
Combination Treatment Strategies to Overcome PARP Inhibitor Resistance

Abstract
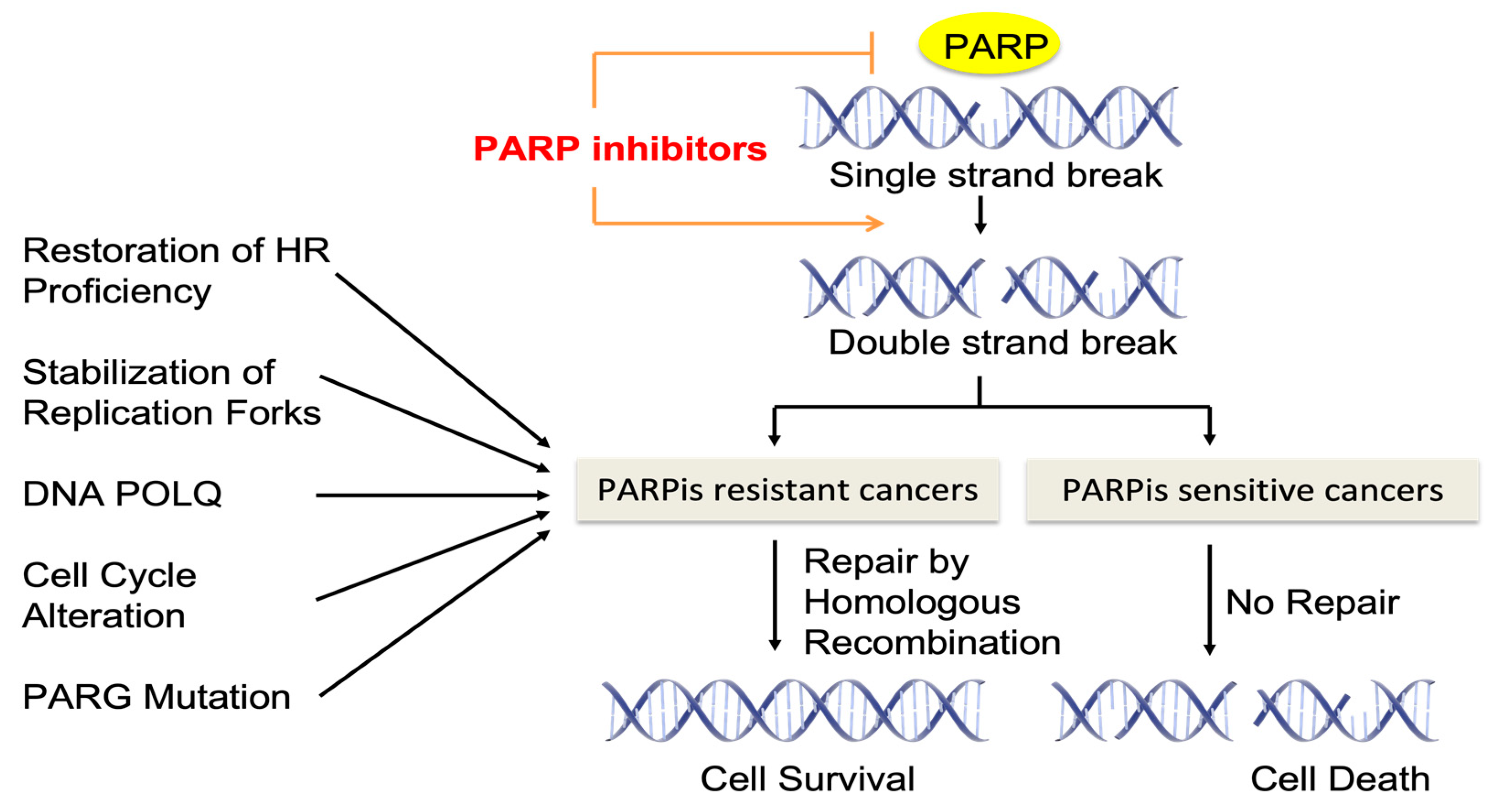
:1. Introduction
2. Mechanisms of DNA-Damaging Responses
3. The Mechanism by which PARP Inhibitors Act
4. Mechanisms of Resistance to PARP Inhibitors
4.1. Restoration of Homologous Recombination Proficiency
4.2. Stabilization of Replication Forks
4.3. DNA POLQ
4.4. Alterations in Cell Cycle Control
4.5. Inhibition of PARP Trapping by PAR Glycohydrolase (PARG) Downregulation
4.6. Increased PARPi Efflux by P-gp Efflux Pumps
4.7. ssDNA Gap Suppression
5. The Feasible Combination Treatment Options to Overcome Resistance to PARP Inhibitors
5.1. PARPis and Chemotherapy Drugs
5.2. PARPis and Immunotherapy
5.3. PARPis and Anti-Angiogenic Agents
5.4. PARPis and PI3K/AKT Pathway Inhibitors
5.5. PARPis and RAS/RAF/MEK Pathway Inhibitors
5.6. PARPis and Epigenetic Drugs
5.7. PARPis and ATR/CHK1 Inhibitors
5.8. PARPis and Other DDR Inhibitors
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sieber, O.M.; Heinimann, K.; Tomlinson, I.P.M. Genomic Instability--the Engine of Tumorigenesis? Nat. Rev. Cancer 2003, 3, 701–708. [Google Scholar] [CrossRef]
- Chartron, E.; Theillet, C.; Guiu, S.; Jacot, W. Targeting Homologous Repair Deficiency in Breast and Ovarian Cancers: Biological Pathways, Preclinical and Clinical Data. Crit. Rev. Oncol. Hematol. 2019, 133, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Brandsma, I.; Fleuren, E.D.G.; Williamson, C.T.; Lord, C.J. Directing the Use of DDR Kinase Inhibitors in Cancer Treatment. Expert. Opin. Investig. Drugs 2017, 26, 1341–1355. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G. The Concept of Synthetic Lethality in the Context of Anticancer Therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsdottir, K.; Ashworth, A. The Roles of BRCA1 and BRCA2 and Associated Proteins in the Maintenance of Genomic Stability. Oncogene 2006, 25, 5864–5874. [Google Scholar] [CrossRef]
- Helleday, T. The Underlying Mechanism for the PARP and BRCA Synthetic Lethality: Clearing up the Misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness Revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Frampton, G.M.; Fichtenholtz, A.; Otto, G.A.; Wang, K.; Downing, S.R.; He, J.; Schnall-Levin, M.; White, J.; Sanford, E.M.; An, P.; et al. Development and Validation of a Clinical Cancer Genomic Profiling Test Based on Massively Parallel DNA Sequencing. Nat. Biotechnol. 2013, 31, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Norquist, B.; Wurz, K.A.; Pennil, C.C.; Garcia, R.; Gross, J.; Sakai, W.; Karlan, B.Y.; Taniguchi, T.; Swisher, E.M. Secondary Somatic Mutations Restoring BRCA1/2 Predict Chemotherapy Resistance in Hereditary Ovarian Carcinomas. J. Clin. Oncol. 2011, 29, 3008–3015. [Google Scholar] [CrossRef] [PubMed]
- Keung, M.Y.; Wu, Y.; Badar, F.; Vadgama, J.V. Response of Breast Cancer Cells to PARP Inhibitors Is Independent of BRCA Status. J. Clin. Med. 2020, 9, 940. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. Genome Maintenance Mechanisms for Preventing Cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef]
- Friedberg, E.C.; Aguilera, A.; Gellert, M.; Hanawalt, P.C.; Hays, J.B.; Lehmann, A.R.; Lindahl, T.; Lowndes, N.; Sarasin, A.; Wood, R.D. DNA Repair: From Molecular Mechanism to Human Disease. DNA Repair 2006, 5, 986–996. [Google Scholar] [CrossRef]
- Helleday, T. Pathways for Mitotic Homologous Recombination in Mammalian Cells. Mutat. Res. 2003, 532, 103–115. [Google Scholar] [CrossRef]
- Stracker, T.H.; Petrini, J.H.J. The MRE11 Complex: Starting from the Ends. Nat. Rev. Mol. Cell Biol. 2011, 12, 90–103. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, X.; Chen, S.; Fernandes, N.; Price, B.D. A Role for the Tip60 Histone Acetyltransferase in the Acetylation and Activation of ATM. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar] [CrossRef]
- Bhatti, S.; Kozlov, S.; Farooqi, A.A.; Naqi, A.; Lavin, M.; Khanna, K.K. ATM Protein Kinase: The Linchpin of Cellular Defenses to Stress. Cell. Mol. Life Sci. 2011, 68, 2977–3006. [Google Scholar] [CrossRef]
- Altmeyer, M.; Lukas, J. To Spread or Not to Spread—Chromatin Modifications in Response to DNA Damage. Curr. Opin. Genet. Dev. 2013, 23, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Cejka, P. DNA End Resection: Nucleases Team Up with the Right Partners to Initiate Homologous Recombination. J. Biol. Chem. 2015, 290, 22931–22938. [Google Scholar] [CrossRef] [PubMed]
