26, 28–30, 34–35, 37–41, 43–45, 48, 51–53, 58, 59, and 61 all showed anti-apoptotic activity by modulating these markers.
The mitochondria are pivotal to keeping apoptotic balance, and disruptions to this balance have been implicated in diseases such as PD and AD [115,148,185]. The PI3K/Akt/GSK-3β pathway also promotes the expression of anti-apoptotic and pro-apoptotic proteins. The kinase PI3K releases phosphatidylinositol-3,4,5-trisphosphate (PIP3), which activates Akt by promoting the translocation of Akt to the plasma membrane [115,148]. Activation of Akt then induces the expression of anti-apoptotic proteins such as Bcl-2, while GSK-3β, on the other hand, is a prerequisite for the activation of p53, which induces the expression of pro-apoptotic proteins such as Bax [185,186,187]. PI3K/Akt also inhibits the expression of serine-threonine kinase GSK-3β, a critical effector of PI3K/Akt cellular signaling and activator of neuronal apoptosis. Activation of the PI3K/Akt pathway ameliorates apoptosis through the phosphorylation of GSK-3β by AKT [185,186,187]. Compounds 7, 19, 27, 30, 31, 35, 40, and 56 demonstrated anti-apoptotic activity by targeting the PI3K/Akt/GSK-3β pathway.
2.3. Mitochondrial Biogenesis and Mitophagy
Mitochondrial homeostasis is maintained by keeping the mitochondrial pool in a cell at a steady state through the simultaneous propagation of new mitochondria (mitochondrial biogenesis or mitogenesis) and the removal of old and damaged mitochondria through a process called mitophagy [188,189]. Mitogenesis is a complex process with at least 150 proteins involved [190,191]. However, there are three key interdependent markers known to play a crucial role in this process: peroxisome proliferator-activated receptor-γ coactivator-1α (PGC1α), silent mating-type information regulation proteins (Sirt), and AMP-activated kinase (AMPK) [110,190,191]. PGC1α is a transcriptional factor that binds to Sirt3 promoters, interacting with Nrf2 to facilitate the upregulation of antioxidants [190]. Sirts can modulate the induction of several markers, such as PGC1α, to enhance the expression of antioxidant enzymes such as SOD [190,192,193]. Compounds 1, 3, 5, 10, 14, 18, 21–23, 27, 28, 30, 31, 36, 41, 43–46, 48, and 54 were reported to modulate the AMPK/ PGC1α/Sirt pathway.
AMPK is inhibited by ATP, and the AMP/ATP ratio gives a sensitive indication of the metabolic state of a cell [194]. AMPK, when activated by mitochondrial ROS, promotes mitophagy, which may lead to a reduction in mitochondrial number, a decrease in ATP levels, and a subsequent increase in AMP/ATP ratio [194,195,196]. The PTEN-induced kinase 1 (PINK1) pathway-Parkin pathway also plays a crucial role in the mitophagy of weak mitochondria and maintenance of mitochondrial homeostasis [196,197,198,199]. Several studies have linked the accumulation of weak mitochondria in cells due to decreased mitophagy to deficient levels of PINK1 and Parkin [196,197]. Compounds quercetin (1), resveratrol (14), and ligustilide (49) were reported to modulate PINK1/Parkin.
2.4. Other Effects of Mitochondrial Modulators
In addition to the biological activities earlier discussed, 33 compounds (1, 3–5, 7, 10, 15, 17, 22–24, 26–32, 39, 41, 43, 46–48, 51, 58, 59) were reported to increase ATP synthesis, 24 (4–7, 10, 11, 13, 14, 16, 17, 23, 24, 28, 30, 34-37, 41, 45, 46, 51, 55, 57) enhance the activity of mitochondria complexes, and 8 (1, 10, 11, 27, 29, 31, 43, 50) restore mitochondrial morphology. In addition to their reported antioxidant activity and increase in MMP, 7,8-dihydroxyflavone (6) and diphenyl diselenide (57) increased the level of mitochondrial complexes, while β-lapachone (47) improved ATP synthesis [141,154]. However, sarain 2 (33) did not show any other activity apart from inhibiting ROS and increasing MMP [114].
Prohibitins (PHB) are evolutionarily conserved proteins ubiquitously expressed in eukaryotic cells and localized in the nucleus, cytosol, and mitochondria [200,201]. Large assemblies of homologous prohibitin members, prohibitin 1 (PHB1) and prohibitin 2 (PHB2), have been identified in the inner mitochondria membrane [201,202]. These mitochondrial PHB subunits play an important role in cell proliferation, mitochondrial biogenesis, mitochondrial dynamics, cell apoptosis, and senescence [200,201,203]. PHB impairments have been reported in aging, cancer, neurodegenerative, kidney, cardiovascular, and metabolic diseases, in which significant loss of mitochondrial function has been proven [200,201,202,203]. The marine natural product, aurilide which binds to PHB1, and a synthetic molecule, fluorizoline, which binds to PHB1 and PHB 2 are known potent PHB-binding compounds which have been reported to induce apoptosis and mitochondrial fragmentation [204,204,205]. Fluorizoline and aurilide have not been included in Table 1 since this review is focused on compounds that are able to mitigate or prevent mitochondrial dysfunction.
2.5. The Standouts
The most outstanding compound of all 61 discussed is perhaps rasagiline (58), a well-known monoamine oxidase inhibitor. Rasagiline is a novel propargylamine and an approved treatment for PD either as a monotherapy or in combination with other treatments such as levodopa [206,207]. Rasagiline was reported to attenuate mitochondrial dysfunction by reducing oxidative stress, preventing MMP collapse, suppressing apoptosis by lowering the release of cytochrome C, and improving ATP synthesis [158]. This is quite interesting because there are well-established links between PD and mitochondrial dysfunction [208], suggesting that rasagiline has multifaceted modes of action as far as the treatment of PD is concerned. The compound was also highly potent, showing excellent activity at a concentration range of 10 μM to 10 nM [158]. Rasagiline prevented ΔΨm collapse [158] and caused a three-fold increase in ATP level at 10 μM and 1 μM as well as a two-fold increase at 100 nM and 10 nM [159]. The compound also completely suppressed cell death at 10 μM and 1 μM by reducing cytochrome c [158,159]. Rasagiline is also currently trialed for treatment in AD, with results from the proof of concept of the phase II trials recently published [209].
Melatonin (48), a nocturnal hormone in the brain produced by the pineal gland, is also approved as Circadin®, a prolonged-release melatonin tablet for the treatment of insomnia, a common comorbidity of neurological disorders [210,211]. Additionally, a randomized phase II clinical trial of melatonin in the treatment of AD was successfully completed in 2013, with patients treated with prolonged-release melatonin showing significantly improved cognitive performance [212]. Melatonin also displayed a significantly better potency compared to the other compounds, second only to rasagiline at the tested concentration of 500 nM [142]. At 500 nM, melatonin significantly reduced ROS and caspase-3 in porcine oocytes treated with rotenone to levels comparable to that of the untreated group and enhanced mitochondrial biogenesis by upregulating PGC1α/SIRT while also increasing MMP and ATP synthesis [142].
The flavonoids, quercetin (1), and resveratrol (14) display the best coverage for biological activities discussed. They both have antioxidant potentials, activate the Nrf2 pathway, improve MMP, reduce apoptosis, and improve mitochondrial biogenesis and ATP synthesis [46,49,76,78]. In addition, Genistein (8) and asiatic acid (38) are by far the most active of the 52 natural products at the cellular level. Genistein reduced oxidative stress and improved MMP in a dose-dependent fashion in the concentration range from 10 pM to 100 nM in cells [66]. Asiatic acid, at 10 nM, caused a 75% decline in ROS generation, a 30–50% decline in pro-apoptotic markers such as Bax, cytochrome c, caspase-3, -6, -8, -9, and a 40% increase in Bcl-2 level when compared to cells treated with rotenone [125].
3. Conclusions and Future Directions
In this review, we have highlighted the multifaceted roles of the mitochondria as well as their relevance in health and disease. We enumerated 61 compounds, 52 of which are natural products, while nine are synthetic compounds. The fact that most of the compounds discussed are natural products underscores the well-known concept that nature holds vastly untapped therapeutic potential and will continue to play a critical role in drug discovery [213]. It cannot be overemphasized that tremendous advances have been made in natural product drug discovery. However, attention to natural products has declined in the past two decades due to challenging isolation and screening techniques, especially in high-throughput assays against molecular targets [213,214]. Despite these challenges, the field remains exceptionally viable, with millions of species unexplored and more metabolites waiting to be discovered [215]. Additionally, there is also the potential to improve current practices and approaches to natural products-based screening through innovative technological advances that are less arduous, time-saving, and with larger yields such as metabolomics, genome mining driven isolation, enhanced microbial culturing and biosynthetic engineering strategies [213].
Although unsurprising, it is noteworthy that 29 of the 61 compounds discussed in this article are phenolic compounds, many of which are considered safe and already available as dietary supplements [216,217]. Notable examples are resveratrol, quercetin, catechin, and kaempferol, but none of these have become approved commercially available drugs [217]. One major challenge with phenolic compounds is their low bioavailability; hence it is difficult to reproduce the in vitro biological activities of phenolic compounds in vivo [216,217,218]. Catechin, for instance, has very low bioavailability when taken orally, with its plasma concentration reported to be about 50 times lower than the concentration required to reproduce levels of bioactivity reported in vitro [218,219]. Studies have shown that absorption of quercetin in humans after ingestion can be as low as 2% [218], curcumin 5% [220,221], and resveratrol 70% but with bioavailability at trace levels (less than 1%) [222,223]. Consequently, there is a need for more in-depth studies into the pharmacokinetics of these compounds with the aim of enhancing their absorption and bioavailability through the development of more effective drug delivery systems such as micelles, liposomes, and nanoparticles. There is also the potential of developing analogous compounds through modification of existing phenolic compounds such that their absorption and bioavailability are improved without loss of bioactivity. Genistein is potentially a good candidate for absorption and bioavailability since it has high efficacy and potency in vitro at concentrations below nanomolar levels to the tune of 10 pM [66]. This may imply that a low concentration of genistein is needed to reproduce its activity in vivo without the problem of low bioavailability and absorption. However, this notion remains a theoretical suggestion and is subject to further investigations.
