



Different Forms of Disorder in NMDA-Sensitive Glutamate Receptor Cytoplasmic Domains Are Associated with Differences in Condensate Formation


 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Protein Purification
2.2. Single Molecule Total Internal Reflection Fluorescence (smTIRF) Microscopy
2.3. Discrete Molecular Dynamics (DMD) Simulations
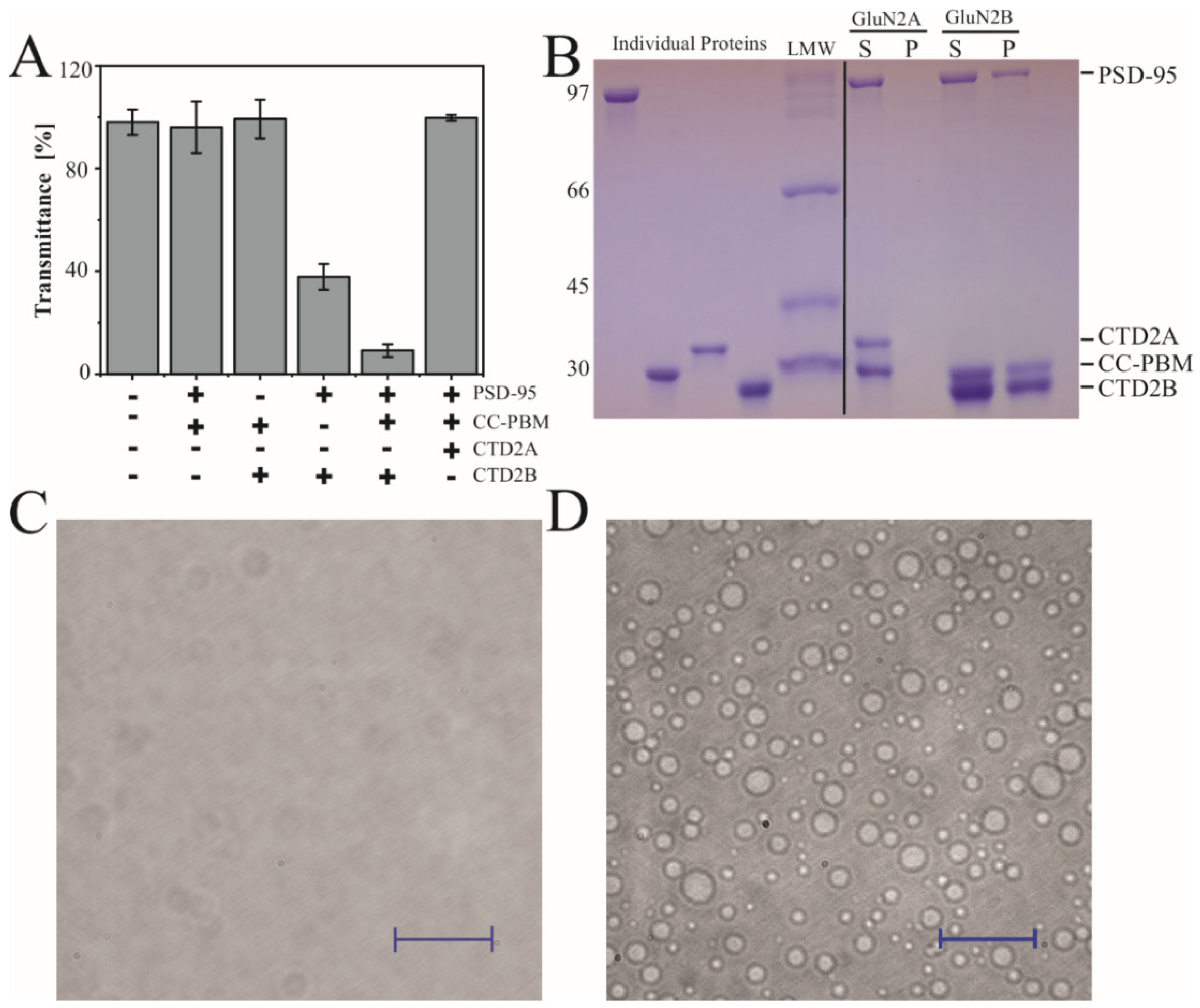
2.4. Measurement of Turbidity
2.5. Sedimentation Analysis of Condensates
2.6. Differential Interference Contrast (DIC) and Fluorescence Microscopy
3. Results
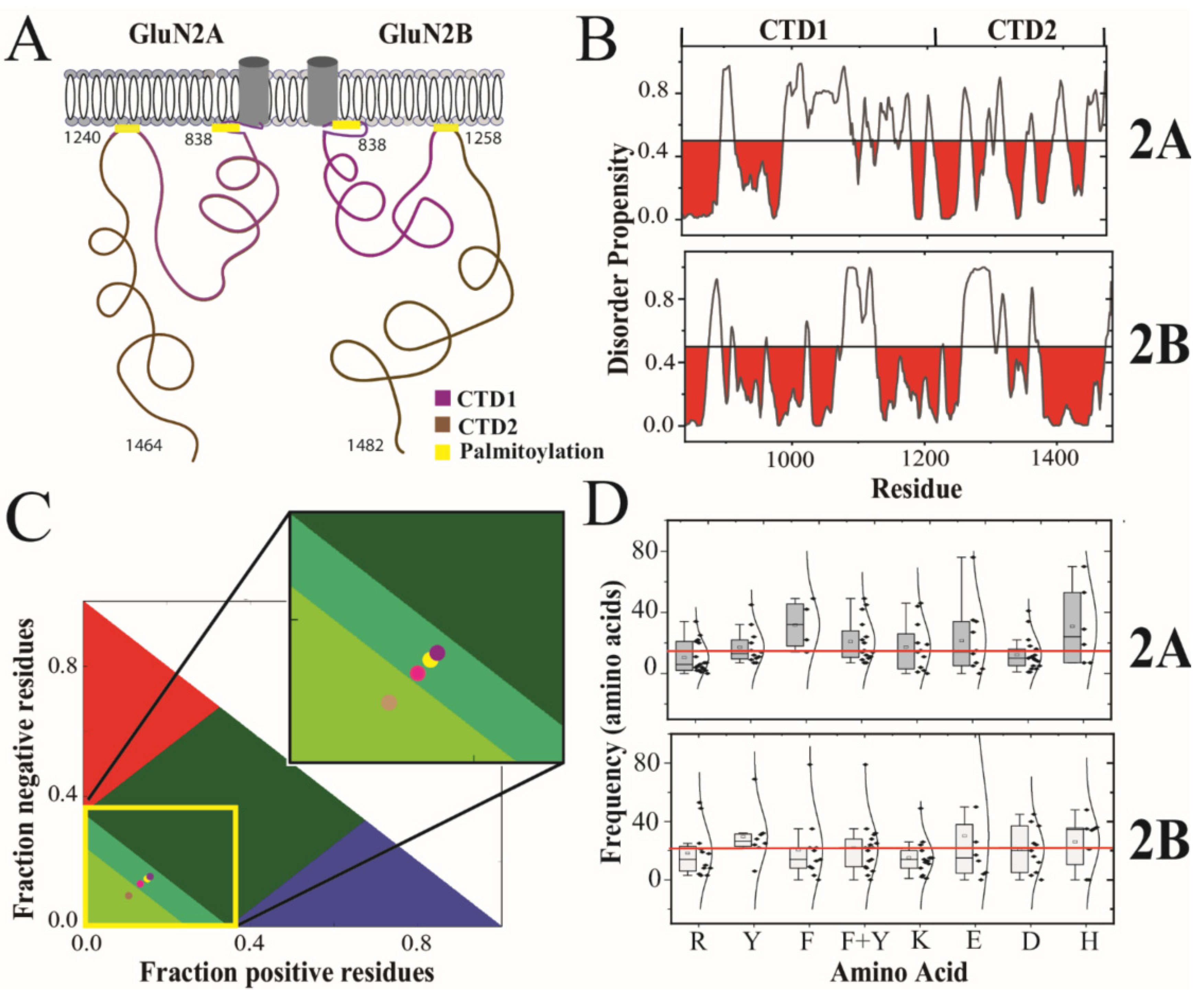
3.1. Primary Sequence Analysis
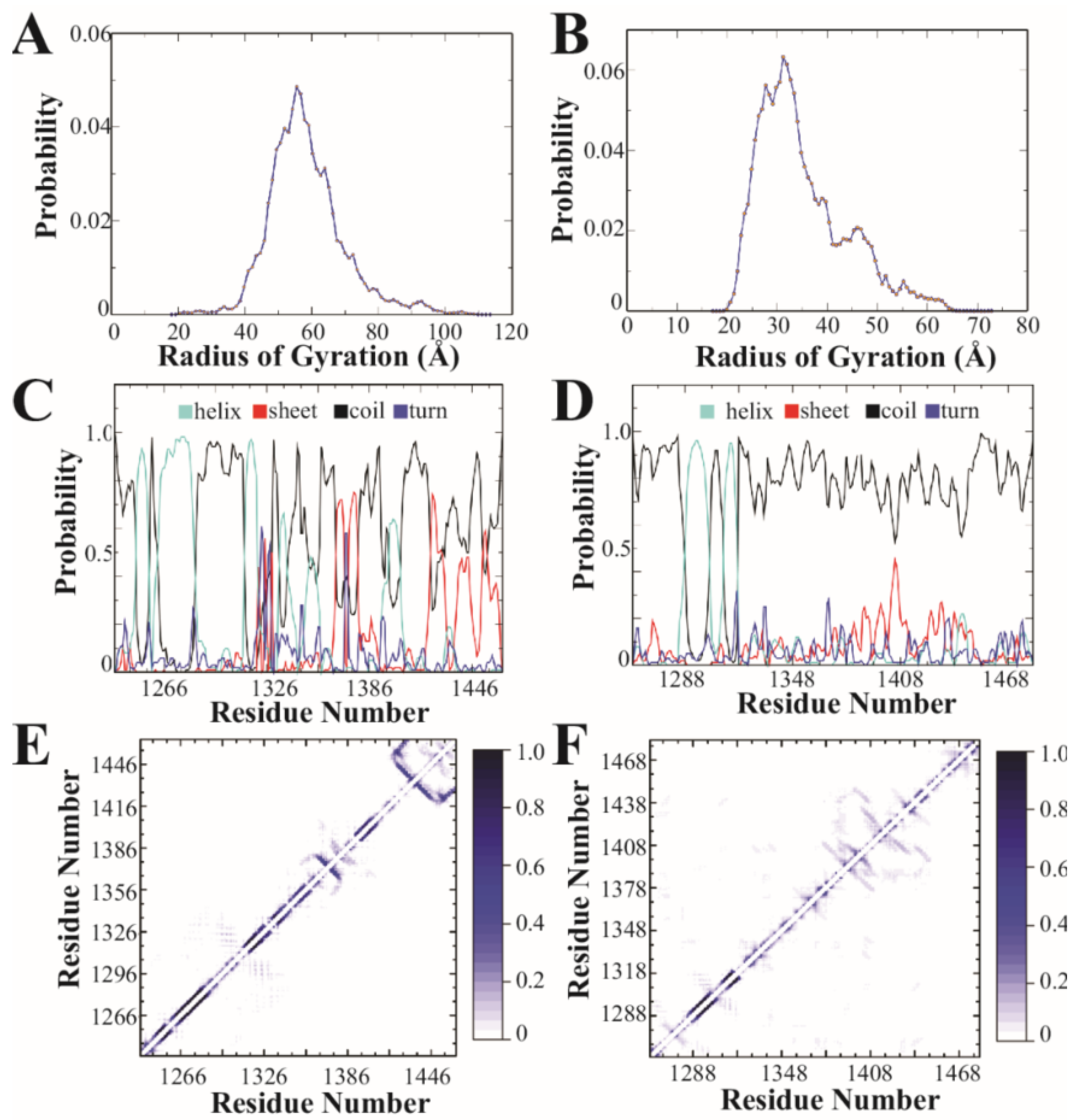
3.2. Discrete Molecular Dynamics
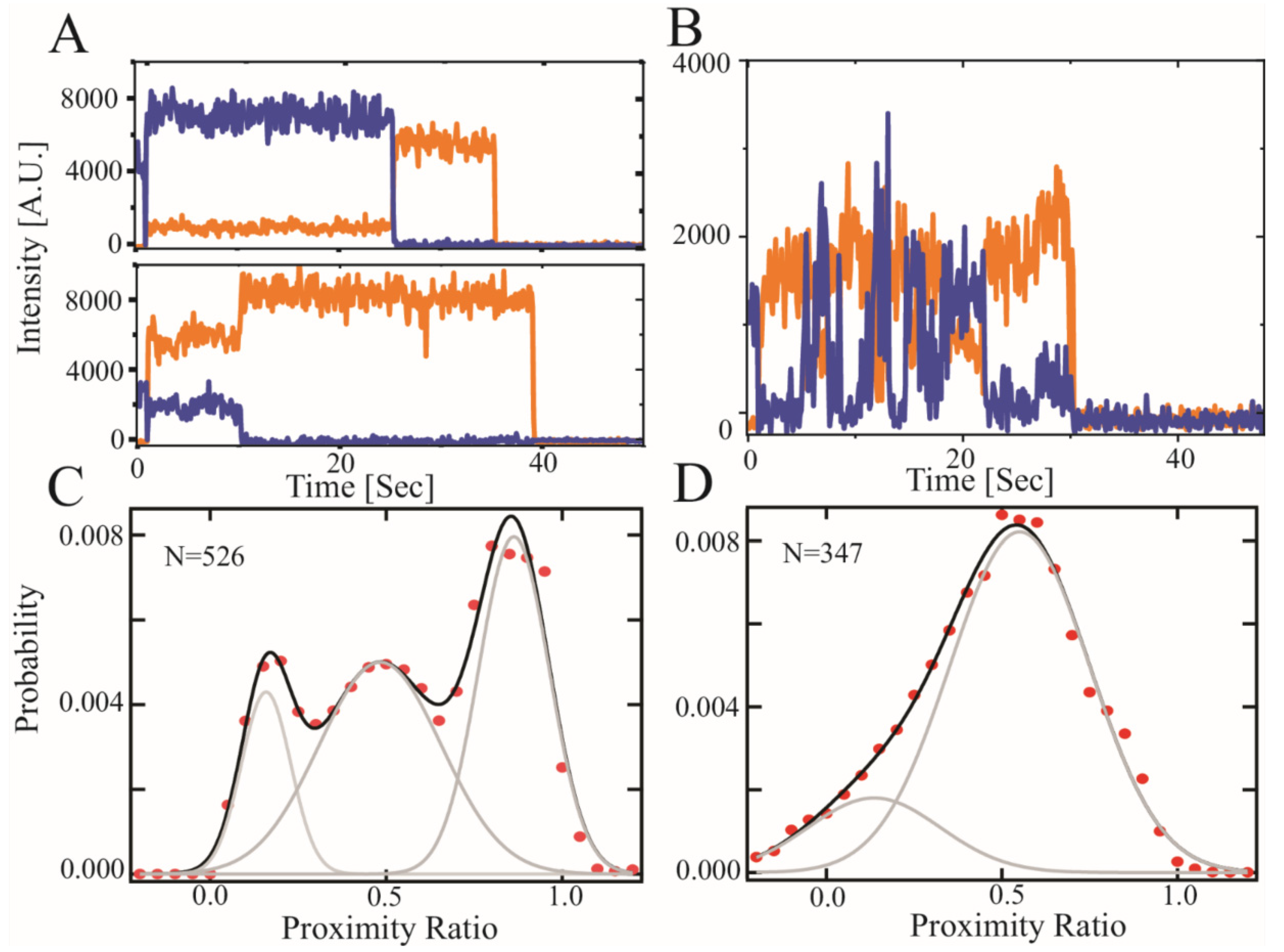
3.3. Single Molecule Fluorescence Resonance Energy Transfer (smFRET)
3.4. Condensate Formation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, F.; Tsien, J.Z. Memory and the NMDA receptors. N Engl. J. Med. 2009, 361, 302–303. [Google Scholar] [CrossRef]
- Newcomer, J.W.; Farber, N.B.; Olney, J.W. NMDA receptor function, memory, and brain aging. Dialogues Clin. Neurosci. 2000, 2, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Furukawa, H.; Wollmuth, L.P.; Gibb, A.J.; Traynelis, S.F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150, 1081–1105. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.B.; Wollmuth, L.P.; Bowie, D.; Furukawa, H.; Menniti, F.S.; Sobolevsky, A.I.; Swanson, G.T.; Swanger, S.A.; Greger, I.H.; Nakagawa, T.; et al. Structure, Function, and Pharmacology of Glutamate Receptor Ion Channels. Pharm. Rev. 2021, 73, 298–487. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Gouaux, E. Structure and symmetry inform gating principles of ionotropic glutamate receptors. Neuropharmacology 2017, 112(Pt A), 11–15. [Google Scholar] [CrossRef]
- Yuan, H.; Hansen, K.B.; Vance, K.M.; Ogden, K.K.; Traynelis, S.F. Control of NMDA receptor function by the NR2 subunit amino-terminal domain. J. Neurosci. 2009, 29, 12045–12058. [Google Scholar] [CrossRef] [PubMed]
- Punnakkal, P.; Jendritza, P.; Kohr, G. Influence of the intracellular GluN2 C-terminal domain on NMDA receptor function. Neuropharmacology 2012, 62, 1985–1992. [Google Scholar] [CrossRef] [PubMed]