- Liu, V.F.; Weaver, D.T. The Ionizing Radiation-Induced Replication Protein A Phosphorylation Response Differs between Ataxia Telangiectasia and Normal Human Cells. Mol. Cell. Biol. 1993, 13, 7222–7231. [Google Scholar] [CrossRef] [PubMed]
- Sy, S.M.H.; Huen, M.S.Y.; Chen, J. PALB2 Is an Integral Component of the BRCA Complex Required for Homologous Recombination Repair. Proc. Natl. Acad. Sci. USA 2009, 106, 7155–7160. [Google Scholar] [CrossRef]
- Godin, S.K.; Sullivan, M.R.; Bernstein, K.A. Novel Insights into RAD51 Activity and Regulation during Homologous Recombination and DNA Replication. Biochem. Cell Biol. 2016, 94, 407–418. [Google Scholar] [CrossRef]
- Bai, P.; Cantó, C. The Role of PARP-1 and PARP-2 Enzymes in Metabolic Regulation and Disease. Cell Metab. 2012, 16, 290–295. [Google Scholar] [CrossRef]
- Richard, I.A.; Burgess, J.T.; O’Byrne, K.J.; Bolderson, E. Beyond PARP1: The Potential of Other Members of the Poly (ADP-Ribose) Polymerase Family in DNA Repair and Cancer Therapeutics. Front. Cell Dev. Biol. 2021, 9, 801200. [Google Scholar] [CrossRef]
- Kim, M.Y.; Zhang, T.; Kraus, W.L. Poly(ADP-Ribosyl)Ation by PARP-1: “PAR-Laying” NAD+ into a Nuclear Signal. Genes. Dev. 2005, 19, 1951–1967. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.-Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef]
- Satoh, M.S.; Lindahl, T. Role of Poly(ADP-Ribose) Formation in DNA Repair. Nature 1992, 356, 356–358. [Google Scholar] [CrossRef]
- Murai, J.; Pommier, Y. PARP Trapping Beyond Homologous Recombination and Platinum Sensitivity in Cancers. Annu. Rev. Cancer Biol. 2019, 3, 131–150. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib Monotherapy in Patients with Advanced Cancer and a Germline BRCA1/2 Mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef]
- Zandarashvili, L.; Langelier, M.-F.; Velagapudi, U.K.; Hancock, M.A.; Steffen, J.D.; Billur, R.; Hannan, Z.M.; Wicks, A.J.; Krastev, D.B.; Pettitt, S.J.; et al. Structural Basis for Allosteric PARP-1 Retention on DNA Breaks. Science 2020, 368, eaax6367. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53BP1 Causes PARP Inhibitor Resistance in Brca1-Mutated Mouse Mammary Tumors. Cancer Discov. 2013, 3, 68–81. [Google Scholar] [CrossRef]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to Therapy Caused by Intragenic Deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-Art Strategies for Targeting the DNA Damage Response in Cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.-Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef]
- Nesic, K.; Kondrashova, O.; Hurley, R.M.; McGehee, C.D.; Vandenberg, C.J.; Ho, G.-Y.; Lieschke, E.; Dall, G.; Bound, N.; Shield-Artin, K.; et al. Acquired RAD51C Promoter Methylation Loss Causes PARP Inhibitor Resistance in High-Grade Serous Ovarian Carcinoma. Cancer Res. 2021, 81, 4709–4722. [Google Scholar] [CrossRef]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 Loss Rescues BRCA1 Deficiency and Is Associated with Triple-Negative and BRCA-Mutated Breast Cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.-T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Álvarez-Quilón, A.; Moatti, N.; Zimmermann, M.; et al. The Shieldin Complex Mediates 53BP1-Dependent DNA Repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 2018, 173, 972–988.e23. [Google Scholar] [CrossRef] [PubMed]
- Dev, H.; Chiang, T.-W.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin Complex Promotes DNA End-Joining and Counters Homologous Recombination in BRCA1-Null Cells. Nat. Cell Biol. 2018, 20, 954–965. [Google Scholar] [CrossRef]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-Strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef]
- Schlacher, K.; Wu, H.; Jasin, M. A Distinct Replication Fork Protection Pathway Connects Fanconi Anemia Tumor Suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.-W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 2017, 68, 414–430.e8. [Google Scholar] [CrossRef]
