
An Update on the Interplay between LRRK2, Rab GTPases and Parkinson’s Disease


Abstract
1. Introduction
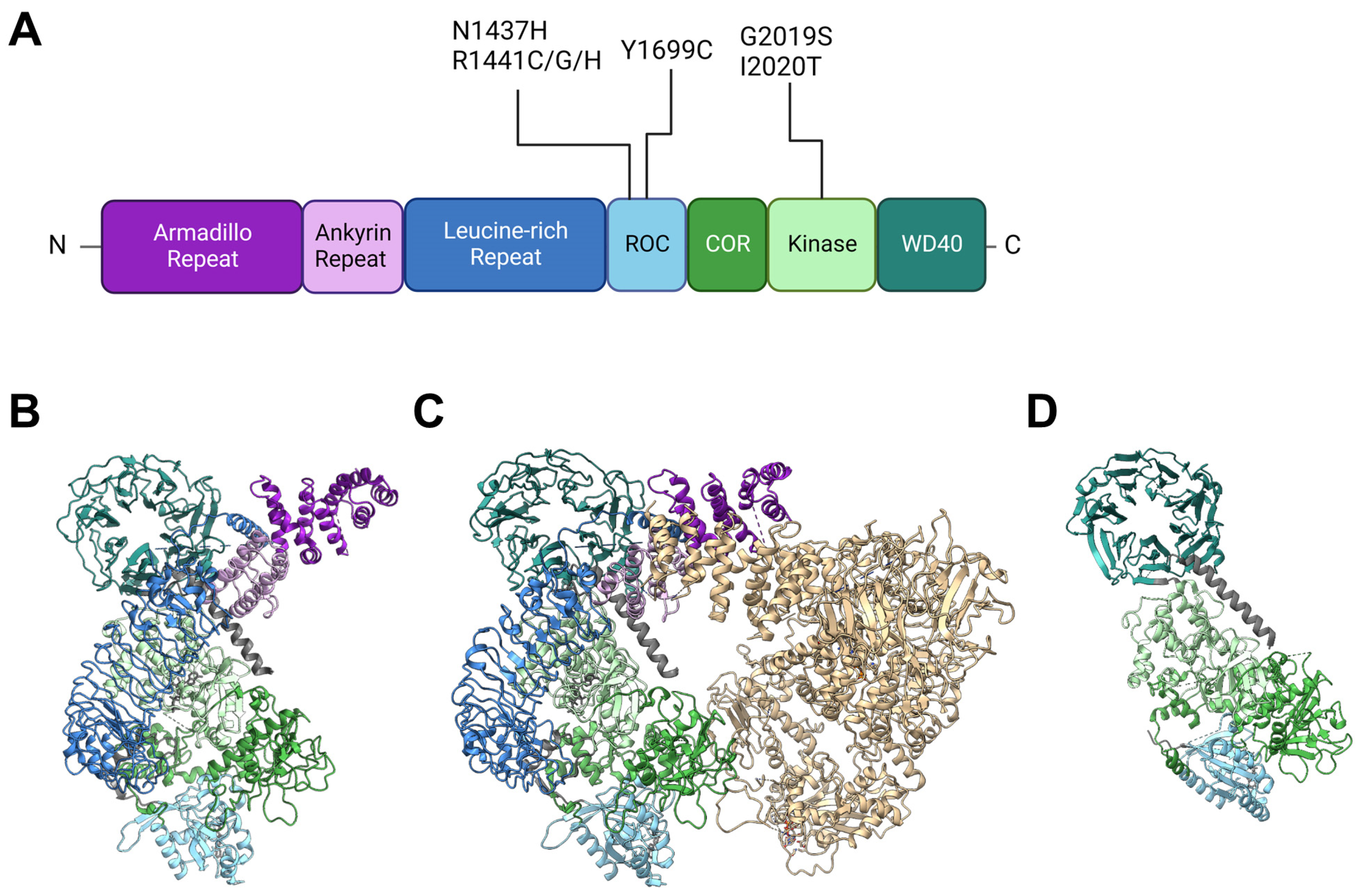
2. Insights from Genetic and Structural Studies of LRRK2

3. Rab GTPases and LRRK2
3.1. Rab GTPases and the Endolysosomal System
3.2. LRRK2 and Substrate Rab GTPases
3.2.1. Rab8 and Rab10
3.2.2. Rab29
3.2.3. Rab12
3.2.4. Rab35
3.2.5. Rab5
3.2.6. Rab3
3.2.7. Rab1
4. Rab Phosphorylation in Relation to PD
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 Cause Autosomal-Dominant Parkinsonism with Pleomorphic Pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Paisán-Ruíz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simón, J.; van der Brug, M.; de Munain, A.L.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the Gene Containing Mutations That Cause PARK8-Linked Parkinson’s Disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, C.; Lewis, P.A. Leucine-Rich Repeat Kinase 2 (LRRK2). Adv. Neurobiol. 2017, 14, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Singh, F.; Ganley, I.G. Parkinson’s Disease and Mitophagy: An Emerging Role for LRRK2. Biochem. Soc. Trans. 2021, 49, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Berwick, D.C.; Heaton, G.R.; Azeggagh, S.; Harvey, K. LRRK2 Biology from Structure to Dysfunction: Research Progresses, but the Themes Remain the Same. Mol. Neurodegener. 2019, 14, 49. [Google Scholar] [CrossRef]
- Boecker, C.A. The Role of LRRK2 in Intracellular Organelle Dynamics. J. Mol. Biol. 2023, 435, 167998. [Google Scholar] [CrossRef]
- Herbst, S.; Gutierrez, M.G. LRRK2 in Infection: Friend or Foe? ACS Infect. Dis. 2019, 5, 809–815. [Google Scholar] [CrossRef]
- Kuwahara, T.; Iwatsubo, T. The Emerging Functions of LRRK2 and Rab GTPases in the Endolysosomal System. Front. Neurosci. 2020, 14, 227. [Google Scholar] [CrossRef]
- Taylor, M.; Alessi, D.R. Advances in Elucidating the Function of Leucine-Rich Repeat Protein Kinase-2 in Normal Cells and Parkinson’s Disease. Curr. Opin. Cell Biol. 2020, 63, 102–113. [Google Scholar] [CrossRef]
- Erb, M.L.; Moore, D.J. LRRK2 and the Endolysosomal System in Parkinson’s Disease. J. Park. Dis. 2020, 10, 1271–1291. [Google Scholar] [CrossRef]
- Norris, A.; Grant, B.D. Endosomal Microdomains: Formation and Function. Curr. Opin. Cell Biol. 2020, 65, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Dalfó, E.; Gómez-Isla, T.; Rosa, J.L.; Bodelón, M.N.; Tejedor, M.C.; Barrachina, M.; Ambrosio, S.; Ferrer, I. Abnormal α-Synuclein Interactions with Rab Proteins in α-Synuclein A30P Transgenic Mice. J. Neuropathol. Exp. Neurol. 2004, 63, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. α-Synuclein Blocks ER-Golgi Traffic and Rab1 Rescues Neuron Loss in Parkinson’s Models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics Reveals That Parkinson’s Disease Kinase LRRK2 Regulates a Subset of Rab GTPases. eLife 2016, 5, e12813. [Google Scholar] [CrossRef] [PubMed]
- Steger, M.; Diez, F.; Dhekne, H.S.; Lis, P.; Nirujogi, R.S.; Karayel, O.; Tonelli, F.; Martinez, T.N.; Lorentzen, E.; Pfeffer, S.R.; et al. Systematic Proteomic Analysis of LRRK2-Mediated Rab GTPase Phosphorylation Establishes a Connection to Ciliogenesis. eLife 2017, 6, e31012. [Google Scholar] [CrossRef]
- Liu, Z.; Bryant, N.; Kumaran, R.; Beilina, A.; Abeliovich, A.; Cookson, M.R.; West, A.B. LRRK2 Phosphorylates Membrane-Bound Rabs and Is Activated by GTP-Bound Rab7L1 to Promote Recruitment to the Trans Golgi Network. Hum. Mol. Genet. 2018, 27, 385–395. [Google Scholar] [CrossRef]
- Madero-Pérez, J.; Fdez, E.; Fernández, B.; Ordóñez, A.J.L.; Ramírez, M.B.; Gómez-Suaga, P.; Waschbüsch, D.; Lobbestael, E.; Baekelandt, V.; Nairn, A.C.; et al. Parkinson Disease-Associated Mutations in LRRK2 Cause Centrosomal Defects via Rab8a Phosphorylation. Mol. Neurodegener. 2018, 13, 3. [Google Scholar] [CrossRef]
- Fujimoto, T.; Kuwahara, T.; Eguchi, T.; Sakurai, M.; Komori, T.; Iwatsubo, T. Parkinson’s Disease-Associated Mutant LRRK2 Phosphorylates Rab7L1 and Modifies Trans-Golgi Morphology. Biochem. Biophys. Res. Commun. 2018, 495, 1708–1715. [Google Scholar] [CrossRef]
- Purlyte, E.; Dhekne, H.S.; Sarhan, A.R.; Gomez, R.; Lis, P.; Wightman, M.; Martinez, T.N.; Tonelli, F.; Pfeffer, S.R.; Alessi, D.R. Rab29 Activation of the Parkinson’s Disease-associated LRRK2 Kinase. EMBO J. 2018, 37, 1–18. [Google Scholar] [CrossRef]
- Funayama, M.; Hasegawa, K.; Kowa, H.; Saito, M.; Tsuji, S.; Obata, F. A New Locus for Parkinson’s Disease (PARK8) Maps to Chromosome 12p11.2–Q13.1. Ann. Neurol. 2002, 51, 296–301. [Google Scholar] [CrossRef]
- Aasly, J.O.; Vilariño-Güell, C.; Dachsel, J.C.; Webber, P.J.; West, A.B.; Haugarvoll, K.; Johansen, K.K.; Toft, M.; Nutt, J.G.; Payami, H.; et al. Novel Pathogenic LRRK2 p.Asn1437His Substitution in Familial Parkinson’s Disease. Mov. Disord. 2010, 25, 2156–2163. [Google Scholar] [CrossRef] [PubMed]
- Mata, I.F.; Kachergus, J.M.; Taylor, J.P.; Lincoln, S.; Aasly, J.; Lynch, T.; Hulihan, M.M.; Cobb, S.A.; Wu, R.-M.; Lu, C.-S.; et al. Lrrk2 Pathogenic Substitutions in Parkinson’s Disease. Neurogenetics 2005, 6, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Kachergus, J.; Mata, I.F.; Hulihan, M.; Taylor, J.P.; Lincoln, S.; Aasly, J.; Gibson, J.M.; Ross, O.A.; Lynch, T.; Wiley, J.; et al. Identification of a Novel LRRK2 Mutation Linked to Autosomal Dominant Parkinsonism: Evidence of a Common Founder across European Populations. Am. J. Hum. Genet. 2005, 76, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Kalogeropulou, A.F.; Purlyte, E.; Tonelli, F.; Lange, S.M.; Wightman, M.; Prescott, A.R.; Padmanabhan, S.; Sammler, E.; Alessi, D.R. Impact of 100 LRRK2 Variants Linked to Parkinson’s Disease on Kinase Activity and Microtubule Binding. Biochem. J. 2022, 479, 1759–1783. [Google Scholar] [CrossRef] [PubMed]