- Petit-Pedrol, M.; Groc, L. Regulation of membrane NMDA receptors by dynamics and protein interactions. J. Cell Biol. 2021, 220, e202006101. [Google Scholar] [CrossRef]
- XiangWei, W.; Jiang, Y.; Yuan, H. De Novo Mutations and Rare Variants Occurring in NMDA Receptors. Curr. Opin. Physiol. 2018, 2, 27–35. [Google Scholar] [CrossRef]
- Mota Vieira, M.; Nguyen, T.A.; Wu, K.; Badger, J.D., 2nd; Collins, B.M.; Anggono, V.; Lu, W.; Roche, K.W. An Epilepsy-Associated GRIN2A Rare Variant Disrupts CaMKIIalpha Phosphorylation of GluN2A and NMDA Receptor Trafficking. Cell Rep. 2020, 32, 108104. [Google Scholar] [CrossRef]
- Liu, S.; Zhou, L.; Yuan, H.; Vieira, M.; Sanz-Clemente, A.; Badger, J.D., 2nd; Lu, W.; Traynelis, S.F.; Roche, K.W. A Rare Variant Identified Within the GluN2B C-Terminus in a Patient with Autism Affects NMDA Receptor Surface Expression and Spine Density. J. Neurosci. 2017, 37, 4093–4102. [Google Scholar] [CrossRef]
- Karakas, E.; Furukawa, H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science 2014, 344, 992–997. [Google Scholar] [CrossRef]
- Lee, C.H.; Lu, W.; Michel, J.C.; Goehring, A.; Du, J.; Song, X.; Gouaux, E. NMDA receptor structures reveal subunit arrangement and pore architecture. Nature 2014, 511, 191–197. [Google Scholar] [CrossRef]
- Regan, M.C.; Romero-Hernandez, A.; Furukawa, H. A structural biology perspective on NMDA receptor pharmacology and function. Curr. Opin. Struct Biol. 2015, 33, 68–75. [Google Scholar] [CrossRef]
- Choi, U.B.; Xiao, S.; Wollmuth, L.P.; Bowen, M.E. Effect of Src kinase phosphorylation on disordered C-terminal domain of N-methyl-D-aspartic acid (NMDA) receptor subunit GluN2B protein. J. Biol. Chem. 2011, 286, 29904–29912. [Google Scholar] [CrossRef]
- Ryan, T.J.; Emes, R.D.; Grant, S.G.N.; Komiyama, N.H. Evolution of NMDA receptor cytoplasmic interaction domains: Implications for organisation of synaptic signalling complexes. BMC Neurosci. 2008, 9, 14. [Google Scholar] [CrossRef]
- Hayashi, T.; Thomas, G.M.; Huganir, R.L. Dual palmitoylation of NR2 subunits regulates NMDA receptor trafficking. Neuron 2009, 64, 213–226. [Google Scholar] [CrossRef]