- Rondinelli, B.; Gogola, E.; Yücel, H.; Duarte, A.A.; van de Ven, M.; van der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 Promotes Degradation of Stalled Replication Forks by Recruiting MUS81 through Histone H3 Trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication Fork Stability Confers Chemoresistance in BRCA-Deficient Cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef]
- Kais, Z.; Rondinelli, B.; Holmes, A.; O’Leary, C.; Kozono, D.; D’Andrea, A.D.; Ceccaldi, R. FANCD2 Maintains Fork Stability in BRCA1/2-Deficient Tumors and Promotes Alternative End-Joining DNA Repair. Cell Rep. 2016, 15, 2488–2499. [Google Scholar] [CrossRef] [PubMed]
- Michl, J.; Zimmer, J.; Buffa, F.M.; McDermott, U.; Tarsounas, M. FANCD2 Limits Replication Stress and Genome Instability in Cells Lacking BRCA2. Nat. Struct. Mol. Biol. 2016, 23, 755–757. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.R.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-Recombination-Deficient Tumours Are Dependent on Polθ-Mediated Repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Zhou, J.; Gelot, C.; Pantelidou, C.; Li, A.; Yücel, H.; Davis, R.E.; Färkkilä, A.; Kochupurakkal, B.; Syed, A.; Shapiro, G.I.; et al. A First-in-Class Polymerase Theta Inhibitor Selectively Targets Homologous-Recombination-Deficient Tumors. Nat. Cancer 2021, 2, 598–610. [Google Scholar] [CrossRef]
- Zatreanu, D.; Robinson, H.M.R.; Alkhatib, O.; Boursier, M.; Finch, H.; Geo, L.; Grande, D.; Grinkevich, V.; Heald, R.A.; Langdon, S.; et al. Polθ Inhibitors Elicit BRCA-Gene Synthetic Lethality and Target PARP Inhibitor Resistance. Nat. Commun. 2021, 12, 3636. [Google Scholar] [CrossRef]
- Bajrami, I.; Frankum, J.R.; Konde, A.; Miller, R.E.; Rehman, F.L.; Brough, R.; Campbell, J.; Sims, D.; Rafiq, R.; Hooper, S.; et al. Genome-Wide Profiling of Genetic Synthetic Lethality Identifies CDK12 as a Novel Determinant of PARP1/2 Inhibitor Sensitivity. Cancer Res. 2014, 74, 287–297. [Google Scholar] [CrossRef]
- Johnson, S.F.; Cruz, C.; Greifenberg, A.K.; Dust, S.; Stover, D.G.; Chi, D.; Primack, B.; Cao, S.; Bernhardy, A.J.; Coulson, R.; et al. CDK12 Inhibition Reverses De Novo and Acquired PARP Inhibitor Resistance in BRCA Wild-Type and Mutated Models of Triple-Negative Breast Cancer. Cell Rep. 2016, 17, 2367–2381. [Google Scholar] [CrossRef]
- Ha, D.-H.; Min, A.; Kim, S.; Jang, H.; Kim, S.H.; Kim, H.-J.; Ryu, H.S.; Ku, J.-L.; Lee, K.-H.; Im, S.-A. Antitumor Effect of a WEE1 Inhibitor and Potentiation of Olaparib Sensitivity by DNA Damage Response Modulation in Triple-Negative Breast Cancer. Sci. Rep. 2020, 10, 9930. [Google Scholar] [CrossRef]
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Guerrero Llobet, S.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 2018, 33, 1078–1093.e12. [Google Scholar] [CrossRef]
- Dias, M.P.; Moser, S.C.; Ganesan, S.; Jonkers, J. Understanding and Overcoming Resistance to PARP Inhibitors in Cancer Therapy. Nat. Rev. Clin. Oncol. 2021, 18, 773–791. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.H.; Zander, S.A.L.; Derksen, P.W.B.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High Sensitivity of BRCA1-Deficient Mammary Tumors to the PARP Inhibitor AZD2281 Alone and in Combination with Platinum Drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef] [PubMed]
- Leitner, I.; Nemeth, J.; Feurstein, T.; Abrahim, A.; Matzneller, P.; Lagler, H.; Erker, T.; Langer, O.; Zeitlinger, M. The Third-Generation P-Glycoprotein Inhibitor Tariquidar May Overcome Bacterial Multidrug Resistance by Increasing Intracellular Drug Concentration. J. Antimicrob. Chemother. 2011, 66, 834–839. [Google Scholar] [CrossRef]
- Christie, E.L.; Pattnaik, S.; Beach, J.; Copeland, A.; Rashoo, N.; Fereday, S.; Hendley, J.; Alsop, K.; Brady, S.L.; Lamb, G.; et al. Multiple ABCB1 Transcriptional Fusions in Drug Resistant High-Grade Serous Ovarian and Breast Cancer. Nat. Commun. 2019, 10, 1295. [Google Scholar] [CrossRef]
- Vaidyanathan, A.; Sawers, L.; Gannon, A.-L.; Chakravarty, P.; Scott, A.L.; Bray, S.E.; Ferguson, M.J.; Smith, G. ABCB1 (MDR1) Induction Defines a Common Resistance Mechanism in Paclitaxel- and Olaparib-Resistant Ovarian Cancer Cells. Br. J. Cancer 2016, 115, 431–441. [Google Scholar] [CrossRef]
- Cantor, S.B. Revisiting the BRCA-Pathway through the Lens of Replication Gap Suppression: “Gaps Determine Therapy Response in BRCA Mutant Cancer”. DNA Repair 2021, 107, 103209. [Google Scholar] [CrossRef] [PubMed]
