
Hydroxyl Radical vs. One-Electron Oxidation Reactivities in an Alternating GC Double-Stranded Oligonucleotide: A New Type Electron Hole Stabilization

 ,
,  ,
,  ,
, 

Abstract
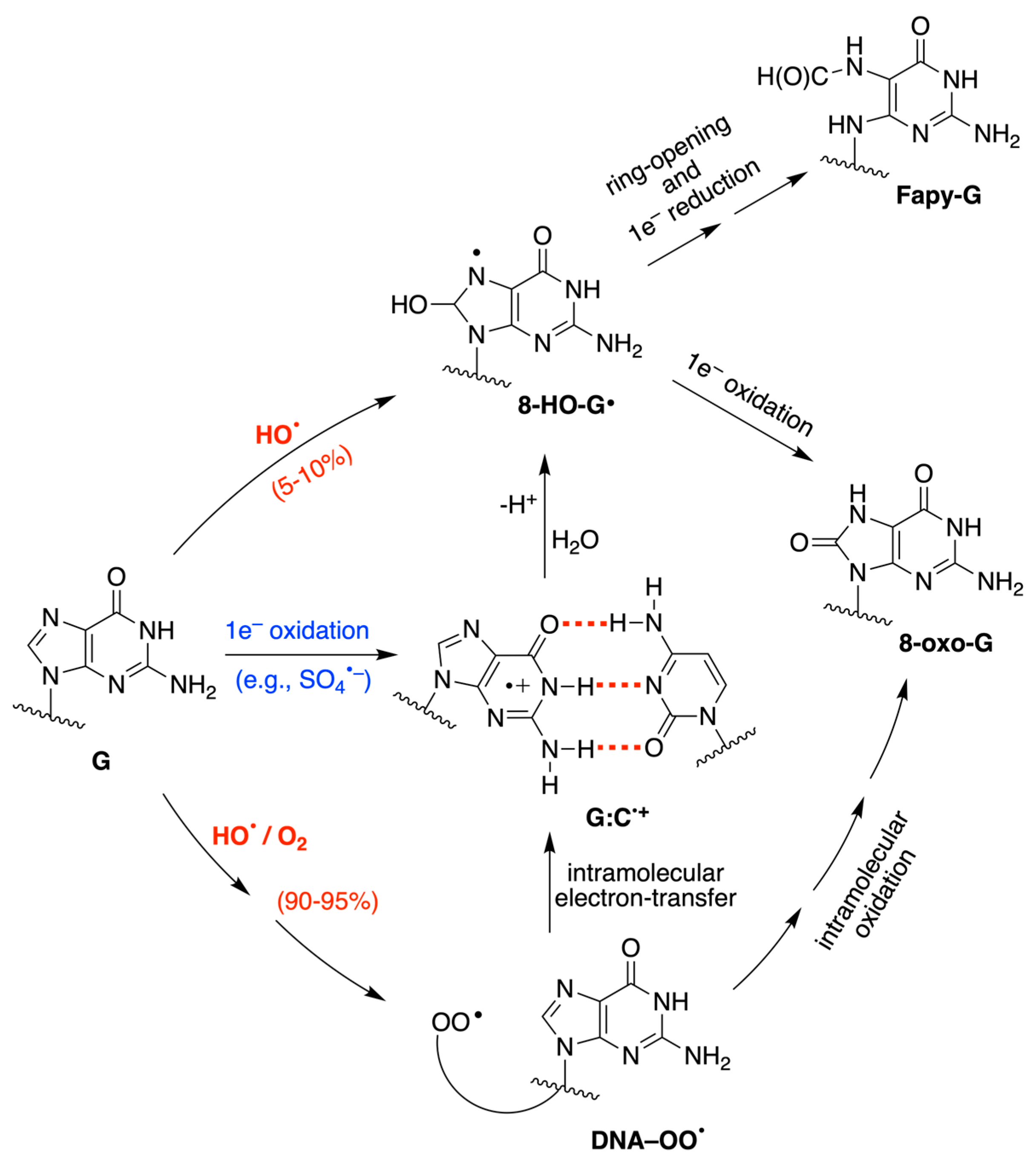
:1. Introduction
2. Materials and Methods
2.1. Preparation of Double-Stranded Oligonucleotide (ds-ODN)
2.2. Pulse Radiolysis
2.3. Computational Details
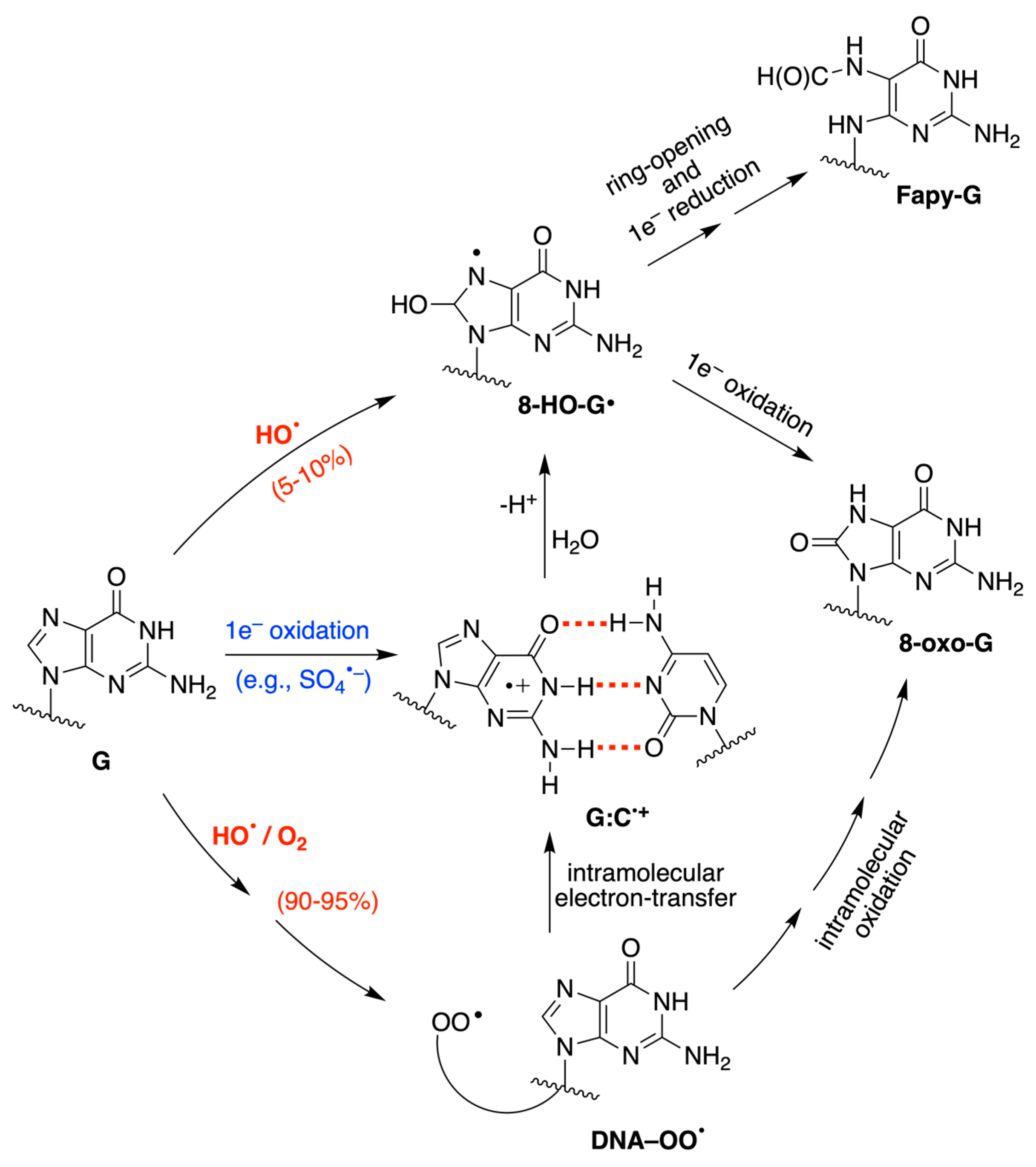
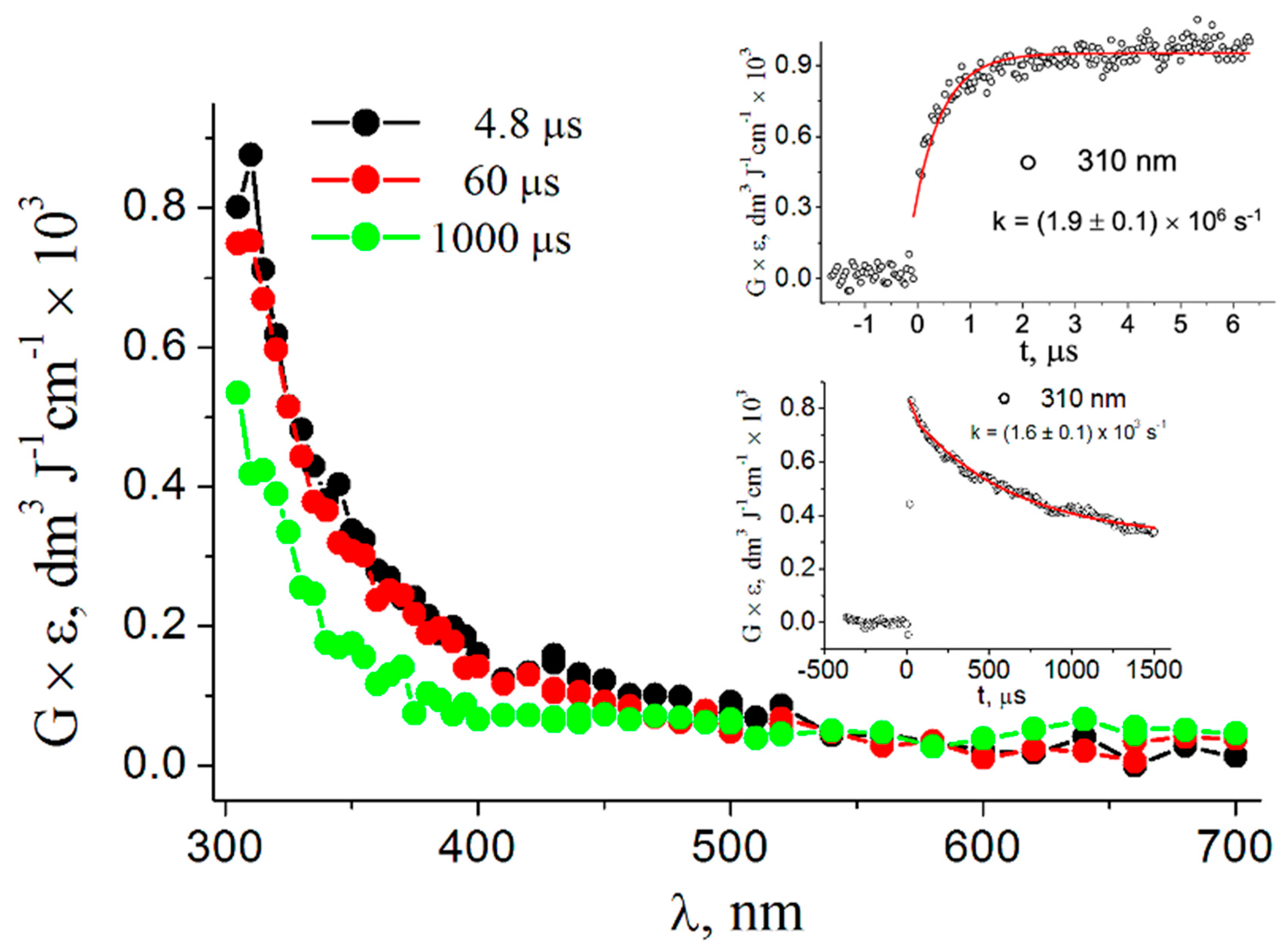
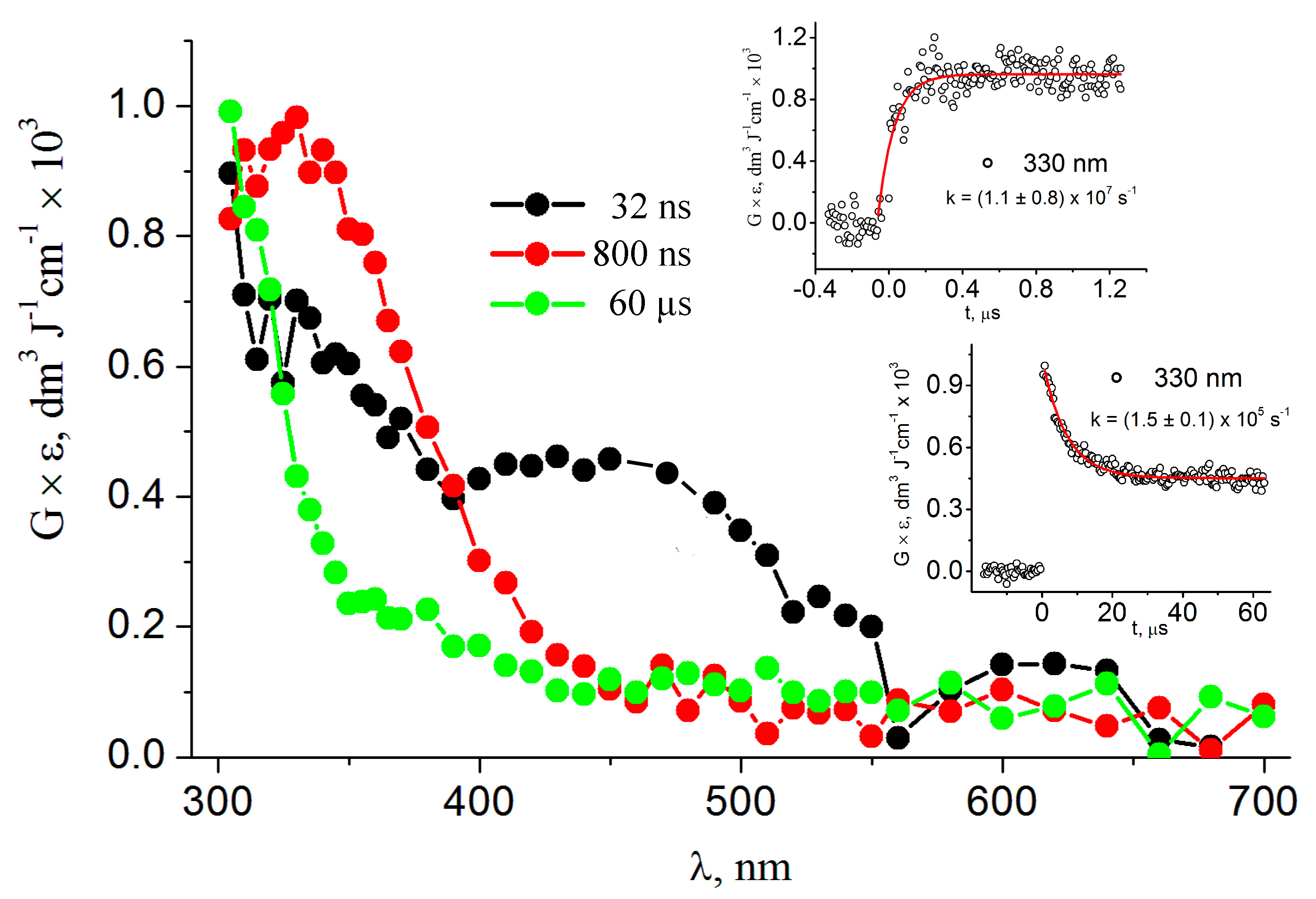
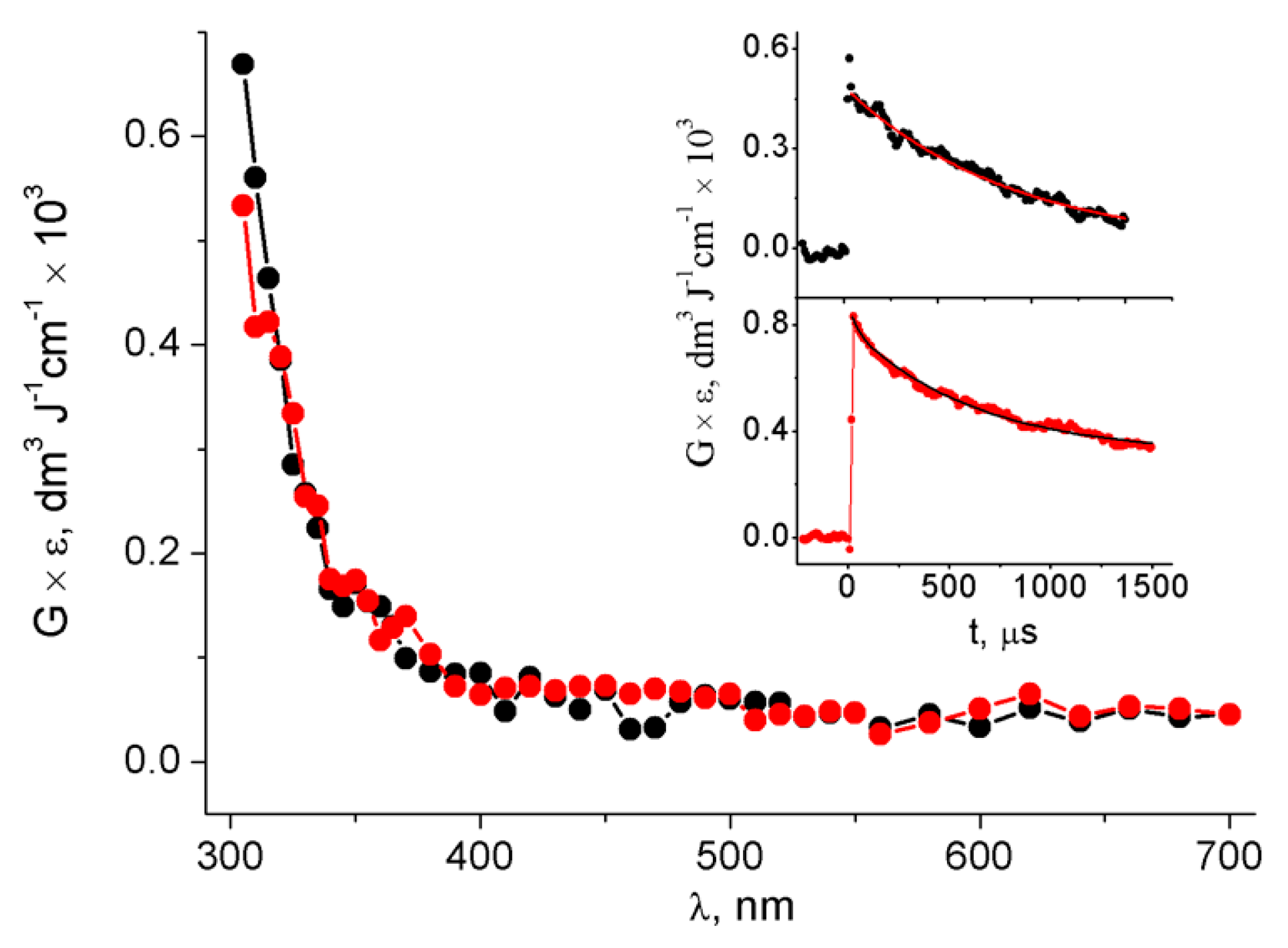
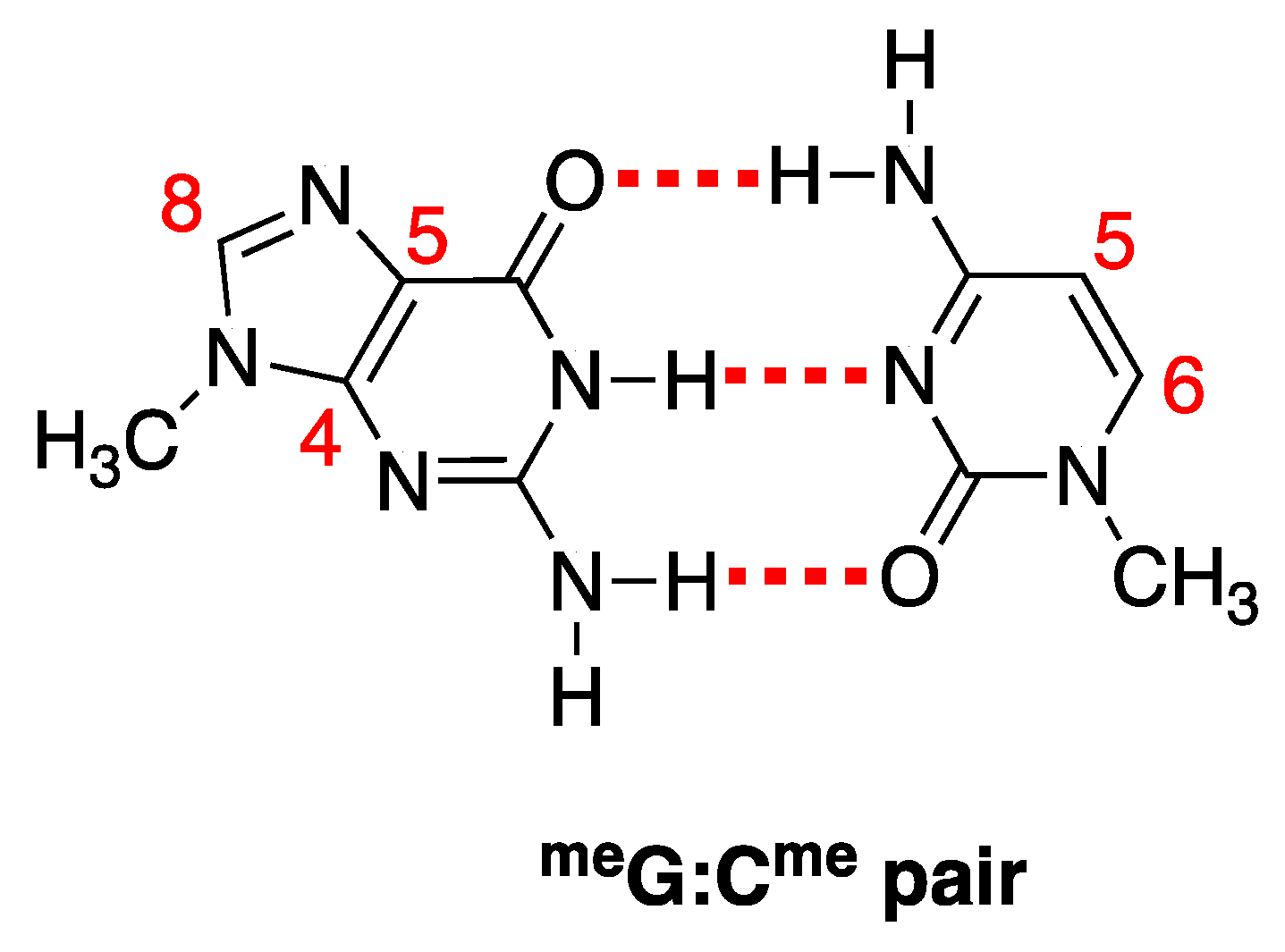
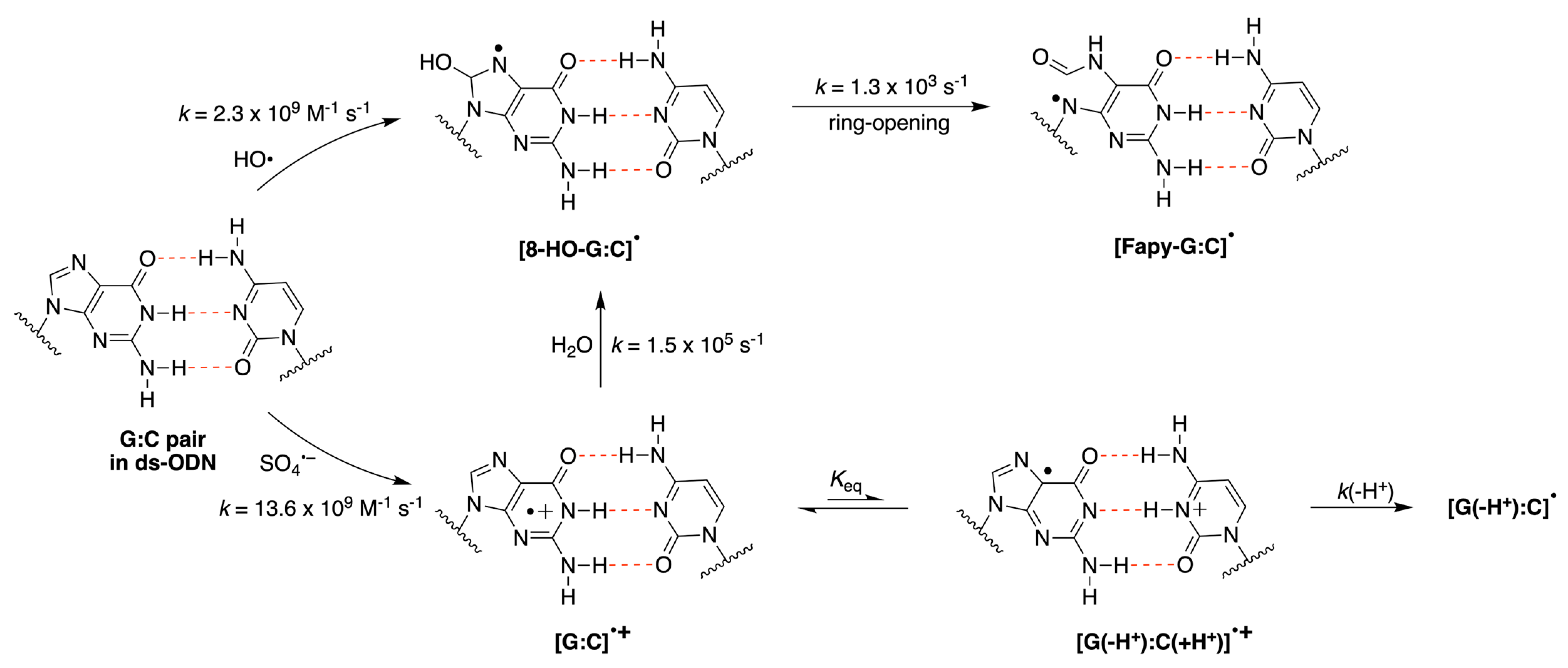
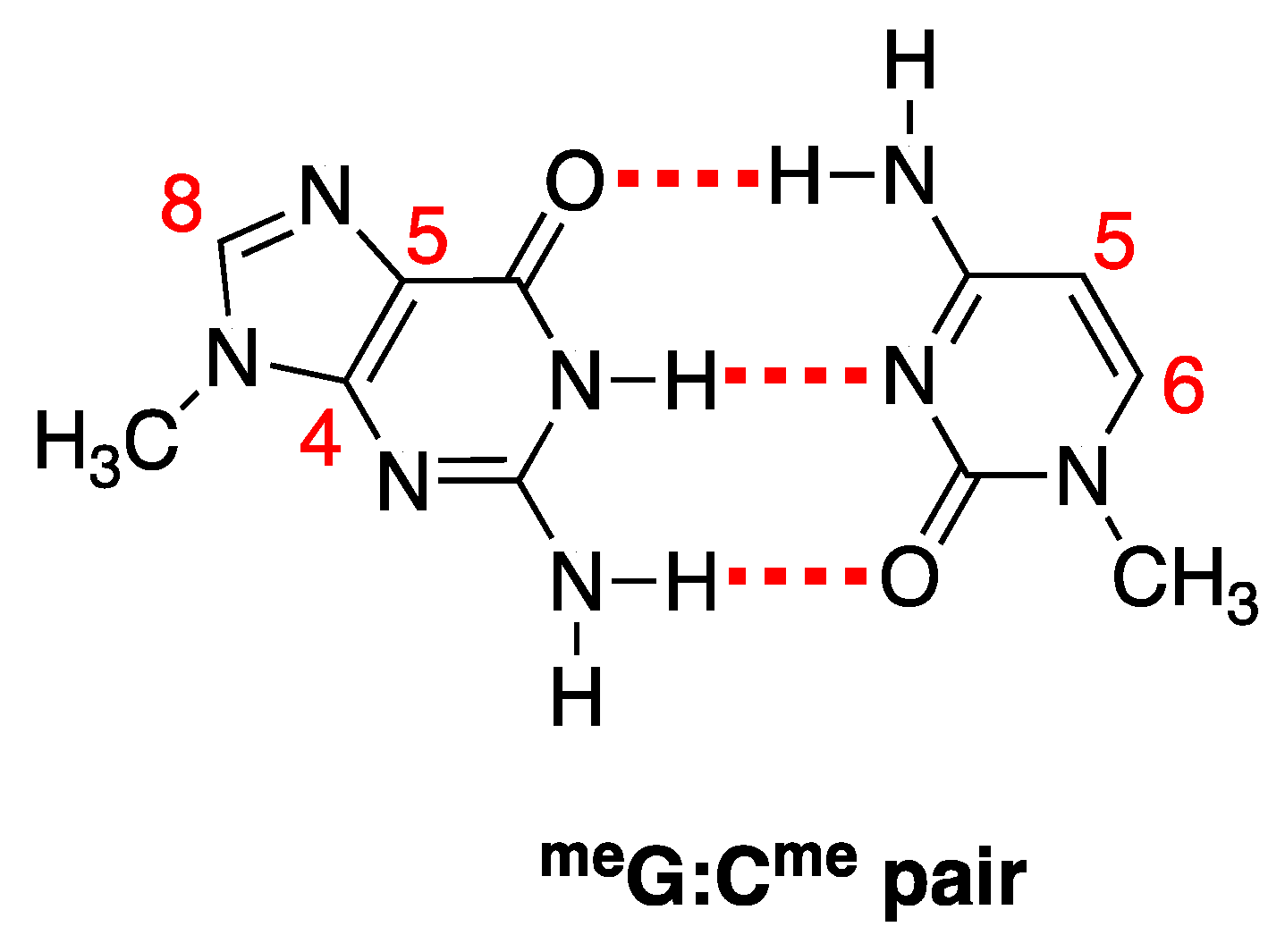
3. Results
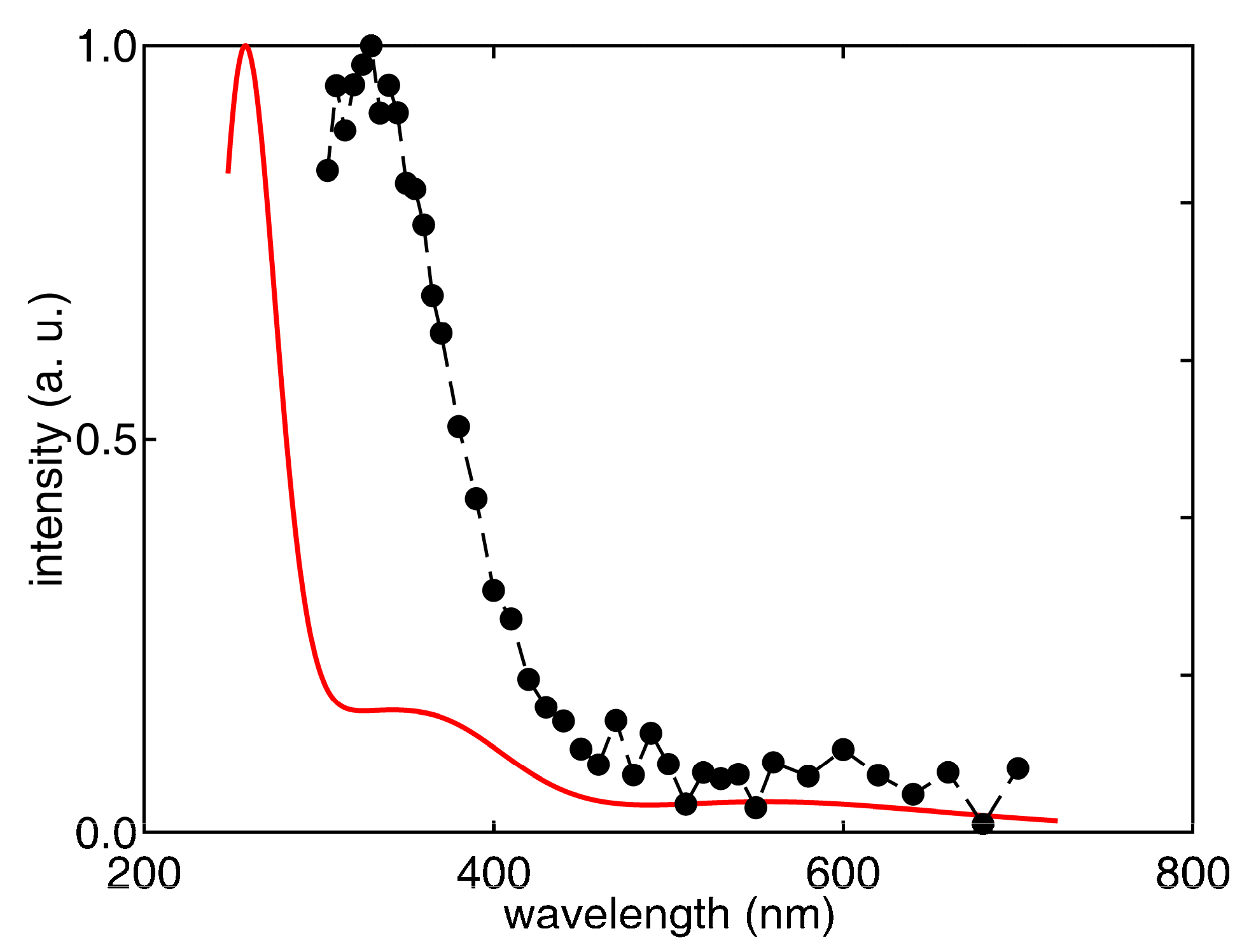
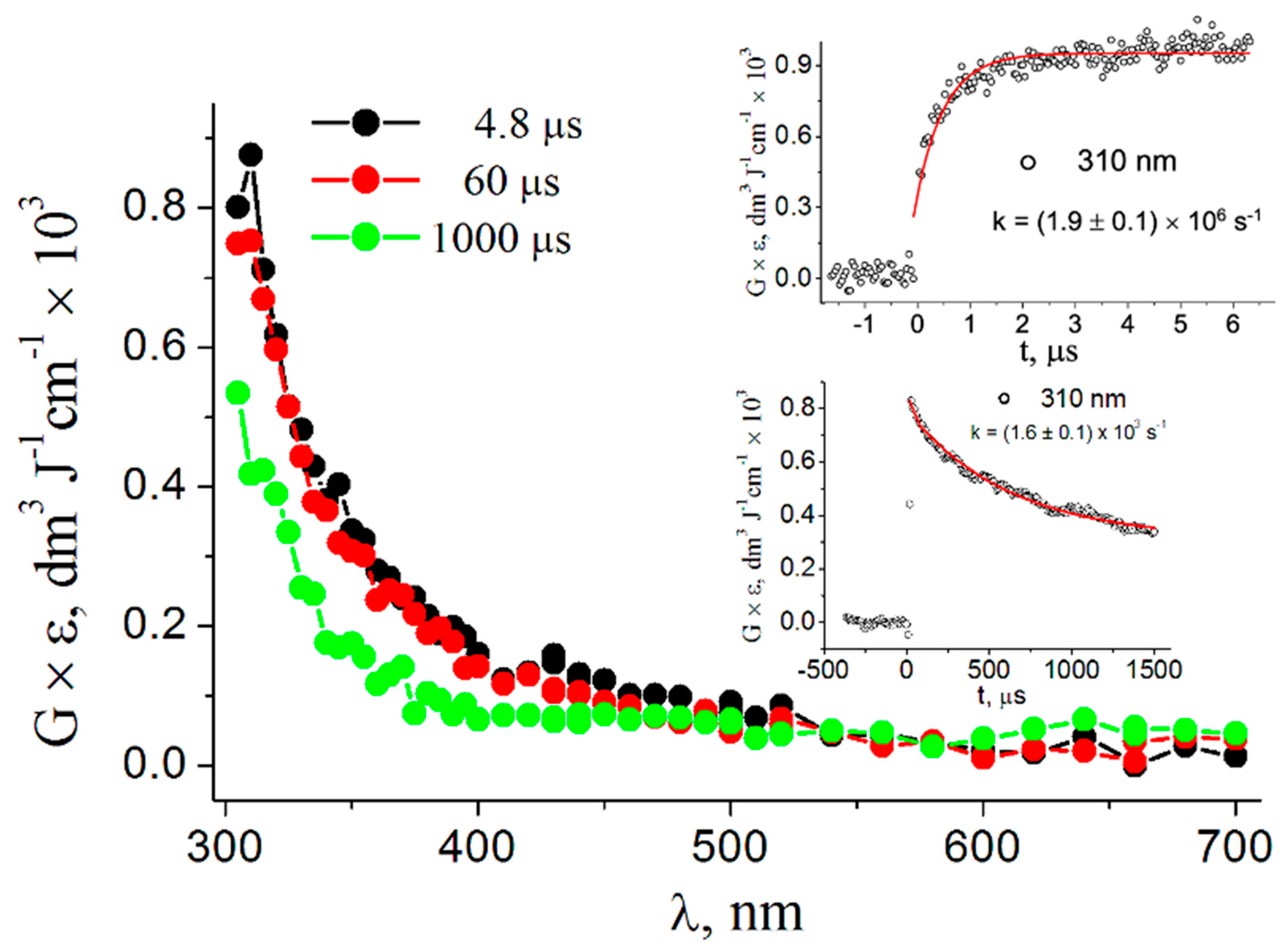
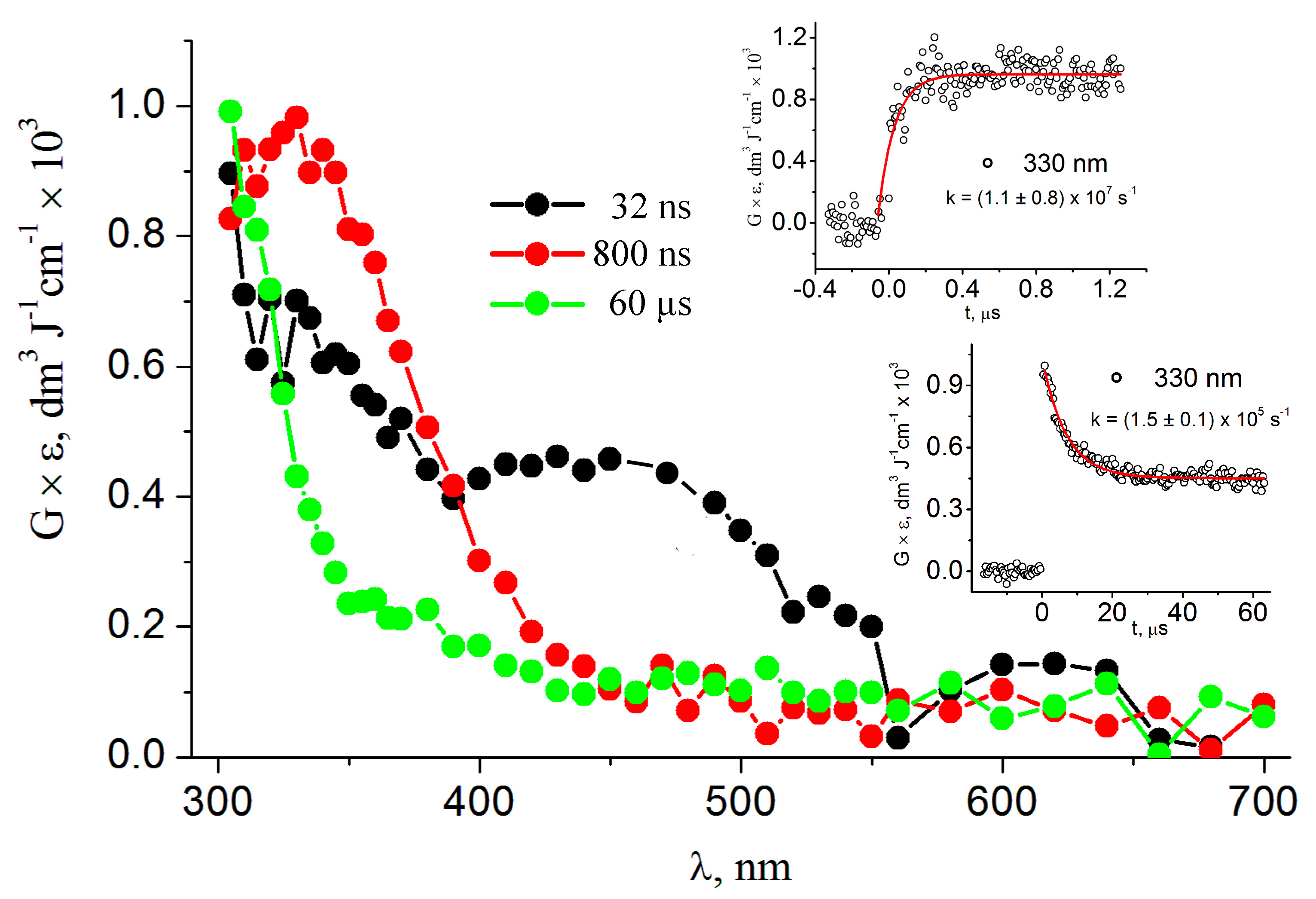
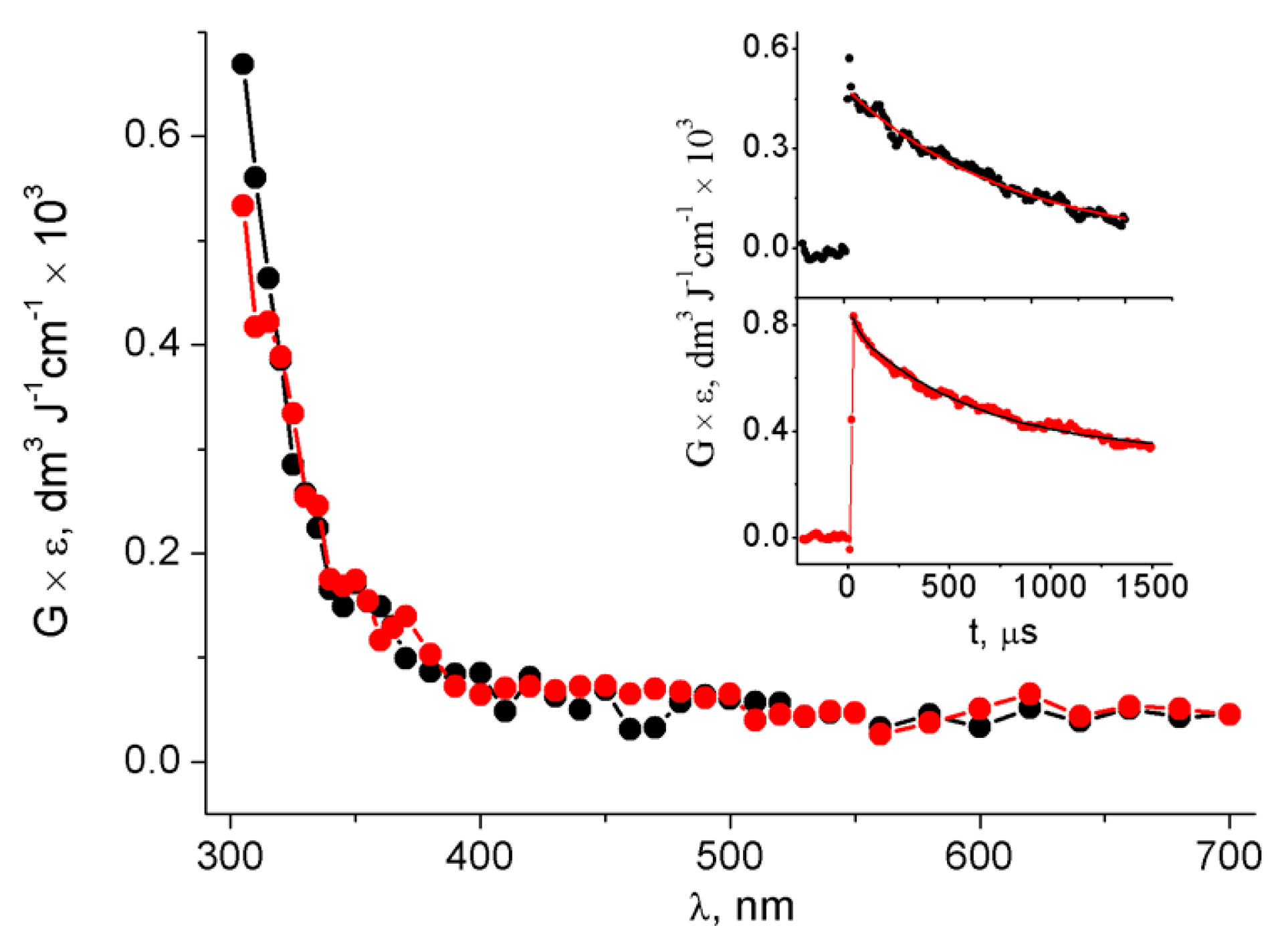
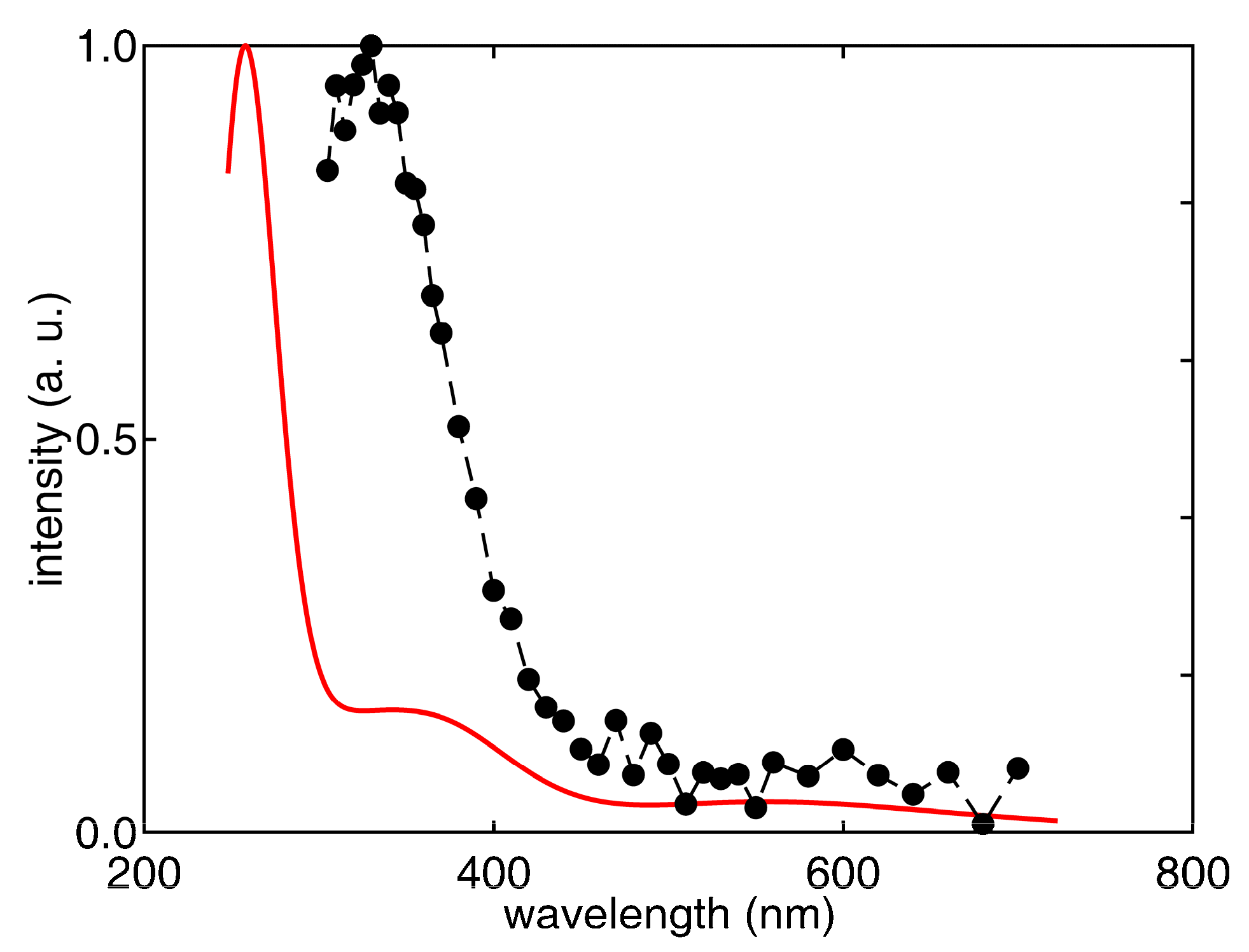
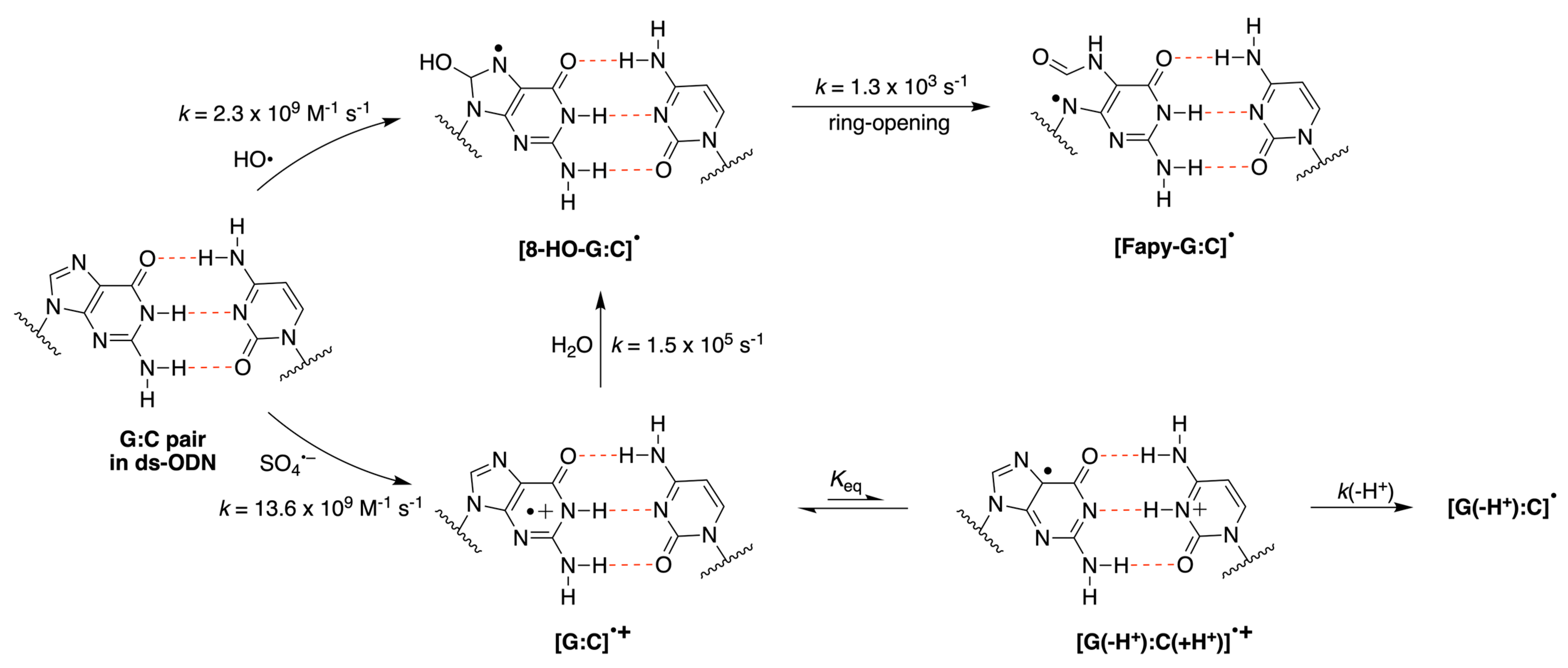
3.1. Pulse Radiolysis Study
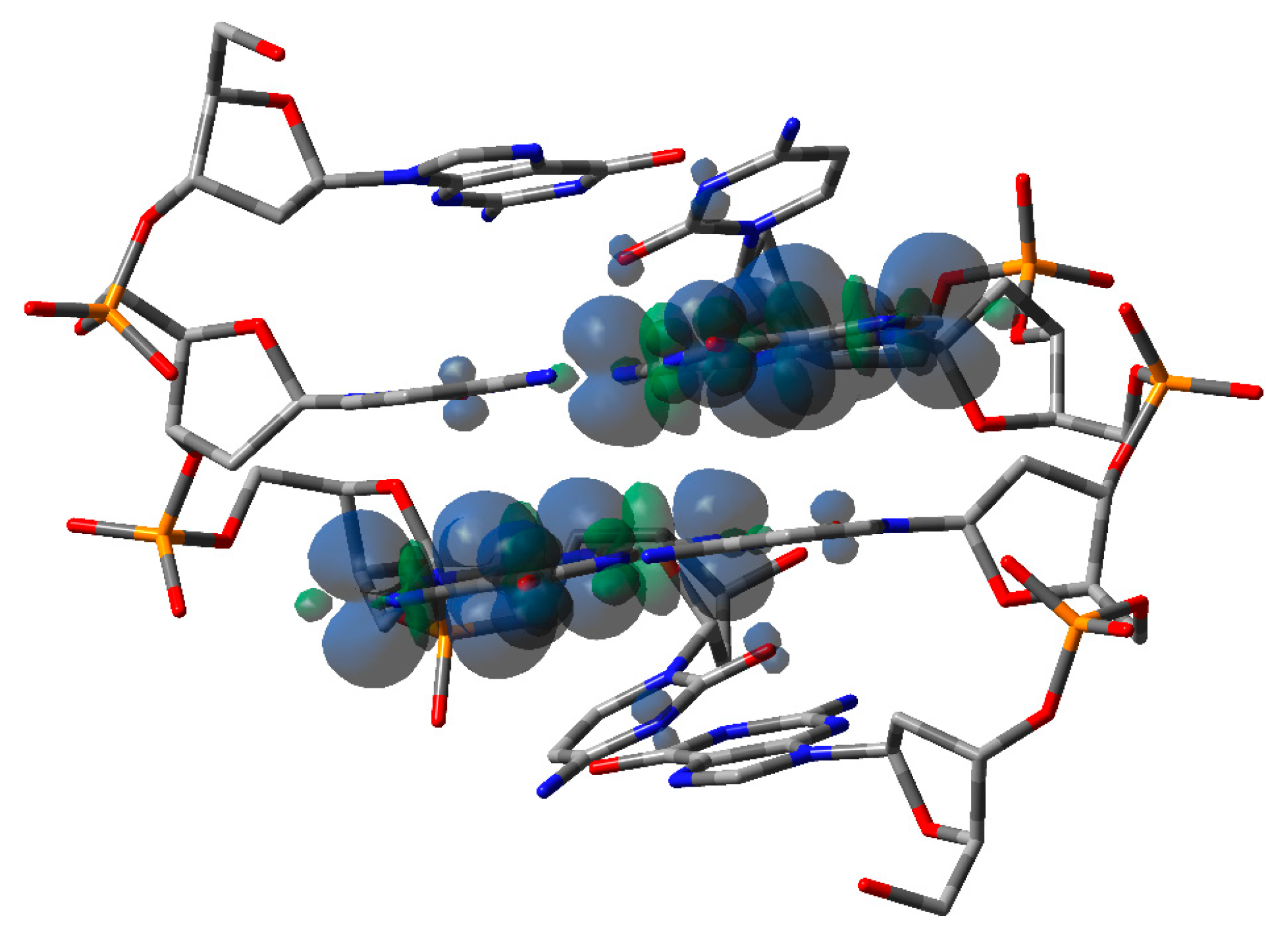
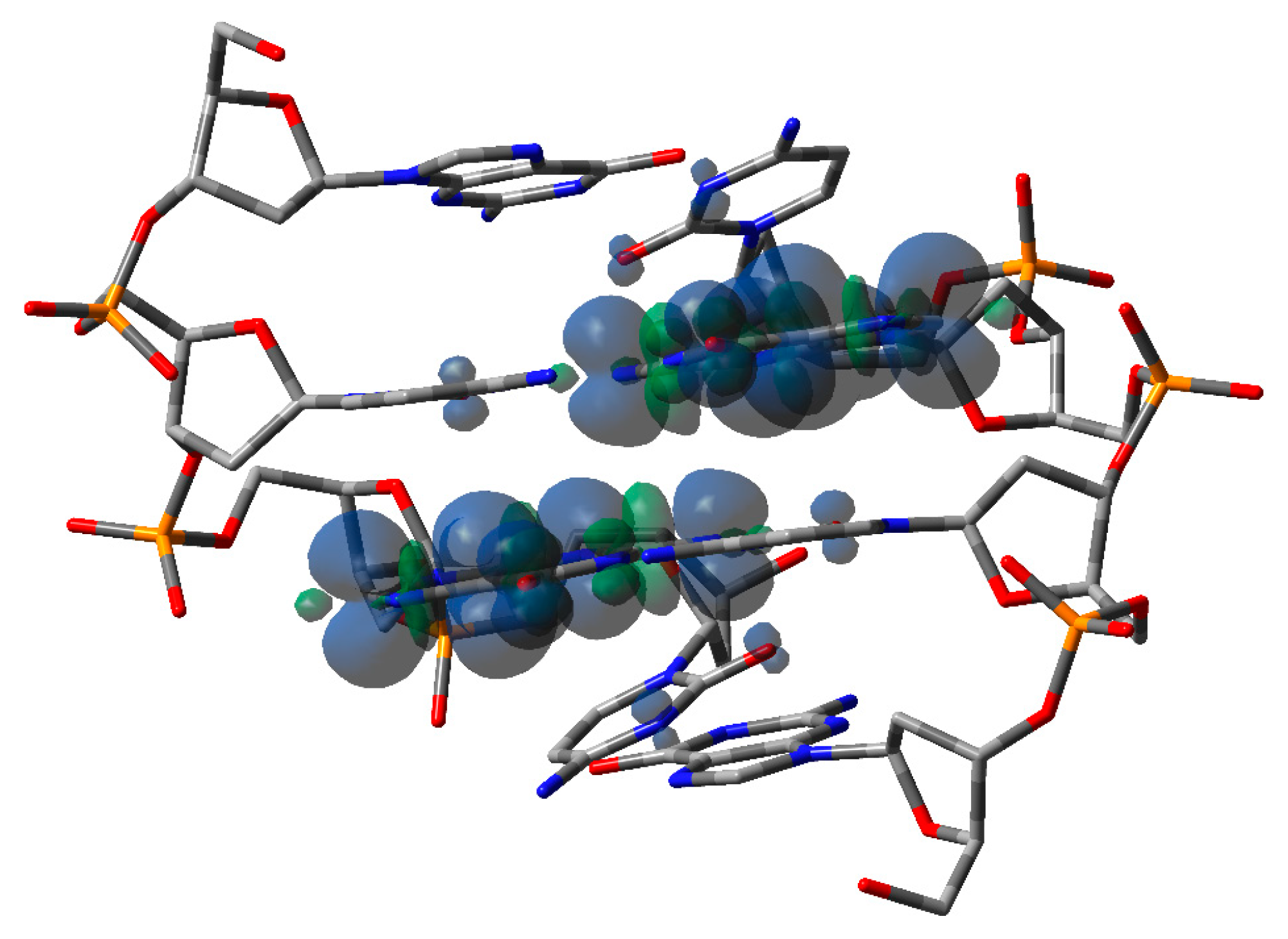
3.2. Theoretical Calculations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell. Biol. 2020, 21, 363–383. [Google Scholar]
- Dizdaroglu, M.; Lloyd, R.S. DNA Damage, DNA Repair and Disease; Royal Society of Chemistry: Croydon, UK, 2021. [Google Scholar]
- Chatgilialoglu, C.; Ferreri, C.; Krokidis, M.G.; Masi, A.; Terzidis, M.A. On the relevance of hydroxyl radical to purine DNA damage. Free Radic. Res. 2021, 55, 384–404. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Adhikary, A.; Dingfelder, M.; Dizdaroglu, M. Hydroxyl radical is a significant player in oxidative DNA damage in vivo. Chem. Soc. Rev. 2021, 50, 8355–8360. [Google Scholar] [CrossRef] [PubMed]
- Von Sonntag, C. Free-Radical-Induced DNA Damage and Its Repair—A Chemical Perspective; Springer Science: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Bergeron, F.; Auvre, F.; Radicella, J.P.; Ravanat, J.-L. HO radicals induce an unexpected high proportion of tandem base lesions refractory to repair by DNA glycosylases. Proc. Natl. Acad. Sci. USA 2010, 107, 5528–5533. [Google Scholar] [CrossRef] [PubMed]
- Ravanat, J.-L.; Breton, J.; Douki, T.; Gasparutto, D.; Grand, A.; Rachidi, W.; Sauvaigo, S. Radiation-mediated formation of complex damage to DNA: A chemical aspect overview. Br. J. Radiol. 2014, 87, 20130715. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Eriksson, L.A.; Krokidis, M.G.; Masi, A.; Wang, S.-D.; Zhang, R. Oxygen dependent purine lesions in double- stranded oligodeoxynucleotides: Kinetic and computational studies highlight the mechanism for 5′,8-cyplopurine formation. J. Am. Chem. Soc. 2020, 142, 5825–5833. [Google Scholar] [CrossRef]