- Choi, U.B.; McCann, J.J.; Weninger, K.R.; Bowen, M.E. Beyond the random coil: Stochastic conformational switching in intrinsically disordered proteins. Structure 2011, 19, 566–576. [Google Scholar] [CrossRef]
- Holehouse, A.S.; Das, R.K.; Ahad, J.N.; Richardson, M.O.; Pappu, R.V. CIDER: Resources to Analyze Sequence-Ensemble Relationships of Intrinsically Disordered Proteins. Biophys. J. 2017, 112, 16–21. [Google Scholar] [CrossRef]
- Das, R.K.; Pappu, R.V. Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. USA 2013, 110, 13392–13397. [Google Scholar] [CrossRef]
- Lin, Y.; Skeberdis, V.A.; Francesconi, A.; Bennett, M.V.; Zukin, R.S. Postsynaptic density protein-95 regulates NMDA channel gating and surface expression. J. Neurosci. 2004, 24, 10138–10148. [Google Scholar] [CrossRef]
- Sun, Y.; Xu, Y.; Cheng, X.; Chen, X.; Xie, Y.; Zhang, L.; Wang, L.; Hu, J.; Gao, Z. The differences between GluN2A and GluN2B signaling in the brain. J. Neurosci. Res. 2018, 96, 1430–1443. [Google Scholar] [CrossRef]
- Ishchenko, Y.; Carrizales, M.G.; Koleske, A.J. Regulation of the NMDA receptor by its cytoplasmic domains: (How) is the tail wagging the dog? Neuropharmacology 2021, 195, 108634. [Google Scholar] [CrossRef]
- Aow, J.; Dore, K.; Malinow, R. Conformational signaling required for synaptic plasticity by the NMDA receptor complex. Proc. Natl. Acad. Sci. USA 2015, 112, 14711–14716. [Google Scholar] [CrossRef]
- Zeng, M.; Shang, Y.; Araki, Y.; Guo, T.; Huganir, R.L.; Zhang, M. Phase Transition in Postsynaptic Densities Underlies Formation of Synaptic Complexes and Synaptic Plasticity. Cell 2016, 166, 1163–1175.e12. [Google Scholar] [CrossRef]
- Zeng, M.; Chen, X.; Guan, D.; Xu, J.; Wu, H.; Tong, P.; Zhang, M. Reconstituted Postsynaptic Density as a Molecular Platform for Understanding Synapse Formation and Plasticity. Cell 2018, 174, 1172–1187.e16. [Google Scholar] [CrossRef]
- Hosokawa, T.; Liu, P.W.; Cai, Q.; Ferreira, J.S.; Levet, F.; Butler, C.; Sibarita, J.B.; Choquet, D.; Groc, L.; Hosy, E.; et al. CaMKII activation persistently segregates postsynaptic proteins via liquid phase separation. Nat. Neurosci. 2021, 24, 777–785. [Google Scholar] [CrossRef]