- Cong, K.; Peng, M.; Kousholt, A.N.; Lee, W.T.C.; Lee, S.; Nayak, S.; Krais, J.; VanderVere-Carozza, P.S.; Pawelczak, K.S.; Calvo, J.; et al. Replication Gaps Are a Key Determinant of PARP Inhibitor Synthetic Lethality with BRCA Deficiency. Mol. Cell 2021, 81, 3128–3144.e7. [Google Scholar] [CrossRef]
- Panzarino, N.J.; Krais, J.J.; Cong, K.; Peng, M.; Mosqueda, M.; Nayak, S.U.; Bond, S.M.; Calvo, J.A.; Doshi, M.B.; Bere, M.; et al. Replication Gaps Underlie BRCA Deficiency and Therapy Response. Cancer Res. 2021, 81, 1388–1397. [Google Scholar] [CrossRef]
- Quinet, A.; Tirman, S.; Jackson, J.; Šviković, S.; Lemaçon, D.; Carvajal-Maldonado, D.; González-Acosta, D.; Vessoni, A.T.; Cybulla, E.; Wood, M.; et al. PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Mol. Cell 2020, 77, 461–474.e9. [Google Scholar] [CrossRef]
- Kang, Z.; Fu, P.; Alcivar, A.L.; Fu, H.; Redon, C.; Foo, T.K.; Zuo, Y.; Ye, C.; Baxley, R.; Madireddy, A.; et al. BRCA2 Associates with MCM10 to Suppress PRIMPOL-Mediated Repriming and Single-Stranded Gap Formation after DNA Damage. Nat. Commun. 2021, 12, 5966. [Google Scholar] [CrossRef]
- FDA Approves Olaparib Tablets for Maintenance Treatment in Ovarian Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-tablets-maintenance-treatment-ovarian-cancer (accessed on 17 August 2017).
- Lee, J.-M.; Hays, J.L.; Annunziata, C.M.; Noonan, A.M.; Minasian, L.; Zujewski, J.A.; Yu, M.; Gordon, N.; Ji, J.; Sissung, T.M.; et al. Phase I/Ib Study of Olaparib and Carboplatin in BRCA1 or BRCA2 Mutation-Associated Breast or Ovarian Cancer with Biomarker Analyses. J. Natl. Cancer Inst. 2014, 106, dju089. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Cibula, D.; Benzaquen, A.O.; Poole, C.; Mathijssen, R.H.J.; Sonke, G.S.; Colombo, N.; Špaček, J.; Vuylsteke, P.; Hirte, H.; et al. Olaparib Combined with Chemotherapy for Recurrent Platinum-Sensitive Ovarian Cancer: A Randomised Phase 2 Trial. Lancet Oncol. 2015, 16, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Hao, M.; Fang, Z.; Ding, J.; Duan, S.; Yi, F.; Wei, Y.; Zhang, W. PARP Inhibitor plus Chemotherapy versus Chemotherapy Alone in Patients with Triple-Negative Breast Cancer: A Systematic Review and Meta-Analysis Based on Randomized Controlled Trials. Cancer Chemother. Pharmacol. 2023, 91, 203–217. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.-K.; Hsu, J.-M.; Hsu, J.L.; Yu, W.-H.; Du, Y.; Lee, H.-H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination with Pembrolizumab in Patients with Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-Label Clinical Trial of Niraparib Combined with Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132–1140. [Google Scholar] [CrossRef]
- Drew, Y.; de Jonge, M.; Hong, S.; Park, Y.; Wolfer, A.; Brown, J.; Ferguson, M.; Gore, M.; Alvarez, R.; Gresty, C.; et al. An Open-Label, Phase II Basket Study of Olaparib and Durvalumab (MEDIOLA): Results in Germline BRCA-Mutated (gBRCAm) Platinum-Sensitive Relapsed (PSR) Ovarian Cancer (OC). Gynecol. Oncol. 2018, 149, 246–247. [Google Scholar] [CrossRef]
- Hardy-Bessard, A.-C.; Moore, K.N.; Mirza, M.R.; Asselain, B.; Redondo, A.; Pfisterer, J.; Pignata, S.; Provencher, D.M.; Cibula, D.; Reyners, A.K.L.; et al. ENGOT-OV44/FIRST Study: A Randomized, Double-Blind, Adaptive, Phase III Study of Standard of Care (SOC) Platinum-Based Therapy ± Dostarlimab Followed by Niraparib ± Dostarlimab Maintenance as First-Line (1L) Treatment of Stage 3 or 4 Ovarian Cancer (OC). J. Clin. Oncol. 2020, 38, TPS6101. [Google Scholar] [CrossRef]
- Fujiwara, K.; Vergote, I.B.; Sehouli, J.; Salutari, V.; Zola, P.; Madry, R.; Wenham, R.M.; Korach, J.; Pautier, P.; Cibula, D.; et al. ENGOT-Ov43/KEYLYNK-001: A Phase III Trial of Pembrolizumab plus Chemotherapy with Olaparib Maintenance for First-Line Treatment of BRCA¬-Nonmutated Advanced Epithelial Ovarian Cancer. Ann. Oncol. 2019, 30, ix89. [Google Scholar] [CrossRef]