All compounds discussed in this review showed antioxidant activities. Hence, it is evident that alleviation of oxidative stress is the primary mechanism and mode of action of mitochondrial modulators, followed by inhibition of apoptosis reported in 45 of the compounds. Only 33 compounds improved ATP synthesis, while 22 improved mitochondrial biogenesis, and 24 enhanced the activity of mitochondrial complexes, suggesting that there is still room further to establish the mitochondria-modulating activities of the untested compounds.
Another issue worth considering in the search for mitochondrial modulators is the complexity of the mitochondria and the fact that mitochondria are tissue-specific organelles [224]. As a consequence of the specificity and heterogeneity of the mitochondria in different tissues, the mitochondria may show different morphology, distinct biochemical properties, and varied interactions with other intracellular organelles [224,225]. Notably, live imaging techniques have been used to show alterations in mitochondrial ROS, calcium homeostasis, membrane potential, and redox state in mitochondria isolated from different cells or tissues [226]. Furthermore, mitochondria in various tissues may also display different responses and sensitivity to molecules [225]. Hence, it might be interesting to consider if these molecules retain their biological activities across various cell lines and tissues and how that might affect the utility of the compounds in treating diseases associated with mitochondria dysfunction.
Author Contributions
E.M. wrote the manuscript and drew the illustrations. L.M., G.D.M., and Y.F. supervised the writing of the manuscript and contributed to the development of its’ contents. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Emmanuel Makinde would like to acknowledge Griffith University for providing Griffith University Postgraduate Research Scholarship and Griffith University International Postgraduate Research Scholarship.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sharma, L.K.; Lu, J.; Bai, Y. Mitochondrial Respiratory Complex I: Structure, Function and Implication in Human Diseases. Curr. Med. Chem. 2009, 16, 1266–1277. [Google Scholar] [PubMed]
- Annesley, S.J.; Fisher, P.R. Mitochondria in Health and Disease. Cells 2019, 8, 680. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Haigis, M.C. The Multifaceted Contributions of Mitochondria to Cellular Metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Nowinski, S.M.; Solmonson, A.; Rusin, S.F.; Maschek, J.A.; Bensard, C.L.; Fogarty, S.; Jeong, M.-Y.; Lettlova, S.; Berg, J.A.; Morgan, J.T.; et al. Mitochondrial Fatty Acid Synthesis Coordinates Oxidative Metabolism in Mammalian Mitochondria. eLife 2020, 9, e58041. [Google Scholar] [CrossRef] [PubMed]
- Janssen, R.J.R.J.; Nijtmans, L.G.; van den Heuvel, L.P.; Smeitink, J.A.M. Mitochondrial Complex I: Structure, Function and Pathology. J. Inherit. Metab. Dis. 2006, 29, 499–515. [Google Scholar] [CrossRef]
- Mimaki, M.; Wang, X.; McKenzie, M.; Thorburn, D.R.; Ryan, M.T. Understanding Mitochondrial Complex I Assembly in Health and Disease. Biochim. Biophys. Acta BBA-Bioenerg. 2012, 1817, 851–862. [Google Scholar] [CrossRef]
- Lenaz, G.; Fato, R.; Genova, M.L.; Bergamini, C.; Bianchi, C.; Biondi, A. Mitochondrial Complex I: Structural and Functional Aspects. Biochim. Biophys. Acta BBA-Bioenerg. 2006, 1757, 1406–1420. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Kausar, S.; Wang, F.; Cui, H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef]
- Lenaz, G.; Bovina, C.; D’aurelio, M.; Fato, R.; Formiggini, G.; Genova, M.L.; Giuliano, G.; Pich, M.M.; Paolucci, U.; Castelli, G.P.; et al. Role of Mitochondria in Oxidative Stress and Aging. Ann. N. Y. Acad. Sci. 2002, 959, 199–213. [Google Scholar] [CrossRef]
- Trushina, E.; Trushin, S.; Hasan, M.F. Mitochondrial Complex I as a Therapeutic Target for Alzheimer’s Disease. Acta Pharm. Sin. B 2022, 12, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, K.; Barba, D.; Castillo, K.; Noboa, L.; Argueta-Zamora, D.; Robayo, P.; Arizaga, E.; Caicedo, A.; Gavilanes, A.W.D. Fighting Parkinson’s Disease: The Return of the Mitochondria. Mitochondrion 2022, 64, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New Insights into the Complex Role of Mitochondria in Parkinson’s Disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Vona, R.; Ascione, B.; Malorni, W.; Straface, E. Mitochondria and Sex-Specific Cardiac Function. In Sex-Specific Analysis of Cardiovascular Function; Kerkhof, P.L.M., Miller, V.M., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2018; pp. 241–256. ISBN 978-3-319-77932-4. [Google Scholar]
- Matigian, N.; Abrahamsen, G.; Sutharsan, R.; Cook, A.L.; Vitale, A.M.; Nouwens, A.; Bellette, B.; An, J.; Anderson, M.; Beckhouse, A.G.; et al. Disease-Specific, Neurosphere-Derived Cells as Models for Brain Disorders. Dis. Model. Mech. 2010, 3, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Bandara, A.B.; Drake, J.C.; Brown, D.A. Complex II Subunit SDHD Is Critical for Cell Growth and Metabolism, Which Can Be Partially Restored with a Synthetic Ubiquinone Analog. BMC Mol. Cell Biol. 2021, 22, 35. [Google Scholar] [CrossRef]
- Rango, M.; Bresolin, N. Brain Mitochondria, Aging, and Parkinson’s Disease. Genes 2018, 9, 250. [Google Scholar] [CrossRef]
- Rangaraju, V.; Lewis, T.L.; Hirabayashi, Y.; Bergami, M.; Motori, E.; Cartoni, R.; Kwon, S.-K.; Courchet, J. Pleiotropic Mitochondria: The Influence of Mitochondria on Neuronal Development and Disease. J. Neurosci. 2019, 39, 8200–8208. [Google Scholar] [CrossRef]
- Faris, R.; Moore, R.A.; Ward, A.; Sturdevant, D.E.; Priola, S.A. Mitochondrial Respiration Is Impaired during Late-Stage Hamster Prion Infection. J. Virol. 2017, 91, e00524-17. [Google Scholar] [CrossRef]
- Kann, O.; Kovács, R. Mitochondria and Neuronal Activity. Am. J. Physiol.-Cell Physiol. 2007, 292, C641–C657. [Google Scholar] [CrossRef]
- Picone, P.; Nuzzo, D.; Caruana, L.; Scafidi, V.; Di Carlo, M. Mitochondrial Dysfunction: Different Routes to Alzheimer’s Disease Therapy. Oxid. Med. Cell. Longev. 2014, 2014, e780179. [Google Scholar] [CrossRef]
- Murtaza, M.; Shan, J.; Matigian, N.; Todorovic, M.; Cook, A.L.; Ravishankar, S.; Dong, L.F.; Neuzil, J.; Silburn, P.; Mackay-Sim, A.; et al. Rotenone Susceptibility Phenotype in Olfactory Derived Patient Cells as a Model of Idiopathic Parkinson’s Disease. PLoS ONE 2016, 11, e0154544. [Google Scholar] [CrossRef]
- Cook, A.L.; Vitale, A.M.; Ravishankar, S.; Matigian, N.; Sutherland, G.T.; Shan, J.; Sutharsan, R.; Perry, C.; Silburn, P.A.; Mellick, G.D.; et al. NRF2 Activation Restores Disease Related Metabolic Deficiencies in Olfactory Neurosphere-Derived Cells from Patients with Sporadic Parkinson’s Disease. PLoS ONE 2011, 6, e21907. [Google Scholar] [CrossRef]
- Mohamed, T.M.; Youssef, M.A.M.; Bakry, A.A.; El-Keiy, M.M. Alzheimer’s Disease Improved through the Activity of Mitochondrial Chain Complexes and Their Gene Expression in Rats by Boswellic Acid. Metab. Brain Dis. 2021, 36, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.-H.; Zhao, X.-Y.; Yao, P.-P.; Xu, D.-E.; Ma, Q.-H. The Mitochondrion: A Potential Therapeutic Target for Alzheimer’s Disease. Neurosci. Bull. 2018, 34, 1127–1130. [Google Scholar] [CrossRef]
- Wada, J.; Nakatsuka, A. Mitochondrial Dynamics and Mitochondrial Dysfunction in Diabetes. Acta Med. Okayama 2016, 70, 151–158. [Google Scholar] [CrossRef]
- Henriksen, E.J.; Diamond-Stanic, M.K.; Marchionne, E.M. Oxidative Stress and the Etiology of Insulin Resistance and Type 2 Diabetes. Free Radic. Biol. Med. 2011, 51, 993–999. [Google Scholar] [CrossRef]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA Mutations, Oxidative Stress, and Apoptosis in Mammalian Aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef]
- Vona, R.; Gambardella, L.; Cittadini, C.; Straface, E.; Pietraforte, D. Biomarkers of Oxidative Stress in Metabolic Syndrome and Associated Diseases. Oxid. Med. Cell. Longev. 2019, 2019, e8267234. [Google Scholar] [CrossRef]