- Vistrup-Parry, M.; Chen, X.; Johansen, T.L.; Bach, S.; Buch-Larsen, S.C.; Bartling, C.R.O.; Ma, C.; Clemmensen, L.S.; Nielsen, M.L.; Zhang, M.; et al. Site-specific phosphorylation of PSD-95 dynamically regulates the postsynaptic density as observed by phase separation. iScience 2021, 24, 103268. [Google Scholar] [CrossRef]
- Wang, J.; Choi, J.M.; Holehouse, A.S.; Lee, H.O.; Zhang, X.; Jahnel, M.; Maharana, S.; Lemaitre, R.; Pozniakovsky, A.; Drechsel, D.; et al. A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 2018, 174, 688–699.e16. [Google Scholar] [CrossRef]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; van den Bosch, L.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef]
- Martin, E.W.; Holehouse, A.S.; Peran, I.; Farag, M.; Incicco, J.J.; Bremer, A.; Grace, C.R.; Soranno, A.; Pappu, R.V.; Mittag, T. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science 2020, 367, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Palay, S.L. Synapses in the central nervous system. J. Biophys. Biochem. Cytol. 1956, 2 (Suppl. 4), 193–202. [Google Scholar] [CrossRef] [PubMed]
- Palade, G. Electron microscope observations of interneuronal and neuromuscular synapses. Anat. Rec 1954, 118, 335–336. [Google Scholar]
- De Robertis, E.; Bennett, H.S. Submicroscopic vesicular component in the synapse. Fed. Proc. 1954, 13, 170. [Google Scholar]
- Wu, X.; Ganzella, M.; Zhou, J.; Zhu, S.; Jahn, R.; Zhang, M. Vesicle Tethering on the Surface of Phase-Separated Active Zone Condensates. Mol. Cell 2021, 81, 13–24.e7. [Google Scholar] [CrossRef]
- Chen, X.; Wu, X.; Wu, H.; Zhang, M. Phase separation at the synapse. Nat. Neurosci. 2020, 23, 301–310. [Google Scholar] [CrossRef]
- Choi, U.B.; Sanabria, H.; Smirnova, T.; Bowen, M.E.; Weninger, K.R. Spontaneous Switching among Conformational Ensembles in Intrinsically Disordered Proteins. Biomolecules 2019, 9, 114. [Google Scholar] [CrossRef]
- McKay, S.; Ryan, T.J.; McQueen, J.; Indersmitten, T.; Marwick, K.F.M.; Hasel, P.; Kopanitsa, M.V.; Baxter, P.S.; Martel, M.A.; Kind, P.C.; et al. The Developmental Shift of NMDA Receptor Composition Proceeds Independently of GluN2 Subunit-Specific GluN2 C-Terminal Sequences. Cell Rep. 2018, 25, 841–851.e4. [Google Scholar] [CrossRef]