- Monk, B.J.; Coleman, R.L.; Fujiwara, K.; Wilson, M.K.; Oza, A.M.; Oaknin, A.; O’Malley, D.M.; Lorusso, D.; Westin, S.N.; Safra, T.; et al. ATHENA (GOG-3020/ENGOT-Ov45): A Randomized, Phase III Trial to Evaluate Rucaparib as Monotherapy (ATHENA-MONO) and Rucaparib in Combination with Nivolumab (ATHENA-COMBO) as Maintenance Treatment Following Frontline Platinum-Based Chemotherapy in Ovarian Cancer. Int. J. Gynecol. Cancer 2021, 31, 1589–1594. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.; Pilat, M.J.P.; Santa-Maria, C.A.; Connolly, R.M.; Roesch, E.E.; Afghahi, A.; Han, H.S.; Nanda, R.; Wulf, G.M.; Assad, H.; et al. Trial in Progress: A Phase II Open-Label, Randomized Study of PARP Inhibition (Olaparib) Either Alone or in Combination with Anti-PD-L1 Therapy (Atezolizumab) in Homologous DNA Repair (HDR) Deficient, Locally Advanced or Metastatic Non-HER2-Positive Breast Cancer. J. Clin. Oncol. 2020, 38, TPS1102. [Google Scholar] [CrossRef]
- Kaplan, A.R.; Gueble, S.E.; Liu, Y.; Oeck, S.; Kim, H.; Yun, Z.; Glazer, P.M. Cediranib Suppresses Homology-Directed DNA Repair through down-Regulation of BRCA1/2 and RAD51. Sci. Transl. Med. 2019, 11, eaav4508. [Google Scholar] [CrossRef]
- Harter, P.; Trillsch, F.; Okamoto, A.; Reuss, A.; Kim, J.-W.; Rubio-Pérez, M.J.; Vardar, M.A.; Scambia, G.; Tredan, O.; Nyvang, G.-B.; et al. Durvalumab with Paclitaxel/Carboplatin (PC) and Bevacizumab (Bev), Followed by Maintenance Durvalumab, Bev, and Olaparib in Patients (Pts) with Newly Diagnosed Advanced Ovarian Cancer (AOC) without a Tumor BRCA1/2 Mutation (Non-tBRCAm): Results from the Randomized, Placebo (Pbo)-Controlled Phase III DUO-O Trial. J. Clin. Oncol. 2023, 41, LBA5506. [Google Scholar] [CrossRef]
- Lee, J.-M.; Moore, R.G.; Ghamande, S.; Park, M.S.; Diaz, J.P.; Chapman, J.; Kendrick, J.; Slomovitz, B.M.; Tewari, K.S.; Lowe, E.S.; et al. Cediranib in Combination with Olaparib in Patients without a Germline BRCA1/2 Mutation and with Recurrent Platinum-Resistant Ovarian Cancer: Phase IIb CONCERTO Trial. Clin. Cancer Res. 2022, 28, 4186–4193. [Google Scholar] [CrossRef]
- Mo, W.; Liu, Q.; Lin, C.C.-J.; Dai, H.; Peng, Y.; Liang, Y.; Peng, G.; Meric-Bernstam, F.; Mills, G.B.; Li, K.; et al. mTOR Inhibitors Suppress Homologous Recombination Repair and Synergize with PARP Inhibitors via Regulating SUV39H1 in BRCA-Proficient Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 1699–1712. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Barry, W.T.; Birrer, M.; Westin, S.N.; Cadoo, K.A.; Shapiro, G.I.; Mayer, E.L.; O’Cearbhaill, R.E.; Coleman, R.L.; Kochupurakkal, B.; et al. Olaparib and α-Specific PI3K Inhibitor Alpelisib for Patients with Epithelial Ovarian Cancer: A Dose-Escalation and Dose-Expansion Phase 1b Trial. Lancet Oncol. 2019, 20, 570–580. [Google Scholar] [CrossRef]
- Shah, P.D.; Wethington, S.L.; Pagan, C.; Latif, N.; Tanyi, J.; Martin, L.P.; Morgan, M.; Burger, R.A.; Haggerty, A.; Zarrin, H.; et al. Combination ATR and PARP Inhibitor (CAPRI): A Phase 2 Study of Ceralasertib plus Olaparib in Patients with Recurrent, Platinum-Resistant Epithelial Ovarian Cancer. Gynecol. Oncol. 2021, 163, 246–253. [Google Scholar] [CrossRef]
- Yap, T.A.; Konstantinopoulos, P.; Grisham, R.N.; Gupta, D.; Wilkinson, G.; Cao, A.; Jeffers, M.; Sharma, N. 494TiP Phase Ib Study of Elimusertib (ATRi; BAY 1895344) in Combination with Niraparib (PARPi) in Patients with Advanced Solid Tumors. Ann. Oncol. 2022, 33, S767–S768. [Google Scholar] [CrossRef]
- Do, K.T.; Hill, S.J.; Kochupurakkal, B.; Supko, J.G.; Gannon, C.; Anderson, A.; Muzikansky, A.; Wolanski, A.; Hedglin, J.; Parmar, K.; et al. Abstract CT232: Phase I Combination Study of the CHK1 Inhibitor Prexasertib (LY2606368) and Olaparib in Patients with High-Grade Serous Ovarian Cancer and Other Advanced Solid Tumors. Cancer Res. 2019, 79, CT232. [Google Scholar] [CrossRef]
- Westin, S.N.; Coleman, R.L.; Fellman, B.M.; Yuan, Y.; Sood, A.K.; Soliman, P.T.; Wright, A.A.; Horowitz, N.S.; Campos, S.M.; Konstantinopoulos, P.A.; et al. EFFORT: EFFicacy Of Adavosertib in Parp ResisTance: A Randomized Two-Arm Non-Comparative Phase II Study of Adavosertib with or without Olaparib in Women with PARP-Resistant Ovarian Cancer. J. Clin. Oncol. 2021, 39, 5505. [Google Scholar] [CrossRef]
- Artios Pharma Ltd. A Phase I/IIa, Open-Label, Multi-Centre Study to Assess the Safety, Tolerability, Pharmacokinetics and Preliminary Efficacy of the DNA Polymerase Theta Inhibitor ART4215 Administered Orally as Monotherapy and in Combination to Patients with Advanced or Metastatic Solid Tumors. Available online: https://clinicaltrials.gov/study/NCT04991480 (accessed on 17 August 2017).