- Douki, T.; Rivière, J.; Cadet, J. DNA tandem lesions containing 8-oxo-7,8-dihydroguanine and formamido residues arise from intramolecular addition of thymine peroxyl radical to guanine. Chem. Res. Toxicol. 2002, 15, 445–454. [Google Scholar] [CrossRef]
- Robert, G.; Wagner, J.R. Tandem lesions arising from 5-(uracilyl)methyl peroxyl radical addition to guanine: Product analysis and mechanistic studies. Chem. Res. Toxicol. 2020, 33, 565–575. [Google Scholar] [CrossRef]
- Dizdaroglu, M.; Kirkali, G.; Jaruga, P. Formamidopyrimidines in DNA: Mechanisms of formation, repair, and biological effects. Free Radic. Biol. Med. 2008, 45, 1610–1621. [Google Scholar]
- Greenberg, M.M. The formamidopyrimidines: Purine lesions formed in competition with 8-oxopurines from oxidative stress. Acc. Chem. Res. 2012, 45, 588–597. [Google Scholar] [CrossRef]
- Ryan, B.J.; Yang, H.; Bacurio, J.H.T.; Smith, M.R.; Basu, A.K.; Marc, M.; Greenberg, M.M.; Freudenthal, B.D. Structural Dynamics of a Common Mutagenic Oxidative DNA Lesion in Duplex DNA and during DNA Replication. J. Am. Chem. Soc. 2022, 144, 8054–8065. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Brett, A.M.; Piedade, J.A.P.; Silva, L.A.; Diculescu, V.C. Voltammetric Determination of All DNA Nucleotides. Anal. Biochem. 2004, 332, 321–329. [Google Scholar] [CrossRef]
- Brotons, A.; Mas, L.A.; Metters, J.P.; Banks, C.E.; Iniesta, J. Voltammetric Behaviour of Free DNA Bases, Methylcytosine and Oligonucleotides at Disposable Screen Printed Graphite Electrode Platforms. Analyst 2013, 138, 5239–5249. [Google Scholar] [CrossRef]
- Pluharová, E.; Slavíček, P.; Jungwirth, P. Modeling Photoionization of Aqueous DNA and Its Components. Acc. Chem. Res. 2015, 48, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Peluso, A.; Caruso, T.; Landi, A.; Capobianco, A. The Dynamics of Hole Transfer in DNA. Molecules 2019, 24, 4044. [Google Scholar] [CrossRef] [PubMed]
- Genereux, J.C.; Barton, J.K. Mechanisms for DNA Charge Transport. Chem. Rev. 2010, 110, 1642–1662. [Google Scholar]
- Kawai, K.; Majima, T. Hole Transfer Kinetics of DNA. Acc. Chem. Res. 2013, 46, 2616–2625. [Google Scholar] [CrossRef]