- Chiu, H.Y.; Tay, E.X.Y.; Ong, D.S.T.; Taneja, R. Mitochondrial Dysfunction at the Center of Cancer Therapy. Antioxid. Redox Signal. 2020, 32, 309–330. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg Effect: Historical Dogma versus Current Understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg Effect: Essential Part of Metabolic Reprogramming and Central Contributor to Cancer Progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.K.; Fang, H.; Liu, J.; Vartak, R.; Deng, J.; Bai, Y. Mitochondrial Respiratory Complex I Dysfunction Promotes Tumorigenesis through ROS Alteration and AKT Activation. Hum. Mol. Genet. 2011, 20, 4605–4616. [Google Scholar] [CrossRef]
- Chatterjee, A.; Mambo, E.; Sidransky, D. Mitochondrial DNA Mutations in Human Cancer. Oncogene 2006, 25, 4663–4674. [Google Scholar] [CrossRef]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial Dysfunction and Mitochondrial Dynamics-The Cancer Connection. Biochim. Biophys. Acta BBA-Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef]
- Sharma, P.; Sampath, H. Mitochondrial DNA Integrity: Role in Health and Disease. Cells 2019, 8, 100. [Google Scholar] [CrossRef]
- Guaragnella, N.; Giannattasio, S.; Moro, L. Mitochondrial Dysfunction in Cancer Chemoresistance. Biochem. Pharmacol. 2014, 92, 62–72. [Google Scholar] [CrossRef]
- Banoth, B.; Cassel, S.L. Mitochondria in Innate Immune Signaling. Transl. Res. 2018, 202, 52–68. [Google Scholar] [CrossRef]
- Tiku, V.; Tan, M.-W.; Dikic, I. Mitochondrial Functions in Infection and Immunity. Trends Cell Biol. 2020, 30, 263–275. [Google Scholar] [CrossRef]
- Andrieux, P.; Chevillard, C.; Cunha-Neto, E.; Nunes, J.P.S. Mitochondria as a Cellular Hub in Infection and Inflammation. Int. J. Mol. Sci. 2021, 22, 11338. [Google Scholar] [CrossRef] [PubMed]
- Brokatzky, D.; Häcker, G. Mitochondria: Intracellular Sentinels of Infections. Med. Microbiol. Immunol. (Berl.) 2022, 211, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Hisata, S.; Choi, A.M.K. The Roles of Mitochondrial Damage-Associated Molecular Patterns in Diseases. Antioxid. Redox Signal. 2015, 23, 1329–1350. [Google Scholar] [CrossRef] [PubMed]
- Koch, R.E.; Josefson, C.C.; Hill, G.E. Mitochondrial Function, Ornamentation, and Immunocompetence. Biol. Rev. 2017, 92, 1459–1474. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-W.; Han, R.; He, H.-J.; Li, J.; Chen, S.-Y.; Gu, Y.; Xie, C. Administration of Quercetin Improves Mitochondria Quality Control and Protects the Neurons in 6-OHDA-Lesioned Parkinson’s Disease Models. Aging 2021, 13, 11738–11751. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.K.; Garabadu, D. Quercetin Exhibits A7nAChR/Nrf2/HO-1-Mediated Neuroprotection Against STZ-Induced Mitochondrial Toxicity and Cognitive Impairments in Experimental Rodents. Neurotox. Res. 2021, 39, 1859–1879. [Google Scholar] [CrossRef]
- Vanani, A.R.; Mahdavinia, M.; Shirani, M.; Alizadeh, S.; Dehghani, M.A. Protective Effects of Quercetin against Oxidative Stress Induced by Bisphenol-A in Rat Cardiac Mitochondria. Environ. Sci. Pollut. Res. 2020, 27, 15093–15102. [Google Scholar] [CrossRef]
- Zhang, Q.; Song, W.; Zhao, B.; Xie, J.; Sun, Q.; Shi, X.; Yan, B.; Tian, G.; Liang, X. Quercetin Attenuates Diabetic Peripheral Neuropathy by Correcting Mitochondrial Abnormality via Activation of AMPK/PGC-1α Pathway in vivo and in vitro. Front. Neurosci. 2021, 15, 636172. [Google Scholar]
- Kuang, L.; Cao, X.; Lu, Z. Baicalein Protects against Rotenone-Induced Neurotoxicity through Induction of Autophagy. Biol. Pharm. Bull. 2017, 40, 1537–1543. [Google Scholar] [CrossRef]
- Lee, I.K.; Kang, K.A.; Zhang, R.; Kim, B.J.; Kang, S.S.; Hyun, J.W. Mitochondria Protection of Baicalein against Oxidative Damage via Induction of Manganese Superoxide Dismutase. Environ. Toxicol. Pharmacol. 2011, 31, 233–241. [Google Scholar] [CrossRef]
- Wang, S.-F.; Liu, L.-F.; Wu, M.-Y.; Cai, C.-Z.; Su, H.; Tan, J.; Lu, J.-H.; Li, M. Baicalein Prevents 6-OHDA/Ascorbic Acid-Induced Calcium-Dependent Dopaminergic Neuronal Cell Death. Sci. Rep. 2017, 7, 8398. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Yang, B.; Qiao, Y.; Zhou, Q.; He, H.; He, M. Kaempferol Protects Mitochondria and Alleviates Damages against Endotheliotoxicity Induced by Doxorubicin. Biomed. Pharmacother. 2020, 126, 110040. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, K.; Prem, P.N.; Boovarahan, S.R.; Sivakumar, B.; Kurian, G.A. FIsetin Preserves Interfibrillar Mitochondria to Protect Against Myocardial Ischemia-Reperfusion Injury. Cell Biochem. Biophys. 2022, 80, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Alikatte, K.; Palle, S.; Rajendra Kumar, J.; Pathakala, N. Fisetin Improved Rotenone-Induced Behavioral Deficits, Oxidative Changes, and Mitochondrial Dysfunctions in Rat Model of Parkinson’s Disease. J. Diet. Suppl. 2021, 18, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh, A.K.; Garg, G.; Rizvi, S.I. Fisetin as a Caloric Restriction Mimetic Protects Rat Brain against Aging Induced Oxidative Stress, Apoptosis and Neurodegeneration. Life Sci. 2018, 193, 171–179. [Google Scholar] [CrossRef]
- Wu, B.; Song, H.; Fan, M.; You, F.; Zhang, L.; Luo, J.; Li, J.; Wang, L.; Li, C.; Yuan, M. Luteolin Attenuates Sepsis-induced Myocardial Injury by Enhancing Autophagy in Mice. Int. J. Mol. Med. 2020, 45, 1477–1487. [Google Scholar] [CrossRef]
- Akhtar, A.; Dhaliwal, J.; Sah, S.P. 7,8-Dihydroxyflavone Improves Cognitive Functions in ICV-STZ Rat Model of Sporadic Alzheimer’s Disease by Reversing Oxidative Stress, Mitochondrial Dysfunction, and Insulin Resistance. Psychopharmacology 2021, 238, 1991–2009. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, S.; Shao, Q.; Li, P.; Sun, Y.; Luo, L.; Yan, X.; Fan, Z.; Hu, J.; Zhao, J.; et al. Brain-Derived Neurotrophic Factor Mimetic, 7,8-Dihydroxyflavone, Protects against Myocardial Ischemia by Rebalancing Optic Atrophy 1 Processing. Free Radic. Biol. Med. 2019, 145, 187–197. [Google Scholar] [CrossRef]
- Kesh, S.; Kannan, R.R.; Balakrishnan, A. Naringenin Alleviates 6-Hydroxydopamine Induced Parkinsonism in SHSY5Y Cells and Zebrafish Model. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2021, 239, 108893. [Google Scholar] [CrossRef]
- de Oliveira, M.R.; Custódio de Souza, I.C.; Fürstenau, C.R. Promotion of Mitochondrial Protection by Naringenin in Methylglyoxal-Treated SH-SY5Y Cells: Involvement of the Nrf2/GSH Axis. Chem. Biol. Interact. 2019, 310, 108728. [Google Scholar] [CrossRef]
- de Oliveira, M.R.; Brasil, F.B.; Andrade, C.M.B. Naringenin Attenuates H2O2-Induced Mitochondrial Dysfunction by an Nrf2-Dependent Mechanism in SH-SY5Y Cells. Neurochem. Res. 2017, 42, 3341–3350. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Wang, H. Naringenin Inhibit the Hydrogen Peroxide-Induced SH-SY5Y Cells Injury Through Nrf2/HO-1 Pathway. Neurotox. Res. 2019, 36, 796–805. [Google Scholar] [CrossRef]
- Luo, M.; Zheng, L.-W.; Wang, Y.-S.; Huang, J.-C.; Yang, Z.-Q.; Yue, Z.-P.; Guo, B. Genistein Exhibits Therapeutic Potential for PCOS Mice via the ER-Nrf2-Foxo1-ROS Pathway. Food Funct. 2021, 12, 8800–8811. [Google Scholar] [CrossRef]
- Qian, Y.; Guan, T.; Huang, M.; Cao, L.; Li, Y.; Cheng, H.; Jin, H.; Yu, D. Neuroprotection by the Soy Isoflavone, Genistein, via Inhibition of Mitochondria-Dependent Apoptosis Pathways and Reactive Oxygen Induced-NF-ΚB Activation in a Cerebral Ischemia Mouse Model. Neurochem. Int. 2012, 60, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Farruggio, S.; Raina, G.; Cocomazzi, G.; Librasi, C.; Mary, D.; Gentilli, S.; Grossini, E. Genistein Improves Viability, Proliferation and Mitochondrial Function of Cardiomyoblasts Cultured in Physiologic and Peroxidative Conditions. Int. J. Mol. Med. 2019, 44, 2298–2310. [Google Scholar] [CrossRef]
- Hua, W.; Li, S.; Luo, R.; Wu, X.; Zhang, Y.; Liao, Z.; Song, Y.; Wang, K.; Zhao, K.; Yang, S.; et al. Icariin Protects Human Nucleus Pulposus Cells from Hydrogen Peroxide-Induced Mitochondria-Mediated Apoptosis by Activating Nuclear Factor Erythroid 2-Related Factor 2. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2020, 1866, 165575. [Google Scholar] [CrossRef]