- Evers, B.; Drost, R.; Schut, E.; de Bruin, M.; van der Burg, E.; Derksen, P.W.B.; Holstege, H.; Liu, X.; van Drunen, E.; Beverloo, H.B.; et al. Selective Inhibition of BRCA2-Deficient Mammary Tumor Cell Growth by AZD2281 and Cisplatin. Clin. Cancer Res. 2008, 14, 3916–3925. [Google Scholar] [CrossRef] [PubMed]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and Prognostic Significance of BRCA1/2-Mutation Status with Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes and Expression of PD-1/PD-L1 in High Grade Serous Ovarian Cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef]
- Drew, Y.; Penson, R.T.; O’Malley, D.M.; Kim, J.-W.; Zimmermann, S.; Roxburgh, P.; Sohn, J.; Stemmer, S.M.; Bastian, S.; Ferguson, M.; et al. 814MO Phase II Study of Olaparib (O) plus Durvalumab (D) and Bevacizumab (B) (MEDIOLA): Initial Results in Patients (Pts) with Non-Germline BRCA-Mutated (Non-gBRCAm) Platinum Sensitive Relapsed (PSR) Ovarian Cancer (OC). Ann. Oncol. 2020, 31, S615–S616. [Google Scholar] [CrossRef]
- Lee, Y.J.; Lim, M.C.; Kim, B.-G.; Ngoi, N.Y.; Choi, C.H.; Park, S.-Y.; Tan, D.S.; Go, Y.; Lee, J.-Y. A Single-Arm Phase II Study of Olaparib Maintenance with Pembrolizumab and Bevacizumab in BRCA Non-Mutated Patients with Platinum-Sensitive Recurrent Ovarian Cancer (OPEB-01). J. Gynecol. Oncol. 2021, 32, e31. [Google Scholar] [CrossRef]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.-M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.; Buss, M.K.; Nattam, S.; Hurteau, J.; et al. Combination Cediranib and Olaparib versus Olaparib Alone for Women with Recurrent Platinum-Sensitive Ovarian Cancer: A Randomised Phase 2 Study. Lancet Oncol. 2014, 15, 1207–1214. [Google Scholar] [CrossRef]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.-M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.J.; Buss, M.K.; Nattam, S.R.; Hurteau, J.; et al. Overall Survival and Updated Progression-Free Survival Outcomes in a Randomized Phase II Study of Combination Cediranib and Olaparib versus Olaparib in Relapsed Platinum-Sensitive Ovarian Cancer. Ann. Oncol. 2019, 30, 551–557. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- González-Martín, A.; Desauw, C.; Heitz, F.; Cropet, C.; Gargiulo, P.; Berger, R.; Ochi, H.; Vergote, I.; Colombo, N.; Mirza, M.R.; et al. Maintenance Olaparib plus Bevacizumab in Patients with Newly Diagnosed Advanced High-Grade Ovarian Cancer: Main Analysis of Second Progression-Free Survival in the Phase III PAOLA-1/ENGOT-Ov25 Trial. Eur. J. Cancer 2022, 174, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Ray-Coquard, I.; Leary, A.; Pignata, S.; Cropet, C.; González-Martín, A.; Marth, C.; Nagao, S.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab First-Line Maintenance in Ovarian Cancer: Final Overall Survival Results from the PAOLA-1/ENGOT-Ov25 Trial. Ann. Oncol. 2023, 34, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Lundqvist, E.; Birrer, M.J.; Christensen, R.D.; Nyvang, G.-B.; Malander, S.; Anttila, M.; Werner, T.L.; Lund, B.; Lindahl, G.; et al. Niraparib plus Bevacizumab versus Niraparib Alone for Platinum-Sensitive Recurrent Ovarian Cancer (NSGO-AVANOVA2/ENGOT-Ov24): A Randomised, Phase 2, Superiority Trial. Lancet Oncol. 2019, 20, 1409–1419. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Y.H.; García-García, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzmán, M.; Grueso, J.; et al. PI3K Inhibition Impairs BRCA1/2 Expression and Sensitizes BRCA-Proficient Triple-Negative Breast Cancer to PARP Inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef]
- Batalini, F.; Xiong, N.; Tayob, N.; Polak, M.; Eismann, J.; Cantley, L.C.; Shapiro, G.I.; Adalsteinsson, V.; Winer, E.P.; Konstantinopoulos, P.A.; et al. Phase 1b Clinical Trial with Alpelisib plus Olaparib for Patients with Advanced Triple-Negative Breast Cancer. Clin. Cancer Res. 2022, 28, 1493–1499. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Gonzalez-Martin, A.; Cruz, F.M.; Friedlander, M.; Glasspool, R.; Lorusso, D.; Marth, C.; Monk, B.J.; Kim, J.-W.; Hinson, P.; et al. EPIK-O/ENGOT-OV61: Alpelisib plus Olaparib vs Cytotoxic Chemotherapy in High-Grade Serous Ovarian Cancer (Phase III Study). Future Oncol. 2022, 18, 3481–3492. [Google Scholar] [CrossRef]
- MD Anderson Cancer Center. A Phase Ib Study of the Oral PARP Inhibitor Niraparib with the Intravenous PI3K Inhibitor Copanlisib for Recurrent Endometrial and Recurrent Ovarian, Primary Peritoneal, or Fallopian Tube Cancer. Available online: https://clinicaltrials.gov/study/NCT03586661 (accessed on 17 August 2017).
- Westin, S.N.; Labrie, M.; Litton, J.K.; Blucher, A.; Fang, Y.; Vellano, C.P.; Marszalek, J.R.; Feng, N.; Ma, X.; Creason, A.; et al. Phase Ib Dose Expansion and Translational Analyses of Olaparib in Combination with Capivasertib in Recurrent Endometrial, Triple-Negative Breast, and Ovarian Cancer. Clin. Cancer Res. 2021, 27, 6354–6365. [Google Scholar] [CrossRef]
- Yap, T.A.; Kristeleit, R.; Michalarea, V.; Pettitt, S.J.; Lim, J.S.J.; Carreira, S.; Roda, D.; Miller, R.; Riisnaes, R.; Miranda, S.; et al. Phase I Trial of the PARP Inhibitor Olaparib and AKT Inhibitor Capivasertib in Patients with BRCA1/2- and Non-BRCA1/2-Mutant Cancers. Cancer Discov. 2020, 10, 1528–1543. [Google Scholar] [CrossRef]
- Sun, C.; Fang, Y.; Yin, J.; Chen, J.; Ju, Z.; Zhang, D.; Chen, X.; Vellano, C.P.; Jeong, K.J.; Ng, P.K.-S.; et al. Rational Combination Therapy with PARP and MEK Inhibitors Capitalizes on Therapeutic Liabilities in RAS Mutant Cancers. Sci. Transl. Med. 2017, 9, eaal5148. [Google Scholar] [CrossRef]
- Vena, F.; Jia, R.; Esfandiari, A.; Garcia-Gomez, J.J.; Rodriguez-Justo, M.; Ma, J.; Syed, S.; Crowley, L.; Elenbaas, B.; Goodstal, S.; et al. MEK Inhibition Leads to BRCA2 Downregulation and Sensitization to DNA Damaging Agents in Pancreas and Ovarian Cancer Models. Oncotarget 2018, 9, 11592–11603. [Google Scholar] [CrossRef]
- MD Anderson Cancer Center. Evaluation of the Combination of Selumetinib and Olaparib in Endometrial, Ovarian and Other Solid Tumors with Ras Pathway Alterations, and Ovarian Tumors with PARP Resistance. Available online: https://clinicaltrials.gov/study/NCT03162627 (accessed on 17 August 2017).