- Zwang, T.J.; Tse, E.C.M.; Barton, J.K. Sensing DNA through DNA Charge Transport. ACS Chem. Biol. 2018, 13, 1799–1809. [Google Scholar] [CrossRef]
- Lewis, F.D.; Young, R.M.; Wasielewski, M.R. Tracking Photoinduced Charge Separation in DNA: From Start to Finish. Acc. Chem. Res. 2018, 51, 1746–1754. [Google Scholar] [CrossRef]
- Caruso, T.; Carotenuto, M.; Vasca, E.; Peluso, A. Direct Experimental Observation of the Effect of the Base Pairing on the Oxidation Potential of Guanine. J. Am. Chem. Soc. 2005, 127, 15040–15041. [Google Scholar] [CrossRef]
- Caruso, T.; Capobianco, A.; Peluso, A. The Oxidation Potential of Adenosine and Adenosine-Thymidine Base-Pair in Chloroform Solution. J. Am. Chem. Soc. 2007, 129, 15347–15353. [Google Scholar] [CrossRef] [PubMed]
- Capobianco, A.; Caruso, T.; Peluso, A. Hole Delocalization over Adenine Tracts in Single Stranded DNA Oligonucleotides. Phys. Chem. Chem. Phys. 2015, 17, 4750–4756. [Google Scholar] [CrossRef] [PubMed]
- Capobianco, A.; Caruso, T.; D’Ursi, A.M.; Fusco, S.; Masi, A.; Scrima, M.; Chatgilialoglu, C.; Peluso, A. Delocalized Hole Domains in Guanine-Rich DNA Oligonucleotides. J. Phys. Chem. B 2015, 119, 5462–5466. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, A.; Khanduri, D.; Sevilla, M.D. Direct observation of the hole protonation state and hole localization site in DNA-oligomers. J. Am. Chem. Soc. 2009, 131, 8614–8619. [Google Scholar] [CrossRef]
- Shukla, L.I.; Adhikary, A.; Pazdro, R.; Becker, D.; Sevilla, M.D. Formation of 8-oxo-7,8-dihydroguanine-radicals in γ-irradiated DNA by multiple one-electron oxidations. Nucl. Acids Res. 2004, 32, 6565–6574. [Google Scholar] [CrossRef]
- Becker, D.; Kumar, A.; Adhikary, A.; Sevilla, M.D. Gamma-and Ion-beam DNA Radiation Damage: Theory and Experiments. In DNA Damage, DNA Repair and Disease; Royal Society of Chemistry: Croydon, UK, 2021; Chapter 31; pp. 426–457. [Google Scholar]
- Barnett, R.N.; Bongiorno, A.; Cleveland, C.L.; Joy, A.; Landman, U.; Schuster, G.B. Oxidative Damage to DNA: Counterion-Assisted Addition of Water to ionized DNA. J. Am. Chem. Soc. 2006, 128, 10795–10800. [Google Scholar] [CrossRef]
- Giese, B.; Spichty, M. Long Distance Charge Transport through DNA: Quantification and Extension of the Hopping Model. ChemPhysChem 2000, 1, 195–198. [Google Scholar] [CrossRef]
- Chatgilialoglu, C. The Two Faces of the Guanyl Radical: Molecular Context and Behavior. Molecules 2021, 26, 3511. [Google Scholar] [CrossRef]