- Zhang, T.; Mu, Y.; Yang, M.; Al Maruf, A.; Li, P.; Li, C.; Dai, S.; Lu, J.; Dong, Q. (+)-Catechin Prevents Methylglyoxal-Induced Mitochondrial Dysfunction and Apoptosis in EA.Hy926 Cells. Arch. Physiol. Biochem. 2017, 123, 121–127. [Google Scholar] [CrossRef]
- Rafiei, H.; Omidian, K.; Bandy, B. Dietary Polyphenols Protect Against Oleic Acid-Induced Steatosis in an in vitro Model of NAFLD by Modulating Lipid Metabolism and Improving Mitochondrial Function. Nutrients 2019, 11, 541. [Google Scholar] [CrossRef]
- Silva Santos, L.F.; Stolfo, A.; Calloni, C.; Salvador, M. Catechin and Epicatechin Reduce Mitochondrial Dysfunction and Oxidative Stress Induced by Amiodarone in Human Lung Fibroblasts. J. Arrhythmia 2017, 33, 220–225. [Google Scholar] [CrossRef]
- Ling, J.; Wu, Y.; Zou, X.; Chang, Y.; Li, G.; Fang, M. (−)-Epicatechin Reduces Neuroinflammation, Protects Mitochondria Function, and Prevents Cognitive Impairment in Sepsis-Associated Encephalopathy. Oxid. Med. Cell. Longev. 2022, 2022, 2657713. [Google Scholar] [CrossRef]
- Wu, Q.; Li, Z.; Lu, X.; Song, J.; Wang, H.; Liu, D.; Guo, D.; Bi, H. Epigallocatechin Gallate Protects the Human Lens Epithelial Cell Survival against UVB Irradiation through AIF/Endo G Signalling Pathways in vitro. Cutan. Ocul. Toxicol. 2021, 40, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Song, J.; Gao, Y.; Zou, Y.; Guo, J.; Zhang, X.; Liu, D.; Guo, D.; Bi, H. Epigallocatechin Gallate Enhances Human Lens Epithelial Cell Survival after UVB Irradiation via the Mitochondrial Signaling Pathway. Mol. Med. Rep. 2022, 25, 87. [Google Scholar] [CrossRef] [PubMed]
- Antunes, M.S.; Ladd, F.V.L.; Ladd, A.A.B.L.; Moreira, A.L.; Boeira, S.P.; Cattelan Souza, L. Hesperidin Protects against Behavioral Alterations and Loss of Dopaminergic Neurons in 6-OHDA-Lesioned Mice: The Role of Mitochondrial Dysfunction and Apoptosis. Metab. Brain Dis. 2021, 36, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Kamaraj, S.; Anandakumar, P.; Jagan, S.; Ramakrishnan, G.; Devaki, T. Hesperidin Attenuates Mitochondrial Dysfunction during Benzo(a)Pyrene-Induced Lung Carcinogenesis in Mice. Fundam. Clin. Pharmacol. 2011, 25, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.N.; Lim, J.H.; Kim, M.Y.; Ban, T.H.; Jang, I.-A.; Yoon, H.E.; Park, C.W.; Chang, Y.S.; Choi, B.S. Resveratrol, an Nrf2 Activator, Ameliorates Aging-Related Progressive Renal Injury. Aging 2018, 10, 83–99. [Google Scholar] [CrossRef]
- Chen, J.; Liu, Q.; Wang, Y.; Guo, Y.; Xu, X.; Huang, P.; Lian, B.; Zhang, R.; Chen, Y.; Ha, Y. Protective Effects of Resveratrol Liposomes on Mitochondria in Substantia Nigra Cells of Parkinsonized Rats. Ann. Palliat. Med. 2021, 10, 2458468. [Google Scholar] [CrossRef]
- Ma, J.; Wang, Z.; Zhao, J.; Miao, W.; Ye, T.; Chen, A. Resveratrol Attenuates Lipopolysaccharides (LPS)-Induced Inhibition of Osteoblast Differentiation in MC3T3-E1 Cells. Med. Sci. Monit. 2018, 24, 2045–2052. [Google Scholar] [CrossRef]
- Dewapriya, P.; Himaya, S.W.A.; Li, Y.-X.; Kim, S.-K. Tyrosol Exerts a Protective Effect against Dopaminergic Neuronal Cell Death in in vitro Model of Parkinson’s Disease. Food Chem. 2013, 141, 1147–1157. [Google Scholar] [CrossRef]
- Hsu, S.-S.; Lin, Y.-S.; Liang, W.-Z. Inhibition of the Pesticide Rotenone-Induced Ca2+ Signaling, Cytotoxicity and Oxidative Stress in HCN-2 Neuronal Cells by the Phenolic Compound Hydroxytyrosol. Pestic. Biochem. Physiol. 2021, 179, 104979. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, L.; Zhu, L.; Jia, X.; Li, X.; Jia, H.; Wang, Y.; Weber, P.; Long, J.; Liu, J. Hydroxytyrosol Protects Retinal Pigment Epithelial Cells from Acrolein-Induced Oxidative Stress and Mitochondrial Dysfunction. J. Neurochem. 2007, 103, 2690–2700. [Google Scholar]
- Naserzadeh, P.; Mehr, S.N.; Sadabadi, Z.; Seydi, E.; Salimi, A.; Pourahmad, J. Curcumin Protects Mitochondria and Cardiomyocytes from Oxidative Damage and Apoptosis Induced by Hemiscorpius Lepturus Venom. Drug Res. 2018, 68, 113–120. [Google Scholar] [CrossRef]
- Uğuz, A.C.; Öz, A.; Nazıroğlu, M. Curcumin Inhibits Apoptosis by Regulating Intracellular Calcium Release, Reactive Oxygen Species and Mitochondrial Depolarization Levels in SH-SY5Y Neuronal Cells. J. Recept. Signal Transduct. 2016, 36, 395–401. [Google Scholar] [CrossRef]
- Han, M.H.; Park, C.; Lee, D.-S.; Hong, S.-H.; Choi, I.-W.; Kim, G.-Y.; Choi, S.H.; Shim, J.-H.; Chae, J.-I.; Yoo, Y.H.; et al. Cytoprotective Effects of Esculetin against Oxidative Stress Are Associated with the Upregulation of Nrf2-Mediated NQO1 Expression via the Activation of the ERK Pathway. Int. J. Mol. Med. 2017, 39, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.; Jiang, B.; Bao, Y.M.; An, L.J. Protocatechuic Acid Suppresses MPP+-Induced Mitochondrial Dysfunction and Apoptotic Cell Death in PC12 Cells. Food Chem. Toxicol. 2006, 44, 1659–1666. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-M.; Jiang, B.; Bao, Y.-M.; An, L.-J. Protocatechuic Acid Inhibits Apoptosis by Mitochondrial Dysfunction in Rotenone-Induced PC12 Cells. Toxicol. In Vitro 2008, 22, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Ya, F.; Li, K.; Chen, H.; Tian, Z.; Fan, D.; Shi, Y.; Song, F.; Xu, X.; Ling, W.; Adili, R.; et al. Protocatechuic Acid Protects Platelets from Apoptosis via Inhibiting Oxidative Stress-Mediated PI3K/Akt/GSK3β Signaling. Thromb. Haemost. 2021, 121, 931–943. [Google Scholar] [CrossRef]
- Wang, X.-J.; Wang, Z.-B.; Xu, J.-X. Effect of Salvianic Acid A on Lipid Peroxidation and Membrane Permeability in Mitochondria. J. Ethnopharmacol. 2005, 97, 441–445. [Google Scholar] [CrossRef]
- Wang, X.-J.; Xu, J.-X. Salvianic Acid A Protects Human Neuroblastoma SH-SY5Y Cells against MPP+-Induced Cytotoxicity. Neurosci. Res. 2005, 51, 129–138. [Google Scholar] [CrossRef]
- Bai, Q.; Wang, Z.; Piao, Y.; Zhou, X.; Piao, Q.; Jiang, J.; Liu, H.; Piao, H.; Li, L.; Song, Y.; et al. Sesamin Alleviates Asthma Airway Inflammation by Regulating Mitophagy and Mitochondrial Apoptosis. J. Agric. Food Chem. 2022, 70, 4921–4933. [Google Scholar] [CrossRef]
- Ding, Y.; Kong, D.; Zhou, T.; Yang, N.; Xin, C.; Xu, J.; Wang, Q.; Zhang, H.; Wu, Q.; Lu, X.; et al. α-Arbutin Protects Against Parkinson’s Disease-Associated Mitochondrial Dysfunction in vitro and in vivo. NeuroMolecular Med. 2020, 22, 56–67. [Google Scholar] [CrossRef]
- Mohammad Khanlou, E.; Atashbar, S.; Kahrizi, F.; Shokouhi Sabet, N.; Salimi, A. Bevacizumab as a Monoclonal Antibody Inhibits Mitochondrial Complex II in Isolated Rat Heart Mitochondria: Ameliorative Effect of Ellagic Acid. Drug Chem. Toxicol. 2022, 45, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Magaña, M.Y.; Vega-García, C.C.; León-Contreras, J.C.; Hernández-Pando, R.; Zazueta, C.; García-Niño, W.R. Ellagic Acid Ameliorates Hexavalent Chromium-Induced Renal Toxicity by Attenuating Oxidative Stress, Suppressing TNF-α and Protecting Mitochondria. Toxicol. Appl. Pharmacol. 2022, 454, 116242. [Google Scholar] [CrossRef] [PubMed]
- Firdaus, F.; Zafeer, M.F.; Waseem, M.; Anis, E.; Hossain, M.M.; Afzal, M. Ellagic Acid Mitigates Arsenic-Trioxide-Induced Mitochondrial Dysfunction and Cytotoxicity in SH-SY5Y Cells. J. Biochem. Mol. Toxicol. 2018, 32, e22024. [Google Scholar] [CrossRef]
- Khodaei, F.; Rashedinia, M.; Heidari, R.; Rezaei, M.; Khoshnoud, M.J. Ellagic Acid Improves Muscle Dysfunction in Cuprizone-Induced Demyelinated Mice via Mitochondrial Sirt3 Regulation. Life Sci. 2019, 237, 116954. [Google Scholar] [CrossRef]
- Ebrahimi, R.; Sepand, M.R.; Seyednejad, S.A.; Omidi, A.; Akbariani, M.; Gholami, M.; Sabzevari, O. Ellagic Acid Reduces Methotrexate-Induced Apoptosis and Mitochondrial Dysfunction via up-Regulating Nrf2 Expression and Inhibiting the IĸBα/NFĸB in Rats. DARU J. Pharm. Sci. 2019, 27, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Kavitha, M.; Manivasagam, T.; Essa, M.M.; Tamilselvam, K.; Selvakumar, G.P.; Karthikeyan, S.; Thenmozhi, J.A.; Subash, S. Mangiferin Antagonizes Rotenone: Induced Apoptosis Through Attenuating Mitochondrial Dysfunction and Oxidative Stress in SK-N-SH Neuroblastoma Cells. Neurochem. Res. 2014, 39, 668–676. [Google Scholar] [CrossRef]