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.-Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of All BRCA1 Copies Predicts Response to the PARP Inhibitor Rucaparib in Ovarian Carcinoma. Nat. Commun. 2018, 9, 3970. [Google Scholar] [CrossRef] [PubMed]
- Lazo, P.A. Targeting Histone Epigenetic Modifications and DNA Damage Responses in Synthetic Lethality Strategies in Cancer? Cancers 2022, 14, 4050. [Google Scholar] [CrossRef] [PubMed]
- Abbotts, R.; Topper, M.J.; Biondi, C.; Fontaine, D.; Goswami, R.; Stojanovic, L.; Choi, E.Y.; McLaughlin, L.; Kogan, A.A.; Xia, L.; et al. DNA Methyltransferase Inhibitors Induce a BRCAness Phenotype That Sensitizes NSCLC to PARP Inhibitor and Ionizing Radiation. Proc. Natl. Acad. Sci. USA 2019, 116, 22609–22618. [Google Scholar] [CrossRef] [PubMed]
- Pulliam, N.; Fang, F.; Ozes, A.R.; Tang, J.; Adewuyi, A.; Keer, H.; Lyons, J.; Baylin, S.B.; Matei, D.; Nakshatri, H.; et al. An Effective Epigenetic-PARP Inhibitor Combination Therapy for Breast and Ovarian Cancers Independent of BRCA Mutations. Clin. Cancer Res. 2018, 24, 3163–3175. [Google Scholar] [CrossRef]
- Muvarak, N.E.; Chowdhury, K.; Xia, L.; Robert, C.; Choi, E.Y.; Cai, Y.; Bellani, M.; Zou, Y.; Singh, Z.N.; Duong, V.H.; et al. Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents—A Potential Therapy for Cancer. Cancer Cell 2016, 30, 637–650. [Google Scholar] [CrossRef]
- Baer, M.R.; Kogan, A.A.; Bentzen, S.M.; Mi, T.; Lapidus, R.G.; Duong, V.H.; Emadi, A.; Niyongere, S.; O’Connell, C.L.; Youngblood, B.A.; et al. Phase I Clinical Trial of DNA Methyltransferase Inhibitor Decitabine and PARP Inhibitor Talazoparib Combination Therapy in Relapsed/Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2022, 28, 1313–1322. [Google Scholar] [CrossRef]
- Chang, J.C. Multicenter Phase I/Ib Trial of Olaparib in Combination with Vorinostat in Patients with Relapsed/Refractory and/or Metastatic Breast Cancer. Available online: https://clinicaltrials.gov/study/NCT03742245 (accessed on 17 August 2017).
- Karnitz, L.M.; Zou, L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin. Cancer Res. 2015, 21, 4780–4785. [Google Scholar] [CrossRef]
- Ngoi, N.Y.L.; Pham, M.M.; Tan, D.S.P.; Yap, T.A. Targeting the Replication Stress Response through Synthetic Lethal Strategies in Cancer Medicine. Trends Cancer 2021, 7, 930–957. [Google Scholar] [CrossRef]
- Kim, H.; George, E.; Ragland, R.L.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.A.; Herlyn, M.; Brown, E.J.; Simpkins, F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR Inhibition Overcomes PARP Inhibitor and Platinum Resistance in Ovarian Cancer Models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef]
- Wethington, S.L.; Shah, P.D.; Martin, L.; Tanyi, J.L.; Latif, N.; Morgan, M.; Torigian, D.A.; Rodriguez, D.; Smith, S.A.; Dean, E.; et al. Combination ATR (Ceralasertib) and PARP (Olaparib) Inhibitor (CAPRI) Trial in Acquired PARP Inhibitor-Resistant Homologous Recombination-Deficient Ovarian Cancer. Clin. Cancer Res. 2023, 29, 2800–2807. [Google Scholar] [CrossRef] [PubMed]
- Do, K.T.; Kochupurakkal, B.; Kelland, S.; de Jonge, A.; Hedglin, J.; Powers, A.; Quinn, N.; Gannon, C.; Vuong, L.; Parmar, K.; et al. Phase 1 Combination Study of the CHK1 Inhibitor Prexasertib and the PARP Inhibitor Olaparib in High-Grade Serous Ovarian Cancer and Other Solid Tumors. Clin. Cancer Res. 2021, 27, 4710–4716. [Google Scholar] [CrossRef] [PubMed]
- Forment, J.V.; O’Connor, M.J. Targeting the Replication Stress Response in Cancer. Pharmacol. Ther. 2018, 188, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Lallo, A.; Frese, K.K.; Morrow, C.J.; Sloane, R.; Gulati, S.; Schenk, M.W.; Trapani, F.; Simms, N.; Galvin, M.; Brown, S.; et al. The Combination of the PARP Inhibitor Olaparib and the WEE1 Inhibitor AZD1775 as a New Therapeutic Option for Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 5153–5164. [Google Scholar] [CrossRef]
- Fang, Y.; McGrail, D.J.; Sun, C.; Labrie, M.; Chen, X.; Zhang, D.; Ju, Z.; Vellano, C.P.; Lu, Y.; Li, Y.; et al. Sequential Therapy with PARP and WEE1 Inhibitors Minimizes Toxicity While Maintaining Efficacy. Cancer Cell 2019, 35, 851–867.e7. [Google Scholar] [CrossRef]

{kind=link}
| Trial Name | Combination Therapy | Phase | Cohort | Start Completion Date | Ref | |
|---|---|---|---|---|---|---|
| PARPi + Chemotherapy | (STUDY41) NCT01081951 | Olaparib + Paclitaxel + Carbopatin | II | Platinum sensitive advanced ovarian cancer | 4 Feb 2010– 29 Dec 2023 | [59] |