- Cadet, J.; Wagner, J.R. Oxidatively generated base damage to cellular DNA by hydroxyl radical and one-electron oxidants: Similarities and differences. Arch. Biochem. Biophys. 2014, 557, 47–54. [Google Scholar]
- Chatgilialoglu, C.; D’Angelantonio, M.; Guerra, M.; Kaloudis, P.; Mulazzani, Q.G. A reevaluation of the ambident reactivity of guanine moiety towards hydroxyl radicals. Angew. Chem. Int. Ed. 2009, 48, 2214–2217. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; D’Angelantonio, M.; Kciuk, G.; Bobrowski, K. New insights into the reaction paths of hydroxyl radicals with 2′-deoxyguanosine. Chem. Res. Toxicol. 2011, 24, 2200–2206. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Caminal, C.; Altieri, A.; Vougioukalakis, G.C.; Mulazzani, Q.G.; Gimisis, T.; Guerra, M. Tautomerism in the guanyl radicals. J. Am. Chem. Soc. 2006, 128, 13796–13805. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Bazzanini, R.; Jimenez, L.B.; Miranda, M.A. (5′S)- and (5′R)-5′,8-cyclo-2′-deoxyguanosine: Mechanistic insights on the 2′-deoxyguanosin-5′-yl radical cyclization. Chem. Res. Toxicol. 2007, 20, 1820–1824. [Google Scholar] [CrossRef] [PubMed]
- Aravindakumar, C.T.; Schuchmann, M.N.; Rao, B.S.M.; von Sonntag, J.; von Sonntag, C. The Reactions of Cytidine and 2′-Deoxycytidine with SO4•– Revisited. Pulse Radiolysis and Product Studies. Org. Biomol. Chem. 2003, 1, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, H.; Yang, C.; Jie, J.; Dai, X.; Zhou, Q.; Liu, K.; Song, D.; Su, H. Degradation of Cytosine Radical Cations in 2′-Deoxycytidine and in i-Motif DNA: Hydrogen-Bonding Guided Pathways. J. Am. Chem. Soc. 2019, 141, 1970–1979. [Google Scholar] [CrossRef]
- Peng, H.; Jie, J.; Mortimer, I.P.; Ma, Z.; Su, H.; Greenberg, M.M. Reactivity and DNA Damage by Independently Generated 2′-Deoxycytidin-N4-yl Radical. J. Am. Chem. Soc. 2021, 143, 14738–14747. [Google Scholar] [CrossRef] [PubMed]
- Arhondakis, S.; Auletta, F.; Torelli, G.; D’Onofrio, G. Base composition and expression level of human genes. Gene 2004, 325, 165–169. [Google Scholar] [CrossRef]
- Bernaola-Galván, P.; Carpena, P.; Gómez-Martín, C.; Oliver, J.L. Compositional Structure of the Genome: A Review. Biology 2023, 12, 849. [Google Scholar]
- Kobayashi, K.; Yamagami, R.; Tagawa, S. Effect of base sequence and deprotonation of guanine cation radical in DNA. J. Phys. Chem. B 2008, 112, 10752–10757. [Google Scholar] [CrossRef]