- Bosgraaf, L.; Haastert, P.J.M.V. Roc, a Ras/GTPase Domain in Complex Proteins. Biochim. Biophys. Acta BBA Mol. Cell Res. 2003, 1643, 5–10. [Google Scholar] [CrossRef]
- Guo, L.; Gandhi, P.N.; Wang, W.; Petersen, R.B.; Wilson-Delfosse, A.L.; Chen, S.G. The Parkinson’s Disease-Associated Protein, Leucine-Rich Repeat Kinase 2 (LRRK2), Is an Authentic GTPase Thatstimulates Kinase Activity. Exp. Cell Res. 2007, 313, 3658–3670. [Google Scholar] [CrossRef]
- Taymans, J.-M.; Vancraenenbroeck, R.; Ollikainen, P.; Beilina, A.; Lobbestael, E.; Maeyer, M.D.; Baekelandt, V.; Cookson, M.R. LRRK2 Kinase Activity Is Dependent on LRRK2 GTP Binding Capacity but Independent of LRRK2 GTP Binding. PLoS ONE 2011, 6, e23207. [Google Scholar] [CrossRef]
- Cogo, S.; Ho, F.Y.; Tosoni, E.; Tomkins, J.E.; Tessari, I.; Iannotta, L.; Montine, T.J.; Manzoni, C.; Lewis, P.A.; Bubacco, L.; et al. The Roc Domain of LRRK2 as a Hub for Protein-Protein Interactions: A Focus on PAK6 and Its Impact on RAB Phosphorylation. Brain Res. 2022, 1778, 147781. [Google Scholar] [CrossRef]
- Mills, R.D.; Liang, L.; Lio, D.S.; Mok, Y.; Mulhern, T.D.; Cao, G.; Griffin, M.; Kenche, V.B.; Culvenor, J.G.; Cheng, H. The Roc-COR Tandem Domain of Leucine-rich Repeat Kinase 2 Forms Dimers and Exhibits Conventional Ras-like GTPase Properties. J. Neurochem. 2018, 147, 409–428. [Google Scholar] [CrossRef]
- Myasnikov, A.; Zhu, H.; Hixson, P.; Xie, B.; Yu, K.; Pitre, A.; Peng, J.; Sun, J. Structural Analysis of the Full-Length Human LRRK2. Cell 2021, 184, 3519–3527.e10. [Google Scholar] [CrossRef]
- Sheng, Z.; Zhang, S.; Bustos, D.; Kleinheinz, T.; Pichon, C.E.; Dominguez, S.L.; Solanoy, H.O.; Drummond, J.; Zhang, X.; Ding, X.; et al. Ser1292 Autophosphorylation Is an Indicator of LRRK2 Kinase Activity and Contributes to the Cellular Effects of PD Mutations. Sci. Transl. Med. 2012, 4, 164ra161. [Google Scholar] [CrossRef]
- Christensen, K.; Hentzer, M.; Oppermann, F.S.; Elschenbroich, S.; Dossang, P.; Thirstrup, K.; Egebjerg, J.; Williamson, D.S.; Smith, G.P. LRRK2 Exonic Variants Associated with Parkinson’s Disease Augment Phosphorylation Levels for LRRK2-Ser1292 and Rab10-Thr73. bioRxiv 2018, 447946. [Google Scholar] [CrossRef]
- Mosavi, L.K.; Cammett, T.J.; Desrosiers, D.C.; Peng, Z. The Ankyrin Repeat as Molecular Architecture for Protein Recognition. Protein Sci. 2004, 13, 1435–1448. [Google Scholar] [CrossRef] [PubMed]
- Tewari, R.; Bailes, E.; Bunting, K.A.; Coates, J.C. Armadillo-Repeat Protein Functions: Questions for Little Creatures. Trends Cell Biol. 2010, 20, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, N.; Takatsuka, S.; Miyashita, H.; Kretsinger, R.H. Leucine Rich Repeat Proteins: Sequences, Mutations, Structures and Diseases. Protein Pept. Lett. 2019, 26, 108–131. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Min, J. Structure and Function of WD40 Domain Proteins. Protein Cell 2011, 2, 202–214. [Google Scholar] [CrossRef]
- Schapira, M.; Tyers, M.; Torrent, M.; Arrowsmith, C.H. WD40 Repeat Domain Proteins: A Novel Target Class? Nat. Rev. Drug Discov. 2017, 16, 773–786. [Google Scholar] [CrossRef]
- Lee, H.; Flynn, R.; Sharma, I.; Haberman, E.; Carling, P.J.; Nicholls, F.J.; Stegmann, M.; Vowles, J.; Haenseler, W.; Wade-Martins, R.; et al. LRRK2 Is Recruited to Phagosomes and Co-Recruits RAB8 and RAB10 in Human Pluripotent Stem Cell-Derived Macrophages. Stem Cell Rep. 2020, 14, 940–955. [Google Scholar] [CrossRef]
- Ito, G.; Utsunomiya-Tate, N. Overview of the Impact of Pathogenic LRRK2 Mutations in Parkinson’s Disease. Biomolecules 2023, 13, 845. [Google Scholar] [CrossRef]
- Deniston, C.K.; Salogiannis, J.; Mathea, S.; Snead, D.M.; Lahiri, I.; Matyszewski, M.; Donosa, O.; Watanabe, R.; Böhning, J.; Shiau, A.K.; et al. Structure of LRRK2 in Parkinson’s Disease and Model for Microtubule Interaction. Nature 2020, 588, 344–349. [Google Scholar] [CrossRef]
- Guaitoli, G.; Gilsbach, B.K.; Raimondi, F.; Gloeckner, C.J. First Model of Dimeric LRRK2: The Challenge of Unrevealing the Structure of a Multidomain Parkinson’s-Associated Protein. Biochem. Soc. Trans. 2016, 44, 1635–1641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Fan, Y.; Ru, H.; Wang, L.; Magupalli, V.G.; Taylor, S.S.; Alessi, D.R.; Wu, H. Crystal Structure of the WD40 Domain Dimer of LRRK2. Proc. Natl. Acad. Sci. USA 2019, 116, 1579–1584. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Buschauer, R.; Böhning, J.; Audagnotto, M.; Lasker, K.; Lu, T.-W.; Boassa, D.; Taylor, S.; Villa, E. The In Situ Structure of Parkinson’s Disease-Linked LRRK2. Cell 2020, 182, 1508–1518.e16. [Google Scholar] [CrossRef]
- Zhu, H.; Tonelli, F.; Alessi, D.R.; Sun, J. Structural Basis of Human LRRK2 Membrane Recruitment and Activation. bioRxiv 2022. [Google Scholar] [CrossRef]
- Greggio, E.; Zambrano, I.; Kaganovich, A.; Beilina, A.; Taymans, J.-M.; Daniëls, V.; Lewis, P.; Jain, S.; Ding, J.; Syed, A.; et al. The Parkinson Disease-Associated Leucine-Rich Repeat Kinase 2 (LRRK2) Is a Dimer That Undergoes Intramolecular Autophosphorylation*. J. Biol. Chem. 2008, 283, 16906–16914. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Lewis, P.A.; Greggio, E.; Sluch, E.; Beilina, A.; Cookson, M.R. Structure of the ROC Domain from the Parkinson’s Disease-Associated Leucine-Rich Repeat Kinase 2 Reveals a Dimeric GTPase. Proc. Natl. Acad. Sci. USA 2008, 105, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Guaitoli, G.; Raimondi, F.; Gilsbach, B.K.; Gómez-Llorente, Y.; Deyaert, E.; Renzi, F.; Li, X.; Schaffner, A.; Jagtap, P.K.A.; Boldt, K.; et al. Structural Model of the Dimeric Parkinson’s Protein LRRK2 Reveals a Compact Architecture Involving Distant Interdomain Contacts. Proc. Natl. Acad. Sci. USA 2016, 113, E4357–E4366. [Google Scholar] [CrossRef]