- Tang, Z.; Lai, C.-C.; Luo, J.; Ding, Y.-T.; Chen, Q.; Guan, Z.-Z. Mangiferin Prevents the Impairment of Mitochondrial Dynamics and an Increase in Oxidative Stress Caused by Excessive Fluoride in SH-SY5Y Cells. J. Biochem. Mol. Toxicol. 2021, 35, e22705. [Google Scholar] [CrossRef]
- Wang, X.-L.; Feng, S.-T.; Wang, Y.-T.; Zhang, N.-N.; Guo, Z.-Y.; Yan, X.; Yuan, Y.-H.; Wang, Z.-Z.; Chen, N.-H.; Zhang, Y. Mangiferin, a Natural Glucoxilxanthone, Inhibits Mitochondrial Dynamin-Related Protein 1 and Relieves Aberrant Mitophagic Proteins in Mice Model of Parkinson’s Disease. Phytomedicine 2022, 104, 154281. [Google Scholar] [CrossRef] [PubMed]
- Worakajit, N.; Thipboonchoo, N.; Chaturongakul, S.; Jutabha, P.; Soontornniyomkij, V.; Tuchinda, P.; Soodvilai, S. Nephroprotective Potential of Panduratin A against Colistin-Induced Renal Injury via Attenuating Mitochondrial Dysfunction and Cell Apoptosis. Biomed. Pharmacother. 2022, 148, 112732. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.-M.; Li, L.-D.; Duan, C.-L.; Li, Y.-J. Neuroprotective Effect of α-Mangostin on Mitochondrial Dysfunction and α-Synuclein Aggregation in Rotenone-Induced Model of Parkinson’s Disease in Differentiated SH-SY5Y Cells. J. Asian Nat. Prod. Res. 2017, 19, 833–845. [Google Scholar] [CrossRef]
- Cai, G.; Lin, F.; Wu, D.; Lin, C.; Chen, H.; Wei, Y.; Weng, H.; Chen, Z.; Wu, M.; Huang, E.; et al. Rosmarinic Acid Inhibits Mitochondrial Damage by Alleviating Unfolded Protein Response. Front. Pharmacol. 2022, 13, 859978. [Google Scholar] [PubMed]
- Diao, J.; Zhao, H.; You, P.; You, H.; Wu, H.; Shou, X.; Cheng, G. Rosmarinic Acid Ameliorated Cardiac Dysfunction and Mitochondrial Injury in Diabetic Cardiomyopathy Mice via Activation of the SIRT1/PGC-1α Pathway. Biochem. Biophys. Res. Commun. 2021, 546, 29–34. [Google Scholar] [CrossRef]
- Han, X.; Han, B.; Zhao, Y.; Li, G.; Wang, T.; He, J.; Du, W.; Cao, X.; Gan, J.; Wang, Z.; et al. Rosmarinic Acid Attenuates Rotenone-Induced Neurotoxicity in SH-SY5Y Parkinson’s Disease Cell Model through Abl Inhibition. Nutrients 2022, 14, 3508. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Han, Y.; Wang, Z.; Chen, T.; Qian, H.; He, J.; Li, J.; Han, B.; Wang, T. Rosmarinic Acid Protects against 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Dopaminergic Neurotoxicity in Zebrafish Embryos. Toxicol. In Vitro 2020, 65, 104823. [Google Scholar] [CrossRef]
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Rathore, A.S.; Dilnashin, H.; Singh, R.; Singh, S.P. Neuroprotective Effect of Chlorogenic Acid on Mitochondrial Dysfunction-Mediated Apoptotic Death of DA Neurons in a Parkinsonian Mouse Model. Oxid. Med. Cell. Longev. 2020, 2020, e6571484. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.-L.; Hung, C.-H.; Chan, S.-H.; Hsieh, P.-L.; Ou, H.-C.; Cheng, Y.-H.; Chu, P.-M. Chlorogenic Acid Protects Against OxLDL-Induced Oxidative Damage and Mitochondrial Dysfunction by Modulating SIRT1 in Endothelial Cells. Mol. Nutr. Food Res. 2018, 62, 1700928. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, L.; Ruan, Z.; Mi, S.; Jiang, M.; Li, X.; Wu, X.; Deng, Z.; Yin, Y. Chlorogenic Acid Ameliorates Intestinal Mitochondrial Injury by Increasing Antioxidant Effects and Activity of Respiratory Complexes. Biosci. Biotechnol. Biochem. 2016, 80, 962–971. [Google Scholar] [CrossRef]
- Li, Z.; Zhu, J.; Wan, Z.; Li, G.; Chen, L.; Guo, Y. Theaflavin Ameliorates Renal Ischemia/Reperfusion Injury by Activating the Nrf2 Signalling Pathway in vivo and in vitro. Biomed. Pharmacother. 2021, 134, 111097. [Google Scholar] [CrossRef]
- Sun, J.; Leng, P.; Li, X.; Guo, Q.; Zhao, J.; Liang, Y.; Zhang, X.; Yang, X.; Li, J. Salvianolic Acid A Promotes Mitochondrial Biogenesis and Mitochondrial Function in 3T3-L1 Adipocytes through Regulation of the AMPK-PGC1α Signalling Pathway. Adipocyte 2022, 11, 562–571. [Google Scholar] [CrossRef]
- Li, X.; Fan, J.; Liu, J.; Liang, L. Salvianolic Acid A Protects Neonatal Cardiomyocytes Against Hypoxia/Reoxygenation-Induced Injury by Preserving Mitochondrial Function and Activating Akt/GSK-3β Signals. Chin. J. Integr. Med. 2019, 25, 23–30. [Google Scholar] [CrossRef]
- Wang, D.; Lu, X.; Wang, E.; Shi, L.; Ma, C.; Tan, X. Salvianolic Acid B Attenuates Oxidative Stress-Induced Injuries in Enterocytes by Activating Akt/GSK3β Signaling and Preserving Mitochondrial Function. Eur. J. Pharmacol. 2021, 909, 174408. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, Y.; Zhang, J.; Yang, G. Salvianolic Acid B Protects against MPP+-Induced Neuronal Injury via Repressing Oxidative Stress and Restoring Mitochondrial Function. NeuroReport 2021, 32, 815–823. [Google Scholar] [CrossRef]
- Alvariño, R.; Alonso, E.; Tribalat, M.-A.; Gegunde, S.; Thomas, O.P.; Botana, L.M. Evaluation of the Protective Effects of Sarains on H2O2-Induced Mitochondrial Dysfunction and Oxidative Stress in SH-SY5Y Neuroblastoma Cells. Neurotox. Res. 2017, 32, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, P.; Vaibhav, K.; Tabassum, R.; Khan, A.; Ishrat, T.; Khan, M.M.; Ahmad, A.; Islam, F.; Safhi, M.M.; Islam, F. Anti-Apoptotic and Anti-Inflammatory Effect of Piperine on 6-OHDA Induced Parkinson’s Rat Model. J. Nutr. Biochem. 2013, 24, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Dutta, M.; Ghosh, A.K.; Mishra, P.; Jain, G.; Rangari, V.; Chattopadhyay, A.; Das, T.; Bhowmick, D.; Bandyopadhyay, D. Protective Effects of Piperine against Copper-Ascorbate Induced Toxic Injury to Goat Cardiac Mitochondria in vitro. Food Funct. 2014, 5, 2252–2267. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, P.; Ali, M.; Salman, M.; Tabassum, H.; Parvez, S. Harnessing the Mitochondrial Integrity for Neuroprotection: Therapeutic Role of Piperine against Experimental Ischemic Stroke. Neurochem. Int. 2021, 149, 105138. [Google Scholar] [CrossRef]
- Nakaso, K.; Ito, S.; Nakashima, K. Caffeine Activates the PI3K/Akt Pathway and Prevents Apoptotic Cell Death in a Parkinson’s Disease Model of SH-SY5Y Cells. Neurosci. Lett. 2008, 432, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Dragicevic, N.; Delic, V.; Cao, C.; Copes, N.; Lin, X.; Mamcarz, M.; Wang, L.; Arendash, G.W.; Bradshaw, P.C. Caffeine Increases Mitochondrial Function and Blocks Melatonin Signaling to Mitochondria in Alzheimer’s Mice and Cells. Neuropharmacology 2012, 63, 1368–1379. [Google Scholar] [CrossRef]
- Kolahdouzan, M.; Hamadeh, M.J. The Neuroprotective Effects of Caffeine in Neurodegenerative Diseases. CNS Neurosci. Ther. 2017, 23, 272–290. [Google Scholar] [CrossRef]
- Mishra, J.; Kumar, A. Improvement of Mitochondrial NAD+/FAD+-Linked State-3 Respiration by Caffeine Attenuates Quinolinic Acid Induced Motor Impairment in Rats: Implications in Huntington’s Disease. Pharmacol. Rep. 2014, 66, 1148–1155. [Google Scholar] [CrossRef]
- Abu Bakar, M.H.; Nor Shahril, N.S.; Mohamad Khalid, M.S.F.; Mohammad, S.; Shariff, K.A.; Karunakaran, T.; Mohd Salleh, R.; Mohamad Rosdi, M.N. Celastrol Alleviates High-Fat Diet-Induced Obesity via Enhanced Muscle Glucose Utilization and Mitochondrial Oxidative Metabolism-Mediated Upregulation of Pyruvate Dehydrogenase Complex. Toxicol. Appl. Pharmacol. 2022, 449, 116099. [Google Scholar] [CrossRef] [PubMed]
- Abu Bakar, M.H.; Shariff, K.A.; Tan, J.S.; Lee, L.K. Celastrol Attenuates Inflammatory Responses in Adipose Tissues and Improves Skeletal Muscle Mitochondrial Functions in High Fat Diet-Induced Obese Rats via Upregulation of AMPK/SIRT1 Signaling Pathways. Eur. J. Pharmacol. 2020, 883, 173371. [Google Scholar] [CrossRef]
- Barakat, B.M.; Ahmed, H.I.; Bahr, H.I.; Elbahaie, A.M. Protective Effect of Boswellic Acids against Doxorubicin-Induced Hepatotoxicity: Impact on Nrf2/HO-1 Defense Pathway. Oxid. Med. Cell. Longev. 2018, 2018, e8296451. [Google Scholar] [CrossRef]
- Nataraj, J.; Manivasagam, T.; Justin Thenmozhi, A.; Essa, M.M. Neuroprotective Effect of Asiatic Acid on Rotenone-Induced Mitochondrial Dysfunction and Oxidative Stress-Mediated Apoptosis in Differentiated SH-SYS5Y Cells. Nutr. Neurosci. 2017, 20, 351–359. [Google Scholar] [CrossRef]