| PARPi + Immunotherapy | (TOPACIO) NCT02657889 | Niraparib + Pembrolizumab | II | Platinum-resistance ovarian cancer (regardless BRCA or HRD status) or BRCA mutated metastatic TNBC | 15 Apr 2016– 17 Sep 2021 | [66,67] |
| (ENGOT-OV44) NCT03602859 | Niraparib + Dostarlimab | III | Stage III or IV nonmucinous Epithelial ovarin cancer | 11 Oct 2018– 22 Jun 2026 | [71] | |
| (MEDIOLA) NCT02734004 | Olaparib + Durvalumab | II | gBRCAm platinum-sensitive ovarian cancer | 28 Oct 2018– 2026 | [68] | |
| (OPEB-1) NCT04361370 | Olaparib + Durvalumab + Bevacizumab | II | Platinum-sensitive recurrent BRCA wt ovarian cancer | 28 Oct 2020– Aug 2026 | [70] | |
| (ATHENA) NCT03522246 | Rucaparib + Nivolumab | III | Newly diagonsed advanced (FIGO stage III-IV) epithelial ovarin cancer. Fallopian Tube diseases. | 14 May 2018– 30 Dec2030 | [72] | |
| (DUO-O) NCT03737643 | Olaparib + Durvalumab | III | Newly diagonsed advanced ovarain cancer (Regardless BRCA status) | 4 Jan 2019– 25 May 2028 | [73] | |
| NCT02849496 | Olaparib + Atezolizumb (Anti-PD-L1) | II | Locally advanced unresectable or metastatic non-HER2 positive breast cancer | 30 Mar 2017– 31 Aug 2023 | [74] | |
| PARPi + Anti-angiogenic agents | NCT01116648 | Olaparib + Cediranib | I/II | Relapsed Platinum sensitive high-grade or endometrioid ovarian cancer | 25 Mar 2010– 31 Oct 2018 | [75] |
| (NRG-GY005) NCT02502266 | Olaparib + Cediranib | II/III | Recurrent Platinum-resistant ovarian cancer, Fallopian Tube diseases. | 3 May 2016– 30 Jun 2024 | [76] | |
| (PAOLA-1) NCT02477644 | Olaparib + Bevacizumab | III | HRD positive high-grade ovarian cancer | 6 May 2015– 22 Mar 2022 | [77,78,79] | |
| NCT02354131 | Niraparib + Bevacizumab | I/II | platinum-sensitive ovarian cancer | 15 Feb 2015– 15 Dec 2021 | [80] | |
| PARPi + PI3K/AKT pathway inhibitors | NCT01623349 | Olaparib + Alpelisib | I | Platinum-resistant ovarian cancer or recurrent triple negative breast cancer (regardless BRCA or HRD status) | Sep 2012– Dec 2020 | [81,82] |
| (EPIK-O) NCT04729387 | Olaparib + Alpelisib | III | Platinum-resistant or refractory high-grade serious epithelial ovarian cancer (HGSOC) with BRCA wt. | 2 Jul 2021– 29 Jul 2025 | [83] | |
| NCT02208375 | Olaparib + Vistusertib or Olaparib + Capivasertib | I/II | Recurrent endometrial, triple negative breast cancer or ovarian cancer | 11 Nov 2014– 20 Jun 2024 | [84] | |
| NCT02338622 | Olaparib + Capivasertib | I | Platinum-resistant HGSOC (regardless BRCA or HRD status) | 31 Mar 2014– 21 Mar 2017 | [85] | |
| NCT03586661 | Niraparib + Copanlisib | I | Recurrent high-grade serous or BRCA mutant ovarian cancer | 29 Apr 2019– 31 Dec 23 | [86] | |
| PARPi + MAPK pathway inhibitors | NCT03162627 | Olaprib + Selumetinib | I/II | PARPi resistant ovarian cancer or Solid tumor with RAS pathway alteration | 4 Aug 2017– 30 Aug 2026 | [87] |
| PARPi + Epigenetic drugs | NCT02878785 | Talazoparib + Decitabine | I/II | Acute Myeloid Leukemia | Aug 2016– 19 Nov 2020 | [88] |
| NCT03742245 | Olaprib + Vorinostat | I | Relapsed metastatic breast cancer | 11 Jun 2019– 1 Sep 2024 | [89] | |
| PARPi + DDR inhibitors | (CAPRI) NCT03462342 | Olaparib + Ceralasertib | II | Platinum-resistant HGSOC | 9 Mar 2018– 31 Dec 2023 | [90] |
| NCT04267939 | Niraparib + Elimsertib | I | Recurrent advanced solid tumors and ovarian cancer. | 26 Feb 2020– 3 Mar 2025 | [91] | |
| NCT03057145 | Olaparib + Prexasertib | I | BRCA mutant PARPi resistant HGSOC | 10 Mar 2017– 9 Jun 2021 | [92] | |
| (EFFORT) NCT03579316 | Olaparib + Adavosertib | II | PARPis resistant ovarian cancer | 7 Dec 2018– 30 Dec 2023 | [93] | |
| NCT04991480 | Talozoparib + ART4215 | I/II | BRCA deficient breast cancer | 13 Sep 2021– Aug 2025 | [94] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soung, Y.-H.; Chung, J. Combination Treatment Strategies to Overcome PARP Inhibitor Resistance. Biomolecules 2023, 13, 1480. https://doi.org/10.3390/biom13101480
Soung Y-H, Chung J. Combination Treatment Strategies to Overcome PARP Inhibitor Resistance. Biomolecules. 2023; 13(10):1480. https://doi.org/10.3390/biom13101480
Chicago/Turabian StyleSoung, Young-Hwa, and Jun Chung. 2023. "Combination Treatment Strategies to Overcome PARP Inhibitor Resistance" Biomolecules 13, no. 10: 1480. https://doi.org/10.3390/biom13101480
APA StyleSoung, Y.-H., & Chung, J. (2023). Combination Treatment Strategies to Overcome PARP Inhibitor Resistance. Biomolecules, 13(10), 1480. https://doi.org/10.3390/biom13101480