- Sejwal, K.; Chami, M.; Rémigy, H.; Vancraenenbroeck, R.; Sibran, W.; Sütterlin, R.; Baumgartner, P.; McLeod, R.; Chartier-Harlin, M.-C.; Baekelandt, V.; et al. Cryo-EM Analysis of Homodimeric Full-Length LRRK2 and LRRK1 Protein Complexes. Sci. Rep. 2017, 7, 8667. [Google Scholar] [CrossRef]
- Snead, D.M.; Matyszewski, M.; Dickey, A.M.; Lin, Y.X.; Leschziner, A.E.; Reck-Peterson, S.L. Structural Basis for Parkinson’s Disease-Linked LRRK2’s Binding to Microtubules. Nat. Struct. Mol. Biol. 2022, 29, 1196–1207. [Google Scholar] [CrossRef]
- Ito, G.; Iwatsubo, T. Re-Examination of the Dimerization State of Leucine-Rich Repeat Kinase 2: Predominance of the Monomeric Form. Biochem. J. 2012, 441, 987–998. [Google Scholar] [CrossRef]
- Kett, L.R.; Boassa, D.; Ho, C.C.-Y.; Rideout, H.J.; Hu, J.; Terada, M.; Ellisman, M.; Dauer, W.T. LRRK2 Parkinson Disease Mutations Enhance Its Microtubule Association. Hum. Mol. Genet. 2012, 21, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Vides, E.G.; Adhikari, A.; Chiang, C.Y.; Lis, P.; Purlyte, E.; Limouse, C.; Shumate, J.L.; Spinola-Lasso, E.; Dhekne, H.S.; Alessi, D.R.; et al. A Feed-Forward Pathway Drives LRRK2 Kinase Membrane Recruitment and Activation. eLife 2022, 11, e79771. [Google Scholar] [CrossRef] [PubMed]
- Dhekne, H.S.; Tonelli, F.; Yeshaw, W.M.; Chiang, C.Y.; Limouse, C.; Jaimon, E.; Purlyte, E.; Alessi, D.R.; Pfeffer, S.R. Genome-Wide Screen Reveals Rab12 GTPase as a Critical Activator of Parkinson’s Disease-Linked LRRK2 Kinase. eLife 2023, 12, e87098. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bondar, V.V.; Davis, O.B.; Maloney, M.T.; Agam, M.; Chin, M.Y.; Ho, A.C.-N.; Ghosh, R.; Leto, D.E.; Joy, D.; et al. Rab12 Is a Regulator of LRRK2 and Its Activation by Damaged Lysosomes. eLife 2023, 12, e87255. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, T.; Funakawa, K.; Komori, T.; Sakurai, M.; Yoshii, G.; Eguchi, T.; Fukuda, M.; Iwatsubo, T. Roles of Lysosomotropic Agents on LRRK2 Activation and Rab10 Phosphorylation. Neurobiol. Dis. 2020, 145, 105081. [Google Scholar] [CrossRef]
- Piccoli, G.; Onofri, F.; Cirnaru, M.D.; Kaiser, C.J.O.; Jagtap, P.; Kastenmüller, A.; Pischedda, F.; Marte, A.; von Zweydorf, F.; Vogt, A.; et al. Leucine-Rich Repeat Kinase 2 Binds to Neuronal Vesicles through Protein Interactions Mediated by Its C-Terminal WD40 Domain. Mol. Cell Biol. 2014, 34, 2147–2161. [Google Scholar] [CrossRef]
- Cullen, P.J.; Steinberg, F. To Degrade or Not to Degrade: Mechanisms and Significance of Endocytic Recycling. Nat. Rev. Mol. Cell Biol. 2018, 19, 679–696. [Google Scholar] [CrossRef]
- Scott, C.C.; Vacca, F.; Gruenberg, J. Endosome Maturation, Transport and Functions. Semin. Cell Dev. Biol. 2014, 31, 2–10. [Google Scholar] [CrossRef]
- Wang, J.; Fedoseienko, A.; Chen, B.; Burstein, E.; Jia, D.; Billadeau, D.D. Endosomal Receptor Trafficking: Retromer and Beyond. Traffic 2018, 19, 578–590. [Google Scholar] [CrossRef]
- Langemeyer, L.; Fröhlich, F.; Ungermann, C. Rab GTPase Function in Endosome and Lysosome Biogenesis. Trends Cell Biol. 2018, 28, 957–970. [Google Scholar] [CrossRef]
- Gruenberg, J. Life in the Lumen: The Multivesicular Endosome. Traffic 2020, 21, 76–93. [Google Scholar] [CrossRef]
- O’Sullivan, M.J.; Lindsay, A.J. The Endosomal Recycling Pathway—At the Crossroads of the Cell. Int. J. Mol. Sci. 2020, 21, 6074. [Google Scholar] [CrossRef]
- Huotari, J.; Helenius, A. Endosome Maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef]
- Campa, C.C.; Margaria, J.P.; Derle, A.; Giudice, M.D.; Santis, M.C.D.; Gozzelino, L.; Copperi, F.; Bosia, C.; Hirsch, E. Rab11 Activity and PtdIns(3)P Turnover Removes Recycling Cargo from Endosomes. Nat. Chem. Biol. 2018, 14, 801–810. [Google Scholar] [CrossRef]
- Bonifacino, J.S.; Rojas, R. Retrograde Transport from Endosomes to the Trans-Golgi Network. Nat. Rev. Mol. Cell Biol. 2006, 7, 568–579. [Google Scholar] [CrossRef]
- Mignogna, M.L.; D’Adamo, P. Critical Importance of RAB Proteins for Synaptic Function. Small GTPase 2018, 9, 145–157. [Google Scholar] [CrossRef]
- Fukuda, M. Rab GTPases: Key Players in Melanosome Biogenesis, Transport, and Transfer. Pigment. Cell Melanoma Res. 2021, 34, 222–235. [Google Scholar] [CrossRef]
- Lis, P.; Burel, S.; Steger, M.; Mann, M.; Brown, F.; Diez, F.; Tonelli, F.; Holton, J.L.; Ho, P.W.; Ho, S.-L.; et al. Development of Phospho-Specific Rab Protein Antibodies to Monitor in Vivo Activity of the LRRK2 Parkinson’s Disease Kinase. Biochem. J. 2018, 475, BCJ20170802. [Google Scholar] [CrossRef]
- Nirujogi, R.S.; Tonelli, F.; Taylor, M.; Lis, P.; Zimprich, A.; Sammler, E.; Alessi, D.R. Development of a Multiplexed Targeted Mass Spectrometry Assay for LRRK2-Phosphorylated Rabs and Ser910/Ser935 Biomarker Sites. Biochem. J. 2021, 478, 299–326. [Google Scholar] [CrossRef]
- Yun, H.J.; Kim, H.; Ga, I.; Oh, H.; Ho, D.H.; Kim, J.; Seo, H.; Son, I.; Seol, W. An Early Endosome Regulator, Rab5b, Is an LRRK2 Kinase Substrate. J. Biochem. 2015, 157, 485–495. [Google Scholar] [CrossRef]
- Ordóñez, A.; Fernández, B.; Fdez, E.; Romo-Lozano, M.; Madero-Pérez, J.; Lobbestael, E.; Baekelandt, V.; Aiastui, A.; Munaín, A.; Melrose, H.L.; et al. RAB8, RAB10 and RILPL1 Contribute to Both LRRK2 Kinase-Mediated Centrosomal Cohesion and Ciliogenesis Deficits. Hum. Mol. Genet. 2019, 28, 3552–3568. [Google Scholar] [CrossRef]
- Madero-Pérez, J.; Fernández, B.; Ordóñez, A.J.L.; Fdez, E.; Lobbestael, E.; Baekelandt, V.; Hilfiker, S. RAB7L1-Mediated Relocalization of LRRK2 to the Golgi Complex Causes Centrosomal Deficits via RAB8A. Front. Mol. Neurosci. 2018, 11, 417. [Google Scholar] [CrossRef]