- Rokhlenko, Y.; Cadet, J.; Geacintov, N.E.; Shafirovich, V. Mechanistic aspects of hydration of guanine radical cations in DNA. J. Am. Chem. Soc. 2014, 136, 5956–5962. [Google Scholar]
- Banyasz, A.; Martínez-Fernández, L.; Improta, R.; Ketola, T.-M.; Balty, C.; Markovitsi, D. Radicals generated in alternating guanine cytosine duplexes by direct absorption of low-energy UV radiation. Phys. Chem. Chem. Phys. 2018, 20, 21381–21389. [Google Scholar] [CrossRef] [PubMed]
- Balanikas, E.; Banyasz, A.; Baldacchino, G.; Markovitsi, D. Populations and Dynamics of Guanine Radicals in DNA strands—Direct versus Indirect Generation. Molecules 2019, 24, 2347. [Google Scholar] [CrossRef] [PubMed]
- Banyasz, A.; Martínez-Fernandez, L.; Balty, C.; Perron, M.; Douki, T.; Improta, R.; Markovitsi, D. Absorption of Low-Energy UV Radiation by Human Telomere G-Quadruplexes Generates Long-Lived Guanine Radical Cations. J. Am. Chem. Soc. 2017, 139, 10561–10568. [Google Scholar] [CrossRef]
- Masi, A.; Sabbia, A.; Ferreri, C.; Manoli, F.; Lai, Y.; Laverde, E.; Liu, Y.; Krokidis, M.G.; Chatgilialoglu, C.; Faraone Mennella, M.R. Diastereomeric Recognition of 5′,8-cyclo-2′-Deoxyadenosine Lesions by Human Poly(ADP-ribose) Polymerase 1 in a Biomimetic Model. Cells 2019, 8, 116. [Google Scholar] [CrossRef] [PubMed]
- Bobrowski, K. Free radicals in chemistry, biology and medicine: Contribution of radiation chemistry. Nukleonika 2005, 50 (Suppl. S3), S67–S76. [Google Scholar]
- Janata, E.; Schuler, R.H. Rate constant for scavenging eaq− in N2O-saturated solutions. J. Phys. Chem. 1982, 86, 2078–2084. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar]
- Lu, X.J.; Olson, W.K. 3DNA: A Versatile, Integrated Software System for the Analysis, Rebuilding and Visualization of Three-dimensional Nucleic-Acid Structures. Nat. Protoc. 2008, 3, 1213–1227. [Google Scholar]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar]
- Capobianco, A.; Velardo, A.; Peluso, A. Single-Stranded DNA Oligonucleotides Retain Rise Coordinates Characteristic of Double Helices. J. Phys. Chem. B 2018, 122, 7978–7989. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A New Hybrid Exchange-Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for hydrated electrons, hydrogen atoms and hydroxyl radicals (OH/O−) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Buxton, G.V. An overview of the radiation chemistry of liquids. In Radiation Chemistry: From Basics to Applications in Material and Life Sciences; Spotheim Maurizot, M., Mostafavi, M., Douki, T., Belloni, J., Eds.; EDP Sciences: Paris, France, 2008; pp. 3–16. [Google Scholar]