- Wyllie, D.J.; Livesey, M.R.; Hardingham, G.E. Influence of GluN2 subunit identity on NMDA receptor function. Neuropharmacology 2013, 74, 4–17. [Google Scholar] [CrossRef]
- Choi, U.B.; Kazi, R.; Stenzoski, N.; Wollmuth, L.P.; Uversky, V.N.; Bowen, M.E. Modulating the intrinsic disorder in the cytoplasmic domain alters the biological activity of the N-methyl-D-aspartate-sensitive glutamate receptor. J. Biol. Chem. 2013, 288, 22506–22515. [Google Scholar] [CrossRef]
- McCann, J.J.; Zheng, L.; Rohrbeck, D.; Felekyan, S.; Kühnemuth, R.; Sutton, R.B.; Seidel, C.A.; Bowen, M.E. Supertertiary structure of the synaptic MAGuK scaffold proteins is conserved. Proc. Natl. Acad. Sci. USA 2012, 109, 15775–15780. [Google Scholar] [CrossRef] [PubMed]
- Choi, U.B.; Weninger, K.R.; Bowen, M.E. Immobilization of proteins for single-molecule fluorescence resonance energy transfer measurements of conformation and dynamics. Methods Mol. Biol. 2012, 896, 3–20. [Google Scholar] [PubMed]
- Yanez Orozco, I.S.; Mindlin, F.A.; Ma, J.; Wang, B.; Levesque, B.; Spencer, M.; Adariani, S.R.; Hamilton, G.; Ding, F.; Bowen, M.E.; et al. Identifying weak interdomain interactions that stabilize the supertertiary structure of the N-terminal tandem PDZ domains of PSD-95. Nat. Commun. 2018, 9, 3724. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Sakia, N.; Dougherty, L.; Guo, Z.; Wu, F.; Mindlin, F.; Lary, J.W.; Cole, J.L.; Ding, F.; Bowen, M.E. Probing Interdomain Linkers and Protein Supertertiary Structure In Vitro and in Live Cells with Fluorescent Protein Resonance Energy Transfer. J. Mol. Biol. 2021, 433, 166793. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, G.L.; Saikia, N.; Basak, S.; Welcome, F.S.; Wu, F.; Kubiak, J.; Zhang, C.; Hao, Y.; Seidel, C.A.M.; Ding, F.; et al. Fuzzy supertertiary interactions within PSD-95 enable ligand binding. Elife 2022, 11, e77242. [Google Scholar] [CrossRef]
- Ding, F.; Tsao, D.; Nie, H.; Dokholyan, N.V. Ab Initio Folding of Proteins with All-Atom Discrete Molecular Dynamics. Structure 2008, 16, 1010–1018. [Google Scholar] [CrossRef]