- Yu, M.; Arshad, M.; Wang, W.; Zhao, D.; Xu, L.; Zhou, L. LRRK2 Mediated Rab8a Phosphorylation Promotes Lipid Storage. Lipids Health Dis. 2018, 17, 34. [Google Scholar] [CrossRef]
- Waschbüsch, D.; Purlyte, E.; Pal, P.; McGrath, E.; Alessi, D.R.; Khan, A.R. Structural Basis for Rab8a Recruitment of RILPL2 via LRRK2 Phosphorylation of Switch 2. Structure 2020, 28, 406–417.e6. [Google Scholar] [CrossRef]
- Mamais, A.; Kluss, J.H.; Bonet-Ponce, L.; Landeck, N.; Langston, R.G.; Smith, N.; Beilina, A.; Kaganovich, A.; Ghosh, M.C.; Pellegrini, L.; et al. Mutations in LRRK2 Linked to Parkinson Disease Sequester Rab8a to Damaged Lysosomes and Regulate Transferrin-Mediated Iron Uptake in Microglia. PLoS Biol. 2021, 19, e3001480. [Google Scholar] [CrossRef]
- Eguchi, T.; Kuwahara, T.; Sakurai, M.; Komori, T.; Fujimoto, T.; Ito, G.; Yoshimura, S.; Harada, A.; Fukuda, M.; Koike, M.; et al. LRRK2 and Its Substrate Rab GTPases Are Sequentially Targeted onto Stressed Lysosomes and Maintain Their Homeostasis. Proc. Natl. Acad. Sci. USA 2018, 115, E9115–E9124. [Google Scholar] [CrossRef]
- Dhekne, H.S.; Yanatori, I.; Vides, E.G.; Sobu, Y.; Diez, F.; Tonelli, F.; Pfeffer, S.R. LRRK2-Phosphorylated Rab10 Sequesters Myosin Va with RILPL2 during Ciliogenesis Blockade. Life Sci. Alliance 2021, 4, e202101050. [Google Scholar] [CrossRef]
- Ordóñez, A.J.L.; Fasiczka, R.; Fernández, B.; Naaldijk, Y.; Fdez, E.; Ramírez, M.B.; Phan, S.; Boassa, D.; Hilfiker, S. The LRRK2 Signaling Network Converges on a Centriolar Phospho-Rab10/RILPL1 Complex to Cause Deficits in Centrosome Cohesion and Cell Polarization. Biol. Open 2022, 11, bio059468. [Google Scholar] [CrossRef]
- Dhekne, H.S.; Yanatori, I.; Gomez, R.C.; Tonelli, F.; Diez, F.; Schüle, B.; Steger, M.; Alessi, D.R.; Pfeffer, S.R. A Pathway for Parkinson’s Disease LRRK2 Kinase to Block Primary Cilia and Sonic Hedgehog Signaling in the Brain. eLife 2018, 7, e40202. [Google Scholar] [CrossRef]
- Sobu, Y.; Wawro, P.S.; Dhekne, H.S.; Yeshaw, W.M.; Pfeffer, S.R. Pathogenic LRRK2 Regulates Ciliation Probability Upstream of Tau Tubulin Kinase 2 via Rab10 and RILPL1 Proteins. Proc. Natl. Acad. Sci. USA 2021, 118, e2005894118. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, E.; Zhao, H.T.; Cole, T.; West, A.B. LRRK2 and Rab10 Coordinate Macropinocytosis to Mediate Immunological Responses in Phagocytes. EMBO J. 2020, 39, e104862. [Google Scholar] [CrossRef] [PubMed]
- Kluss, J.H.; Beilina, A.; Williamson, C.D.; Lewis, P.A.; Cookson, M.R.; Bonet-Ponce, L. Lysosomal Positioning Regulates Rab10 Phosphorylation at LRRK2+ Lysosomes. Proc. Natl. Acad. Sci. USA 2022, 119, e2205492119. [Google Scholar] [CrossRef]
- Bonet-Ponce, L.; Beilina, A.; Williamson, C.D.; Lindberg, E.; Kluss, J.H.; Saez-Atienzar, S.; Landeck, N.; Kumaran, R.; Mamais, A.; Bleck, C.K.E.; et al. LRRK2 Mediates Tubulation and Vesicle Sorting from Lysosomes. Sci. Adv. 2020, 6, eabb2454. [Google Scholar] [CrossRef] [PubMed]
- Ysselstein, D.; Nguyen, M.; Young, T.J.; Severino, A.; Schwake, M.; Merchant, K.; Krainc, D. LRRK2 Kinase Activity Regulates Lysosomal Glucocerebrosidase in Neurons Derived from Parkinson’s Disease Patients. Nat. Commun. 2019, 10, 5570. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, M.; Gong, Y.; Tang, J.; Shen, J.; Xu, L.; Dang, B.; Chen, G. Inhibition of LRRK2-Rab10 Pathway Improves Secondary Brain Injury After Surgical Brain Injury in Rats. Front. Surg. 2022, 8, 749310. [Google Scholar] [CrossRef]
- Ito, K.; Araki, M.; Katai, Y.; Nishimura, Y.; Imotani, S.; Inoue, H.; Ito, G.; Tomita, T. Pathogenic LRRK2 Compromises the Subcellular Distribution of Lysosomes in a Rab12-RILPL1-dependent Manner. FASEB J. 2023, 37, e22930. [Google Scholar] [CrossRef]
- Komori, T.; Kuwahara, T.; Fujimoto, T.; Sakurai, M.; Koyama-Honda, I.; Fukuda, M.; Iwatsubo, T. Phosphorylation of Rab29 at Ser185 Regulates Its Localization and Role in the Lysosomal Stress Response in Concert with LRRK2. J. Cell Sci. 2023, 136, jcs261003. [Google Scholar] [CrossRef]
- Bae, E.-J.; Kim, D.-K.; Kim, C.; Mante, M.; Adame, A.; Rockenstein, E.; Ulusoy, A.; Klinkenberg, M.; Jeong, G.R.; Bae, J.R.; et al. LRRK2 Kinase Regulates α-Synuclein Propagation via RAB35 Phosphorylation. Nat. Commun. 2018, 9, 3465. [Google Scholar] [CrossRef]
- Homma, Y.; Hiragi, S.; Fukuda, M. Rab Family of Small GTPases: An Updated View on Their Regulation and Functions. FEBS J. 2021, 288, 36–55. [Google Scholar] [CrossRef]
- Ito, G.; Katsemonova, K.; Tonelli, F.; Lis, P.; Baptista, M.; Shpiro, N.; Duddy, G.; Wilson, S.; Ho, P.; Ho, S.-L.; et al. Phos-Tag Analysis of Rab10 Phosphorylation by LRRK2: A Powerful Assay for Assessing Kinase Function and Inhibitors. Biochem. J. 2016, 473, 2671–2685. [Google Scholar] [CrossRef]
- Fan, Y.; Howden, A.J.; Sarhan, A.R.; Lis, P.; Ito, G.; Martinez, T.N.; Brockmann, K.; Gasser, T.; Alessi, D.R.; Sammler, E.M. Interrogating Parkinson’s Disease LRRK2 Kinase Pathway Activity by Assessing Rab10 Phosphorylation in Human Neutrophils. Biochem. J. 2017, 475, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Gomez, R.C.; Wawro, P.; Lis, P.; Alessi, D.R.; Pfeffer, S.R. Membrane Association but Not Identity Is Required for LRRK2 Activation and Phosphorylation of Rab GTPases. J. Cell Biol. 2019, 218, 4157–4170. [Google Scholar] [CrossRef] [PubMed]
- Korecka, J.A.; Thomas, R.; Hinrich, A.J.; Moskites, A.M.; Macbain, Z.K.; Hallett, P.J.; Isacson, O.; Hastings, M.L. Splice-Switching Antisense Oligonucleotides Reduce LRRK2 Kinase Activity in Human LRRK2 Transgenic Mice. Mol. Ther. Nucleic Acids 2020, 21, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Iannotta, L.; Biosa, A.; Kluss, J.H.; Tombesi, G.; Kaganovich, A.; Cogo, S.; Plotegher, N.; Civiero, L.; Lobbestael, E.; Baekelandt, V.; et al. Divergent Effects of G2019S and R1441C LRRK2 Mutations on LRRK2 and Rab10 Phosphorylations in Mouse Tissues. Cells 2020, 9, 2344. [Google Scholar] [CrossRef]