- Zhang, R.; Eriksson, L.A. Effects of OH Radical Addition on Proton Transfer in the Guanine-Cytosine Base Pair. J Phys. Chem. B 2007, 111, 6571–6576. [Google Scholar] [CrossRef] [PubMed]
- Cerón-Carrasco, J.P.; Jacquemin, D. Interplay between hydroxyl radical attack and H-bond stability in guanine–cytosine. RCS Adv. 2012, 2, 11867–11875. [Google Scholar] [CrossRef]
- Li, M.; Diao, L.; Liao, X.; Kou, L.; Lu, W. DFT study on addition reaction mechanism of guanine-cytosine base pair with OH radical. J. Phys. Org. Chem. 2015, 28, 437–444. [Google Scholar]
- Ambrosio, F.; Miceli, G.; Pasquarello, A. Redox levels in aqueous solution: Effect of van der Waals interactions and hybrid functionals. Chem. Phys. 2015, 143, 244508. [Google Scholar] [CrossRef]
- Adriaanse, C.; Cheng, J.; Chau, V.; Sulpizi, M.; VandeVondele, J.; Sprik, M. Aqueous Redox Chemistry and the Electronic Band Structure of Liquid Water. J. Phys. Chem. Lett. 2012, 3, 3411–3415. [Google Scholar] [CrossRef]
- Capobianco, A.; Russo, A.; Lattanzi, A.; Peluso, A. On the Mechanism of Asymmetric Epoxidation of Enones Catalyzed by α,α-L-Diarylprolinols: A Theoretical Insight. Adv. Synth. Catal. 2012, 354, 2789–2796. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, Q.; Fang, Y.; Chen, W.; Kirchon, A.A.; Baci, M.; Feng, M.; Sharma, V.K.; Zhou, H.-C. 7—Metal-organic frameworks for capture and degradation of organic pollutants. In Metal-Organic Frameworks (MOFs) for Environmental Applications; Ghosh, S.K., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 203–229. [Google Scholar]
- Fukuzumi, S.; Miyao, H.; Ohkubo, K.; Suenobu, T. Electron-Transfer Oxidation Properties of DNA Bases and DNA Oligomers. J. Phys. Chem. A 2005, 109, 3285–3294. [Google Scholar]
- Capobianco, A.; Carotenuto, M.; Caruso, T.; Peluso, A. The Charge-Transfer Band of an Oxidized Watson-Crick Guanosine-Cytidine Complex. Angew. Chem. Int. Ed. 2009, 48, 9526–9528. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sevilla, M.D. Excited States of One-Electron Oxidized Guanine-Cytosine Base Pair Radicals: A Time Dependent Density Functional Theory Study. J. Phys. Chem. A 2019, 123, 3098–3108. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.A.; Mishra, A.K.; Young, R.M.; Brown, K.E.; Wasielewski, M.R.; Lewis, F.D. Direct Observation of the Hole Carriers in DNA Photoinduced Charge Transport. J. Am. Chem. Soc. 2016, 138, 5491–5494. [Google Scholar] [CrossRef]