- Shirvanyants, D.; Ding, F.; Tsao, D.; Ramachandran, S.; Dokholyan, N.V. Discrete Molecular Dynamics: An Efficient and Versatile Simulation Method for Fine Protein Characterization. J. Phys. Chem. B 2012, 116, 8375–8382. [Google Scholar] [CrossRef]
- Tokunaga, M.; Imamoto, N.; Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods 2008, 5, 159–161. [Google Scholar] [CrossRef]
- Craven, S.E.; El-Husseini, A.E.; Bredt, D.S. Synaptic targeting of the postsynaptic density protein PSD-95 mediated by lipid and protein motifs. Neuron 1999, 22, 497–509. [Google Scholar] [CrossRef]
- El-Husseini, A.E.; Craven, S.E.; Chetkovich, D.M.; Firestein, B.L.; Schnell, E.; Aoki, C.; Bredt, D.S. Dual palmitoylation of PSD-95 mediates its vesiculotubular sorting, postsynaptic targeting, and ion channel clustering. J. Cell Biol. 2000, 148, 159–172. [Google Scholar] [CrossRef]
- Mao, A.H.; Crick, S.L.; Vitalis, A.; Chicoine, C.L.; Pappu, R.V. Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 8183–8188. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Weathers, E.A.; Paulaitis, M.E.; Woolf, T.B.; Hoh, J.H. Reduced amino acid alphabet is sufficient to accurately recognize intrinsically disordered protein. FEBS Lett. 2004, 576, 348–352. [Google Scholar] [CrossRef]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are "natively unfolded" proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Tomasso, M.E.; Tarver, M.J.; Devarajan, D.; Whitten, S.T. Hydrodynamic Radii of Intrinsically Disordered Proteins Determined from Experimental Polyproline II Propensities. PLoS Comput Biol. 2016, 12, e1004686. [Google Scholar] [CrossRef]
- Saikia, N.; Yanez-Orozco, I.S.; Qiu, R.; Hao, P.; Milikisiyants, S.; Ou, E.; Hamilton, G.L.; Weninger, K.R.; Smirnova, T.I.; Sanabria, H.; et al. Integrative structural dynamics probing of the conformational heterogeneity in synaptosomal-associated protein 25. Cell Rep. Phys. Sci. 2021, 2, 100616. [Google Scholar] [CrossRef]
- Gopich, I.V.; Szabo, A. FRET efficiency distributions of multistate single molecules. J. Phys. Chem. B 2010, 114, 15221–15226. [Google Scholar] [CrossRef]