- Kelly, K.; Chang, A.; Hastings, L.; Abdelmotilib, H.; West, A.B. Genetic Background Influences LRRK2-Mediated Rab Phosphorylation in the Rat Brain. Brain Res. 2021, 1759, 147372. [Google Scholar] [CrossRef]
- Nazish, I.; Arber, C.; Piers, T.M.; Warner, T.T.; Hardy, J.A.; Lewis, P.A.; Pocock, J.M.; Bandopadhyay, R. Abrogation of LRRK2 Dependent Rab10 Phosphorylation with TLR4 Activation and Alterations in Evoked Cytokine Release in Immune Cells. Neurochem. Int. 2021, 147, 105070. [Google Scholar] [CrossRef]
- Keeney, M.; Hoffman, E.; Greenamyre, J.; Maio, R.D. Measurement of LRRK2 Kinase Activity by Proximity Ligation Assay. Bio-protocol 2021, 11, e4140. [Google Scholar] [CrossRef]
- Tasegian, A.; Singh, F.; Ganley, I.G.; Reith, A.D.; Alessi, D.R. Impact of Type II LRRK2 Inhibitors on Signaling and Mitophagy. Biochem. J. 2021, 478, 3555–3573. [Google Scholar] [CrossRef]
- Kluss, J.H.; Bonet-Ponce, L.; Lewis, P.A.; Cookson, M.R. Directing LRRK2 to Membranes of the Endolysosomal Pathway Triggers RAB Phosphorylation and JIP4 Recruitment. Neurobiol. Dis. 2022, 170, 105769. [Google Scholar] [CrossRef]
- Atashrazm, F.; Hammond, D.; Perera, G.; Bolliger, M.F.; Matar, E.; Halliday, G.M.; Schüle, B.; Lewis, S.J.G.; Nichols, R.J.; Dzamko, N. LRRK2-mediated Rab10 Phosphorylation in Immune Cells from Parkinson’s Disease Patients. Mov. Disord. 2019, 34, 406–415. [Google Scholar] [CrossRef]
- Fan, Y.; Tonelli, F.; Padmanabhan, S.; Baptista, M.A.S.; Riley, L.; Smith, D.; Marras, C.; Howden, A.; Alessi, D.R.; Sammler, E. Human Peripheral Blood Neutrophil Isolation for Interrogating the Parkinson’s Associated LRRK2 Kinase Pathway by Assessing Rab10 Phosphorylation. J. Vis. Exp. 2020, e58956. [Google Scholar] [CrossRef]
- Karayel, Ö.; Tonelli, F.; Winter, S.V.; Geyer, P.E.; Fan, Y.; Sammler, E.M.; Alessi, D.R.; Steger, M.; Mann, M. Accurate MS-Based Rab10 Phosphorylation Stoichiometry Determination as Readout for LRRK2 Activity in Parkinson’s Disease. Mol. Cell Proteom. 2020, 19, 1546–1560. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kelly, K.; Brotchie, J.M.; Koprich, J.B.; West, A.B. Exosome Markers of LRRK2 Kinase Inhibition. npj Park. Dis. 2020, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Bright, J.M.; Carlisle, H.J.; Toda, A.M.A.; Murphy, M.; Molitor, T.P.; Wren, P.; Andruska, K.M.; Liu, E.; Barlow, C. Differential Inhibition of LRRK2 in Parkinson’s Disease Patient Blood by a G2019S Selective LRRK2 Inhibitor. Mov. Disord. 2021, 36, 1362–1371. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Negrou, E.; Maloney, M.T.; Bondar, V.V.; Andrews, S.V.; Montalban, M.; Llapashtica, C.; Maciuca, R.; Nguyen, H.; Solanoy, H.; et al. Understanding LRRK2 Kinase Activity in Preclinical Models and Human Subjects through Quantitative Analysis of LRRK2 and PT73 Rab10. Sci. Rep. 2021, 11, 12900. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Nirujogi, R.S.; Garrido, A.; Ruiz-Martínez, J.; Bergareche-Yarza, A.; Mondragón-Rezola, E.; Vinagre-Aragón, A.; Croitoru, I.; Pagola, A.G.; Markinez, L.P.; et al. R1441G but Not G2019S Mutation Enhances LRRK2 Mediated Rab10 Phosphorylation in Human Peripheral Blood Neutrophils. Acta Neuropathol. 2021, 142, 475–494. [Google Scholar] [CrossRef]
- Jennings, D.; Huntwork-Rodriguez, S.; Henry, A.G.; Sasaki, J.C.; Meisner, R.; Diaz, D.; Solanoy, H.; Wang, X.; Negrou, E.; Bondar, V.V.; et al. Preclinical and Clinical Evaluation of the LRRK2 Inhibitor DNL201 for Parkinson’s Disease. Sci. Transl. Med. 2022, 14, eabj2658. [Google Scholar] [CrossRef]
- Petropoulou-Vathi, L.; Simitsi, A.; Valkimadi, P.-E.; Kedariti, M.; Dimitrakopoulos, L.; Koros, C.; Papadimitriou, D.; Papadimitriou, A.; Stefanis, L.; Alcalay, R.N.; et al. Distinct Profiles of LRRK2 Activation and Rab GTPase Phosphorylation in Clinical Samples from Different PD Cohorts. npj Park. Dis. 2022, 8, 73. [Google Scholar] [CrossRef]
- Wang, S.; Unnithan, S.; Bryant, N.; Chang, A.; Rosenthal, L.S.; Pantelyat, A.; Dawson, T.M.; Al-Khalidi, H.R.; West, A.B. Elevated Urinary Rab10 Phosphorylation in Idiopathic Parkinson Disease. Mov. Disord. 2022, 37, 1454–1464. [Google Scholar] [CrossRef]
- Chua, C.E.L.; Tang, B.L. Rab 10—A Traffic Controller in Multiple Cellular Pathways and Locations. J. Cell. Physiol. 2018, 233, 6483–6494. [Google Scholar] [CrossRef]
- Peränen, J. Rab8 GTPase as a Regulator of Cell Shape. Cytoskeleton 2011, 68, 527–539. [Google Scholar] [CrossRef]
- Taymans, J.-M.; Mutez, E.; Sibran, W.; Vandewynckel, L.; Deldycke, C.; Bleuse, S.; Marchand, A.; Sarchione, A.; Leghay, C.; Kreisler, A.; et al. Alterations in the LRRK2-Rab Pathway in Urinary Extracellular Vesicles as Parkinson’s Disease and Pharmacodynamic Biomarkers. npj Park. Dis. 2023, 9, 21. [Google Scholar] [CrossRef]
- Huber, L.A.; Pimplikar, S.; Parton, R.G.; Virta, H.; Zerial, M.; Simons, K. Rab8, a Small GTPase Involved in Vesicular Traffic between the TGN and the Basolateral Plasma Membrane. J. Cell Biol. 1993, 123, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Huber, L.A.; de Hoop, M.J.; Dupree, P.; Zerial, M.; Simons, K.; Dotti, C. Protein Transport to the Dendritic Plasma Membrane of Cultured Neurons Is Regulated by Rab8p. J. Cell Biol. 1993, 123, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Huber, L.A.; Dupree, P.; Dotti, C.G. A Deficiency of the Small GTPase Rab8 Inhibits Membrane Traffic in Developing Neurons. Mol. Cell. Biol. 1995, 15, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Homma, Y.; Fukuda, M. Rabin8 Regulates Neurite Outgrowth in Both GEF Activity–Dependent and –Independent Manners. Mol. Biol. Cell. 2016, 27, 2107–2118. [Google Scholar] [CrossRef]