- Cong, C.; Kluwe, L.; Li, S.; Liu, X.; Liu, Y.; Liu, H.; Gui, W.; Liu, T.; Xu, L. Paeoniflorin Inhibits Tributyltin Chloride-Induced Apoptosis in Hypothalamic Neurons via Inhibition of MKK4-JNK Signaling Pathway. J. Ethnopharmacol. 2019, 237, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Liu, J.; Yu, M.; Xia, M.; Zhang, Y.; Sun, X.; Xu, Y.; Cui, X. Paeoniflorin Ameliorates BiPN by Reducing IL6 Levels and Regulating PARKIN-Mediated Mitochondrial Autophagy. Drug Des. Devel. Ther. 2022, 16, 2241–2259. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhu, L.; Zhu, X.; Zhang, K.; Huang, B.; Zhang, J.; Zhang, Y.; Zhu, L.; Zhou, B.; Zhou, F. Protective Effect of Paeoniflorin on Aβ25–35-Induced SH-SY5Y Cell Injury by Preventing Mitochondrial Dysfunction. Cell. Mol. Neurobiol. 2014, 34, 227–234. [Google Scholar] [CrossRef]
- Balakrishnan, R.; Elangovan, N.; Mohankumar, T.; Nataraj, J.; Manivasagam, T.; Thenmozhi, A.J.; Essa, M.M.; Akbar, M.; Khan, M.A.S. Isolongifolene Attenuates Rotenone-Induced Mitochondrial Dysfunction, Oxidative Stress and Apoptosis. Front. Biosci.-Sch. 2018, 10, 248–261. [Google Scholar] [CrossRef]
- Balakrishnan, R.; Vijayraja, D.; Mohankumar, T.; Manimaran, D.; Ganesan, P.; Choi, D.-K.; Elangovan, N. Isolongifolene Mitigates Rotenone-Induced Dopamine Depletion and Motor Deficits through Anti-Oxidative and Anti-Apoptotic Effects in a Rat Model of Parkinson’s Disease. J. Chem. Neuroanat. 2021, 112, 101890. [Google Scholar] [CrossRef]
- Qu, M.; Ni, Y.; Guo, B.; Feng, X.; Jiang, Z. Lycopene Antagonizes Lead Toxicity by Reducing Mitochondrial Oxidative Damage and Mitochondria-mediated Apoptosis in Cultured Hippocampal Neurons. MedComm 2020, 1, 228–239. [Google Scholar] [CrossRef]
- Jang, Y.; Choo, H.; Lee, M.J.; Han, J.; Kim, S.J.; Ju, X.; Cui, J.; Lee, Y.L.; Ryu, M.J.; Oh, E.S.; et al. Auraptene Mitigates Parkinson’s Disease-Like Behavior by Protecting Inhibition of Mitochondrial Respiration and Scavenging Reactive Oxygen Species. Int. J. Mol. Sci. 2019, 20, 3409. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Jang, Y.; Zhu, J.; Namgung, E.; Go, D.; Seo, C.; Ju, X.; Cui, J.; Lee, Y.L.; Kang, H.; et al. Auraptene Enhances Junction Assembly in Cerebrovascular Endothelial Cells by Promoting Resilience to Mitochondrial Stress through Activation of Antioxidant Enzymes and MtUPR. Antioxidants 2021, 10, 475. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Mo, W.; Feng, J.; Li, J.; Yu, Q.; Li, S.; Zhang, J.; Chen, K.; Ji, J.; Dai, W.; et al. Astaxanthin Attenuates Hepatic Damage and Mitochondrial Dysfunction in Non-alcoholic Fatty Liver Disease by Up-regulating the FGF21/PGC-1α Pathway. Br. J. Pharmacol. 2020, 177, 3760–3777. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Yang, P.; Gao, T.; Liu, M.; Li, X. Allicin Attenuates Myocardial Apoptosis, Inflammation and Mitochondrial Injury during Hypoxia-Reoxygenation: An in vitro Study. BMC Cardiovasc. Disord. 2021, 21, 200. [Google Scholar] [CrossRef]
- Lv, R.; Du, L.; Lu, C.; Wu, J.; Ding, M.; Wang, C.; Mao, N.; Shi, Z. Allicin Protects against H2O2-Induced Apoptosis of PC12 Cells via the Mitochondrial Pathway. Exp. Ther. Med. 2017, 14, 2053–2059. [Google Scholar] [CrossRef]
- Han, Y.-S.; Lee, J.H.; Lee, S.H. Fucoidan Suppresses Mitochondrial Dysfunction and Cell Death against 1-Methyl-4-Phenylpyridinum-Induced Neuronal Cytotoxicity via Regulation of PGC-1α Expression. Mar. Drugs 2019, 17, 518. [Google Scholar] [CrossRef]
- Díaz-Resendiz, K.J.G.; Covantes-Rosales, C.E.; Benítez-Trinidad, A.B.; Navidad-Murrieta, M.S.; Razura-Carmona, F.F.; Carrillo-Cruz, C.D.; Frias-Delgadillo, E.J.; Pérez-Díaz, D.A.; Díaz-Benavides, M.V.; Zambrano-Soria, M.; et al. Effect of Fucoidan on the Mitochondrial Membrane Potential (ΔΨm) of Leukocytes from Patients with Active COVID-19 and Subjects That Recovered from SARS-CoV-2 Infection. Mar. Drugs 2022, 20, 99. [Google Scholar] [CrossRef]
- Lei, P.; Tian, S.; Teng, C.; Huang, L.; Liu, X.; Wang, J.; Zhang, Y.; Li, B.; Shan, Y. Sulforaphane Improves Lipid Metabolism by Enhancing Mitochondrial Function and Biogenesis in vivo and in vitro. Mol. Nutr. Food Res. 2019, 63, 1800795. [Google Scholar] [CrossRef]
- Tian, S.; Lei, P.; Zhang, J.; Sun, Y.; Li, B.; Shan, Y. Sulforaphane Balances Ca2+ Homeostasis Injured by Excessive Fat via Mitochondria-Associated Membrane (MAM). Mol. Nutr. Food Res. 2021, 65, 2001076. [Google Scholar] [CrossRef]
- Jeong, M.H.; Kim, J.H.; Seo, K.; Kwak, T.H.; Park, W.J. β-Lapachone Attenuates Mitochondrial Dysfunction in MELAS Cybrid Cells. Biochem. Biophys. Res. Commun. 2014, 454, 417–422. [Google Scholar] [CrossRef]
- Niu, Y.-J.; Zhou, W.; Nie, Z.-W.; Shin, K.-T.; Cui, X.-S. Melatonin Enhances Mitochondrial Biogenesis and Protects against Rotenone-Induced Mitochondrial Deficiency in Early Porcine Embryos. J. Pineal Res. 2020, 68, e12627. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Tian, L.; Liu, J.; Wu, Q.; Wang, N.; Wang, G.; Wang, Y.; Seto, S. Ligustilide Ameliorates Hippocampal Neuronal Injury after Cerebral Ischemia Reperfusion through Activating PINK1/Parkin-Dependent Mitophagy. Phytomedicine 2022, 101, 154111. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yao, X.; Liu, Z.; Zhang, H.; Li, W.; Li, Z.; Wang, G.-L.; Pang, J.; Lin, Y.; Xu, Z.; et al. Protective Effects of Xyloketal B against MPP+-Induced Neurotoxicity in Caenorhabditis Elegans and PC12 Cells. Brain Res. 2010, 1332, 110–119. [Google Scholar] [CrossRef]
- Zhao, J.; Li, L.; Ling, C.; Li, J.; Pang, J.-Y.; Lin, Y.-C.; Liu, J.; Huang, R.; Wang, G.-L.; Pei, Z.; et al. Marine Compound Xyloketal B Protects PC12 Cells against OGD-Induced Cell Damage. Brain Res. 2009, 1302, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Shokoohinia, Y.; Hosseinzadeh, L.; Moieni-Arya, M.; Mostafaie, A.; Mohammadi-Motlagh, H.-R. Osthole Attenuates Doxorubicin-Induced Apoptosis in PC12 Cells through Inhibition of Mitochondrial Dysfunction and ROS Production. BioMed Res. Int. 2014, 2014, 156848. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, L.; Feng, F.; Yuan, H.; Gao, D.; Fu, L.; Fei, Z. Osthole Attenuates Spinal Cord Ischemia–Reperfusion Injury through Mitochondrial Biogenesis–Independent Inhibition of Mitochondrial Dysfunction in Rats. J. Surg. Res. 2013, 185, 805–814. [Google Scholar] [CrossRef]
- Anupama, N.; Preetha Rani, M.R.; Shyni, G.L.; Raghu, K.G. Glucotoxicity Results in Apoptosis in H9c2 Cells via Alteration in Redox Homeostasis Linked Mitochondrial Dynamics and Polyol Pathway and Possible Reversal with Cinnamic Acid. Toxicol. In Vitro 2018, 53, 178–192. [Google Scholar] [CrossRef]
- Zhang, B.; Zhao, J.; Wang, Z.; Xu, L.; Liu, A.; Du, G. DL0410 Attenuates Oxidative Stress and Neuroinflammation via BDNF/TrkB/ERK/CREB and Nrf2/HO-1 Activation. Int. Immunopharmacol. 2020, 86, 106729. [Google Scholar] [CrossRef]
- Kang, L.; Liu, S.; Li, J.; Tian, Y.; Xue, Y.; Liu, X. The Mitochondria-Targeted Anti-Oxidant MitoQ Protects against Intervertebral Disc Degeneration by Ameliorating Mitochondrial Dysfunction and Redox Imbalance. Cell Prolif. 2020, 53, e12779. [Google Scholar] [CrossRef]
- Fang, F.; Liu, G. Protective Effects of Compound FLZ, a Novel Synthetic Analogue of Squamosamide, on β-Amyloid-Induced Rat Brain Mitochondrial Dysfunction in vitro. Acta Pharmacol. Sin. 2009, 30, 522–529. [Google Scholar] [CrossRef][Green Version]
- Yang, H.; Wang, L.; Zang, C.; Yang, X.; Bao, X.; Shang, J.; Zhang, Z.; Liu, H.; Ju, C.; Li, F.; et al. Squamosamide Derivative FLZ Diminishes Aberrant Mitochondrial Fission by Inhibiting Dynamin-Related Protein 1. Front. Pharmacol. 2021, 12, 588003. [Google Scholar] [PubMed]
- Jang, W.B.; Park, J.H.; Ji, S.T.; Lee, N.K.; Kim, D.Y.; Kim, Y.J.; Jung, S.Y.; Kang, S.; Lamichane, S.; Lamichane, B.D.; et al. Cytoprotective Roles of a Novel Compound, MHY-1684, against Hyperglycemia-Induced Oxidative Stress and Mitochondrial Dysfunction in Human Cardiac Progenitor Cells. Oxid. Med. Cell. Longev. 2018, 2018, e4528184. [Google Scholar] [CrossRef]