- Gopich, I.V.; Szabo, A. Single-molecule FRET with diffusion and conformational dynamics. J. Phys. Chem. B 2007, 111, 12925–12932. [Google Scholar] [CrossRef]
- Zheng, W.; Zerze, G.H.; Borgia, A.; Mittal, J.; Schuler, B.; Best, R.B. Inferring properties of disordered chains from FRET transfer efficiencies. J. Chem. Phys. 2018, 148, 123329. [Google Scholar] [CrossRef]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Mitrea, D.M.; Chandra, B.; Ferrolino, M.C.; Gibbs, E.B.; Tolbert, M.; White, M.R.; Kriwacki, R.W. Methods for Physical Characterization of Phase-Separated Bodies and Membrane-less Organelles. J. Mol. Biol. 2018, 430, 4773–4805. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Ryan, T.J.; Kopanitsa, M.V.; Indersmitten, T.; Nithianantharajah, J.; Afinowi, N.O.; Pettit, C.; Stanford, L.E.; Sprengel, R.; Saksida, L.M.; Bussey, T.J.; et al. Evolution of GluN2A/B cytoplasmic domains diversified vertebrate synaptic plasticity and behavior. Nat. Neurosci. 2013, 16, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, G.J.; Wen, H.; Helou, M.; Liu, B.; Zheng, W.; Popescu, G.K. Cross-subunit interactions that stabilize open states mediate gating in NMDA receptors. Proc. Natl. Acad. Sci. USA 2021, 118, e2007511118. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Wen, H.; Iacobucci, G.J.; Popescu, G.K. Probing the Structural Dynamics of the NMDA Receptor Activation by Coarse-Grained Modeling. Biophys. J. 2017, 112, 2589–2601. [Google Scholar] [CrossRef]
- Pang, X.; Zhou, H.X. Structural modeling for the open state of an NMDA receptor. J. Struct. Biol. 2017, 200, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Sinitskiy, A.V.; Pande, V.S. Computer Simulations Predict High Structural Heterogeneity of Functional State of NMDA Receptors. Biophys. J. 2018, 115, 841–852. [Google Scholar] [CrossRef]
- Hardingham, G. NMDA receptor C-terminal signaling in development, plasticity, and disease. F1000Res 2019, 8. [Google Scholar] [CrossRef]
- Cohen, R.S.; Blomberg, F.; Berzins, K.; Siekevitz, P. The structure of postsynaptic densities isolated from dog cerebral cortex. I. Overall morphology and protein composition. J. Cell Biol. 1977, 74, 181–203. [Google Scholar] [CrossRef]