- Urrutia, P.J.; Bodaleo, F.; Bórquez, D.A.; Homma, Y.; Rozes-Salvador, V.; Villablanca, C.; Conde, C.; Fukuda, M.; González-Billault, C. Tuba Activates Cdc42 during Neuronal Polarization Downstream of the Small GTPase Rab8a. J. Neurosci. 2021, 41, 1636–1649. [Google Scholar] [CrossRef]
- Stypulkowski, E.; Feng, Q.; Joseph, I.; Farrell, V.; Flores, J.; Yu, S.; Sakamori, R.; Sun, J.; Bandyopadhyay, S.; Das, S.; et al. Rab8 Attenuates Wnt Signaling and Is Required for Mesenchymal Differentiation into Adipocytes. J. Biol. Chem. 2021, 296, 100488. [Google Scholar] [CrossRef]
- Sato, T.; Iwano, T.; Kunii, M.; Matsuda, S.; Mizuguchi, R.; Jung, Y.; Hagiwara, H.; Yoshihara, Y.; Yuzaki, M.; Harada, R.; et al. Rab8a and Rab8b Are Essential for Several Apical Transport Pathways but Insufficient for Ciliogenesis. J. Cell Sci. 2013, 127, 422–431. [Google Scholar] [CrossRef]
- Yin, G.; da Fonseca, T.L.; Eisbach, S.E.; Anduaga, A.M.; Breda, C.; Orcellet, M.L.; Szegő, É.M.; Guerreiro, P.; Lázaro, D.F.; Braus, G.H.; et al. α-Synuclein Interacts with the Switch Region of Rab8a in a Ser129 Phosphorylation-Dependent Manner. Neurobiol. Dis. 2014, 70, 149–161. [Google Scholar] [CrossRef][Green Version]
- Corbier, C.; Sellier, C. C9ORF72 Is a GDP/GTP Exchange Factor for Rab8 and Rab39 and Regulates Autophagy. Small GTPases 2017, 8, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Mamais, A.; Sanyal, A.; Fajfer, A.; Zykoski, C.G.; Guldin, M.; Riley-DiPaolo, A.; Subrahmanian, N.; Gibbs, W.; Lin, S.; LaVoie, M.J. The LRRK2 Kinase Substrates Rab8a and Rab10 Contribute Complementary but Distinct Disease-Relevant Phenotypes in Human Neurons. bioRxiv 2023. [Google Scholar] [CrossRef]
- Li, Z.; Schulze, R.J.; Weller, S.G.; Krueger, E.W.; Schott, M.B.; Zhang, X.; Casey, C.A.; Liu, J.; Stöckli, J.; James, D.E.; et al. A Novel Rab10-EHBP1-EHD2 Complex Essential for the Autophagic Engulfment of Lipid Droplets. Sci. Adv. 2016, 2, e1601470. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Bleimling, N.; Vetter, I.R.; Goody, R.S. The Mechanism of Activation of the Actin Binding Protein EHBP1 by Rab8 Family Members. Nat. Commun. 2020, 11, 4187. [Google Scholar] [CrossRef] [PubMed]
- Simón-Sánchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-Wide Association Study Reveals Genetic Risk Underlying Parkinson’s Disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-Wide Association Study Identifies Common Variants at Four Loci as Genetic Risk Factors for Parkinson’s Disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef]
- Pihlstrøm, L.; Rengmark, A.; Bjørnarå, K.A.; Dizdar, N.; Fardell, C.; Forsgren, L.; Holmberg, B.; Larsen, J.P.; Linder, J.; Nissbrandt, H.; et al. Fine Mapping and Resequencing of the PARK16 Locus in Parkinson’s Disease. J. Hum. Genet. 2015, 60, 357–362. [Google Scholar] [CrossRef]
- MacLeod, D.A.; Rhinn, H.; Kuwahara, T.; Zolin, A.; Di Paolo, G.; McCabe, B.D.; Marder, K.S.; Honig, L.S.; Clark, L.N.; Small, S.A.; et al. RAB7L1 Interacts with LRRK2 to Modify Intraneuronal Protein Sorting and Parkinson’s Disease Risk. Neuron 2013, 77, 425–439. [Google Scholar] [CrossRef]
- Beilina, A.; Rudenko, I.N.; Kaganovich, A.; Civiero, L.; Chau, H.; Kalia, S.K.; Kalia, L.V.; Lobbestael, E.; Chia, R.; Ndukwe, K.; et al. Unbiased Screen for Interactors of Leucine-Rich Repeat Kinase 2 Supports a Common Pathway for Sporadic and Familial Parkinson Disease. Proc. Natl. Acad. Sci. USA 2014, 111, 2626–2631. [Google Scholar] [CrossRef]
- Kuwahara, T.; Inoue, K.; D’Agati, V.D.; Fujimoto, T.; Eguchi, T.; Saha, S.; Wolozin, B.; Iwatsubo, T.; Abeliovich, A. LRRK2 and RAB7L1 Coordinately Regulate Axonal Morphology and Lysosome Integrity in Diverse Cellular Contexts. Sci. Rep. 2016, 6, 29945. [Google Scholar] [CrossRef]
- Unapanta, A.; Shavarebi, F.; Porath, J.; Shen, Y.; Balen, C.; Nguyen, A.; Tseng, J.; Leong, W.S.; Liu, M.; Lis, P.; et al. Endogenous Rab38 Regulates LRRK2’s Membrane Recruitment and Substrate Rab Phosphorylation in Melanocytes. J. Biol. Chem. 2023, 299, 105192. [Google Scholar] [CrossRef] [PubMed]
- Helip-Wooley, A.; Thoene, J.G. Sucrose-Induced Vacuolation Results in Increased Expression of Cholesterol Biosynthesis and Lysosomal Genes. Exp. Cell Res. 2004, 292, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Kalogeropulou, A.F.; Freemantle, J.B.; Lis, P.; Vides, E.G.; Polinski, N.K.; Alessi, D.R. Endogenous Rab29 Does Not Impact Basal or Stimulated LRRK2 Pathway Activity. Biochem. J. 2020, 477, 4397–4423. [Google Scholar] [CrossRef]
- Wang, S.; Ma, Z.; Xu, X.; Wang, Z.; Sun, L.; Zhou, Y.; Lin, X.; Hong, W.; Wang, T. A Role of Rab29 in the Integrity of the Trans-Golgi Network and Retrograde Trafficking of Mannose-6-Phosphate Receptor. PLoS ONE 2014, 9, e96242. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Ríos, P.; Romo-Lozano, M.; Fernández, B.; Fdez, E.; Hilfiker, S. Distinct Roles for RAB10 and RAB29 in Pathogenic LRRK2-Mediated Endolysosomal Trafficking Alterations. Cells 2020, 9, 1719. [Google Scholar] [CrossRef]
- Matsui, T.; Itoh, T.; Fukuda, M. Small GTPase Rab12 Regulates Constitutive Degradation of Transferrin Receptor. Traffic 2011, 12, 1432–1443. [Google Scholar] [CrossRef]
- Rydell, G.E.; Renard, H.; Garcia-Castillo, M.; Dingli, F.; Loew, D.; Lamaze, C.; Römer, W.; Johannes, L. Rab12 Localizes to Shiga Toxin-Induced Plasma Membrane Invaginations and Controls Toxin Transport. Traffic 2014, 15, 772–787. [Google Scholar] [CrossRef]