- de Oliveira, J.; Moreira, E.L.G.; Mancini, G.; Hort, M.A.; Latini, A.; Ribeiro-do-Valle, R.M.; Farina, M.; da Rocha, J.B.T.; de Bem, A.F. Diphenyl Diselenide Prevents Cortico-Cerebral Mitochondrial Dysfunction and Oxidative Stress Induced by Hypercholesterolemia in LDL Receptor Knockout Mice. Neurochem. Res. 2013, 38, 2028–2036. [Google Scholar] [CrossRef] [PubMed]
- Dobrachinski, F.; da Silva, M.H.; Tassi, C.L.C.; de Carvalho, N.R.; Dias, G.R.M.; Golombieski, R.M.; da Silva Loreto, É.L.; da Rocha, J.B.T.; Fighera, M.R.; Soares, F.A.A. Neuroprotective Effect of Diphenyl Diselenide in a Experimental Stroke Model: Maintenance of Redox System in Mitochondria of Brain Regions. Neurotox. Res. 2014, 26, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Quispe, R.L.; Jaramillo, M.L.; Galant, L.S.; Engel, D.; Dafre, A.L.; Teixeira da Rocha, J.B.; Radi, R.; Farina, M.; de Bem, A.F. Diphenyl Diselenide Protects Neuronal Cells against Oxidative Stress and Mitochondrial Dysfunction: Involvement of the Glutathione-Dependent Antioxidant System. Redox Biol. 2019, 20, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Straliotto, M.R.; Hort, M.A.; Fiuza, B.; Rocha, J.B.T.; Farina, M.; Chiabrando, G.; de Bem, A.F. Diphenyl Diselenide Modulates OxLDL-Induced Cytotoxicity in Macrophage by Improving the Redox Signaling. Biochimie 2013, 95, 1544–1551. [Google Scholar] [CrossRef]
- Akao, Y.; Maruyama, W.; Shimizu, S.; Yi, H.; Nakagawa, Y.; Shamoto-Nagai, M.; Youdim, M.B.H.; Tsujimoto, Y.; Naoi, M. Mitochondrial Permeability Transition Mediates Apoptosis Induced by N-Methyl(R)Salsolinol, an Endogenous Neurotoxin, and Is Inhibited by Bcl-2 and Rasagiline, N-Propargyl-1(R)-Aminoindan. J. Neurochem. 2002, 82, 913–923. [Google Scholar] [CrossRef]
- Naoi, M.; Maruyama, W.; Yi, H. Rasagiline Prevents Apoptosis Induced by PK11195, a Ligand of the Outer Membrane Translocator Protein (18 KDa), in SH-SY5Y Cells through Suppression of Cytochrome c Release from Mitochondria. J. Neural Transm. 2013, 120, 1539–1551. [Google Scholar] [CrossRef]
- Youdim, M.B.H.; Bar Am, O.; Yogev-Falach, M.; Weinreb, O.; Maruyama, W.; Naoi, M.; Amit, T. Rasagiline: Neurodegeneration, Neuroprotection, and Mitochondrial Permeability Transition. J. Neurosci. Res. 2005, 79, 172–179. [Google Scholar] [CrossRef]
- Chau, K.Y.; Cooper, J.M.; Schapira, A.H.V. Rasagiline Protects against Alpha-Synuclein Induced Sensitivity to Oxidative Stress in Dopaminergic Cells. Neurochem. Int. 2010, 57, 525–529. [Google Scholar] [CrossRef][Green Version]
- Colle, D.; Santos, D.B.; Hartwig, J.M.; Godoi, M.; Engel, D.F.; de Bem, A.F.; Braga, A.L.; Farina, M. Succinobucol, a Lipid-Lowering Drug, Protects Against 3-Nitropropionic Acid-Induced Mitochondrial Dysfunction and Oxidative Stress in SH-SY5Y Cells via Upregulation of Glutathione Levels and Glutamate Cysteine Ligase Activity. Mol. Neurobiol. 2016, 53, 1280–1295. [Google Scholar] [CrossRef]
- Rehfeldt, S.C.H.; Laufer, S.; Goettert, M.I. A Highly Selective in vitro JNK3 Inhibitor, FMU200, Restores Mitochondrial Membrane Potential and Reduces Oxidative Stress and Apoptosis in SH-SY5Y Cells. Int. J. Mol. Sci. 2021, 22, 3701. [Google Scholar] [CrossRef]
- Jayaraj, R.L.; Tamilselvam, K.; Manivasagam, T.; Elangovan, N. Neuroprotective Effect of CNB-001, a Novel Pyrazole Derivative of Curcumin on Biochemical and Apoptotic Markers Against Rotenone-Induced SK-N-SH Cellular Model of Parkinson’s Disease. J. Mol. Neurosci. 2013, 51, 863–870. [Google Scholar] [CrossRef]
- Jayaraj, R.L.; Elangovan, N.; Manigandan, K.; Singh, S.; Shukla, S. CNB-001 a Novel Curcumin Derivative, Guards Dopamine Neurons in MPTP Model of Parkinson’s Disease. BioMed Res. Int. 2014, 2014, e236182. [Google Scholar] [CrossRef]
- Fernandez-Panchon, M.S.; Villano, D.; Troncoso, A.M.; Garcia-Parrilla, M.C. Antioxidant Activity of Phenolic Compounds: From in vitro Results to in vivo Evidence. Crit. Rev. Food Sci. Nutr. 2008, 48, 649–671. [Google Scholar] [CrossRef]
- Rahman, M.M.; Rahaman, M.S.; Islam, M.R.; Rahman, F.; Mithi, F.M.; Alqahtani, T.; Almikhlafi, M.A.; Alghamdi, S.Q.; Alruwaili, A.S.; Hossain, M.S.; et al. Role of Phenolic Compounds in Human Disease: Current Knowledge and Future Prospects. Molecules 2021, 27, 233. [Google Scholar] [CrossRef]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The SH-SY5Y Cell Line in Parkinson’s Disease Research: A Systematic Review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef]
- Xie, H.; Hu, L.; Li, G. SH-SY5Y Human Neuroblastoma Cell Line: In vitro Cell Model of Dopaminergic Neurons in Parkinson’s Disease. Chin. Med. J. 2010, 123, 1086–1092. [Google Scholar] [CrossRef]
- Lopes, F.M.; Schröder, R.; da Frota, M.L.C., Jr.; Zanotto-Filho, A.; Müller, C.B.; Pires, A.S.; Meurer, R.T.; Colpo, G.D.; Gelain, D.P.; Kapczinski, F.; et al. Comparison between Proliferative and Neuron-like SH-SY5Y Cells as an in vitro Model for Parkinson Disease Studies. Brain Res. 2010, 1337, 85–94. [Google Scholar] [CrossRef]
- Yarmohammadi, F.; Wallace Hayes, A.; Najafi, N.; Karimi, G. The Protective Effect of Natural Compounds against Rotenone-Induced Neurotoxicity. J. Biochem. Mol. Toxicol. 2020, 34, e22605. [Google Scholar] [CrossRef]
- Copeland, W.C.; Longley, M.J. Mitochondrial Genome Maintenance in Health and Disease. DNA Repair 2014, 19, 190–198. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Bagheri, H.; Ghasemi, F.; Barreto, G.E.; Rafiee, R.; Sathyapalan, T.; Sahebkar, A. Effects of Curcumin on Mitochondria in Neurodegenerative Diseases. BioFactors 2020, 46, 5–20. [Google Scholar] [CrossRef]
- Holmström, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 Impacts Cellular Bioenergetics by Controlling Substrate Availability for Mitochondrial Respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef]
- Itoh, K.; Ye, P.; Matsumiya, T.; Tanji, K.; Ozaki, T. Emerging Functional Cross-Talk between the Keap1-Nrf2 System and Mitochondria. J. Clin. Biochem. Nutr. 2015, 56, 91–97. [Google Scholar] [CrossRef]
- Brand, M.D.; Nicholls, D.G. Assessing Mitochondrial Dysfunction in Cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef]
- Katwal, G.; Baral, D.; Fan, X.; Weiyang, H.; Zhang, X.; Ling, L.; Xiong, Y.; Ye, Q.; Wang, Y. SIRT3 a Major Player in Attenuation of Hepatic Ischemia-Reperfusion Injury by Reducing ROS via Its Downstream Mediators: SOD2, CYP-D, and HIF-1α. Oxid. Med. Cell. Longev. 2018, 2018, e2976957. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Corona, J.C.; Duchen, M.R. Impaired Mitochondrial Homeostasis and Neurodegeneration: Towards New Therapeutic Targets? J. Bioenerg. Biomembr. 2015, 47, 89–99. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Izzo, V.; Bravo-San Pedro, J.M.; Sica, V.; Kroemer, G.; Galluzzi, L. Mitochondrial Permeability Transition: New Findings and Persisting Uncertainties. Trends Cell Biol. 2016, 26, 655–667. [Google Scholar] [CrossRef]
- Friberg, H.; Ferrand-Drake, M.; Bengtsson, F.; Halestrap, A.P.; Wieloch, T. Cyclosporin A, but Not FK 506, Protects Mitochondria and Neurons against Hypoglycemic Damage and Implicates the Mitochondrial Permeability Transition in Cell Death. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 5151–5159. [Google Scholar] [CrossRef]
- Seaton, T.A.; Cooper, J.M.; Schapira, A.H.V. Cyclosporin Inhibition of Apoptosis Induced by Mitochondrial Complex I Toxins. Brain Res. 1998, 809, 12–17. [Google Scholar] [CrossRef]
- Abate, M.; Festa, A.; Falco, M.; Lombardi, A.; Luce, A.; Grimaldi, A.; Zappavigna, S.; Sperlongano, P.; Irace, C.; Caraglia, M.; et al. Mitochondria as Playmakers of Apoptosis, Autophagy and Senescence. Semin. Cell Dev. Biol. 2020, 98, 139–153. [Google Scholar] [CrossRef]
- Chamcheu, J.C.; Roy, T.; Uddin, M.B.; Banang-Mbeumi, S.; Chamcheu, R.-C.N.; Walker, A.L.; Liu, Y.-Y.; Huang, S. Role and Therapeutic Targeting of the PI3K/Akt/MTOR Signaling Pathway in Skin Cancer: A Review of Current Status and Future Trends on Natural and Synthetic Agents Therapy. Cells 2019, 8, 803. [Google Scholar] [CrossRef]
- Kitagishi, Y.; Nakanishi, A.; Ogura, Y.; Matsuda, S. Dietary Regulation of PI3K/AKT/GSK-3β Pathway in Alzheimer’s Disease. Alzheimers Res. Ther. 2014, 6, 35. [Google Scholar] [CrossRef]