- Petersen, J.D.; Chen, X.; Vinade, L.; Dosemeci, A.; Lisman, J.E.; Reese, T.S. Distribution of postsynaptic density (PSD)-95 and Ca2+/calmodulin-dependent protein kinase II at the PSD. J. Neurosci. 2003, 23, 11270–11278. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Sequence | Kappa | FCR | NCPR | Hydropathy |
|---|---|---|---|---|---|
| GluN2A | 838–1464 | 0.158 | 0.264 | 0.018 | 3.5 |
| GluN2B | 838–1482 | 0.183 | 0.245 | 0.025 | 3.6 |
| CTD1A | 873–1211 | 0.138 | 0.286 | 0.009 | 3.3 |
| CTD1B | 874–1212 | 0.176 | 0.292 | 0.009 | 3.4 |
| CTD2A | 1243–1462 | 0.204 | 0.261 | 0.009 | 3.6 |
| CTD2B | 1250–1482 | 0.221 | 0.206 | 0.026 | 3.8 |
| Mean | Width | <RDA> | Mean | Width | <RDA> | Mean | Width | <RDA> | |
|---|---|---|---|---|---|---|---|---|---|
| CTD2A | 0.21 ± 0.04 | 0.11 ± 0.01 | 8.4 ± 0.6 | 0.46 ± 0.08 | 0.17 ± 0.01 | 6.0 ± 0.6 | 0.85 ± 0.01 | 0.15 ± 0.02 | 3.5 ± 0.1 |
| CTD2B | 0.2 ± 0.01 | 0.33 ± 0.03 | 8.6 ± 0.2 | 0.55 ± 0.05 | 0.28 ± 0.03 | 5.3 ± 0.3 | NA | NA | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basak, S.; Saikia, N.; Kwun, D.; Choi, U.B.; Ding, F.; Bowen, M.E. Different Forms of Disorder in NMDA-Sensitive Glutamate Receptor Cytoplasmic Domains Are Associated with Differences in Condensate Formation. Biomolecules 2023, 13, 4. https://doi.org/10.3390/biom13010004
Basak S, Saikia N, Kwun D, Choi UB, Ding F, Bowen ME. Different Forms of Disorder in NMDA-Sensitive Glutamate Receptor Cytoplasmic Domains Are Associated with Differences in Condensate Formation. Biomolecules. 2023; 13(1):4. https://doi.org/10.3390/biom13010004
Chicago/Turabian StyleBasak, Sujit, Nabanita Saikia, David Kwun, Ucheor B. Choi, Feng Ding, and Mark E. Bowen. 2023. "Different Forms of Disorder in NMDA-Sensitive Glutamate Receptor Cytoplasmic Domains Are Associated with Differences in Condensate Formation" Biomolecules 13, no. 1: 4. https://doi.org/10.3390/biom13010004
APA StyleBasak, S., Saikia, N., Kwun, D., Choi, U. B., Ding, F., & Bowen, M. E. (2023). Different Forms of Disorder in NMDA-Sensitive Glutamate Receptor Cytoplasmic Domains Are Associated with Differences in Condensate Formation. Biomolecules, 13(1), 4. https://doi.org/10.3390/biom13010004