- Wang, J.; Lau, P.K.; Li, C.W.; Guo, Y. The Clathrin Adaptor Complex-1 and Rab12 Regulate Post-Golgi Trafficking of WT Epidermal Growth Factor Receptor (EGFR). J. Biol. Chem. 2023, 299, 102979. [Google Scholar] [CrossRef]
- Kluss, J.H.; Mazza, M.C.; Li, Y.; Manzoni, C.; Lewis, P.A.; Cookson, M.R.; Mamais, A. Preclinical Modeling of Chronic Inhibition of the Parkinson’s Disease Associated Kinase LRRK2 Reveals Altered Function of the Endolysosomal System in Vivo. Mol. Neurodegener. 2021, 16, 17. [Google Scholar] [CrossRef]
- Ito, G.; Tomita, T.; Utsunomiya-Tate, N. LRRK2-Mediated Phosphorylation and Thermal Stability of Rab12 Are Regulated by Bound Nucleotides. Biochem. Biophys. Res. Commun. 2023, 667, 43–49. [Google Scholar] [CrossRef]
- Klinkert, K.; Echard, A. Rab35 GTPase: A Central Regulator of Phosphoinositides and F-actin in Endocytic Recycling and Beyond. Traffic 2016, 17, 1063–1077. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-I.; Kong, C.; Su, X.; Stahl, P.D. Rab5 Isoforms Differentially Regulate the Trafficking and Degradation of Epidermal Growth Factor Receptors*. J. Biol. Chem. 2009, 284, 30328–30338. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-I.; Schauer, K.; Kong, C.; Harding, A.R.; Goud, B.; Stahl, P.D. Rab5 Isoforms Orchestrate a “Division of Labor” in the Endocytic Network; Rab5C Modulates Rac-Mediated Cell Motility. PLoS ONE 2014, 9, e90384. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.; Weyer, Y.; Fink, M.J.; Müller, M.; Weys, S.; Bindreither, M.; Teis, D. Regulation of Rab5 Isoforms by Transcriptional and Post-transcriptional Mechanisms in Yeast. FEBS Lett. 2017, 591, 2803–2815. [Google Scholar] [CrossRef]
- Sung, J.Y.; Kim, J.; Paik, S.R.; Park, J.H.; Ahn, Y.S.; Chung, K.C. Induction of Neuronal Cell Death by Rab5A-Dependent Endocytosis of α-Synuclein*. J. Biol. Chem. 2001, 276, 27441–27448. [Google Scholar] [CrossRef]
- Maekawa, T.; Sasaoka, T.; Azuma, S.; Ichikawa, T.; Melrose, H.L.; Farrer, M.J.; Obata, F. Leucine-Rich Repeat Kinase 2 (LRRK2) Regulates α-Synuclein Clearance in Microglia. BMC Neurosci. 2016, 17, 77. [Google Scholar] [CrossRef]
- Heo, H.Y.; Kim, K.-S.; Seol, W. Coordinate Regulation of Neurite Outgrowth by LRRK2 and Its Interactor, Rab5. Exp. Neurobiol. 2010, 19, 97–105. [Google Scholar] [CrossRef]
- Lang, J. Molecular Mechanisms and Regulation of Insulin Exocytosis as a Paradigm of Endocrine Secretion. Eur. J. Biochem. 1999, 259, 3–17. [Google Scholar] [CrossRef]
- Geppert, M.; Bolshakov, V.Y.; Siegelbaum, S.A.; Takei, K.; Camilli, P.D.; Hammer, R.E.; Südhof, T.C. The Role of Rab3A in Neurotransmitter Release. Nature 1994, 369, 493–497. [Google Scholar] [CrossRef]
- Geppert, M.; Goda, Y.; Stevens, C.F.; Südhof, T.C. The Small GTP-Binding Protein Rab3A Regulates a Late Step in Synaptic Vesicle Fusion. Nature 1997, 387, 810–814. [Google Scholar] [CrossRef]
- Riedel, D.; Antonin, W.; Fernandez-Chacon, R.; de Toledo, G.A.; Jo, T.; Geppert, M.; Valentijn, J.A.; Valentijn, K.; Jamieson, J.D.; Südhof, T.C.; et al. Rab3D Is Not Required for Exocrine Exocytosis but for Maintenance of Normally Sized Secretory Granules. Mol. Cell Biol. 2002, 22, 6487–6497. [Google Scholar] [CrossRef] [PubMed]
- Schlüter, O.M.; Schmitz, F.; Jahn, R.; Rosenmund, C.; Südhof, T.C. A Complete Genetic Analysis of Neuronal Rab3 Function. J. Neurosci. 2004, 24, 6629–6637. [Google Scholar] [CrossRef] [PubMed]
- Persoon, C.M.; Hoogstraaten, R.I.; Nassal, J.P.; van Weering, J.R.T.; Kaeser, P.S.; Toonen, R.F.; Verhage, M. The RAB3-RIM Pathway Is Essential for the Release of Neuromodulators. Neuron 2019, 104, 1065–1080.e12. [Google Scholar] [CrossRef] [PubMed]
- Encarnação, M.; Espada, L.; Escrevente, C.; Mateus, D.; Ramalho, J.; Michelet, X.; Santarino, I.; Hsu, V.W.; Brenner, M.B.; Barral, D.C.; et al. A Rab3a-Dependent Complex Essential for Lysosome Positioning and Plasma Membrane Repair. J. Cell Biol. 2016, 213, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Raffaniello, R.D. Rab3 Proteins and Cancer: Exit Strategies. J. Cell. Biochem. 2021, 122, 1295–1301. [Google Scholar] [CrossRef]
- Dalfó, E.; Barrachina, M.; Rosa, J.L.; Ambrosio, S.; Ferrer, I. Abnormal α-Synuclein Interactions with Rab3a and Rabphilin in Diffuse Lewy Body Disease. Neurobiol. Dis. 2004, 16, 92–97. [Google Scholar] [CrossRef]
- Chen, R.H.C.; Wislet-Gendebien, S.; Samuel, F.; Visanji, N.P.; Zhang, G.; Marsilio, D.; Langman, T.; Fraser, P.E.; Tandon, A. α-Synuclein Membrane Association Is Regulated by the Rab3a Recycling Machinery and Presynaptic Activity. J. Biol. Chem. 2013, 288, 7438–7449. [Google Scholar] [CrossRef]
- Lv, G.; Ko, M.S.; Das, T.; Eliezer, D. Molecular and Functional Interactions of Alpha-Synuclein with Rab3a. J. Biol. Chem. 2022, 298, 102239. [Google Scholar] [CrossRef]
- Petridi, S.; Middleton, A.C.; Ugbode, C.; Fellgett, A.; Covill, L.; Elliott, C. In Vivo Visual Screen for Dopaminergic Rab ↔ LRRK2-G2019S Interactions in Drosophila Discriminates Rab10 from Rab3. G3 Genes Genomes Genet. 2020, 10, 1903–1914. [Google Scholar] [CrossRef]
- Hatoyama, Y.; Homma, Y.; Hiragi, S.; Fukuda, M. Establishment and Analysis of Conditional Rab1 and Rab5 Knockout Cells by Using the Auxin-Inducible Degron System. J. Cell Sci. 2021, 134, jcs259184. [Google Scholar] [CrossRef]
- Tomás, M.; Martínez-Alonso, E.; Martínez-Martínez, N.; Cara-Esteban, M.; Martínez-Menárguez, J.A. Fragmentation of the Golgi Complex of Dopaminergic Neurons in Human Substantia Nigra: New Cytopathological Findings in Parkinson’s Disease. Histol. Histopathol. 2020, 36, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Winslow, A.R.; Chen, C.-W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. α-Synuclein Impairs Macroautophagy: Implications for Parkinson’s Disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-Z.; Li, X.-X.; Zhang, Y.-J.; Rodriguez-Rodriguez, L.; Xiang, M.-Q.; Wang, H.-Y.; Zheng, X.F.S. Rab1 in Cell Signaling, Cancer and Other Diseases. Oncogene 2016, 35, 5699–5704. [Google Scholar] [CrossRef]