- Biasutto, L.; Szabo’, I.; Zoratti, M. Mitochondrial Effects of Plant-Made Compounds. Antioxid. Redox Signal. 2011, 15, 3039–3059. [Google Scholar] [CrossRef]
- Yoon, J.C.; Ng, A.; Kim, B.H.; Bianco, A.; Xavier, R.J.; Elledge, S.J. Wnt Signaling Regulates Mitochondrial Physiology and Insulin Sensitivity. Genes Dev. 2010, 24, 1507–1518. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Metabolic Control of Mitochondrial Biogenesis through the PGC-1 Family Regulatory Network. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2011, 1813, 1269–1278. [Google Scholar] [CrossRef]
- Viña, J.; Gomez-Cabrera, M.C.; Borras, C.; Froio, T.; Sanchis-Gomar, F.; Martinez-Bello, V.E.; Pallardo, F.V. Mitochondrial Biogenesis in Exercise and in Ageing. Adv. Drug Deliv. Rev. 2009, 61, 1369–1374. [Google Scholar] [CrossRef]
- Chen, M.; Cui, Y.; Li, H.; Luan, J.; Zhou, X.; Han, J. Icariin Promotes the Osteogenic Action of BMP2 by Activating the CAMP Signaling Pathway. Molecules 2019, 24, 3875. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-E.; Chen, J.; Lou, Z. DBC1 Is a Negative Regulator of SIRT1. Nature 2008, 451, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Pelosse, M.; Cottet-Rousselle, C.; Bidan, C.M.; Dupont, A.; Gupta, K.; Berger, I.; Schlattner, U. Synthetic Energy Sensor AMPfret Deciphers Adenylate-Dependent AMPK Activation Mechanism. Nat. Commun. 2019, 10, 1038. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.-M.; Jung, Y.-K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Liu, Q.; Gao, W.; Sehgal, S.A.; Wu, H. The Multifaceted Regulation of Mitophagy by Endogenous Metabolites. Autophagy 2022, 18, 1216–1239. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Eaten Alive: A History of Macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin Mitochondrial Quality Control: A Source of Regional Vulnerability in Parkinson’s Disease. Mol. Neurodegener. 2020, 15, 20. [Google Scholar] [CrossRef]
- Salazar, C.; Ruiz-Hincapie, P.; Ruiz, L.M. The Interplay among PINK1/PARKIN/Dj-1 Network during Mitochondrial Quality Control in Cancer Biology: Protein Interaction Analysis. Cells 2018, 7, 154. [Google Scholar] [CrossRef]
- Merkwirth, C.; Langer, T. Prohibitin Function within Mitochondria: Essential Roles for Cell Proliferation and Cristae Morphogenesis. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2009, 1793, 27–32. [Google Scholar] [CrossRef]
- Signorile, A.; Sgaramella, G.; Bellomo, F.; De Rasmo, D. Prohibitins: A Critical Role in Mitochondrial Functions and Implication in Diseases. Cells 2019, 8, 71. [Google Scholar] [CrossRef]
- Thuaud, F.; Ribeiro, N.; Nebigil, C.G.; Désaubry, L. Prohibitin Ligands in Cell Death and Survival: Mode of Action and Therapeutic Potential. Chem. Biol. 2013, 20, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.-T.; Chen, P.; Ouyang, R.-Y.; Song, L. Multifaceted Role of Prohibitin in Cell Survival and Apoptosis. Apoptosis 2015, 20, 1135–1149. [Google Scholar] [CrossRef]
- Núñez-Vázquez, S.; Sánchez-Vera, I.; Saura-Esteller, J.; Cosialls, A.M.; Noisier, A.F.M.; Albericio, F.; Lavilla, R.; Pons, G.; Iglesias-Serret, D.; Gil, J. NOXA Upregulation by the Prohibitin-Binding Compound Fluorizoline Is Transcriptionally Regulated by Integrated Stress Response-Induced ATF3 and ATF4. FEBS J. 2021, 288, 1271–1285. [Google Scholar] [CrossRef]
- Sato, S.; Murata, A.; Orihara, T.; Shirakawa, T.; Suenaga, K.; Kigoshi, H.; Uesugi, M. Marine Natural Product Aurilide Activates the OPA1-Mediated Apoptosis by Binding to Prohibitin. Chem. Biol. 2011, 18, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Stocchi, F.; Fossati, C.; Torti, M. Rasagiline for the Treatment of Parkinson’s Disease: An Update. Expert Opin. Pharmacother. 2015, 16, 2231–2241. [Google Scholar] [CrossRef] [PubMed]
- Nayak, L.; Henchcliffe, C. Rasagiline in Treatment of Parkinson’s Disease. Neuropsychiatr. Dis. Treat. 2008, 4, 23–32. [Google Scholar]
- Naoi, M.; Maruyama, W.; Shamoto-Nagai, M. Rasagiline and Selegiline Modulate Mitochondrial Homeostasis, Intervene Apoptosis System and Mitigate α-Synuclein Cytotoxicity in Disease-Modifying Therapy for Parkinson’s Disease. J. Neural Transm. 2020, 127, 131–147. [Google Scholar] [CrossRef]
- Matthews, D.C.; Ritter, A.; Thomas, R.G.; Andrews, R.D.; Lukic, A.S.; Revta, C.; Kinney, J.W.; Tousi, B.; Leverenz, J.B.; Fillit, H.; et al. Rasagiline Effects on Glucose Metabolism, Cognition, and Tau in Alzheimer’s Dementia. Alzheimers Dement. Transl. Res. Clin. Interv. 2021, 7, e12106. [Google Scholar] [CrossRef]
- Mayer, G.; Happe, S.; Evers, S.; Hermann, W.; Jansen, S.; Kallweit, U.; Muntean, M.-L.; Pöhlau, D.; Riemann, D.; Saletu, M.; et al. Insomnia in Neurological Diseases. Neurol. Res. Pract. 2021, 3, 15. [Google Scholar] [CrossRef]
- Quera-Salva, M.-A.; Claustrat, B. Mélatonine: Aspects physiologiques et pharmacologiques en relation avec le sommeil, intérêt d’une forme galénique à libération prolongée (Circadin®) dans l’insomnie. L’Encéphale 2018, 44, 548–557. [Google Scholar] [CrossRef]
- Wade, A.G.; Farmer, M.; Harari, G.; Fund, N.; Laudon, M.; Nir, T.; Frydman-Marom, A.; Zisapel, N. Add-on Prolonged-Release Melatonin for Cognitive Function and Sleep in Mild to Moderate Alzheimer’s Disease: A 6-Month, Randomized, Placebo-Controlled, Multicenter Trial. Clin. Interv. Aging 2014, 9, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The Re-Emergence of Natural Products for Drug Discovery in the Genomics Era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Howes, M.-J.R.; Quave, C.L.; Collemare, J.; Tatsis, E.C.; Twilley, D.; Lulekal, E.; Farlow, A.; Li, L.; Cazar, M.-E.; Leaman, D.J.; et al. Molecules from Nature: Reconciling Biodiversity Conservation and Global Healthcare Imperatives for Sustainable Use of Medicinal Plants and Fungi. Plants People Planet 2020, 2, 463–481. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural Products in Drug Discovery: Advances and Opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Rasouli, H.; Farzaei, M.H.; Khodarahmi, R. Polyphenols and Their Benefits: A Review. Int. J. Food Prop. 2017, 20, 1700–1741. [Google Scholar] [CrossRef]
- Di Lorenzo, C.; Colombo, F.; Biella, S.; Stockley, C.; Restani, P. Polyphenols and Human Health: The Role of Bioavailability. Nutrients 2021, 13, 273. [Google Scholar] [CrossRef]
- Muller, A.G.; Sarker, S.D.; Saleem, I.Y.; Hutcheon, G.A. Delivery of Natural Phenolic Compounds for the Potential Treatment of Lung Cancer. DARU J. Pharm. Sci. 2019, 27, 433–449. [Google Scholar] [CrossRef]
- Chu, K.O.; Pang, C.C.P. Pharmacokinetics and Disposition of Green Tea Catechins. In Pharmacokinetics and Adverse Effects of Drugs; Malangu, N., Ed.; IntechOpen: Rijeka, Croatia, 2018; ISBN 978-1-78923-139-7. [Google Scholar]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of Curcumin: Problems and Promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef]
- Yang, K.-Y.; Lin, L.-C.; Tseng, T.-Y.; Wang, S.-C.; Tsai, T.-H. Oral Bioavailability of Curcumin in Rat and the Herbal Analysis from Curcuma Longa by LC–MS/MS. J. Chromatogr. B 2007, 853, 183–189. [Google Scholar] [CrossRef]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E.; Walle, U.K. High Absorption but Very Low Bioavailability of Oral Resveratrol in Humans. Drug Metab. Dispos. 2004, 32, 1377–1382. [Google Scholar] [CrossRef]
- Berman, A.Y.; Motechin, R.A.; Wiesenfeld, M.Y.; Holz, M.K. The Therapeutic Potential of Resveratrol: A Review of Clinical Trials. Npj Precis. Oncol. 2017, 1, 1–9. [Google Scholar] [CrossRef]
- Herbers, E.; Kekäläinen, N.J.; Hangas, A.; Pohjoismäki, J.L.; Goffart, S. Tissue Specific Differences in Mitochondrial DNA Maintenance and Expression. Mitochondrion 2019, 44, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Margreiter, R. Heterogeneity of Mitochondria and Mitochondrial Function within Cells as Another Level of Mitochondrial Complexity. Int. J. Mol. Sci. 2009, 10, 1911–1929. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Hermann, M.; Saks, V.; Hengster, P.; Margreiter, R. The Cell-Type Specificity of Mitochondrial Dynamics. Int. J. Biochem. Cell Biol. 2009, 41, 1928–1939. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).