- Capobianco, A.; Caruso, T.; Celentano, M.; La Rocca, M.V.; Peluso, A. Proton Transfer in Oxidized Adenosine Self-Aggregates. J. Chem. Phys. 2013, 139, 145101. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, H.; Saito, I. Theoretical Studies of GG-Specific Photocleavage of DNA via Electron Transfer: Significant Lowering of Ionization Potential and 5′-Localization of HOMO of Stacked GG Bases in B-Form DNA. J. Am. Chem. Soc. 1996, 118, 7063–7068. [Google Scholar] [CrossRef]
- Capobianco, A.; Landi, A.; Peluso, A. Modeling DNA Oxidation in Water. Phys. Chem. Chem. Phys. 2017, 19, 13571–13578. [Google Scholar] [CrossRef]
- Conwell, E.M.; Bloch, S.M.; McLaughlin, P.M.; Basko, D.M. Duplex Polarons in DNA. J. Am. Chem. Soc. 2007, 129, 9175–9181. [Google Scholar] [CrossRef]
- Schüssler, H.; Navaratnam, S.; Distel, L. Rate constants for the reactions of DNA with hydrated electrons and with OH-radicals. Radiat. Phys. Chem. 2005, 73, 163–168. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Krokidis, M.G.; Masi, A.; Barata-Vallejo, S.; Ferreri, C.; Terzidis, M.A.; Szreder, T.; Bobrowski, K. New insights into the reaction paths of hydroxyl radicals with purine moieties in DNA and double-stranded oligonucleotides. Molecules 2019, 24, 3860. [Google Scholar] [CrossRef]
- Rokhlenko, Y.; Geacintov, N.E.; Shafirovich, V. Lifetimes and reaction pathways of guanine radical cations and neutral guanine radicals in an oligonucleotide in aqueous solutions. J. Am. Chem. Soc. 2012, 134, 4955–4962. [Google Scholar] [CrossRef]
- Steenken, S. Purine bases, nucleosides, and nucleotides: Aqueous solution redox chemistry and transformation reactions of their radical cations and e– and OH adducts. Chem. Rev. 1989, 89, 503–520. [Google Scholar] [CrossRef]
- Kobayashi, K.; Tagawa, S. Direct Observation of Guanine Radical Cation Deprotonation in Duplex DNA Using Pulse Radiolysis. J. Am. Chem. Soc. 2003, 125, 10213–10218. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
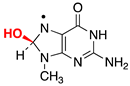 |  | 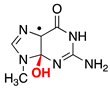 | 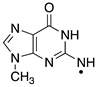 | 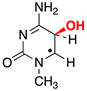 | 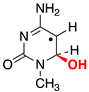 | 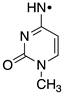 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ΔE = −31.7 | ΔE = −9.9 | ΔE = −15.0 | ΔE = −21.7 | ΔE = −17.3 | ΔE = −18.6 | ΔE = −9.4 | |||||||