- Martínez-Menárguez, J.Á.; Martínez-Alonso, E.; Cara-Esteban, M.; Tomás, M. Focus on the Small GTPase Rab1: A Key Player in the Pathogenesis of Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 12087. [Google Scholar] [CrossRef]
- Homma, Y.; Kinoshita, R.; Kuchitsu, Y.; Wawro, P.S.; Marubashi, S.; Oguchi, M.E.; Ishida, M.; Fujita, N.; Fukuda, M. Comprehensive Knockout Analysis of the Rab Family GTPases in Epithelial Cells. J. Cell Biol. 2019, 218, 2035–2050. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.S.; Sobu, Y.; Dhekne, H.S.; Tonelli, F.; Berndsen, K.; Alessi, D.R.; Pfeffer, S.R. Pathogenic LRRK2 Control of Primary Cilia and Hedgehog Signaling in Neurons and Astrocytes of Mouse Brain. eLife 2021, 10, e67900. [Google Scholar] [CrossRef]
- Pajarillo, E.; Kim, S.; Digman, A.; Dutton, M.; Son, D.-S.; Aschner, M.; Lee, E. The Role of Microglial LRRK2 Kinase in Manganese-Induced Inflammatory Neurotoxicity via NLRP3 Inflammasome and RAB10-Mediated Autophagy Dysfunction. J. Biol. Chem. 2023, 299, 104879. [Google Scholar] [CrossRef]
- Maio, R.D.; Hoffman, E.K.; Rocha, E.M.; Keeney, M.T.; Sanders, L.H.; Miranda, B.R.D.; Zharikov, A.; Laar, A.V.; Stepan, A.F.; Lanz, T.A.; et al. LRRK2 Activation in Idiopathic Parkinson’s Disease. Sci. Transl. Med. 2018, 10, eaar5429. [Google Scholar] [CrossRef]
- Fraser, K.B.; Rawlins, A.B.; Clark, R.G.; Alcalay, R.N.; Standaert, D.G.; Liu, N.; Consortium, P.D.B.P.; West, A.B. Ser(P)-1292 LRRK2 in Urinary Exosomes Is Elevated in Idiopathic Parkinson’s Disease. Mov. Disord. 2016, 31, 1543–1550. [Google Scholar] [CrossRef]
- Boecker, C.A.; Goldsmith, J.; Dou, D.; Cajka, G.G.; Holzbaur, E.L.F. Increased LRRK2 Kinase Activity Alters Neuronal Autophagy by Disrupting the Axonal Transport of Autophagosomes. Curr. Biol. 2021, 31, 2140–2154.e6. [Google Scholar] [CrossRef]
- Kingwell, K. LRRK2-Targeted Parkinson Disease Drug Advances into Phase III. Nat. Rev. Drug Discov. 2023, 22, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Jennings, D.; Huntwork-Rodriguez, S.; Vissers, M.F.J.M.; Daryani, V.M.; Diaz, D.; Goo, M.S.; Chen, J.J.; Maciuca, R.; Fraser, K.; Mabrouk, O.S.; et al. LRRK2 Inhibition by BIIB122 in Healthy Participants and Patients with Parkinson’s Disease. Mov. Disord. 2023, 38, 386–398. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Model of LRRK2 | Method | Part of LRRK2 Analyzed | Interactions | Resolution | PDB ID | Year | Ref. |
|---|---|---|---|---|---|---|---|
| homodimer | Crystallography | LRRK2 ROC domain | Dimerization of ROC domain | 2.0 Å | 2ZEJ | 2008 | [46] |
| homodimer | Negative-stain EM | Full-length Strep/FLAG-LRRK2 | - | 22 Å | N/A | 2016 | [47] |
| homodimer | Cryo-EM | Full-length 3xFLAG-LRRK2 | - | 16 Å | N/A | 2017 | [48] |
| homodimer | Crystallography | LRRK2 WD40 domain | Dimerization of WD40 domain | 2.6 Å | 6DLO/ 6DLP | 2019 | [42] |
| filamentous | Cryo-ET | Full-length LRRK2 (I2020T) | COR:COR WD40:WD40 | 14 Å | 6XR4 | 2020 | [43] |
| filamentous | Cryo-EM | LRRK2 RCKW 1 | COR:COR WD40:WD40 | 3.5 Å | 6VNO (6VP6/6VP7 /6VP8) | 2020 | [40] |
| monomer homodimer | Cryo-EM | Full-length LRRK2 | COR:COR | 3.7 Å 3.5 Å | 7LHW 7LHT | 2021 | [30] |
| monomer dimer tetramer (all with Rab29) | Cryo-EM | Full-length LRRK2 | LRRK2:Rab29 COR:COR WD40:kinase | 3.5 Å | N/A | 2022 | [44] |
| filamentous | Cryo-EM | LRRK2 RCKW 1 | COR:COR WD40:WD40 | 5.0 Å | 7THY/ 7THZ | 2022 | [49] |
| Rab | Phosphorylation Site by LRRK2 ([68] Unless Otherwise Noted) | Effect of Phosphorylation |
|---|---|---|
| Rab1 | Thr75 [69] | Unknown |
| Rab3 | Thr86 | Unknown |
| Rab5 | Ser84, Thr6 (in vitro [70]) | Delay in EGFR degradation [70] |
| Rab8 | Thr72 | Inhibition of ciliogenesis and centrosome cohesion [17,71,72] Decreased binding with Optineurin [73] Increased binding with RILPL2 [74] Mistrafficking of cargo to damaged lysosomes [75] Suppression of enlargement of stressed lysosomes [76] Activation of LRRK2 [52] |
| Rab10 | Thr73 | Inhibition of ciliogenesis and centrosome cohesion [15,71,77,78,79,80] Decreased binding to EHBP1L1 [81] Induction of LYTL by recruitment of JIP4 [82,83] Extracellular release of lysosomal contents under lysosomal stress [76] Lowers GCase activity [84] Induction of apoptosis after injury [85] |
| Rab12 | Ser106 | Perinuclear clustering of lysosomes via increase in RILPL1 binding [86] Activation and recruitment of LRRK2 [53,54] |
| Rab29 | Thr71, Ser72 | Regulation of trans-Golgi morphology [18] Recruitment of Rab29 itself to stressed lysosomes [87] |
| Rab35 | Thr72 | Induction of LYTL by recruitment of JIP4 [83] Increase in α-synuclein propagation [88] |
| Rab43 | Thr82 | Unknown |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komori, T.; Kuwahara, T. An Update on the Interplay between LRRK2, Rab GTPases and Parkinson’s Disease. Biomolecules 2023, 13, 1645. https://doi.org/10.3390/biom13111645
Komori T, Kuwahara T. An Update on the Interplay between LRRK2, Rab GTPases and Parkinson’s Disease. Biomolecules. 2023; 13(11):1645. https://doi.org/10.3390/biom13111645
Chicago/Turabian StyleKomori, Tadayuki, and Tomoki Kuwahara. 2023. "An Update on the Interplay between LRRK2, Rab GTPases and Parkinson’s Disease" Biomolecules 13, no. 11: 1645. https://doi.org/10.3390/biom13111645
APA StyleKomori, T., & Kuwahara, T. (2023). An Update on the Interplay between LRRK2, Rab GTPases and Parkinson’s Disease. Biomolecules, 13(11), 1645. https://doi.org/10.3390/biom13111645