| λ, nm | f | λ, nm | f | λ, nm | f | λ, nm | f | λ, nm | f | λ, nm | f | λ, nm | f |
| 430 | 0.019 | 535 | 0.008 | 481 | 0.007 | 595 | 0.006 | 420 | 0.065 | 808 | 0.003 | 1788 | 0.003 |
| 397 | 0.021 | 483 | 0.074 | 437 | 0.009 | 570 | 0.001 | 346 | 0.024 | 632 | 0.015 | 874 | 0.033 |
| 386 | 0.002 | 402 | 0.033 | 399 | 0.005 | 567 | 0.079 | 313 | 0.002 | 488 | 0.021 | 338 | 0.028 |
| 376 | 0.040 | 381 | 0.006 | 377 | 0.014 | 482 | 0.007 | 312 | 0.028 | 413 | 0.002 | 270 | 0.011 |
| 353 | 0.002 | 363 | 0.036 | 362 | 0.054 | 447 | 0.011 | 373 | 0.001 | 265 | 0.108 | ||
| 339 | 0.010 | 360 | 0.053 | 356 | 0.017 | 350 | 0.056 | 368 | 0.001 | ||||
| 325 | 0.041 | 340 | 0.005 | 350 | 0.059 | 309 | 0.085 | 324 | |||||
| 309 | 0.073 | 328 | 0.003 | 347 | 0.038 | ||||||||
| 308 | 0.019 | 321 | 0.011 | 337 | 0.034 | ||||||||
| Substrate | k(HO•), M−1 s−1 | k(SO4•−), M−1 s−1 |
|---|---|---|
| 2′-dGuo 1 | 5.7 × 109 | 4.1 × 109 |
| 2′-dCyd 1 | 5.6 × 109 | 1.6 × 109 |
| G:C pair in ds-ODN 2 | 2.3 × 109 | 13.6 × 109 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masi, A.; Capobianco, A.; Bobrowski, K.; Peluso, A.; Chatgilialoglu, C. Hydroxyl Radical vs. One-Electron Oxidation Reactivities in an Alternating GC Double-Stranded Oligonucleotide: A New Type Electron Hole Stabilization. Biomolecules 2023, 13, 1493. https://doi.org/10.3390/biom13101493
Masi A, Capobianco A, Bobrowski K, Peluso A, Chatgilialoglu C. Hydroxyl Radical vs. One-Electron Oxidation Reactivities in an Alternating GC Double-Stranded Oligonucleotide: A New Type Electron Hole Stabilization. Biomolecules. 2023; 13(10):1493. https://doi.org/10.3390/biom13101493
Chicago/Turabian StyleMasi, Annalisa, Amedeo Capobianco, Krzysztof Bobrowski, Andrea Peluso, and Chryssostomos Chatgilialoglu. 2023. "Hydroxyl Radical vs. One-Electron Oxidation Reactivities in an Alternating GC Double-Stranded Oligonucleotide: A New Type Electron Hole Stabilization" Biomolecules 13, no. 10: 1493. https://doi.org/10.3390/biom13101493
APA StyleMasi, A., Capobianco, A., Bobrowski, K., Peluso, A., & Chatgilialoglu, C. (2023). Hydroxyl Radical vs. One-Electron Oxidation Reactivities in an Alternating GC Double-Stranded Oligonucleotide: A New Type Electron Hole Stabilization. Biomolecules, 13(10), 1493. https://doi.org/10.3390/biom13101493